

Abstract

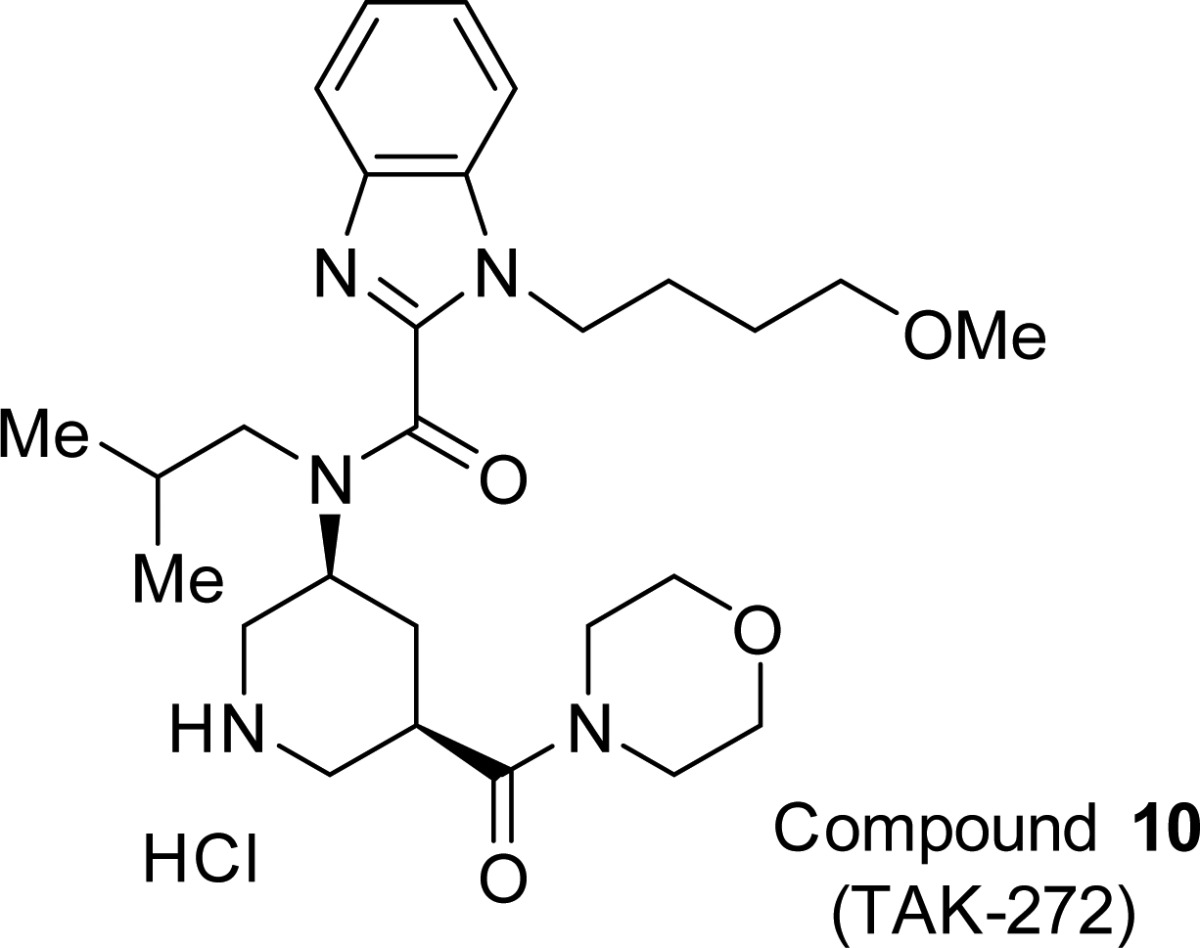
The aspartic proteinase renin is an attractive target for the treatment of hypertension and cardiovascular/renal disease such as chronic kidney disease and heart failure. We introduced an S1′ site binder into the lead compound 1 guided by structure-based drug design (SBDD), and further optimization of physicochemical properties led to the discovery of benzimidazole derivative 10 (1-(4-methoxybutyl)-N-(2-methylpropyl)-N-[(3S,5R)-5-(morpholin-4-yl)carbonylpiperidin-3-yl]-1H-benzimidazole-2-carboxamide hydrochloride, TAK-272) as a highly potent and orally active renin inhibitor. Compound 10 demonstrated good oral bioavailability (BA) and long-lasting efficacy in rats. Compound 10 is currently in clinical trials.
Keywords: Renin inhibitor, TAK-272, benzimidazole, hypertension, SBDD, dTg rat
The renin–angiotensin–aldosterone system (RAAS) is a well-known pathway regulating blood pressure and body fluid homeostasis.1 Several agents that block the RAAS cascade are available and effective for the control of blood pressure.2 A number of clinical studies using angiotensin-converting-enzyme inhibitors and angiotensin-II-receptor blockers revealed that treatment of RAAS cascade blockers is beneficial for organ protection such as diabetic nephropathy and heart failure besides antihypertensive effect.3 The aspartic proteinase renin is the rate-limiting and the first step of the RAAS cascade and considered as an attractive therapeutic strategy to block the whole RAAS cascade.4,5 Despite renin being discovered as a hypertensive substance more than 100 years ago6 and the numerous efforts made to discover potent renin inhibitors in many pharmaceutical companies since early 1980s, no product has been launched over more than two decades.7−9 In 2007, aliskiren was approved as the first orally bioavailable direct renin inhibitor for the treatment of hypertension.10,11
We have already found an attractive lead compound 1 with an IC50 value of 4.6 nM against human plasma renin by our fragment-based (FBDD) and structure-based drug design (SBDD) efforts.12 The X-ray cocrystal structure of compound 1 bound to renin was examined in detail as the starting point for our next round of SBDD efforts (Figure 1). The contiguous lipophilic S3 and S1 sites are occupied by the tert-butylpyrimidine and isobutyl moieties with the flap β-hairpin in a closed conformation.13,14 The piperidine NH group makes a salt bridge with the residues of Asp32 and Asp215, and the S3sp site is occupied by the furfurylamine moiety. The amino group at the pyrimidine 4-position forms a hydrogen bonding interaction with backbone of Gly217. However, the S1′ site was not utilized by our renin inhibitor 1 in contrast to aliskiren.10,11 Additionally, it has been demonstrated by others that occupation of the S1′ site can lead to enhancement of potency.13,14 Thus, we investigated the introduction of additional substituents directed toward the S1′ site to enhance potency.
Figure 1.

X-ray crystal structure (left) and binding mode (right) of 1 with renin. Our optimization strategy is indicated by the arrow.
PK studies revealed that compound 1 has better oral bioavailability in rats than aliskiren (1, 13.8%; aliskiren, 2.4%).10−12 The reasons for this difference were thought to arise from improved physiochemical characteristics such as MW (414 and 552), topological polar surface area (TPSA) (82 and 146), and number of rotatable bonds (nRB) (9 and 20, respectively) which are known as important predictors for BA.15,16 However, compound 1 possesses the furan moiety, which could potentially be oxidized by CYP to form toxic metabolites.17,18 We sought to replace this moiety with metabolically stable substituent through chemical modification.
On the basis of this analysis, our optimization of compound 1 began with the introduction of a variety of substituents at the piperidine 3-position directed toward the S1′ site to increase renin inhibitory activity.13,14,19 Due to the low MW of the compound 1 (414), we thought that the additional substituent could be added without deleterious effects on PK (Figure 1). In particular, sp3-rich substituents that could increase receptor/ligand complementarity and enhance potency and selectivity against off-target effects were employed.20 Subsequently, modification of metabolically liable groups, such as the furfurylamine moiety, was undertaken. Finally, guided by physicochemical parameters, optimization of substituents binding at the S3 and S3sp sites was conducted to discover orally bioavailable renin inhibitors. In this letter, we describe the discovery of a novel, potent, and orally active renin inhibitor 10, which has been selected as a clinical candidate.
The synthesis of compounds 2–10 is detailed in the Supporting Information. Compounds 1–10 were evaluated for the inhibition of recombinant human renin (rh-renin). Inhibitory activity of these compounds against endogenous renin in human plasma (human plasma renin activity, hPRA) was also measured and is shown as IC50 values. The ligand efficiency (LE = (rh-renin pIC50)/(heavy atom count)) and ligand lipophilicity efficiency (LLE = (rh-renin pIC50) – clogD (pH 7.4)) are also shown.21−23 LLE is an important parameter for estimating the potential of the binding interaction without the contribution of lipophilicity.
In order to enhance the potency, a methoxycarbonyl group was introduced at the piperidine 3-position, which was expected to interact with the S1′ site surrounded by both lipophilic and hydrophilic amino acids. Additionally, this functionality was thought to be easily modifiable for latter optimization. The structure–activity relationships of stereoisomers at the piperidine 3- and 5-positions was investigated in detail (Table 1). The cis-racemic mixture 2 showed more potent renin inhibitory activity than that of trans-racemic mixture 3. Among the cis-isomers, the 3R,5S-enantiomer 5 was about 30-fold more potent toward renin than its enantiomer 4, measured either in rh-renin assay (IC50 = 0.039 nM vs 1.2 nM) or in hPRA assay (IC50 = 0.35 nM vs >10 nM). These results indicated that the stereochemistry at the piperidine 3- and 5-positions is important in determining renin inhibitory activity. Compared to 1, the renin inhibitory activity of 5 was increased dramatically by introducing the methoxycarbonyl group on the piperidine ring with moderate increase in MW (increased by 58). Although LE values generally decrease with increase in molecular size, compound 5 retained the same LE value as that of compound 1, indicating that the methoxycarbonyl group efficiently interacts at the S1′ site. This conclusion was also supported by the increase in LLE value from compound 1 (6.3) to 5 (7.1) despite the increase in clogD value (3.0 and 3.3, respectively).
Table 1. In Vitro Activities of 1–5, along with Their clogP, LE, and LLEa.

IC50 values shown as the mean values of duplicate measurements.
IC50 values and 95% confidence limits are calculated from the concentration–response curves generated by GraphPad Prism.
Modification of the metabolically liable furan moiety of compound 5 and optimization of the methoxycarbonyl moiety were then undertaken (Table 2). In the course of these modifications, the methoxypropylamino group was found as an alternative S3sp site binder with a 10-fold decrease in renin inhibitory activity relative to the furfurylamine moiety (6, hPRA IC50 = 5.2 nM). The related methoxypropyloxy side chain is a well-known binding motif of the renin S3sp site, present in aliskiren and in other renin inhibitors.12,15,16 Next, optimization of the methoxycarbonyl moiety of compound 6 was conducted. While carbamoyl derivative 7 showed comparable renin inhibitory activity to 6, morpholinocarbonyl derivative 8 exhibited potent inhibitory activity (hPRA IC50 = 0.79 nM) as well as 5, suggesting that occupation of the S1′ site would be effective for obtaining strong potency in hPRA assay, since introduction of the morpholinamide might induce the decrease of the human plasma protein binding.
Table 2. In Vitro Activities of 5–8, along with Their clogP, LE, and LLEa.

IC50 values shown as the mean values of duplicate measurements.
IC50 values and 95% confidence limits are calculated from the concentration–response curves generated by GraphPad Prism.
The X-ray cocrystal structure of compound 8 bound to renin was obtained and revealed that the piperidine and aminopyrimidine moieties are bound to renin protein in the same manner as observed for compound 1(12) and that the methoxypropylamino moiety was indeed located in the S3sp site (Figure 2). The morpholinocarbonyl group occupies the S1′ site, as we predicted.13,14,19 Furthermore, its carbonyl oxygen atom forms a hydrogen bond with the backbone amino group of Ser76 on the flap being in a closed conformation.13,14 These interactions would be expected to contribute to the improvement in renin inhibitory activity.
Figure 2.
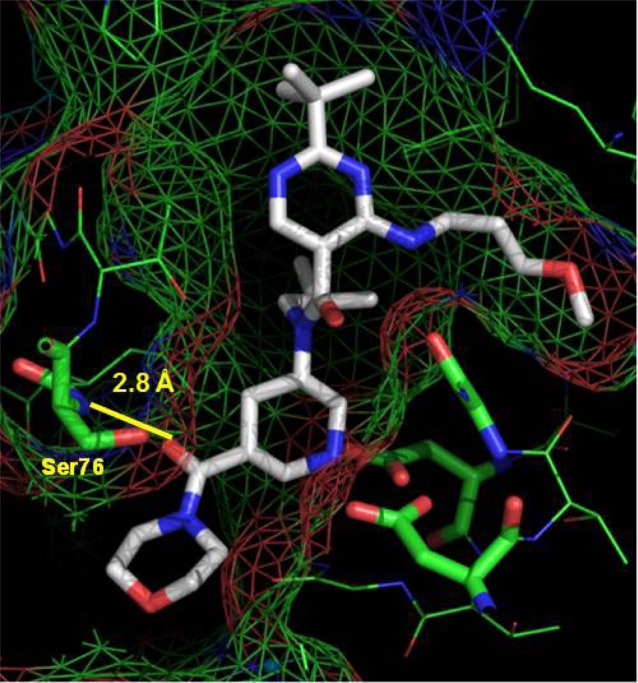
X-ray crystal structure of 8 with renin.
A rat cassette dosing study indicated that compound 8 possessed poor oral exposure (F, <1%; AUCpo, not detected; CL 5539 mL/h/kg; Table 3). The physicochemical properties of 8 were analyzed to improve its poor oral absorption. Compared to orally bioavailable compound 1, the MW and TPSA values of 8 are notably higher. We assumed that improvement of oral absorption could be achieved by control of these physicochemical properties. However, to occupy each binding site, a somewhat higher MW is required for this scaffold. Thus, reduction of the TPSA value was alternatively performed by converting the methoxypropylamino moiety into the corresponding carbon analogue. The n-hexyl derivative 9 showed improved F, AUCpo, and CL values and markedly reduced TPSA value with a 20-fold decrease in renin inhibitory activity (Table 3). This decrease in renin inhibitory activity was interpreted as arising due to the loss of interaction with Gly217 observed in compound 8.
Table 3. In Vitro Activities and PK Profiles of 8–9a.

IC50 values shown as the mean values of duplicate measurements.
IC50 values and 95% confidence limits are calculated from the concentration–response curves generated by GraphPad Prism.
Rat cassette dosing at 0.1 mg/kg, i.v., and 1 mg/kg, p.o. (n = 3).
F means bioavailability.
Not detected.
On the basis of the observations discussed above, further investigation was focused on the S3 and S3sp site binding moieties in order to recover decreased potency and improve the PK profiles of compound 9. Extensive modifications involving the introduction of the fused and nonfused heteroaromatic ring as replacements for the t-butylpyrimidine scaffold led to the discovery of benzimidazole derivative 10 and will be reported elsewhere.
Compound 10 showed potent inhibitory activity against human renin (IC50 = 2.1 nM) in hPRA assay and excellent selectivity against other aspartic proteases, such as pepsin (IC50 > 10 μM) and cathepsin D (IC50 > 10 μM). As shown in Table 4, compound 10 exhibited a dramatically improved PK profile in rats (F = 25.2%). The antihypertensive efficacy of 10 was examined using human-angiotensinogen and human-renin double transgenic rats (dTg rat) (Figure 3). Compound 10 (3 and 10 mg/kg, p.o.) exhibited potent and long-lasting antihypertensive efficacy in a dose-dependent manner. At the time point of 5 h after oral administration, the antihypertensive efficacy of compound 10 at a dosage of 3 mg/kg was comparable to that of aliskiren at a dosage of 75 mg/kg; the antihypertensive efficacy of compound 10 at a dosage of 10 mg/kg also exceeded that of aliskiren at a dosage of 75 mg/kg at both 5 and 24 h after administration. These studies raised expectations of the potential clinical efficacy of compound 10 and encouraged us to investigate this compound further as a candidate for clinical development as a new agent for treating cardiovascular disease.
Table 4. PK Profile of Compound 10 in Rata.
| iv | po | ||
|---|---|---|---|
| t1/2 (h) | 1.3 | t1/2 (h) | 3.3 |
| MRT (h) | 1.1 | Cmax (μg/mL) | 0.017 |
| CL (mL/h/kg) | 2335.7 | Tmax (μg/mL) | 0.5 |
| Vd(ss)(mL/kg) | 2530.3 | AUC0–48 h (μg h/mL) | 0.107 |
| Fb (%) | 25.2 | ||
Compound 10 was administered to rats at 0.2 mg/kg, i.v., or 1 mg/kg, p.o. (n = 3).
F means bioavailability.
Figure 3.
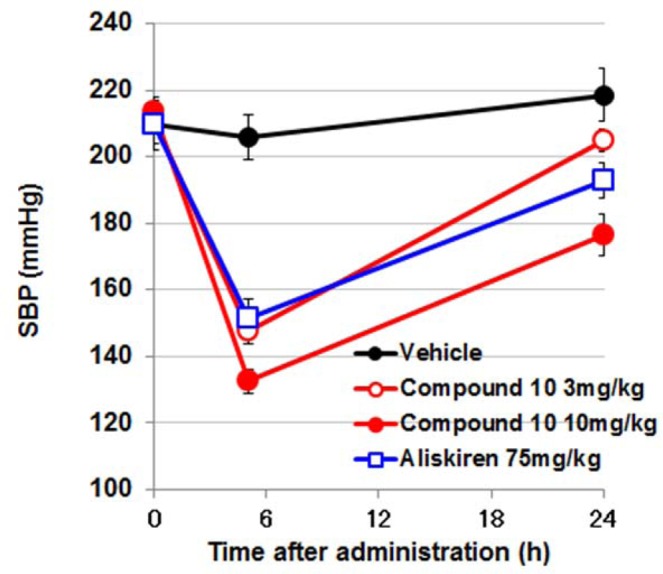
Antihypertensive effect of compound 10 (3 and 10 mg/kg, po) and aliskiren (75 mg/kg, po) in dTg rat model. Data represent the systolic blood pressure (SBP) measured at 5 and 24 h after administration.
In conclusion, we discovered compound 10, a structurally novel renin inhibitor showing potent renin inhibitory activity, in vivo efficacy in dTg rat model, and a favorable PK profile in rats. In the course of our modifications, the S1′ site was utilized for the enhancement of potency by the introduction of sp3-rich substituents into the piperidine ring. Optimization of each binding element aimed at improving physicochemical properties succeeded in improving the PK profile. Compound 10 is currently undergoing human clinical trials.
Acknowledgments
The authors thank Keiji Kubo for supervising the research; Dr. Tsuyoshi Maekawa for helpful discussions; Masato Kitayama for providing the salt formation procedure of TAK-272; Takuya Ebihara, Fumihiro Jinno, and Yoshihiko Tagawa for performing the PK study of TAK-272; Katsuhiko Miwa for analyses of enantiomeric excess of the key intermediate; the DMPK group at Takeda Pharmaceutical Company for the rat cassette dosing experiment in Table 3; and Kengo Okada, Hideyuki Oki, Weston Lane, and Bi-Ching Sang for molecular biology, protein expression, crystallization, and X-ray data collection support highlighted in Figure 2. We thank the staff of the Berkeley Center for Structural Biology (BCSB), Lawrence Berkeley National Laboratory, which operates Advanced Light Source beamline 5.0.3, for their support. BCSB is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Glossary
ABBREVIATIONS
- RAAS
renin–angiotensin–aldosterone system
- BA (F)
bioavailability
- nRB
number of rotatable bonds
- TPSA
topological polar surface area
- rh-renin
recombinant human renin
- hPRA
human plasma renin activity
- LE
ligand efficiency
- LLE
ligand-lipophilicity efficiency
- SBP
systolic blood pressure
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00251.
Experimental procedures and characterization, biological assay protocols (PDF)
Accession Codes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank with codes 5KOQ for 1 and 5KOS for 8.
Author Present Address
§ Sapphire Energy, Inc., 9363 Towne Centre Drive, San Diego, California 92121, United States.
Author Contributions
Y.I., H.T., Y.F., and T.K. contributed design and synthesis of compounds; R.K., Y.K., K.K., and M.K. contributed in vitro and in vivo study; G.S. and C.B. contributed X-ray structures.
The authors declare no competing financial interest.
Supplementary Material
References
- MacGregor G. A.; Markandu N. D.; Roulston J. E.; Jones J. C.; Morton J. J. Maintenance of blood pressure by the renin–angiotensin system in normal man. Nature 1981, 291, 329–331. 10.1038/291329a0. [DOI] [PubMed] [Google Scholar]
- Zaman M. A.; Oparil S.; Calhoun D. A. Drugs Targeting the Renin-Angiotensin-Aldosterone System. Nat. Rev. Drug Discovery 2002, 1, 621–636. 10.1038/nrd873. [DOI] [PubMed] [Google Scholar]
- Roscioni S. S.; Lambers Heerspink H. J.; de Zeeuw D. The effect of RAAS blockade on the progression of diabetic nephropathy. Nat. Rev. Nephrol. 2014, 10, 77–87. 10.1038/nrneph.2013.251. [DOI] [PubMed] [Google Scholar]
- Haberman A. B. Renin Inhibitors: A Novel Approach to Hypertension. SPECTRUM Therapy Markets and Emerging Technologies 2006, 6, 1–16. [Google Scholar]
- Mende C. W. Application of direct renin inhibition to chronic kidney diseases. Cardiovasc. Drugs Ther. 2010, 24, 139–149. 10.1007/s10557-010-6232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigerstedt R.; Bergman P. G. Niere und Kreislauf. Skand. Arch. Physiol. 1898, 8, 223–271. 10.1111/j.1748-1716.1898.tb00272.x. [DOI] [Google Scholar]
- Webb R. L.; Schiering N.; Sedrani R.; Maibaum J. Direct renin inhibitors as a new therapy for hypertension. J. Med. Chem. 2010, 53, 7490–7520. 10.1021/jm901885s. [DOI] [PubMed] [Google Scholar]
- Yokokawa F.; Maibaum J. Recent advances in the discovery of non-peptidic direct renin inhibitors as antihypertensives. Expert Opin. Ther. Pat. 2008, 18, 581–602. 10.1517/13543776.18.6.581. [DOI] [Google Scholar]
- Yokokawa F. Recent progress on the discovery of non-peptidic direct renin inhibitors for the clinical management of hypertension. Expert Opin. Drug Discovery 2013, 8, 673–690. 10.1517/17460441.2013.791279. [DOI] [PubMed] [Google Scholar]
- Maibaum J.; Stutz S.; Göschke R.; Rigollier P.; Yamaguchi Y.; Cumin F.; Rahuel J.; Baum H.-P.; Cohen N.-C.; Schnell C. R.; Fuhrer W.; Gruetter M. G.; Schilling W.; Wood J. M. Structural modification of the P2′ position of 2,7-dialkyl-substituted 5(S)- amino-4(S)-hydroxy-8-phenyl-octanecarboxamides: the discovery of aliskiren, a potent nonpeptide human renin inhibitor active after once daily dosing in marmosets. J. Med. Chem. 2007, 50, 4832–4844. 10.1021/jm070316i. [DOI] [PubMed] [Google Scholar]
- Vaidyanathan S.; Jarugula V.; Dieterich H. A.; Howard D.; Dole W. P. Clinical pharmacokinetics and pharmacodynamics of aliskiren. Clin. Pharmacokinet. 2008, 47, 515–531. 10.2165/00003088-200847080-00002. [DOI] [PubMed] [Google Scholar]
- Imaeda Y.; Tawada M.; Suzuki S.; Tomimoto M.; Kondo M.; Tarui N.; Sanada T.; Kanagawa R.; Snell G.; Behnke C. A.; Kubo K.; Kuroita T.. Structure-based design of a new series of N-(piperidin-3-yl)pyrimidine-5-carboxamides as renin inhibitors. Bioorg. Med. Chem., published online September 13, 2016; DOI: 10.1016/j.bmc.2016.09.030. [DOI] [PubMed]
- Ostermann N.; Ruedisser S.; Ehrhardt C.; Breitenstein W.; Marzinzik A.; Jacoby E.; Vangrevelinghe E.; Ottl J.; Klumpp M.; Hartwieg J. C.; Cumin F.; Hassiepen U.; Trappe J.; Sedrani R.; Geisse S.; Gerhartz B.; Richert P.; Francotte E.; Wagner T.; Krömer M.; Kosaka T.; Webb R. L.; Rigel D. F.; Maibaum J.; Baeschlin D. K. A novel class of oral direct renin inhibitors: highly potent 3,5-disubstituted piperidines bearing a tricyclic p3-p1 pharmacophore. J. Med. Chem. 2013, 56, 2196–2206. 10.1021/jm301706j. [DOI] [PubMed] [Google Scholar]
- Ehara T.; Irie O.; Kosaka T.; Kanazawa T.; Breitenstein W.; Grosche P.; Ostermann N.; Suzuki M.; Kawakami S.; Konishi K.; Hitomi Y.; Toyao A.; Gunji H.; Cumin F.; Schiering N.; Wagner T.; Rigel D. F.; Webb R. L.; Maibaum J.; Yokokawa F. Structure-based design of substituted piperidines as a new class of highly efficacious oral direct Renin inhibitors. ACS Med. Chem. Lett. 2014, 5, 787–792. 10.1021/ml500137b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Dalvie D. K.; Kalgutkar A. S.; Khojasteh-bakht S. C.; Obach R. S.; O’Donnell J. P. Biotransformation reactions of five-membered aromatic heterocyclic rings. Chem. Res. Toxicol. 2002, 15, 269–299. 10.1021/tx015574b. [DOI] [PubMed] [Google Scholar]
- Baer B. R.; Rettie A. E.; Henne K. R. Bioactivation of 4-ipomeanol by CYP4B1: adduct characterization and evidence for an enedial intermediate. Chem. Res. Toxicol. 2005, 18, 855–864. 10.1021/tx0496993. [DOI] [PubMed] [Google Scholar]
- Mori Y.; Ogawa Y.; Mochizuki A.; Nakamura Y.; Sugita C.; Miyazaki S.; Tamaki K.; Matsui Y.; Takahashi M.; Nagayama T.; Nagai Y.; Inoue S.; Nishi T. Design and discovery of new (3S,5R)-5-[4-(2-chlorophenyl)-2,2-dimethyl-5-oxopiperazin-1-yl]piperidine-3-carboxamides as potent renin inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 7677–7682. 10.1016/j.bmcl.2012.09.103. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humbelt C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Bembenek S. D.; Tounge B. A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. 10.1016/j.bmcl.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Ryckmans T.; Edwards M. P.; Horne V. A.; Correia A. M.; Owen D. R.; Thompson L. R.; Tran I.; Tutt M. F.; Young T. Rapid assessment of a novel series of selective CB2 agonists using parallel synthesis protocols: A Lipophilic Efficiency (LipE) analysis. Bioorg. Med. Chem. Lett. 2009, 19, 4406–4409. 10.1016/j.bmcl.2009.05.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.