

Abstract
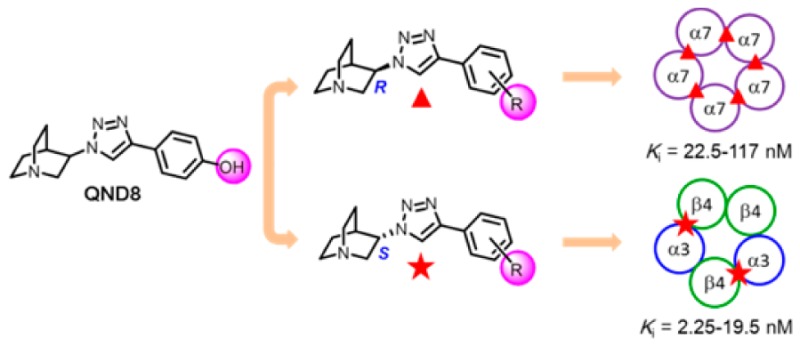
The novel quinuclidine anti-1,2,3-triazole derivatives T1–T6 were designed based on the structure of QND8. The binding studies revealed that the stereochemistry at the C3 position of the quinuclidine scaffold plays an important role in the nAChR subtype selectivity. Whereas the (R)-enantiomers are selective to α7 over α4β2 (by factors of 44–225) and to a smaller degree over α3β4 (3–33), their (S)-counterparts prefer α3β4 over α4β2 (62–237) as well as over α7 (5–294). The (R)-derivatives were highly selective to α7 over α3β4 subtypes compared to (RS)- and (R)-QND8. The (S)-enantiomers are 5–10 times more selective to α4β2 than their (R) forms. The overall strongest affinity is observed for the (S)-enantiomer binding to α3β4 (Ki, 2.25–19.5 nM) followed by their (R)-counterpart binding to α7 (Ki, 22.5–117 nM), with a significantly weaker (S)-enantiomer binding to α4β2 (Ki, 414–1980 nM) still above the very weak respective (R)-analogue affinity (Ki, 5059–10436 nM).
Keywords: Nicotinic acetylcholine receptor; positron emission tomography; quinuclidine anti-1,2,3-triazole; click chemistry
Nicotinic acetylcholine receptors (nAChRs) are members of the Cys-loop receptor superfamily of ligand-gated ion channels that mediate fast synaptic transmission in the central (CNS) and peripheral (PNS) nervous systems.1−4 They are pentameric structures of homomeric or heteromeric subunits symmetrically arranged around a central ion channel. There are 16 homologous mammalian nAChR subunits encoded by this multigene family. These subunits combine to form many different nAChR subtypes with various expression patterns, diverse functional properties, and different pharmacological characteristics.1 Currently, there are at least 12 known nAChR subunits (α2−α10 and β2−β4) expressed in the brain.4 The most predominant nAChRs found in the brain, associated with cognition, memory formation, and behavior, are the α4β2 and α7 subtypes.3 The α4β2 nAChR in mammalian brain accounts for the majority of the high affinity sites for nicotine4 and is mainly found in the thalamus, cortical regions, striatum, and colliculi, whereas the distribution pattern of the α7 nAChR with high affinity for α-bungarotoxin is rather diffuse with focally high density in thalamus and rather low expression in the cerebellum.3 A further important receptor subtype in the central nervous system (CNS) is the α3β4 nAChR because of its involvement in drug addiction and depression pathways.5 In contrast to α4β2 and α7, this subtype is mainly expressed in the autonomic ganglia and only in selected brain regions (subsets of neurons in the medial habenula, nucleus interpeduncularis, dorsal medulla, pineal gland, and retina).4 The nAChRs are well recognized as key receptors involved not only in a variety of neuropsychiatric and neurodegenerative disorders, including Parkinson’s disease (PD), attention deficit hyperactivity disorder (ADHD), schizophrenia, Alzheimer’s disease (AD), depression, epilepsy, and nicotine and drug addiction6−12 but also in cancer.13,14 The multiplicity of nAChR subtypes, their localization in the CNS, and periphery along with their pharmacological, physiological, and kinetic properties afford the opportunities to develop novel subtype-specific nAChR ligands for treatment of these diseases.14,15
Several compounds have been designed and synthesized based on known nAChR ligand recognition components covering the protonated amine that triggers the cation−π interaction, the hydrogen bond acceptor, and the hydrophobic stabilization. The availability of aromatic moieties in this hydrophobic part may participate in favorable electron−π interactions, which can be either electrostatic or dispersive. The tertiary quinuclidine amine is one of the most promising core structures used for the design and development of α7 nAChR ligands (Figure 1A). Besides the core structure, the effective pharmacophore linkers at position 3 of the quinuclidine scaffold are ether, carbamate, urea, amide, sulfonamide, oxazole, oxadiazole, and triazole.16,17 Another approach in the design of α7 ligands was the synthesis of conformational restricted analogues such as oxazolidinone AR-R17779.16 Among them, the compounds containing the quinuclidine anti-1,2,3-triazole skeleton bind with high affinity and selectivity to the α7 nAChR.18 This hydrophobic motif in particular plays an important role in receptor subtype recognition through the interaction with amino acid residues of the complementary face, which has been demonstrated as an essential part for ligand selectivity among various nAChR subtypes.19−21 We recently reported three series of novel potent and selective α7 nAChR agonists featuring the quinuclidine anti-1,2,3-triazole containing molecules (IND8 and QND8) that demonstrated cognitive enhancement in mice.22 There is not only the pharmacological benefit of using the anti-1,2,3-triazole as the linker, but the triazole moiety itself is stable and largely resistant to metabolic degradation.23,24
Figure 1.
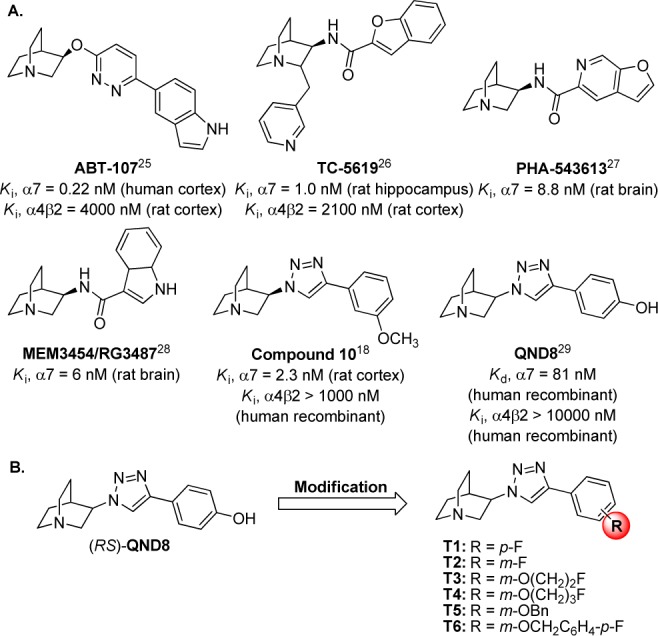
Structure of some nAChR ligands containing quinuclidine scaffold (A)18,25−29 and design strategy (B).
In the present design, four different approaches were applied to the lead compound, (RS)-QND8 (Figure 1B) to improve the potency and selectivity profiles at the most important neuronal nAChR subtypes (α7, α3β4, α4β2). First, the chirality at position 3 of the quinuclidine scaffold was investigated by individual analysis of the two enantiomers of the racemic compounds. Quinuclidine-containing compounds are well recognized for nAChR interaction and the selectivity of their (R)-enantiomers for α7 nAChR. However, detailed information with regard to their (S)-enantiomers was missing at the beginning of this study.18,29 Thus, both enantiomers were prepared to investigate the influence of chirality on the selectivity as well as the affinity for nAChR. Second, the orientation of the hydrophobic aromatic ring was explored by altering the substitution from para to meta. With regard to α7 nAChR, the binding affinity of quinuclidine derivatives was found to prefer the meta-phenyl substitution, compared to the para-position.18 The m-phenyl substitution was included to verify this notion. Third, the hydrophobic side chains were extended to enhance the binding affinity and selectivity, based on the increase in size as well as the lipophilicity for receptor binding. The expansion of the structure was conducted via an ether linkage (R in Figure 1B), where the phenol of QND8 resides. Finally, the fluorine atoms were introduced into the structure for further development as potential positron emission tomography (PET) radioligands for imaging the nAChR in the brain.
Herein we report the synthesis and the binding affinity of quinuclidine derivatives T1–T6 (RS, R, S) at neuronal α7, α3β4, and α4β2 nAChR subtypes. Promising compounds of high α3β4 binding affinity and good selectivity toward other nAChRs were further screened in functional assays using HEK cells expressing human nAChR subtypes.
The general procedure for the preparation of the designed compounds started with the synthesis of the azide building block by a diazo transfer reaction and synthesis of terminal alkynes through the Sonogashira cross-coupling reaction, followed by the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction resulting in the formation of the final anti-1,2,3-triazole-containing ligands (Scheme 1). The diazo transfer reaction for the preparation of azidoquinuclidine compounds (RS)-8a, (R)-8b, and (S)-8c were achieved, respectively, by using 3-aminoquinuclidine dihydrochloride (RS)-7a, (R)-7b, and (S)-7c interacted with freshly prepared trifluoromethanesulfonyl azide (TfN3). The 3-ethynylphenol 11 was used as a starting material for the Williamson etherification with 1-fluoro-2-iodoethane, 1-fluoro-3-iodopropane, benzyl chloride, and 4-fluorobenzyl chloride to afford the 3-ethynyl-phenylethers 12, 13, 14, and 15, respectively. The final compounds (RS)-T1, (R)-T1, and (S)-T1 were synthesized by the reaction of the respective azidoquinuclidines (RS)-8a, (R)-8b, and (S)-8c with 4-fluoro-phenylacetylene 9 under CuAAC conditions. Cycloaddition of these azidoquinuclidines with 3-fluorophenyl-acetylene 10 yielded the final compounds (RS)-T2, (R)-T2, and (S)-T2. CuAAC or click reactions between 8a–c and terminal alkyne 12 resulted in the formation of its corresponding T3 compounds. The remaining quinuclidine anti-1,2,3-triazoles T4–T6 were prepared by the same synthetic route as described above.
Scheme 1. Synthesis of Azide (A), Terminal Alkynes (B), and the Quinuclidine anti-1,2,3-Triazole Containing Molecules (C).

Reagents and conditions: (a) TfN3, K2CO3, CuSO4·5H2O, CH3OH, H2O, rt, overnight; (b) 1-fluoro-2-iodoethane for 12 and 1-fluoro-3-iodopropane for 13, K2CO3, CH3CN, 70 °C, 10 h; (c) benzyl chloride for 14 and 4-fluorobenzyl chloride for 15, K2CO3, CH3CN, 80 °C, 6 h; (d) 5 mol % CuSO4·5H2O, 10 mol % sodium ascorbate, t-BuOH, H2O, rt, overnight.
The blood–brain barrier (BBB) penetration of CNS drugs depends on several physicochemical properties including molecular weight (MW), pKa, hydrophobicity (log P and log D), and topological polar surface area (TPSA). These parameters were calculated for all synthesized compounds using the ACD/ADME Suite software, and the predicted values fall within the ranges of CNS drug-like properties. Calculated log BB values of ≥−1.0 indicate sufficient BBB permeation of all compounds30 (Supplemental Table S1). Additional support for a good BBB penetration of the compounds is provided by calculated pKa values in the range of 7.5–10.5, which furthermore suggest sufficient protonation of the compounds in the CNS for cation−π or other electrostatic interactions with conserved amino acid residues or residues comprising electron-rich aromatic moieties in the binding sites of nAChRs.
The in vitro binding affinities of all modified compounds for the nicotinic receptor subtypes were examined in competitive radioligand displacement studies using [3H]methyllycaconitine and [3H]epibatidine as selective radioligands for human α7 nAChR as well as human α4β2 and α3β4 nAChRs, respectively (Supporting Information). The binding affinities of the compounds tested are presented in Table 1. The racemic compounds showed Ki values at α7 nAChR in the range of 73–133 nM. In addition, all of them bound with higher affinity toward α3β4 nAChR. Replacement of the hydroxyl group of the lead compound QND8 by a fluorine atom decreases the binding affinity on α7 as the Ki value of (RS)-T1 at α7 nAChR is 7 times higher than that of (RS)-QND8 (73 vs 10 nM). However, this modified compound showed better selectivity at α7 over α3β4.
Table 1. In Vitro Binding Affinities of 21 Quinuclidine anti-1,2,3-Triazole Derivatives Towards nAChRsa.
| inhibition
constant Ki in nM; mean ± SD |
selectivityb (inverse of respective Ki ratio) |
||||||
|---|---|---|---|---|---|---|---|
| R | compd | α7c | α3β4d | α4β2d | α7 vs α3β4 | α7 vs α4β2 | α3β4 vs α4β2 |
| p-F | (RS)-T1 | 72.8 ± 13 | 8.50 ± 0.50 | 449 ± 127 | 1/8.6 | 6.2 | 53 |
| (R)-T1 | 73.0 ± 15 | 1010 ± 162 | 10436 ± 1943 | 14 | 143 | 10 | |
| (S)-T1 | 174.5 ± 66 | 3.09 ± 0.10 | 515 ± 64 | 1/56 | 3.0 | 167 | |
| m-F | (RS)-T2 | 133 ± 40 | 5.24 ± 0.35 | 748 ± 114 | 1/25 | 5.6 | 143 |
| (R)-T2 | 117 ± 4 | 362 ± 27 | 5201 ± 412 | 3.1 | 44 | 14 | |
| (S)-T2 | 660.5 ± 139 | 2.25 ± 0.42 | 519 ± 20 | 1/294 | 1/1.3 | 231 | |
| m-O(CH2)2F | (RS)-T3 | 98.7 ± 39 | 20.9 ± 0.7 | 1962 ± 228 | 1/4.7 | 20 | 94 |
| (R)-T3 | 38.8 ± 8 | 558 ± 34 | 7050 ± 200 | 14 | 182 | 13 | |
| (S)-T3 | 74.9 ± 20 | 11.8 ± 0.3 | 1262 ± 187 | 1/6.3 | 17 | 107 | |
| m-O(CH2)3F | (RS)-T4 | 74.6 ± 14 | 44.4 ± 8.0 | 3894 ± 252 | 1/1.7 | 52 | 88 |
| (R)-T4 | 62.3 ± 10 | 1628 ± 11 | 9010 ± 5,034 | 26 | 145 | 5.5 | |
| (S)-T4 | 96.9 ± 17 | 19.5 ± 0.4 | 1980 ± 117 | 1/5.0 | 20 | 102 | |
| m-OBn | (RS)-T5 | 91.3 ± 9 | 7.57 ± 2.9 | 668 ± 7 | 1/12 | 7.3 | 88 |
| (R)-T5 | 22.5 ± 9 | 631 ± 206 | 5059 ± 374 | 28 | 225 | 8.0 | |
| (S)-T5 | 279 ± 31 | 6.67 ± 0.7 | 414 ± 59 | 1/42 | 1.5 | 62 | |
| m-OCH2C6H4F | (RS)-T6 | 127 ± 5 | 13.9 ± 2.8 | 1,013 ± 107 | 1/9.1 | 8.0 | 73 |
| (R)-T6 | 33.2 ± 7 | 1090 ± 163 | 6392 ± 230 | 33 | 193 | 5.9 | |
| (S)-T6 | 149 ± 42 | 7.17 ± 1.2 | 537 ± 11 | 1/21 | 3.6 | 75 | |
| p-OH | (RS)-QND8 | 9.61 ± 1.47 | 3.44 ± 0.04 | 627 ± 52 | 1/2.8 | 65 | 182 |
| (R)-QND8 | 10.9 ± 1.42 | 138 ± 0 | 7389 ± 42 | 12.7 | 678 | 54 | |
| (S)-QND8 | 29.3 ± 0.18 | 2.48 ± 0.04 | 461 ± 89 | 1/11.8 | 16 | 186 | |
Binding affinities are represented by the inhibition constant Ki determined for individual compounds in two separate experiments each performed as triplicate.
Selectivity is reported in terms of the reciprocal of the ratio of the respective Ki values.
Human α7 nAChR in stably transfected SH-SY5Y cells, with radiotracer [3H]methyllycaconitine (0.5–1 nM); Kd = 2.0 nM.
Human α4β2 and α3β4 nAChR in stably transfected HEK-293 cells, with radiotracer [3H]epibatidine (0.5–1 nM); Kd = 0.025 nM for hα4β2 nAChR; Kd = 0.117 nM for hα3β4 nAChR.
The most important finding was the significant dependence of the nAChR subtype affinity on the handedness of the ligands. The chirality of the binding sites of the different nAChR subtypes offers a rationale for the enantioselective affinity profiles of chiral ligands for α7, α4β2, and α3β4 nAChRs. More specifically, the experimentally determined affinities (Table 1) reveal the following pattern: first, all (R)-enantiomers showed a significant selectivity for α7 over α4β2 and to a lesser degree also over α3β4. They bind with low nanomolar affinity at α7, with high nanomolar affinity at α3β4, and with micromolar affinities at α4β2. The resulting selectivity values in the range of 3.1–33 and 44–225 for α7 vs α3β4 and α4β2, respectively, indicate preference for the α7 nAChR binding site. Atypically, (R)-T2 provides only weak preference for α7 over α3β4 (selectivity of 3.1). Second, all (S)-enantiomers are highly selective for α3β4 over α4β2 and mostly to a lesser degree also over α7. They bind with low nanomolar affinity at α3β4 and with high nanomolar to micromolar affinities at α7 and α4β2. The selectivity values of α3β4 vs α7 and α4β2 were estimated in the range of 5–294 and 62–231 and indicate preference of the (S)-enantiomers of quinuclidine-anti-1,2,3-triazole containing nAChR ligands for the binding site of α3β4 nAChR. Compound T2 plays again a special role, with its (S)-enantiomer showing the overall highest selectivity (294) for α3β4 over α7. Notably AT-1001 previously reported as a high affinity and selective α3β4 nAChR antagonist31 also falls into this category, although with slightly lower affinity and selectivity than T2. However, two other recently reported ligands for the α3β4 nAChR, CP-601932(32) and an anabaseine analogue,33 showed modest selectivity of binding to α3β4 over α4β2 and α3β4 over α7, respectively (Figure 2). Third, the preference of the (S)-enantiomers for α3β4 over α4β2 (selectivity 62–231) is more pronounced than the preference of the (R)-enantiomers for α7 over α3β4 (selectivity 3–33). Fourthly, the overall highest binding affinities for the herein assessed nAChR subtypes were observed for binding of the (S)-enantiomers to α3β4 (Ki = 2.25–19.5 nM) followed by the binding of the (R)-enantiomers to α7 (Ki= 22.5–117 nM). Concerning binding to α4β2, the affinity of the (S)-enantiomers is higher than of the (R)-enantiomers (414–1980 vs 5059–10436 nM).
Figure 2.

Structures of selected α3β4 nAChR ligands.
By more detailed investigation of the interaction of the herein presented ligands with α7 nAChR, the fluorine-substituted enantiomers (R)-T1 and(R)-T2, bearing F, at p- and m-position of the phenyl ring, respectively, showed binding profiles comparable to their racemic mixture with (R)-T1 being 3-fold more selective for α7 over α4β2 than (R)-T2 (factors 143 vs 44). The binding affinity increases about 2-fold when replacing the (R)-T4 phenyl substituent m-O(CH2)3F (Ki = 62.3 nM) by the smaller and less hydrophobic m-O(CH2)2F group ((R)-T3, Ki = 38.8 nM) as well as by the larger and more hydrophobic groups m-OBn ((R)-T5, Ki = 22.5 nM) and m-OCH2C6H4F ((R)-T6, Ki = 33.2 nM). A possible explanation would be that the respective variation in Ki might be triggered by the presence or absence of a spatially suitable electron donor functionality of the ligand (e.g., as H-bond acceptor), either through its F lone pair electrons (T4) or through the benzylic π-electrons (T5, T6).
For the corresponding (S)-enantiomers, (S)-T2 shows a 5-fold higher selectivity for α3β4 over α7 in comparison to (S)-T1. Regarding further substituents, the following pattern is observed for the affinity of the (S)-enantiomers to α3β4 nAChR. Here, m-O(CH2)2F ((S)-T3 with an Ki of 11.8 nM) is again ∼2-fold more potent than m-O(CH2)3F ((S)-T4, Ki = 19.5 nM), but now the two phenyl-containing (S)-T5 (Ki = 6.67 nM) and (S)-T6 (Ki = 7.17 nM) yield a further ∼2-fold increase in binding affinity. These results support the suggestion that a ligand π-electron donor capability at a site distant from the quinuclidine triazole moiety may contribute to stabilizing the ligand–receptor complex. Interestingly, (S)-T2 and (S)-T1 as the two most promising α3β4 ligands (Ki = 2.25 vs 3.09 nM) are the only derivatives with an F-substituted phenyl ring, where m-F (S)-T2 yields the overall highest α3β4 selectivity over both α7 and α4β2 as discussed above. The significant difference in the selectivity profiles of the F-substituted isomers (S)-T1 and (S)-T2 suggests the following tentative explanation. In (S)-T1, the electronegative F is in p-position to the electron-rich triazole, the latter of which may partly compensate the electron shift from the benzene ring to F. By contrast, the respective conjugative substitution pattern is missing in (S)-T2, thus probably resulting in a stronger loss of π-electron density through inductive charge transfer to m-F. From this viewpoint it is likely that an electron-poor character of the benzylic moiety in compound (S)-T2 triggers for a strong α3β4 selectivity, with Ki-based selectivity of 294 (over α7) and 231 (over α4β2), respectively. Interestingly, it was apparent that (S)-enantiomers were more selective to α3β4 (47–326 times) and α4β2 nAChR (4.5–20 times) than their corresponding (R)-counterparts. Up until now, the selective binding of (S)-quinuclidine based compounds on α3β4 and α4β2 subtypes have never been reported before.
Four compounds with good affinity (Ki < 10 nM), which are (S)-enantiomers of T1, T2, T5, and T6, were evaluated for the binding at related receptors, these four compounds had no significant effect on N-muscle type nAChR (2–12% inhibition at 1 μM). For the 5-HT3 receptor, (S)-enantiomers of T1, T2, T5, and T6 were tested at 100 nM, all except (S)-T6 showed no significant binding at the 5-HT3 receptor.
To see whether the designed compounds are agonist as QND8 or antagonist as AT-1001, the functional assays of the potential compounds T1 and T2 in both (R) and (S) forms were performed using SH-SY5Y cells stably expressing human α7 nAChR and HEK 293 cells stably expressing human α3β4 nAChR. The screening results indicated that (R)-T1, (R)-T2, (S)-T1, and (S)-T2 are α7 nAChR agonists (Supplemental Figure S1). Compounds (R)-T2 and (S)-T1 are α3β4 nAChR agonists, whereas (R)-T1 and (S)-T2 are α3β4 nAChR antagonists (Supplemental Figure S2). However, the selectivity cannot be concluded due to the high testing concentration (10 μM) in the screening assay for functional effects.
Overall, according to the binding data of all novel analogues presented herein, the stereocenter at C3 position of the quinuclidine motif has significant impact on the selectivity of modified compounds to nAChR subtypes. (R)-Enantiomers bind preferably toward α7, while (S)-enantiomers were more selective for the α3β4 nAChR. Among all, (R)-T5 was the most favorable ligand for the α7 nAChR, (S)-T2 and (S)-T1 for the α3β4 nAChR with almost equal affinities, and (although significantly less strong) (S)-T5 was the most promising α4β2 nAChR ligand.
In conclusion, we have successfully designed two potent compounds (S)-T1 and (S)-T2 that bind selectively to α3β4 nAChR over α7 nAChR. Among the (R)- and (S)-enantiomers of the quinuclidine-based compounds, the binding affinities of (S)-enantiomers are higher compared to their (R) counterparts. This finding may have implications for the treatment of several brain diseases such as drug addiction and dependence. Compounds (R)-QND8 and (R)-T6, which displayed higher affinity for the α7 nAChR and higher selectivity toward the α3β4 nAChR than its parent (RS)-QND8 might be useful candidate molecules in further drug development for the treatment of cognitive disorders. In addition, the introduction of a fluorine substituent in the structure makes it possible to develop the designed compounds as PET radioligands for brain imaging to localize the α3β4 and α7 nAChR subtypes in the brain and monitor drug efficacy.
Glossary
ABBREVIATIONS
- nAChR
nicotinic acetylcholine receptor
- PET
positron emission tomography
- CuAAC
copper-catalyzed azide–alkyne cycloaddition
- PD
Parkinson’s disease
- ADHD
attention deficit hyperactivity disorder
- AD
Alzheimer’s disease
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00146.
Experimental procedures and analytical data (PDF)
This work was supported by Thailand Research Fund (TRF) through the Royal Golden Jubilee Ph.D. Program (Grant No.PHD/0272/2552) to J.S. and O.V., BRG 5780016 to S.C., the Office of the High Education Commission, Thailand; Thailand Research Fund (TRF) and Faculty of Pharmacy, Mahidol University (IRG578007) to O.V.
The authors declare no competing financial interest.
Supplementary Material
References
- Dani J. A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19. 10.1016/bs.irn.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley K. T.; Pandya A. A.; Yakel J. L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. 10.1016/j.tips.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust P.; Deuther-Conrad W.; Donat C. K.; Barthel H.; Riss P.; Paterson L.; Hoepping A.; Sabri O.; Cumming P.. Preclinical aspects of nicotinic acetylcholine receptor imaging. In PET and SPECT of Neurobiological Systems; Dierckx R. A. J. O., Ed.; Springer: Berlin Heidelberg, 2014; pp 465–512. [Google Scholar]
- Gotti C.; Clementi F.; Fornari A.; Gaimarri A.; Guiducci S.; Manfredi I.; Moretti M.; Pedrazzi P.; Pucci L.; Zoli M. Structural and functional diversity of native brain neuronal nicotinic receptors. Biochem. Pharmacol. 2009, 78, 703–711. 10.1016/j.bcp.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Rahman S.; Engleman E. A.; Bell R. L. Nicotinic receptor modulation to treat alcohol and drug dependence. Front. Neurosci. 2015, 8, 426. 10.3389/fnins.2014.00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posadas I.; Lopez-Hernandez B.; Cena V. Nicotinic receptors in neurodegeneration. Curr. Neuropharmacol. 2013, 11, 298–314. 10.2174/1570159X11311030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyohara J.; Hashimoto K. α7 Nicotinic receptor agonists: potential therapeutic drugs for treatment of cognitive impairments in Schizophrenia and Alzheimer’s Disease. Open Med. Chem. J. 2010, 4, 37–56. 10.2174/1874104501004020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani J. A.; Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Wilens T. E.; Decker M. W. Neuronal nicotinic receptor agonists for the treatment of attention-deficit/hyperactivity disorder: focus on cognition. Biochem. Pharmacol. 2007, 74, 1212–1223. 10.1016/j.bcp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick S. D.; Maisonneuve I. M.; Kitchen B. A.; Fleck M. W. Antagonism of a3b4 nicotinic receptors as a strategy to reduce opioid and stimulant self-administration. Eur. J. Pharmacol. 2002, 438, 99–105. 10.1016/S0014-2999(02)01284-0. [DOI] [PubMed] [Google Scholar]
- Haydar S. N.; Dunlop J. Neuronal nicotinic acetylcholine receptors - targets for the development of drugs to treat cognitive impairment associated with schizophrenia and Alzheimer’s disease. Curr. Top. Med. Chem. 2010, 10, 144–152. 10.2174/156802610790410983. [DOI] [PubMed] [Google Scholar]
- Taraschenko O. D.; Panchal V.; Maisonneuve I. M.; Glick S. D. Is antagonism of a3b4 nicotinic receptors a strategy to reduce morphine dependence?. Eur. J. Pharmacol. 2005, 513, 207–218. 10.1016/j.ejphar.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Zhao Y. The oncogenic functions of nicotinic acetylcholine receptors. J. Oncol. 2016, 2016, 1. 10.1155/2016/9650481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucchietto V.; Crespi A.; Fasoli F.; Clementi F.; Gotti C. Neuronal acetylcholine nicotinic receptors as new targets for lung cancer treatment. Curr. Pharm. Des. 2016, 22, 2160–2169. 10.2174/1381612822666160203144114. [DOI] [PubMed] [Google Scholar]
- Gotti C.; Zoli M.; Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Pin F.; Vercouillie J.; Ouach A.; Mavel S.; Gulhan Z.; Chicheri G.; Jarry C.; Massip S.; Deloye J. B.; Guilloteau D.; Suzenet F.; Chalon S.; Routier S. Design of a7 nicotinic acetylcholine receptor ligands in quinuclidine, tropane and quinazoline series. Chemistry, molecular modeling, radiochemistry, in vitro and in rats evaluations of a [18F] quinuclidine derivative. Eur. J. Med. Chem. 2014, 82, 214–224. 10.1016/j.ejmech.2014.04.057. [DOI] [PubMed] [Google Scholar]
- Mazurov A. A.; Speake J. D.; Yohannes D. Discovery and development of a7 nicotinic acetylcholine receptor modulators. J. Med. Chem. 2011, 54, 7943–7961. 10.1021/jm2007672. [DOI] [PubMed] [Google Scholar]
- Ouach A.; Pin F.; Bertrand E.; Vercouillie J.; Gulhan Z.; Mothes C.; Deloye J. B.; Guilloteau D.; Suzenet F.; Chalon S.; Routier S. Design of a7 nicotinic acetylcholine receptor ligands using the (het)Aryl-1,2,3-triazole core: Synthesis, in vitro evaluation and SAR studies. Eur. J. Med. Chem. 2016, 107, 153–164. 10.1016/j.ejmech.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Cohen B. N.; Figl A.; Quick M. W.; Labarca C.; Davidson N.; Lester H. A. Regions of b2 and b4 responsible for differences between the steady state dose-response relationships of the a3b2 and a3b4 neuronal nicotinic receptors. J. Gen. Physiol. 1995, 105, 745–764. 10.1085/jgp.105.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker M. J.; Beck A.; Luetje C. W. Neuronal nicotinic receptor b2 and b4 subunits confer large differences in agonist binding affinity. Mol. Pharmacol. 1998, 54, 1132–1139. [PubMed] [Google Scholar]
- Luetje C. W.; Patrick J. Both a- and b-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J. Neurosci. 1991, 11, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunrungvichian K.; Boonyarat C.; Fokin V. V.; Taylor P.; Vajragupta O. Cognitive improvements in a mouse model with substituted 1,2,3-triazole agonists for nicotinic acetylcholine receptors. ACS Chem. Neurosci. 2015, 6, 1331–1340. 10.1021/acschemneuro.5b00059. [DOI] [PubMed] [Google Scholar]
- Keck T. M.; Banala A. K.; Slack R. D.; Burzynski C.; Bonifazi A.; Okunola-Bakare O. M.; Moore M.; Deschamps J. R.; Rais R.; Slusher B. S. Using click chemistry toward novel 1, 2, 3-triazole-linked dopamine D3 receptor ligands. Bioorg. Med. Chem. 2015, 23, 4000–4012. 10.1016/j.bmc.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M.; Robert V.; Galibert M.; Madinier J.-B.; Marceau P.; Dardente H.; Decourt C.; De Roux N.; Lomet D.; Delmas A. F. Rational design of triazololipopeptides analogs of kisspeptin inducing a long-lasting increase of gonadotropins. J. Med. Chem. 2015, 58, 3459–3470. 10.1021/jm5019675. [DOI] [PubMed] [Google Scholar]
- Malysz J.; Anderson D. J.; Gronlien J. H.; Ji J.; Bunnelle W. H.; Haakerud M.; Thorin-Hagene K.; Ween H.; Helfrich R.; Hu M.; Gubbins E.; Gopalakrishnan S.; Puttfarcken P. S.; Briggs C.; Li J.; Meyer M. D.; Dyhring T.; Ahring P. K.; Nielsen E. O.; Peters D.; Timmermann D. B.; Gopalakrishnan M. In vitro pharmacological characterization of a novel selective a7 neuronal nicotinic acetylcholine receptor agonist ABT-107. J. Pharmacol. Exp. Ther. 2010, 334, 863–874. 10.1124/jpet.110.167072. [DOI] [PubMed] [Google Scholar]
- Hauser T. A.; Kucinski A.; Jordan K. G.; Gatto G. J.; Wersinger S. R.; Hesse R. A.; Stachowiak E. K.; Stachowiak M. K.; Papke R. L.; Lippiello P. M.; Bencherif M. TC-5619: an alpha7 neuronal nicotinic receptor-selective agonist that demonstrates efficacy in animal models of the positive and negative symptoms and cognitive dysfunction of schizophrenia. Biochem. Pharmacol. 2009, 78, 803–812. 10.1016/j.bcp.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishka D. G.; Walker D. P.; Yates K. M.; Reitz S. C.; Jia S.; Myers J. K.; Olson K. L.; Jacobsen E. J.; Wolfe M. L.; Groppi V. E.; Hanchar A. J.; Thornburgh B. A.; Cortes-Burgos L. A.; Wong E. H.; Staton B. A.; Raub T. J.; Higdon N. R.; Wall T. M.; Hurst R. S.; Walters R. R.; Hoffmann W. E.; Hajos M.; Franklin S.; Carey G.; Gold L. H.; Cook K. K.; Sands S. B.; Zhao S. X.; Soglia J. R.; Kalgutkar A. S.; Arneric S. P.; Rogers B. N. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the a7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure--activity relationship. J. Med. Chem. 2006, 49, 4425–4436. 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Wallace T. L.; Callahan P. M.; Tehim A.; Bertrand D.; Tombaugh G.; Wang S.; Xie W.; Rowe W. B.; Ong V.; Graham E.; Terry A. V. Jr.; Rodefer J. S.; Herbert B.; Murray M.; Porter R.; Santarelli L.; Lowe D. A. RG3487, a novel nicotinic a7 receptor partial agonist, improves cognition and sensorimotor gating in rodents. J. Pharmacol. Exp. Ther. 2011, 336, 242–253. 10.1124/jpet.110.171892. [DOI] [PubMed] [Google Scholar]
- Arunrungvichian K.; Fokin V. V.; Vajragupta O.; Taylor P. Selectivity optimization of substituted 1,2,3-triazoles as a7 nicotinic acetylcholine receptor agonists. ACS Chem. Neurosci. 2015, 6, 1317–1330. 10.1021/acschemneuro.5b00058. [DOI] [PubMed] [Google Scholar]
- Ghose A. K.; Herbertz T.; Hudkins R. L.; Dorsey B. D.; Mallamo J. P. Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem. Neurosci. 2012, 3, 50–68. 10.1021/cn200100h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toll L.; Zaveri N. T.; Polgar W. E.; Jiang F.; Khroyan T. V.; Zhou W.; Xie X. S.; Stauber G. B.; Costello M. R.; Leslie F. M. AT-1001: a high affinity and selective a3b4 nicotinic acetylcholine receptor antagonist blocks nicotine self-administration in rats. Neuropsychopharmacology 2012, 37, 1367–1376. 10.1038/npp.2011.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S.; Steensland P.; Simms J. A.; Holgate J.; Coe J. W.; Hurst R. S.; Shaffer C. L.; Lowe J.; Rollema H.; Bartlett S. E. Partial agonists of the a3b4* neuronal nicotinic acetylcholine receptor reduce ethanol consumption and seeking in rats. Neuropsychopharmacology 2011, 36, 603–615. 10.1038/npp.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera C.; Quadri M.; Sciaccaluga M.; Pome D. Y.; Fasoli F.; De Amici M.; Fucile S.; Gotti C.; Dallanoce C.; Grazioso G. Modification of the anabaseine pyridine nucleus allows achieving binding and functional selectivity for the a3b4 nicotinic acetylcholine receptor subtype. Eur. J. Med. Chem. 2016, 108, 392–405. 10.1016/j.ejmech.2015.11.045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.