

Abstract

A series of trisubstituted hydroxylactams was identified as potent enzymatic and cellular inhibitors of human lactate dehydrogenase A. Utilizing structure-based design and physical property optimization, multiple inhibitors were discovered with <10 μM lactate IC50 in a MiaPaca2 cell line. Optimization of the series led to 29, a potent cell active molecule (MiaPaca2 IC50 = 0.67 μM) that also possessed good exposure when dosed orally to mice.
Keywords: Lactate dehydrogenase, tumor metabolism, glycolysis, X-ray crystal structure, structure-based design
Tumor cells often show signs of metabolic alteration when compared to normal cells.1 Instead of the mitochondrial tricarboxylic acid (TCA) cycle utilized by healthy cells, they often rely on glycolysis for energy generation,2 even in the presence of normal oxygen levels.3 Lactate dehydrogenase A (LDHA) is critical to this process by catalyzing the conversion of pyruvate to lactate in the final step of glycolysis.4 Overexpression of LDHA is found in many types of cancer cells,5−8 and shRNA mediated LDHA depletion results in the inhibition of tumor growth in glycolytically dependent cancer cell lines, xenografts, and genetically engineered murine models.7,8 Together, these facts have focused attention on the therapeutic potential of LDHA inhibition for the treatment of cancer. A recent report from our laboratories identified the lactate dehydrogenase B isoform (LDHB) as an essential gene for triple negative breast cancer tumors.9 Given this linkage, we sought a pan-LDH inhibitor, if possible, in order to maximize potential efficacy across the broadest set of tumors.
A number of LDHA inhibitors have recently been disclosed by several academic and industrial groups;10−16 however, they have been hampered primarily by a lack of cellular activity. An LDHA inhibitor with excellent cellular potency was disclosed, but poor pharmacokinetics prevented its evaluation in vivo.15 Our group has recently reported LDHA inhibitors containing a variety of scaffolds.17−21 To date, they have suffered from a lack of cellular activity, despite relatively potent biochemical inhibition. For example, 3,6-disubstituted dihydropyrones and diketones, although possessing low double digit nanomolar IC50s in vitro, failed to show activity against glycolytic cell lines.18,21 This has been attributed to poor permeability and high plasma protein binding, presumably due to the acidic core of the compounds imparting poor physical properties. Herein we report the optimization of a series of hydroxylactam inhibitors and the resulting impact on LDHA biochemical and cellular potency.
In order to further optimize this general class of dicarbonyl LDHA inhibitors, we sought to modify our medicinal chemistry approach in two ways. The first was to replace the diketone and dihydropyrone cores with a hydroxylactam. Previous matched pair comparison of these three scaffolds demonstrated that the hydroxylactam, exemplified by 1, was the least acidic of the set (pKa = 4.1, versus 2.4 and 3.0, respectively, for the dihydropyrone and diketone), while maintaining biochemical potency.18 We hypothesized that this pKa modulation could lead to improved physical properties of the compounds, leading to improved permeability and reduced plasma protein binding, yielding cell active compounds if biochemical potency could be improved as well.
Second, overlay of the crystal structures of two previously disclosed diketone compounds19 (Figure 1) suggested that simultaneous substitution of the axial and equatorial positions of the core might be possible. The resulting diphenyl substituted compound (2) indicated that this substitution pattern was tolerated and led to an 8-fold improvement in biochemical potency (Table 1). We therefore explored phenyl isosteres (3, 4) and found that a 3-thiophene (4) further improved potency by 4-fold. Encouraged by these results, we held the thiophene constant at R2 while exploring a variety of substituents on the phenyl ring at R1 (5–14). Compounds were synthesized and tested as racemic mixtures for synthetic convenience. We held the 3-position on the hydroxylactam core constant, as this group had proven ideal in previous optimization of the series.18,19 2-Pyridyl substitution (5) resulted in the complete loss of activity, and 2-fluorophenyl (6) reduced potency slightly. 3-Bromophenyl (7) improved LDHA potency to 82 nM; however, the selectivity for LDHA over LDHB increased from ∼4–5-fold to 10-fold. This was not desired as we sought a pan-LDH inhibitor. Meta amino and hydroxy substitution (8 and 9, respectively) yielded roughly equipotent analogues, each improving inhibition slightly. Para substitution resulted in more variable SAR. Electron withdrawing groups such as cyano (10) diminished affinity to 700 nM, while electron donating substituents (11–14) maintained or improved potency. Except for 7, this set of compounds maintained a ∼4–5 fold selectivity for LDHA over LDHB relative to that of 1.
Figure 1.
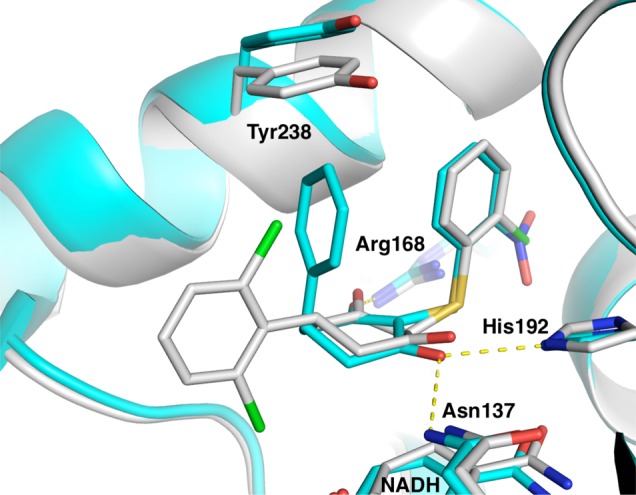
Overlay of previously disclosed X-ray structures of LDHA/diketone-containing inhibitor complexes 4QO7 (cyan) and 4QO8 (white).19 Hydrogen bonds from 4QO7 are shown as yellow dashed lines.
Table 1. Structure–Activity Relationships for Trisubstituted Hydroxylactamsa.

| compd | R1 | R2 | LDHA IC50 (μM) | LDHB IC50 (μM) |
|---|---|---|---|---|
| 1 | Ph | H | 4 | 18 |
| 2 | Ph | Ph | 0.49 | 2.2 |
| 3 | Ph | 4-thiazolyl | 0.40 | 2.3 |
| 4 | Ph | 3-thiophenyl | 0.12 | 0.74 |
| 5 | 2-Pyridyl | 3-thiophenyl | >10 | >10 |
| 6 | (2-F)Ph | 3-thiophenyl | 0.36 | 1.8 |
| 7 | (3-Br)Ph | 3-thiophenyl | 0.082 | 0.8 |
| 8 | (3-NH2)Ph | 3-thiophenyl | 0.058 | 0.20 |
| 9 | (3-OH)Ph | 3-thiophenyl | 0.042 | 0.27 |
| 10 | (4-CN)Ph | 3-thiophenyl | 0.70 | 1.1 |
| 11 | (4-cyclopropyl)Ph | 3-thiophenyl | 0.10 | 0.48 |
| 12 | (4-OMe)Ph | 3-thiophenyl | 0.065 | 0.21 |
| 13 | (4-NMe2)Ph | 3-thiophenyl | 0.015 | 0.055 |
| 14 | (4-OH)Ph | 3-thiophenyl | 0.014 | 0.057 |
LDHA and LDHB enzymatic assay values are the geometric mean of at least 2 separate runs as previously described.19
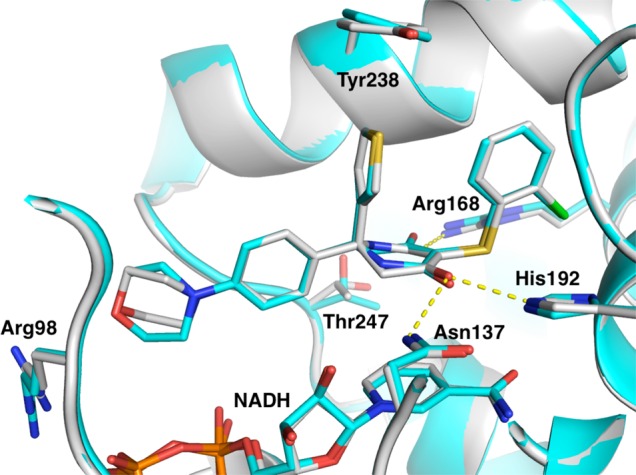
An X-ray cocrystal structure of 9 bound to LDHA (Figure 2) confirmed our hypothesis that simultaneous axial and equatorial substitution could be accommodated by the protein. The hydroxylactam core maintains the core interactions previously observed for this series.18,19 The lactam carbonyl hydrogen bonds the catalytic Arg168 and the hydroxyl bridges His192 and Asn137. There is no hydrogen bond between the lactam NH and the protein, although it points toward Thr247. The inhibitor stacks on top of the NADH cofactor, occupying only the pyruvate binding site. We have previously demonstrated that this class of compounds is noncompetitive with NADH binding in the LDHA active site.17 The thiophene substituent occupies the axial position, forming an intramolecular edge-to-face π–π interaction with the 2-chlorophenyl ring and a second edge-to-face π–π interaction with Tyr238. The 3-hydroxyphenyl ring occupies an equatorial position relative to the hydroxylactam core, stacking against Ile241. The phenol OH is hydrogen bonded to a water, which is in turn involved in an extensive hydrogen bonding network with the backbone NH and carbonyl of Thr247 and a phosphate oxygen of NADH. Intriguingly, despite this highly coordinated water interaction, 9 is only 3-fold more potent against LDHA than the parent compound 4.
Figure 2.

Compound 9 (cyan) cocrystallized with LDHA (white) [PDB: 5IXS]. The NADH cofactor is shown in green sticks, the crystallographic water as a red sphere, and hydrogen bonds are yellow dashed lines. Crystallization was carried out as previously disclosed.21 Additional details can be found in the Supporting Information.
Arg98 is just beyond the para-position of the phenyl ring, with space available in between it and the inhibitor. Given the excellent potency of analogues with para-polar substituents (13, 14), it was hypothesized that polar groups in this region might be interacting with this arginine and be responsible for the increased potency. This arginine is in a structurally variable region of LDHA, differing among individual subunits of the tetrameric crystallographic asymmetric unit.
In order to probe this area, we explored further substitution at the meta- and para-positions of the phenyl ring (Table 2). Direct linkage of a morpholine (15) or ether (19, 20) at the meta-position maintained LDHA potency, while amino linkages to cyclic alkyl or aryl substituents (16–18) improved LDHA potency to a single digit nanomolar level. Unfortunately, selectivity against LDHB increased to ∼10–20-fold, in opposition to our goal of an inhibitor with pan-LDH potency. Direct substitution of piperidines and piperazines in the para-position led to LDHA potencies in the low to mid double digit nanomolar range (21–28). A hydrogen bond donor in the 4-position of these heteroalkyl rings (22) was slightly inferior to either carbon (21, 25, 26) or hydrogen bond acceptors at the same position in the ring (24, 27, 28). This is consistent with the potential interaction of these groups with Arg98. Of particular note is morpholine-substituted phenyl 28. In addition to its 9 nM potency, it has only a 3-fold potency shift between LDHA and LDHB, one of the smallest ratios observed in this series of biochemically potent compounds. We separated 28 into its enantiomers and found that the R enantiomer 29 was 18-fold more potent than S enantiomer 30.
Table 2. Structure–Activity Relationships for 3- and 4-Position Phenyl Modifications on Trisubstituted Hydroxylactamsa.
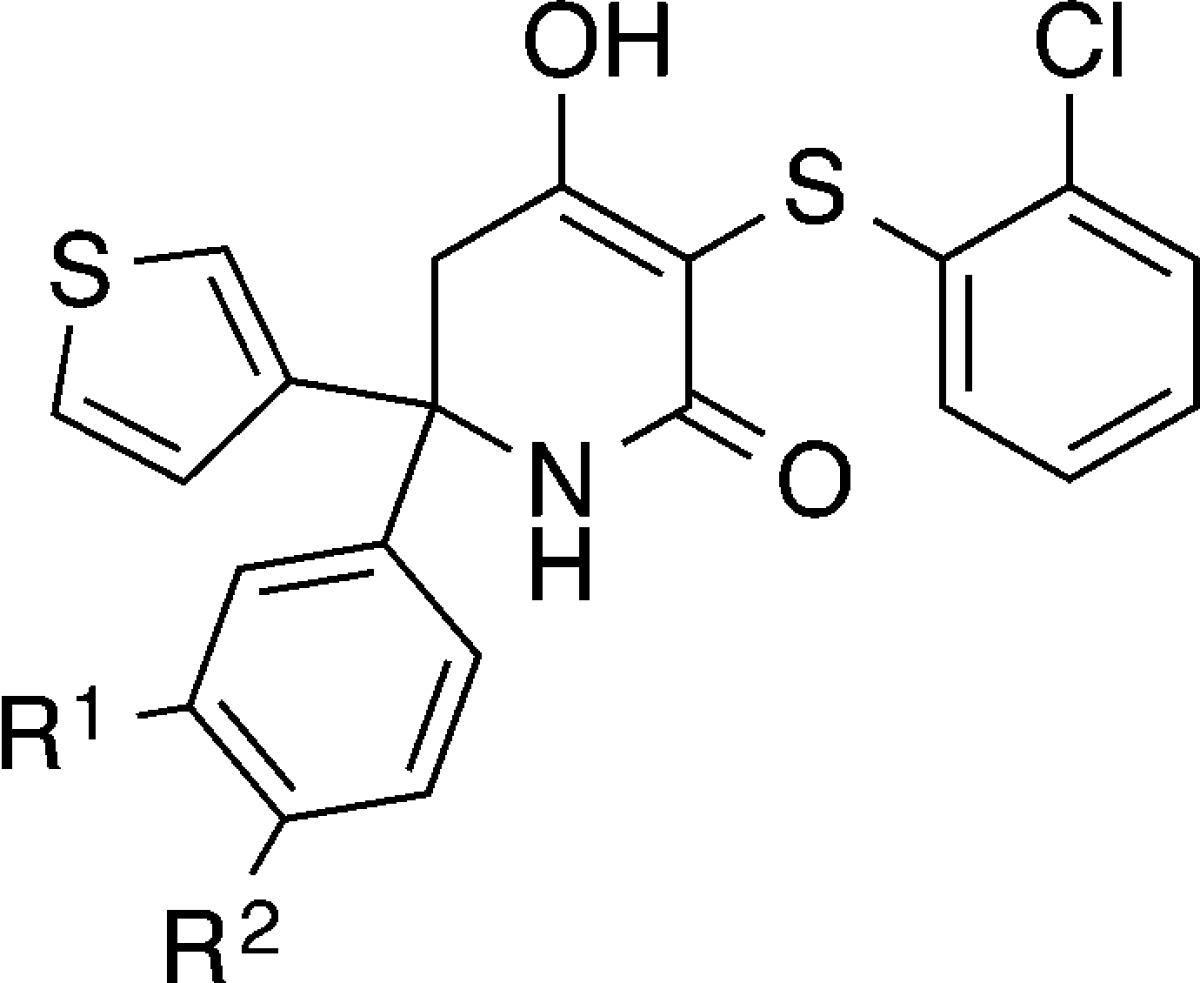
| compd | R1 | R2 | LDHA IC50 (μM) | LDHB IC50 (μM) |
|---|---|---|---|---|
| 15 | 1-morpholino | H | 0.080 | 0.25 |
| 16 | NH-THP | H | 0.009 | 0.16 |
| 17 | NH-cyclohexyl | H | 0.004 | 0.077 |
| 18 | NH-(4-F)Ph | H | 0.003 | 0.035 |
| 19 | O-THP | H | 0.069 | 0.66 |
| 20 | O-(4-F)Ph | H | 0.037 | 0.19 |
| 21 | H | cyclohexyl | 0.025 | 0.18 |
| 22 | H | 1-piperazinyl | 0.052 | 0.78 |
| 23 | H | 1-(4-oxetane)piperazinyl | 0.047 | 0.39 |
| 24 | H | 1-(4-OMe)piperidinyl | 0.016 | 0.075 |
| 25 | H | 1-piperidinyl | 0.009 | 0.049 |
| 26 | H | 1-pyrrolidinyl | 0.007 | 0.050 |
| 27 | H | 1-(4-acetyl)piperazinyl | 0.005 | 0.035 |
| 28 | H | 1-morpholino | 0.009 | 0.030 |
| 29-R | H | 1-morpholino | 0.003 | 0.005 |
| 30-S | H | 1-morpholino | 0.055 | 0.14 |
Compounds are racemic mixtures, except where indicated.
We were able to solve crystal structures of each enantiomer bound to LDHA. An overlay of the two structures (Figure 3) reveals they have the same binding mode in terms of substituent disposition. Interestingly, the hydroxylactam core is rotated 180° in these structures (exchanging the positions of the lactam NH and unsubstituted sp3 carbon). More potent isomer 29 positions the lactam pointing toward Thr247. Although the distance and Thr rotomer do not support a direct hydrogen bond, this conformation has the least potential clashes with the protein. In contrast, 30 points the lactam NH toward an NADH hydroxyl and Asn137, but again is not close enough to make direct hydrogen bonds. The sp3 carbon on the other side of the core is pointed toward the Thr247 OH, a less favorable interaction, consistent with the reduced LDHA potency observed for this compound. This absolute stereochemistry assignment of 29 as R and 30 as S is also supported by the small molecule crystal structure of the more potent enantiomer from a related pair of compounds (see Supporting Information). As expected, the terminal morpholine ring is pointing toward the guanidinium group of Arg98; however, the geometry is not appropriate for hydrogen bond formation. Instead, it interacts via a polar interaction between the morpholine oxygen and the positive charge of the arginine residue.
Figure 3.
Overlay of the crystal structures 29 (white) [PDB: 4ZVV] and 30 (cyan) [PDB: 5IXY] bound to LDHA. Hydrogen bonds are shown as yellow dashed lines.
Encouraged by the favorable enzymatic potency of these inhibitors, we tested selected analogues in a MiaPaca2 cell line for the inhibition of lactate production (Table 3; 21, 25, 29). All showed measurable inhibition, with IC50s ranging from 0.67–25 μM. Given the submicromolar cellular potency of 29, additional close-in analogues around the morpholine ring functionality were prepared (31–36).
Table 3. Structure–Activity Relationships for 4-Phenyl Modifications on Trisubstituted Hydroxylactamsa.
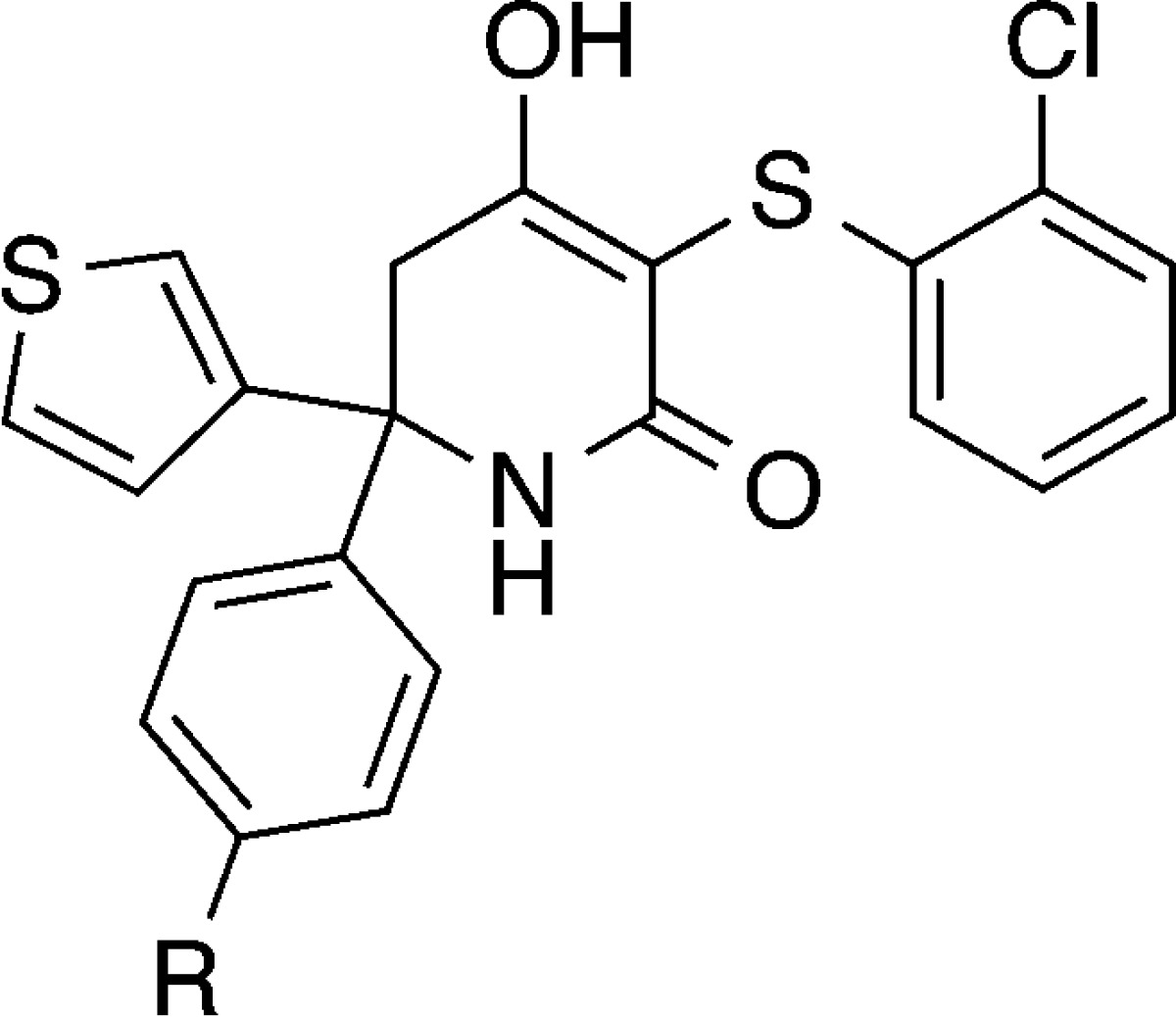
| compd | R | LDHA IC50 (μM) | LDHB IC50 (μM) | MiaPaca2 Lactate IC50 (μM) | LogD7.4 |
|---|---|---|---|---|---|
| 21 | cyclohexyl | 0.025 | 0.18 | 25 | 3.5 |
| 24 | 1-(4-OMe)piperidinyl | 0.016 | 0.075 | 2.9 | 1.2 |
| 29-R | 1-morpholino | 0.003 | 0.005 | 0.67 | 0.6 |
| 31 | 2,6-dimethylmorpholino | 0.027 | 0.14 | 3.9 | 1.3 |
| 32 | 3-oxomorpholino | 0.021 | 0.080 | >50 | 0.4 |
| 33 | 2,2-dimethylmorpholino | 0.015 | 0.070 | 5.1 | 1.3 |
| 34 | 1,1-dioxidothiomorpholino | 0.013 | 0.046 | >50 | ND |
| 35 | 1-(4-spiro-oxetanyl)piperidinyl | 0.017 | 0.14 | 9.9 | 0.7 |
| 36 | 1(4-cyano)piperidinyl | 0.010 | 0.060 | 6.1 | 0.8 |
None of these alternatives improved either the enzymatic or cellular potency relative to 28 or 29. Adding polarity to the morpholine ring via a carbonyl (32) or sulfone (34) resulted in a complete loss of cellular potency.
By switching the core from a diketone or dihydropyrone to a hydroxylactam, we had hoped the reduced acidity would help improve the physical properties of the compounds, yielding molecules with improved permeability and plasma protein binding. We measured the MDCK permeability and mouse plasma protein binding for a subset of the compounds and found several trends (Table 4). MDCK permeability was moderate/high for the smaller, less functionalized compounds 2 and 4. Addition of a saturated or substituted aromatic ring onto the phenyl through an NH linkage (16 and 18) increased the number of hydrogen bond donors to three, resulting in lower permeability and hints of greater efflux. The additional lipophilicity of 18, as measured by LogD7.4, likely led to an increase of protein binding to >99.9% when compared to 2. Direct attachment of a heteroalkyl ring (24, 29, 31) reduced the hydrogen bond donor count and, when combined with the neutral or decrease of LogD7.4 attributable to the morpholine (29, 31), restored moderate MDCK permeability. High plasma protein binding was observed on all compounds containing either a saturated or aromatic fifth ring, with the exception of 29, which had mouse plasma protein binding of 99.1%. This lower protein binding may be one of the contributors to the superior cellular potency observed for this compound, as the cellular assay conditions contain 10% FBS. Compound 29 inhibited proliferation in 37 of 347 cancer cell lines tested at a potency cut off of 5 μM (Figure S1). Interestingly, two chondrosarcoma (bone) cancer cell lines that harbored IDH1 mutations were found to be especially sensitive to 29 (IC50 = 0.8 μM). IDH1 catalyzes the conversion of mitochondrial isocitrate to α-ketoglutarate, so IDH1 mutant tumors may have decreased mitochondrial fitness and increased dependency on glycolysis.
Table 4. Physicochemical Property Effects on Permeability and Protein Binding.
| compd | TPSA (Å2) | HBD | LogD7.4 | MDCKa Papp A:B (10–6 cm/s)/ratio (A:B/B:A) | mouse PPB (%) |
|---|---|---|---|---|---|
| 2 | 46 | 2 | 1.4 | 9.4/0.5 | 99.3 |
| 4 | 46 | 2 | 0.8 | 7.4/1.0 | ND |
| 16 | 67 | 3 | –0.3 | 0.5/1.8 | ND |
| 18 | 58 | 3 | 2.4 | 1.0/2.8 | >99.9 |
| 25 | 49 | 2 | 2.1 | 1.7/0.9 | >99.9 |
| 29-R | 58 | 2 | 0.6 | 3.4/0.9 | 99.1 |
| 31 | 58 | 2 | 1.3 | 6.0/1.4 | >99.9 |
The MDCK assay23 was used as an indicator of cell permeability.
Compound 29 proved to possess the optimal combination of strong enzymatic and MiaPaca2 cellular potency, moderate permeability, and reduced plasma protein binding compared to other potent compounds in this series. It was stable in liver microsomes with predicted hepatic clearances (Clp) of 3.3, 8.1, and 14 mL/min/kg, based on human, rat, and mouse microsomes, respectively. We carried out in vivo pharmacokinetic experiments in mice with 29 and were gratified to observe that this compound presented a low Clp, consistent with the predicted hepatic Cl, and also had high bioavailability when dosed orally at 5 mg/kg (Table 5; concentration vs time plots in Figure S2). At higher oral doses, ranging from 50 to 200 mg/kg, 29 displayed greater exposure (Table 5 and Figure S3), confirming the possibility of 29 serving as a tool compound for in vivo efficacy studies in mice.
Table 5. Pharmacokinetic Parameters Following IV and PO Administrations of 29 to Mice.
| route | dose (mg/kg) | AUC (μM·h) | Cmax (μM) | F (%) | T1/2 (h) | Vss (L/kg) | Clp (mL/min/kg) |
|---|---|---|---|---|---|---|---|
| iv | 1 | 1.5 | 2.5 | 1.0 | 1.4 | 21.1 | |
| po | 5 | 5.1 | 1.7 | 70 | |||
| po | 50 | 125 | 60.7 | ||||
| po | 100 | 250 | 224 | ||||
| po | 200 | 403 | 440 |
Inhibitors were synthesized as shown in Scheme 1. Condensation of ketones 37a–i with racemic tert-butylsulfinamide gave sulfinamides 38a–i. Subsequent addition of methyl 3-oxobutanoate to 38a–i afforded ketoesters 39a–i, which were then subjected to acid deprotection, resulting in amine intermediates 40a–i. Base-mediated cyclization yielded ketolactams 41a–i, which were then condensed with bis(2-chlorophenyl)disulfide to yield final products 3–7, 11, 12, 19, and 42. In the case where R1 was 3-bromophenyl (7), Buchwald cross-coupling reactions with substituted amines or phenols yielded inhibitors 8, 9, 15–18, and 20. Under similar conditions, 42, where R1 was 4-bromophenyl, yielded inhibitors 10, 13, 14, 22–28, and 31–36. Suzuki coupling of 42 with cyclohex-1-en-1-ylboronic acid and subsequent hydrogenation afforded 21. For compounds 29 and 30, racemic compound 28 was resolved by chiral SFC to afford the single stereoisomers. Absolute stereochemistry was assigned based on the small molecule X-ray crystal structure of 29 (Figure S4 and CIF).
Scheme 1. Synthesis of Substituted Hydroxylactam Compounds.

Compounds 9 and 14 were formed as byproducts in the phenol and amine coupling reactions.
In summary, structure-based design was utilized to create a novel series of substituted hydroxylactam LDHA inhibitors. These compounds combined the occupancy of multiple sites that had not been filled simultaneously by previous inhibitors, along with a less acidic hydroxylactam core. This strategy afforded a dramatic improvement in enzymatic potency as well as inhibitors displaying LDHA inhibition in MiaPaca2 cells. Compound 29 has submicromolar MiaPaca2 potency and inhibits proliferation in 11% of cancer cell lines in a broad panel. Moreover, 29 combines potent activity with modest permeability, lower plasma protein binding, and good in vivo exposure when dosed orally to mice.
To our knowledge, 29 is the first example of an LDHA inhibitor possessing both submicromolar cellular potency and adequate pharmacokinetic properties that make it suitable for use as an in vivo tool compound. Reports on potency in cells, mechanism of action, tumor growth inhibition, and efficacy characteristics of this compound are forthcoming from our laboratories.24
Acknowledgments
We thank Drs. Krista Bowman and Jiansheng Wu and their respective research groups for performing protein expression and purification activities. We also thank the Genentech Small Molecule analytical group for their support. We thank the Genentech gCell group for cell line banking and maintenance, and members of the Genentech Cell Line Screening Initiative and Dr. Richard Bourgon for assistance on cell-based drug screens. We thank Crystallographic Consulting, LLC for diffraction data collection. We acknowledge the use of synchrotron X-ray sources at the Advanced Light Source and the Stanford Synchrotron Radiation Lightsource supported by the Department of Energy’s Office of Science under contracts DE-AC02-05CH11231 and DE-AC02-76SF00515, respectively. We also acknowledge NIH Shared Instrumentation Grant S10-RR027172 for the purchase of the diffractometer used for small molecule crystal data collection.
Glossary
ABBREVIATIONS
- LDHA
lactate dehydrogenase A
- LDHB
lactate dehydrogenase B
- PPB
plasma protein binding
- SFC
supercritical fluid chromatography
- MDCK
Madin–Darby canine kidney
- AUC
area under the curve
- Vss
steady-state volume of distribution
- T1/2
half-life
- Clp
plasma clearance
- Cmax
maximum concentration
- F
oral bioavailability
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00190.
The authors declare no competing financial interest.
Supplementary Material
References
- Galluzzi L.; Kepp O.; Vander Heiden M. G.; Kroemer G. Metabolic Targets for Cancer Therapy. Nat. Rev. Drug Discovery 2013, 12 (11), 829–846. 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the Origin of Cancer Cells. Science 1956, 123 (3191), 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M. G.; Cantley L. C.; Thompson C. B. Understanding the Warburg Effect: the Metabolic Requirements of Cell Proliferation. Science 2009, 324 (5930), 1029–1033. 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salway J. G.Metabolism at a Glance; John Wiley & Sons, 2013. [Google Scholar]
- Koukourakis M. I.; Giatromanolaki A.; Sivridis E. Tumour and Angiogenesis Research Group. Lactate Dehydrogenase Isoenzymes 1 and 5: Differential Expression by Neoplastic and Stromal Cells in Non-Small Cell Lung Cancer and Other Epithelial Malignant Tumors. Tumor Biol. 2003, 24 (4), 199–202. 10.1159/000074430. [DOI] [PubMed] [Google Scholar]
- Koukourakis M. I.; Giatromanolaki A.; Panteliadou M.; Pouliliou S. E.; Chondrou P. S.; Mavropoulou S.; Sivridis E. Lactate Dehydrogenase 5 Isoenzyme Overexpression Defines Resistance of Prostate Cancer to Radiotherapy. Br. J. Cancer 2014, 110 (9), 2217–2223. 10.1038/bjc.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin V. R.; St-Pierre J.; Leder P. Attenuation of LDH-a Expression Uncovers a Link Between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9 (6), 425–434. 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- Xie H.; Hanai J.-I.; Ren J.-G.; Kats L.; Burgess K.; Bhargava P.; Signoretti S.; Billiard J.; Duffy K. J.; Grant A.; Wang X.; Lorkiewicz P. K.; Schatzman S.; Bousamra M.; Lane A. N.; Higashi R. M.; Fan T. W. M.; Pandolfi P. P.; Sukhatme V. P.; Seth P. Targeting Lactate Dehydrogenase--a Inhibits Tumorigenesis and Tumor Progression in Mouse Models of Lung Cancer and Impacts Tumor-Initiating Cells. Cell Metab. 2014, 19 (5), 795–809. 10.1016/j.cmet.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan M.; Benner J.; Schilsky R.; Epstein D.; Woosley R.; Friend S.; Sidransky D.; Geoghegan C.; Kessler D. An Accelerated Pathway for Targeted Cancer Therapies. Nat. Rev. Drug Discovery 2011, 10 (2), 79–80. 10.1038/nrd3360. [DOI] [PubMed] [Google Scholar]
- Rani R.; Kumar V. Recent Update on Human Lactate Dehydrogenase Enzyme 5 (hLDH5) Inhibitors: a Promising Approach for Cancer Chemotherapy. J. Med. Chem. 2015, 59, 487. 10.1021/acs.jmedchem.5b00168. [DOI] [PubMed] [Google Scholar]
- Le A.; Cooper C. R.; Gouw A. M.; Dinavahi R.; Maitra A.; Deck L. M.; Royer R. E.; Vander Jagt D. L.; Semenza G. L.; Dang C. V. Inhibition of Lactate Dehydrogenase a Induces Oxidative Stress and Inhibits Tumor Progression. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (5), 2037–2042. 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmann A.; Zech S. G.; Li F.; Zhou T.; Squillace R. M.; Commodore L.; Greenfield M. T.; Lu X.; Miller D. P.; Huang W.-S.; Qi J.; Thomas R. M.; Wang Y.; Zhang S.; Dodd R.; Liu S.; Xu R.; Xu Y.; Miret J. J.; Rivera V.; Clackson T.; Shakespeare W. C.; Zhu X.; Dalgarno D. C. Fragment Growing and Linking Lead to Novel Nanomolar Lactate Dehydrogenase Inhibitors. J. Med. Chem. 2013, 56 (3), 1023–1040. 10.1021/jm3014844. [DOI] [PubMed] [Google Scholar]
- Ward R. A.; Brassington C.; Breeze A. L.; Caputo A.; Critchlow S.; Davies G.; Goodwin L.; Hassall G.; Greenwood R.; Holdgate G. A.; Mrosek M.; Norman R. A.; Pearson S.; Tart J.; Tucker J. A.; Vogtherr M.; Whittaker D.; Wingfield J.; Winter J.; Hudson K. Design and Synthesis of Novel Lactate Dehydrogenase a Inhibitors by Fragment-Based Lead Generation. J. Med. Chem. 2012, 55 (7), 3285–3306. 10.1021/jm201734r. [DOI] [PubMed] [Google Scholar]
- Granchi C.; Roy S.; Giacomelli C.; Macchia M.; Tuccinardi T.; Martinelli A.; Lanza M.; Betti L.; Giannaccini G.; Lucacchini A.; Funel N.; León L. G.; Giovannetti E.; Peters G. J.; Palchaudhuri R.; Calvaresi E. C.; Hergenrother P. J.; Minutolo F. Discovery of N-Hydroxyindole-Based Inhibitors of Human Lactate Dehydrogenase Isoform a (LDH-a) as Starvation Agents Against Cancer Cells. J. Med. Chem. 2011, 54 (6), 1599–1612. 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]
- Billiard J.; Dennison J. B.; Briand J.; Annan R. S.; Chai D.; Colón M.; Dodson C. S.; Gilbert S. A.; Greshock J.; Jing J.; Lu H.; McSurdy-Freed J. E.; Orband-Miller L. A.; Mills G. B.; Quinn C. J.; Schneck J. L.; Scott G. F.; Shaw A. N.; Waitt G. M.; Wooster R. F.; Duffy K. J. Quinoline 3-Sulfonamides Inhibit Lactate Dehydrogenase a and Reverse Aerobic Glycolysis in Cancer Cells. Cancer Metab. 2013, 1, 19. 10.1186/2049-3002-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-Y.; Feng Y.; Chen J.-Y.; Deng H. Identification of a Potent Inhibitor Targeting Human Lactate Dehydrogenase a and Its Metabolic Modulation for Cancer Cell Line. Bioorg. Med. Chem. Lett. 2015, 26, 72. 10.1016/j.bmcl.2015.11.025. [DOI] [PubMed] [Google Scholar]
- Dragovich P. S.; Fauber B. P.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Ge H.; Giannetti A. M.; Hunsaker T.; Labadie S.; Liu Y.; Malek S.; Pan B.; Peterson D.; Pitts K.; Purkey H. E.; Sideris S.; Ultsch M.; VanderPorten E.; Wei B.; Xu Q.; Yen I.; Yue Q.; Zhang H.; Zhang X. Identification of Substituted 2-Thio-6-Oxo-1,6-Dihydropyrimidines as Inhibitors of Human Lactate Dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23 (11), 3186–3194. 10.1016/j.bmcl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- Fauber B. P.; Dragovich P. S.; Chen J.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Labadie S.; Malek S.; Peterson D.; Purkey H. E.; Robarge K.; Sideris S.; Ultsch M.; Wei B.; Yen I.; Yue Q.; Zhou A. Identification of 3,6-Disubstituted Dihydropyrones as Inhibitors of Human Lactate Dehydrogenase. Bioorg. Med. Chem. Lett. 2014, 24 (24), 5683–5687. 10.1016/j.bmcl.2014.10.067. [DOI] [PubMed] [Google Scholar]
- Dragovich P. S.; Fauber B. P.; Boggs J.; Chen J.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Ge H.; Giannetti A. M.; Hunsaker T.; Labadie S.; Li C.; Liu Y.; Liu Y.; Ma S.; Malek S.; Peterson D.; Pitts K. E.; Purkey H. E.; Robarge K.; Salphati L.; Sideris S.; Ultsch M.; VanderPorten E.; Wang J.; Wei B.; Xu Q.; Yen I.; Yue Q.; Zhang H.; Zhang X.; Zhou A. Identification of Substituted 3-Hydroxy-2-Mercaptocyclohex-2-Enones as Potent Inhibitors of Human Lactate Dehydrogenase. Bioorg. Med. Chem. Lett. 2014, 24 (16), 3764–3771. 10.1016/j.bmcl.2014.06.076. [DOI] [PubMed] [Google Scholar]
- Fauber B. P.; Dragovich P. S.; Chen J.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Giannetti A. M.; Hunsaker T.; Labadie S.; Liu Y.; Liu Y.; Malek S.; Peterson D.; Pitts K.; Sideris S.; Ultsch M.; VanderPorten E.; Wang J.; Wei B.; Yen I.; Yue Q. Identification of 2-Amino-5-Aryl-Pyrazines as Inhibitors of Human Lactate Dehydrogenase. Bioorg. Med. Chem. Lett. 2013, 23 (20), 5533–5539. 10.1016/j.bmcl.2013.08.060. [DOI] [PubMed] [Google Scholar]
- Labadie S.; Dragovich P. S.; Chen J.; Fauber B. P.; Boggs J.; Corson L. B.; Ding C. Z.; Eigenbrot C.; Ge H.; Ho Q.; Lai K. W.; Ma S.; Malek S.; Peterson D.; Purkey H. E.; Robarge K.; Salphati L.; Sideris S.; Ultsch M.; VanderPorten E.; Wei B.; Xu Q.; Yen I.; Yue Q.; Zhang H.; Zhang X.; Zhou A. Optimization of 5-(2,6-Dichlorophenyl)-3-Hydroxy-2-Mercaptocyclohex-2-Enones as Potent Inhibitors of Human Lactate Dehydrogenase. Bioorg. Med. Chem. Lett. 2015, 25 (1), 75–82. 10.1016/j.bmcl.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Lin B.; Pease J. H. A Novel Method for High Throughput Lipophilicity Determination by Microscale Shake Flask and Liquid Chromatography Tandem Mass Spectrometry. Comb. Chem. High Throughput Screening 2013, 16 (10), 817–825. 10.2174/1386207311301010007. [DOI] [PubMed] [Google Scholar]
- Irvine J. D.; Takahashi L.; Lockhart K.; Cheong J.; Tolan J. W.; Selick H. E.; Grove J. R. MDCK (Madin-Darby Canine Kidney) Cells: a Tool for Membrane Permeability Screening. J. Pharm. Sci. 1999, 88 (1), 28–33. 10.1021/js9803205. [DOI] [PubMed] [Google Scholar]
- Boudreau A.; Purkey H. E.; Hitz A.; Robarge K.; Peterson D.; Labadie S.; Kwong M.; Hong R.; Gao M.; Del Nagro C.; et al. Metabolic Plasticity Underpins Innate and Acquired Resistance to LDHA Inhibition. Nat. Chem. Bio. 2016, 10.1038/nchembio.2143. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.