

Abstract

Abnormalities in the JAK/STAT signaling pathway lead to many diseases such as immunodeficiency, inflammation, and cancer. Herein, we designed and synthesized a series of 4-amino-(1H)-pyrazole derivatives as potent JAKs inhibitors for cancer treatment. Results from in vitro protein kinase inhibition experiments indicated that compounds 3a–f and 11b are potent JAKs inhibitors. For example, the IC50 values of compound 3f against JAK1, JAK2, and JAK3 were 3.4, 2.2, and 3.5 nM, respectively. In cell culture experiments, compound 3f showed potent antiproliferative activity against various cell lines (PC-3, HEL, K562, MCF-7, and MOLT4) at low micromolar levels, while compound 11b showed selective cytotoxicity at submicromolar levels against HEL (IC50: 0.35 μM) and K562 (IC50: 0.37 μM) cell lines. It is worth noting that both 3f and 11b showed more potent antiproliferative activities than the approved JAKs inhibitor Ruxolitinib.
Keywords: JAKs, Inhibitors, 4-Amino-(1H)-pyrazole, Anticancer
The JAK/STAT signaling pathway plays critical roles in immunity, hematopoiesis, and cell growth.1 Abnormalities in the JAK/STAT signaling pathway lead to many diseases such as immunodeficiency, inflammation, and cancers.2 Constitutive activations of JAKs are correlated to oncogenesis. Dysregulation of JAK2 is discovered in patients with myeloproliferative neoplasms and childhood T cell acute lymphoblastic leukemia.3 Several hematologic malignancies including malignant lymphoma and myeloproliferative disorders are associated with mutations of JAK2.4 Some cytokines and growth factors bind to their receptors and then stimulate JAKs for the phosporylation of STAT3, which is a potential target for anticancer therapy.5 Persistent activation of the JAK/STAT3 signaling promotes proliferation and survival of tumor cells.6 Thus, the JAK/STAT signaling pathway is a promising antitumor target.
Inhibitors of JAKs are widely explored for treatment of immunodeficiency, inflammation, and cancer. Among the synthetic JAK inhibitors for the treatment of cancer identified to date (Figure 1), Ruxolitinib (Incyte’s Jakafi) was approved by the US Food and Drug Administration (FDA) in 2013 for the treatment of myelofibrosis, a rare bone marrow cancer. Besides, it is in phase III clinical trials for the treatment of metastatic pancreatic cancer and phase II clinical trials for the treatment of multiple myeloma, leukemia, and colon cancer.7 There are several other JAKs inhibitors in clinical trials for cancer treatment (Figure 1). JAK2 inhibitor AZD1480 can inhibit STAT5 signaling in prostate cancer cells and then effectively inhibit castration-resistant growth of prostate cancer.8 JAK2 inhibitor TG101348 blocks JAK/STAT signaling leading to suppression of proliferation and induction of apoptosis and is used for the treatment of myelofibrosis.9 JAKs inhibitors Momelotinib and Pacritinib are both developed for the treatment of myelofibrosis.10
Figure 1.

Approved or clinical JAK inhibitors for cancer treatment.
In our previous work, we discovered a series of 4-aminopyrazole derivatives as novel and potent JAK inhibitors (Figure 2).11 Structure–activity relationship (SAR) studies showed that R1 group modifications in these analogues did not influence their JAK inhibition significantly.11 Such results indicate that the R1 side chain is not crucial to the interaction between these 4-aminopyrazole derivatives and JAKs proteins. Herein, using 1 as a lead, a series of 4-amino-(1H)-pyrazole derivatives were designed, synthesized, and evaluated as potent JAKs inhibitors. First, removing the R1 side chain of compound 1 led to compound 3a, which exhibited improved JAKs kinase inhibition at 10 μM. Then further structural modifications of 3a led to pyrimidine-based 4-amino-(1H)-pyrazole derivatives, quinazoline-based 4-amino-(1H)-pyrazole derivatives, and 7H-pyrrolo[2,3-d] pyrimidine-based 4-amino-(1H)-pyrazole derivatives (Figure 2).
Figure 2.
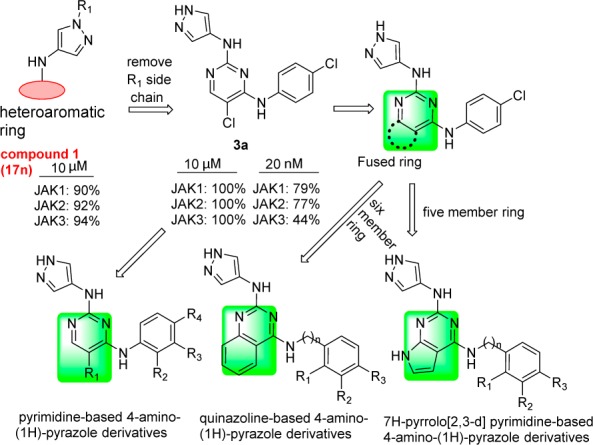
Design of 4-amino-(1H)-pyrazole derivatives as JAKs inhibitor.
Pyrimidine-based 4-amino-(1H)-pyrazole derivatives 3a–m were synthesized as described in Scheme 1. Generally, the reaction of 5-substituted-2,4-dichloropyrimidine with various aromatic amines under acidic (HCl) or basic (DIPEA, Et3N, Na2CO3) conditions led to the intermediates 2a–m, which was then reacted with 1H-pyrazol-4-amine using TFA as a catalyst at high temperature to give target molecules 3a–m.12
Scheme 1. Synthesis of Pyrimidine-Based 4-Amino-(1H)-pyrazole Derivatives 3a–m.

Conditions and reagents: (a) For compounds 2a, 2b, and 2d, DIPEA, DMF, r.t., 16 h. For compound 2c, Na2CO3, EtOH, r.t. overnight. For compound 2e, DIPEA, NMP, 100 °C, 12 h. For compounds 2f and 2g, Et3N, EtOH, 50–80 °C, overnight. For compound 2h, HCl, H2O, r.t., 5 days. For compounds 2i and 2l, HCl, H2O, 50 °C, 5 h. For compound 2j, DIPEA, i-PrOH, 60 °C, overnight. For compounds 2k and 2m, H2O/CH3OH = 3:1, 50 °C, 5 h. (b) TFA, n-BuOH, 120 °C, 1 h, microwave (MW irradiation).
Quinazoline-based 4-amino-(1H)-pyrazole derivatives 6a–f were synthesized as shown in Scheme 2. The synthetic routes of derivatives 6a–f were similar to that of Scheme 1. Intermediates 5a–f were obtained from reaction of 2,4-dichloroquinazoline with various amines under basic condition of CH3COONa.13 Then, 5a–f reacted with 1H-pyrazol-4-amine at high temperature to give target molecules 6a–f.
Scheme 2. Synthesis of Quinazoline-Based 4-Amino-(1H)-pyrazole Derivatives 6a–f.

Conditions and reagents: (a) CH3COONa, THF/H2O = 3:1, r.t. to 60 °C, overnight. (b) n-BuOH, 120 °C, 1 h, microwave (MW irradiation).
7H-Pyrrolo[2,3-d] pyrimidine-based 4-amino-(1H)-pyrazole derivatives 11a–d were synthesized as shown in Scheme 3. The starting material 2,4-dichloro-7H-pyrrolo[2,3-d] pyrimidine was protected by 4-methylbenzenesulfonyl chloride14 to give compound 8, which was reacted with various amines, leading to intermediates 9a–d, under basic conditions at high temperature. Intermediates 9a–d reacted with 1H-pyrazol-4-amine using TFA as a catalyst at high temperature to produce intermediates 10a–d. Ts-deprotection of 10a–d yielded target molecules 11a–d.15
Scheme 3. Synthesis of 7H-Pyrrolo[2,3-d]pyrimidine-Based 4-Amino-(1H)-pyrazole Derivatives 11a–d.

Conditions and reagents: (a), TsCl, Et3N, DMAP, CH2Cl2. r.t., 5 h. (b) DIPEA, n-BuOH, 100 °C, overnight. (c) TFA, n-BuOH, 120 °C, 1 h, microwave (MW irradiation). (d) Cs2CO3, H2O/CH3OH/dioxane = 1:3:3 (v/v/v), 80 °C, 6 h.
The 4-amino-(1H)-pyrazole derivatives were screened for their in vitro kinase inhibitory activities toward JAK1, JAK2, and JAK3 at 10, 1, and 0.1 μM and 40 and 20 nM. Because we are only interested in compounds with nanomolar inhibition activities, the final screening was done at 20 nM. Staurosporine (a prototypical ATP-competitive kinase inhibitor; IC50: JAK1 3 nM, JAK2 2 nM, JAK3 1 nM) and Ruxolitinib (an approved JAK inhibitor; inhibition at 20 nM: JAK1 97%, JAK2 99%, JAK3 95%) were used as positive controls.16 All the inhibition results were shown in Figures 3–6.
Figure 3.
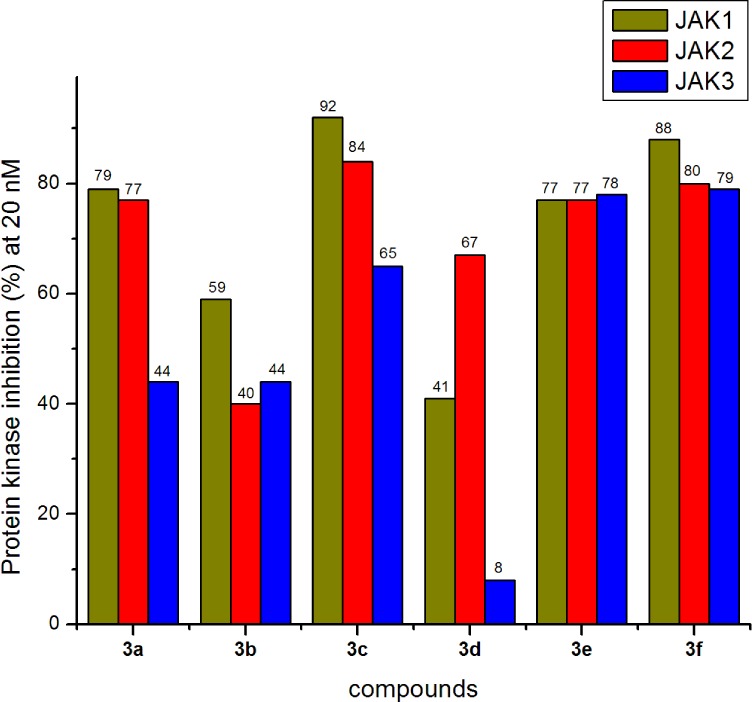
In vitro inhibitory activity against JAK1, JAK2, and JAK3.
Figure 6.

In vitro inhibitory activity against JAK1, JAK2, and JAK3.
Results in Figure 3 showed that compounds 3a–3f exhibited remarkable inhibitory activities against JAK1, JAK2, and JAK3 at 20 nM with the exception of compound 3d, which was not active against JAK3 at 20 nM. For example, at 20 nM, compound 3f inhibited protein kinase activities of 88%, 80%, and 79% against JAK1, JAK2, and JAK3 respectively. Further evaluation revealed that the IC50 values of 3f against JAK1, JAK2, and JAK3 were 3.4, 2.2, and 3.5 nM, respectively. Generally, different substituents on the phenyl ring were well tolerated.
Results in Figure 4 showed that replacing the Cl group on pyrimidine ring with other groups, such as H or F could lead to reduced JAKs inhibition. For example, compounds 3g and 3k were much less potent than 3a (Figure 3). Taking the results in Figure 3 and Figure 4 together, we conclude that R1 group on pyrimidine ring contributed much more to JAKs inhibition than R2, R3, and R4 groups on the phenyl ring.
Figure 4.

In vitro inhibitory activity against JAK1, JAK2, and JAK3.
From the data shown in Figure 5, we could see that quinazoline-based 4-amino-(1H)-pyrazole derivatives 6a–f almost completely lost their inhibitory activities toward JAKs at 20 nM. This indicated that a large fused ring, such as quinazoline, was not beneficial to binding with JAKs.
Figure 5.
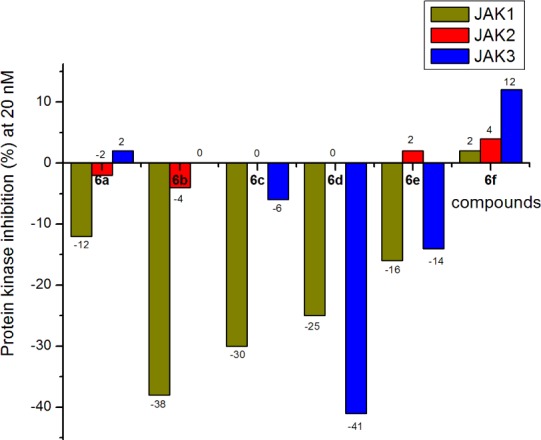
In vitro inhibitory activity against JAK1, JAK2, and JAK3.
Comparing the compounds in Figure 6 with compounds in Figure 3, we could see that 7H-pyrrolo[2,3-d] pyrimidine-based 4-amino-(1H)-pyrazole derivatives (Figure 6) showed moderate JAKs inhibition at 20 nM, but less potent than pyrimidine-based 4-amino-(1H)-pyrazole derivatives (Figure 3). For example, compounds 11a, 11b, and 11c in Figure 6 were less potent than 3a, 3b, and 3d in Figure 3, respectively.
It was reported that mutation in the JH2 pseudokinase domain of the Janus kinase 2 gene (JAK2 V617F) existed in HEL (human erythroleukemia) cell line.17 Therefore, all target compounds were screened against HEL cell line at the concentration of 5 μM to evaluate their in vitro anticancer activities. Results in Figure 7 showed that among these analogues, compounds 3a–f and 11a–d exhibited superior antiproliferative activities against HEL cell line (indicated by the red column) than the other compounds we synthesized. These data were generally consistent with their JAKs inhibitory potency.
Figure 7.

Activity screening against HEL cell line at the concentration of 5 μM. The plates containing compounds and cells were incubated for 48 h in MTT assay.
Considering their potent JAKs inhibitory activities and antiproliferative potency against the HEL cell line, ten compounds (3a–f, 3k, 11b, 11d, and 6d) were chosen for further antiproliferative evaluation against human prostate cancer PC-3, human breast cancer MCF-7, human erythroleukemia HEL, human myelogenous leukemia K562, and human lymphoid leukemia MOLT4 cell lines. Ruxolitinib was used as a positive control. The results in Table 1 showed that most of the ten compounds possessed potent anticancer activity in vitro. Among these compounds, 3a, 3c, 3e, and 3f were cytotoxic to all five tested cell lines, while 11b exhibited remarkably selective cytotoxicity to HEL (IC50: 0.35 μM) and K562 (IC50: 0.37 μM). It is worth emphasizing that, though less potent than Ruxolitinib in JAK inhibition, most of our compounds exhibited more potent cytotoxicity than Ruxolitinib (Table 1), indicating that our compounds might have off-target effects. Therefore, representative compounds 3f and 11b were evaluated against 14 other cancer related kinases. The results in Figure 8 showed that at 20 nM compound 3f was active against a number of kinases including Flt-3, VEGFR-2, PDGFRα, and TYK2, while compound 11b exhibited very good selectivity against JAK2 and JAK3 over the other tested kinases. These results could explain why 3f were cytotoxic to all five cell lines, while 11b was more selective against JAK/STAT pathway promoted cell lines, such as HEL18,19 and K562.20−22 However, our kinase panel screening results still could not explain why 11b were more cytotoxic than Ruxolitinib. Further anticancer mechanism research of 11b is warranted.
Table 1. Inhibitory Activities of Compounds Against Tumor Cell Lines.
| IC50a (μM) |
|||||
|---|---|---|---|---|---|
| compd | PC-3 | MCF-7 | HEL | K562 | MOLT4 |
| 3a | 2.57 ± 0.22 | 1.93 ± 0.02 | 1.53 ± 0.15 | 1.70 ± 0.27 | 1.37 ± 0.23 |
| 3b | 5.38 ± 0.62 | 3.66 ± 1.29 | 5.93 ± 0.01 | >8.3b | >5 |
| 3c | 1.03 ± 0.25 | 1.87 ± 0.01 | 1.18 ± 0.15 | 1.86 ± 0.29 | 3.28 ± 0.45 |
| 3d | 2.30 ± 0.98 | NDc | 1.76 ± 0.24 | 2.08 ± 0.33 | ND |
| 3e | 1.13 ± 0.08 | 1.10 ± 0.01 | 1.24 ± 0.19 | 1.29 ± 0.21 | 1.26 ± 0.15 |
| 3f | 1.08 ± 0.05 | 1.33 ± 0.42 | 1.08 ± 0.06 | 0.77 ± 0.05 | 1.61 ± 0.35 |
| 3k | 10.38 ± 0.97 | ND | 3.96 ± 1.05 | 3.79 ± 0.86 | ND |
| 11b | 4.47 ± 1.29 | >5 | 0.35 ± 0.07 | 0.37 ± 0.11 | >5 |
| 11d | 13.52 ± 1.98 | >5 | ND | 3.72 ± 0.71 | >5 |
| 6d | ND | >5 | 9.71 ± 0.99 | >8.3 | ND |
| Ruxolitinib | >5 | >5 | 2.62 ± 0.19 | 10.3 ± 0.3 | 15.8 ± 1.4 |
IC50 are mean of two or three experiments, and standard deviation is given.
IC50 value of this compound is larger than 8.3 or 5.
ND, not determined.
Figure 8.

Selectivity profile of compounds 3f and 11b on 14 protein kinases at 20 nM.
To investigate the binding mode of these 4-amino-(1H)-pyrazole derivatives in JAK2, the most potent compound 3f was docked into the ATP binding pocket of JAK2 using SYBYL-X 2.0 (Figure 9). The results shown in Figure 9 suggested that the −NH and =N moieties of pyrazole in compound 3f could form hydrogen bonds with GLU930 and LEU932. These hydrogen interactions were the crucial part for the protein kinase inhibition. The −NH moiety of pyrazole as hydrogen donor was necessary for the improvement of binding to JAKs. This maybe the reason why the kinase inhibitory activities of 4-amino-(1H)-pyrazole derivatives were much more potent than their parent 4-amino-pyrazole derivatives. For example, compound 3a reported here is much more potent than its parent 17n (compound 1) reported in our previous research.11 Additional docking with the compound that has a fused phenyl ring (compound 6d) was also studied. Docking results showed compounds 3f and 6d share a similar binding mode. To understand why compound 6d showed weak activity, a molecular dynamics (MD) simulation and a molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) energy decomposition calculation experiment were conducted. Our result showed that the fused phenyl ring and the side chain of ASP939 gave a 1.50 kcal/mol negative energy contribution in binding (Figure 10). Besides, MD simulation indicated that the fused ring position is solvent accessible: it is exposed to water (Figure 11). These two reasons may explain why adding an additional large hydrophobic ring, such as a fused phenyl ring, does not favor binding.
Figure 9.

Docking of compound 3f with JAK2 (PDB code 3FUP).
Figure 10.
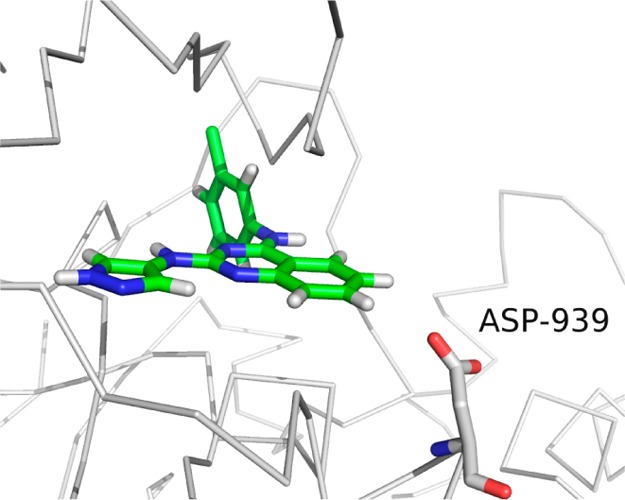
Docking of compound 6d with JAK2 (PDB code 3FUP).
Figure 11.

Fused phenyl ring is exposed to water in MD simulation. Water molecules within 4 Å of compound 6d were selected for visualization (spheres).
In summary, a series of 4-amino-(1H)-pyrazole derivatives as potent JAKs inhibitors were designed, synthesized, and evaluated. In enzyme inhibition screenings, many of the compounds reached IC50 values below 20 nM. In cell-based assays, compound 3f showed potent cytotoxicity against a wide variety of cell lines at micromolar levels. Compound 11b possessed very potent cytotoxicity against HEL and K562 cell lines with IC50 values in the submicromolar range, which were over 10-fold lower than in PC-3, MCF-7, and MOLT4 cell lines. Further kinase panel screening results revealed that compound 3f is a pan-kinase inhibitor, while 11b is a highly selective JAK2 and JAK3 inhibitor, which could be used as lead compound for further structural optimizations to find more potent and selective JAKs inhibitors. Moreover, considering the discrepancy between JAKs inhibition and cytotoxicity when compared with Ruxolitinib, more detailed mechanistic studies are warranted for 11b.
Acknowledgments
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number ACI-1053575. We also gratefully acknowledge the use of Orion that is supported by Georgia State University’s Research Solutions.
Glossary
ABBREVIATIONS
- JAK
Janus kinase
- STAT
signal transducers and activators of transcription
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- TFA
trifluoroacetic acid
- DMF
dimethylformamide
- NMP
N-methyl-2-pyrrolidone
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00247.
Experimental section and characterization data (HRMS, 1H NMR) for new compounds (PDF)
This work was supported by the National High-Tech R&D Program of China (863 Program) (Grant No. 2014AA020523), National Natural Science Foundation of China (Grant Nos. 21302111, 81373282, 21172134), Young Scholars Program of Shandong University (YSPSDU, Grant NO. 2016WLJH33), and Major Project of Science and Technology of Shandong Province (2015ZDJS04001).
The authors declare no competing financial interest.
Supplementary Material
References
- Clark J. D.; Flanagan M. E.; Telliez J. B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–38. 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- Minegishi Y.; Karasuyama H. Defects in Jak-STAT-mediated cytokine signals cause hyper-IgE syndrome: lessons from a primary immunodeficiency. Int. Immunol. 2009, 21, 105–12. 10.1093/intimm/dxn134. [DOI] [PubMed] [Google Scholar]
- Xiong H.; Zhang Z. G.; Tian X. Q.; Sun D. F.; Liang Q. C.; Zhang Y. J.; Lu R.; Chen Y. X.; Fang J. Y. Inhibition of JAK1, 2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces tumor cell invasion in colorectal cancer cells. Neoplasia 2008, 10, 287–97. 10.1593/neo.07971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Curry J. E.; Barber K.; Beer P. A.; Graham B.; Lyons J. F.; Richardson C. J.; Scott M. A.; Smyth T.; Squires M. S.; Thompson N. T.; Green A. R.; Wallis N. G. AT9283, a potent inhibitor of the Aurora kinases and Jak2, has therapeutic potential in myeloproliferative disorders. Br. J. Haematol. 2010, 150, 46–57. 10.1111/j.1365-2141.2010.08175.x. [DOI] [PubMed] [Google Scholar]
- Inghirami G.; Chiarle R.; Simmons W. J.; Piva R.; Schlessinger K.; Levy D. E. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle 2005, 4, 1131–3. 10.4161/cc.4.9.1985. [DOI] [PubMed] [Google Scholar]
- Liu L.; Nam S.; Tian Y.; Yang F.; Wu J.; Wang Y.; Scuto A.; Polychronopoulos P.; Magiatis P.; Skaltsounis L.; Jove R. 6-Bromoindirubin-3′-oxime inhibits JAK/STAT3 signaling and induces apoptosis of human melanoma cells. Cancer Res. 2011, 71, 3972–9. 10.1158/0008-5472.CAN-10-3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan J. C.; Verstovsek S. Overcoming treatment challenges in myelofibrosis and polycythemia vera: the role of ruxolitinib. Cancer Chemother. Pharmacol. 2016, 77, 1125–42. 10.1007/s00280-016-3012-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L.; Liao Z.; Hoang D. T.; Dagvadorj A.; Gupta S.; Blackmon S.; Ellsworth E.; Talati P.; Leiby B.; Zinda M.; Lallas C. D.; Trabulsi E. J.; McCue P.; Gomella L.; Huszar D.; Nevalainen M. T. Pharmacologic inhibition of Jak2-Stat5 signaling By Jak2 inhibitor AZD1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 5658–74. 10.1158/1078-0432.CCR-13-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardanani A.; Gotlib J. R.; Jamieson C.; Cortes J. E.; Talpaz M.; Stone R. M.; Silverman M. H.; Gilliland D. G.; Shorr J.; Tefferi A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J. Clin. Oncol. 2011, 29, 789–96. 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S.; Goh K. C.; Novotny-Diermayr V.; Hu C. Y.; Hentze H.; Tan Y. C.; Madan B.; Amalini C.; Loh Y. K.; Ong L. C.; William A. D.; Lee A.; Poulsen A.; Jayaraman R.; Ong K. H.; Ethirajulu K.; Dymock B. W.; Wood J. W. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–9. 10.1038/leu.2011.148. [DOI] [PubMed] [Google Scholar]
- Liang X. W.; Huang Y. X.; Zang J.; Gao Q. W.; Xu W. F.; Wang B. H.; Zhang Y. J. Design, synthesis and preliminary biological evaluation of 4-aminopyrazole derivatives as novel and potent JAKs inhibitors. Bioorg. Med. Chem. 2016, 24, 2660–72. 10.1016/j.bmc.2016.04.030. [DOI] [PubMed] [Google Scholar]
- Breslin H. J.; Lane B. M.; Ott G. R.; Ghose A. K.; Angeles T. S.; Albom M. S.; Cheng M.; Wan W.; Haltiwanger R. C.; Wells-Knecht K. J.; Dorsey B. D. Design, synthesis, and anaplastic lymphoma kinase (ALK) inhibitory activity for a novel series of 2,4,8,22-tetraazatetracyclo[14.3.1.1(3), (7).1(9),(1)(3)]docosa-1(20),3(22),4,6,9(21),10,12,16,18-nonaene macrocycles. J. Med. Chem. 2012, 55, 449–64. 10.1021/jm201333e. [DOI] [PubMed] [Google Scholar]
- Van Horn K. S.; Burda W. N.; Fleeman R.; Shaw L. N.; Manetsch R. Antibacterial activity of a series of N2,N4-disubstituted quinazoline-2,4-diamines. J. Med. Chem. 2014, 57, 3075–93. 10.1021/jm500039e. [DOI] [PubMed] [Google Scholar]
- Song Y. H.; Xu Q.; Bauer S. M.; Jia Z. Z.. Preparation of pyrimidine, pyrrolopyrimidine and purine-based analogs as inhibitors of protein kinases. PCT Int. Appl.2009, WO 2009131687A2.
- Su Q.; Ioannidis S.; Chuaqui C.; Almeida L.; Alimzhanov M.; Bebernitz G.; Bell K.; Block M.; Howard T.; Huang S.; Huszar D.; Read J. A.; Rivard Costa C.; Shi J.; Su M.; Ye M.; Zinda M. Discovery of 1-methyl-1H-imidazole derivatives as potent Jak2 inhibitors. J. Med. Chem. 2014, 57, 144–58. 10.1021/jm401546n. [DOI] [PubMed] [Google Scholar]
- Peng H.; Liao H. F. Staurosporine induces megakaryocytic differentiation through the upregulation of JAK/Stat3 signaling pathway. Ann. Hematol. 2011, 90, 1017–29. 10.1007/s00277-011-1186-3. [DOI] [PubMed] [Google Scholar]
- Weber A.; Borghouts C.; Brendel C.; Moriggl R.; Delis N.; Brill B.; Vafaizadeh V.; Groner B. Stat5 Exerts Distinct, Vital Functions in the Cytoplasm and Nucleus of Bcr-Abl+ K562 and Jak2(V617F)+ HEL Leukemia Cells. Cancers 2015, 7, 503–37. 10.3390/cancers7010503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quentmeier H. JAK2 V617F tyrosine kinase mutation in cell lines derived from myeloproliferative disorders. Leukemia 2006, 20 (3), 471–476. 10.1038/sj.leu.2404081. [DOI] [PubMed] [Google Scholar]
- Walters D. K. Phosphoproteomic analysis of AML cell lines identifies leukemic oncogenes. Leuk. Res. 2006, 30 (9), 1097–1104. 10.1016/j.leukres.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Dalgıç C. T. Investigating the Role of JAK/STAT Pathway on Dasatinib-Induced Apoptosis for CML Cell Model K562. Clinical Lymphoma, Myeloma & Leukemia 2015, 15 (1), 161–6. 10.1016/j.clml.2015.02.012. [DOI] [PubMed] [Google Scholar]
- de Groot Rolf P. STAT5 Activation by BCR-Abl Contributes to Transformation of K562 Leukemia Cells. Blood 1999, 94 (3), 1108–1112. [PubMed] [Google Scholar]
- Kindler T. In BCR-ABL-positive cells, STAT-5 tyrosine-phosphorylation integrates signals induced by imatinib mesylate and Ara-C. Leukemia 2003, 17, 999–1009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.