

Abstract

Lysophosphatidic acid (LPA) evokes various physiological responses through a series of G protein-coupled receptors known as LPA1–6. A high throughput screen against LPA1 gave compound 7a as a hit. The subsequent optimization of 7a led to ONO-7300243 (17a) as a novel, potent LPA1 antagonist, which showed good efficacy in vivo. The oral dosing of 17a at 30 mg/kg led to reduced intraurethral pressure in rats. Notably, this compound was equal in potency to the α1 adrenoceptor antagonist tamsulosin, which is used in clinical practice to treat dysuria with benign prostatic hyperplasia (BPH). In contrast to tamsulosin, compound 17a had no impact on the mean blood pressure at this dose. These results suggest that LPA1 antagonists could be used to treat BPH without affecting the blood pressure. Herein, we report the hit-to-lead optimization of a unique series of LPA1 antagonists and their in vivo efficacy.
Keywords: LPA1 antagonist, GPCR, benign prostatic hyperplasia, SAR, hit-to-lead optimization
Lysophosphatidic acids (LPAs) are a bioactive class of phospholipids, which are produced by autotaxin from lysophosphatidylcholine in blood.1−3 LPAs exerts a wide range of cellular responses, including intracellular Ca2+ mobilization, cell proliferation, cell survival, and cell motility. Based on their interesting properties, LPAs have been implicated in a wide range of complex physiological responses, including the contraction of smooth muscle, demyelization, wound healing, coagulation, and immunological competence.4 Furthermore, these physiological functions can be related to a number of pathophysiological responses, such as cancer,5 neuropathic pain,6 and fibrosis.7 The biological effects of LPAs are mediated through G protein-coupled receptors (GPCRs). Six LPA receptors (LPA1–6) have been identified and characterized to date. LPA1,2,3 (previously known EDG-2, 4, 7) belong to the EDG family of proteins because of their high sequence homology.8,9
Benign prostatic hyperplasia (BPH) is a chronic disease that affects around 30% of males over the age of 60 and is accompanied by the enlargement of prostate and difficulty urinating. Although the exact causes of BPH are not fully understood, the prostatic tissue mass and the smooth muscle tone are thought to be two of the major components of BPH. One option for the treatment of BPH is to reduce the prostatic smooth muscle tone using α1 adrenoceptor antagonists such as tamsulosin or doxazosin, which are currently used in clinical practice to contract the urethra.10,11 We previously reported that LPAs can induce the contraction of the urethra via LPA1. Notably, LPAs have been reported to exhibit similar levels of contractile potency to the α1 adrenoceptor agonist phenylephrine.12,13 It is therefore envisaged that LPA1 antagonist will show good potential for the treatment of BPH.
Several LPA1 antagonists have been reported to date, and these compounds can be divided into two different categories: (i) lipid-like molecules containing a phosphoric acid moiety and a long alkenyl chain; and (ii) nonlipid-like small molecules (Figure 1). Compounds belonging to the first group generally exhibit poor bioavailability because of their high hydrophobicity. Furthermore, the behavior of these compounds can readily switch from agonist to antagonist depending upon the nature of the polar headgroup, making the design of antagonists particularly challenging. Most of the nonlipid small molecules developed to date as LPA1 antagonists are phenylethoxy carbamoyl derivatives, such as Ki16425, which was the first reported compound to exhibit LPA1,3 dual antagonist activities.14 Several analogues of Ki16425 have been developed bearing a similar phenylethoxy carbamoyl moiety to the parent compound but with a wide variety of different five-membered rings to improve their subtype selectivity and/or in vivo efficacy.14−18 It is noteworthy that some of these compounds are currently being evaluated in clinical trials for the treatment of idiopathic pulmonary fibrosis (IPF). Sanofi Aventis reported a structurally unique indan-2-carboxylic acid-containing antagonist 1. This compound 1 is most likely an analogue of SAR100842, which is currently in Phase IIa trials for systemic sclerosis.18
Figure 1.

Structures of known LPA1 antagonists.
At the beginning of our LPA1 antagonist program, the only small molecule to have been reported in the literature as an LPA receptor antagonist was Ki16425 with no SAR data.14 To obtain a good starting point, we carried out a high-throughput screening (HTS) campaign using a Chinese hamster ovarian (CHO) cell line stably expressing the human LPA1 receptor. In this way, we identified compound 7a as a weak LPA1 antagonist (Table 1). Herein, we report the identification of a novel series of LPA1 antagonists and the results of our structure and activity relationship (SAR) studies for the hit-to-lead optimization of this series for the treatment of BPH.
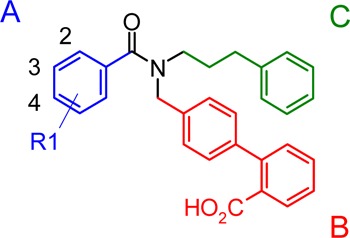
Table 1. Structure–Activity Relationship for Different Substituents on Section A.
| compd | R1 | LPA1 IC50 (μM)a |
|---|---|---|
| 7a | 4-MeO | 4.4 (6.9–3.1) |
| 7b | H | 12.1 (15.0–9.7) |
| 7c | 2-MeO | 3.3 (4.0–2.8) |
| 7d | 3-MeO | 1.3 (1.9–0.9) |
| 7e | 3-F | 5.5 (6.5–4.6) |
| 7f | 3-Me | 1.7 (2.3–1.3) |
| 7g | 3,5-di-MeO | 0.26 (0.32–0.21) |
| 7h | 3,4-di-MeO | 3.7 (4.8–2.8) |
| 7i | 3,4,5-tri-MeO | 0.39 (0.65–0.24) |
| 7j | 3,5-di-Me | 0.26 (0.31–0.21) |
IC50 values were determined by nonlinear regression analysis of the dose–response curves (5 points) generated using GraphPad Prism ver.5.04 with 95% confidence intervals in parentheses.
Our initial exploration of the HTS hit compound 7a involved the modification of section A (blue in Table 1) to identify the optimum substituent at this position (Table 1). A variety of different substituted benzene rings were evaluated at this position, and the results are summarized in Table 1. The removal of the methoxy group from the benzoyl group resulted in a 2-fold decrease in the potency. Among the monosubstituted compounds (7a, 7c–f), the 3-methoxy analogue 7d is the most potent with an IC50 of 1.3 μM. Among the di- and trisubstituted compounds tested in the current study, the 3,5-dimethoxy analogue 7g and 3,5-dimethyl analogue 7j were found to be the most potent. Interestingly, the corresponding 3,4,5-trimethoxy analogue 7i was equipotent with compound 7g, whereas the mono- and disubstituted analogues 7c and 7h showed much lower antagonist activities. These results therefore highlight the importance of having electron-donating substituents at the 3- and 5-positions of the benzene ring for good potency.
We subsequently explored section B of hit compound 7a by varying the substituents attached to the pendent benzoyl moiety. When the benzoic acid substituent from the original hit was replaced with phenylacetic acid (8) or dihydrocinnamic acid (9), the potency remained the same. In contrast, the introduction of an ether linker to give a biphenyl group (10) led to an increase in the antagonist activity (IC50 = 0.089 μM). Furthermore, the ortho- and meta-substituted carboxylic acid analogues 10 and 13 were more potent than the corresponding para-substituted derivatives (IC50 = 0.14 μM for 13, 44% inhibition at 1 μM for 14). These results therefore suggested that the carboxylic acid group needed to be placed in a specific position to interact as effectively as possible with specific basic residues on the target protein. An X-ray structure of LPA1 bound to an antagonist was recently published in the literature.19
Docking studies were conducted using the X-ray crystal structure LPA1 (PDB code 4z34)19 to explore the binding mode of these prototype compounds (10, 13, and 14) and the importance of the positioning of the carboxylic group (Figure 2A–C). The results revealed that the phenyl ring of section A most likely formed a π-stacking interaction with the phenyl ring of section C. This phenyl ring of section C has a van der Waals contact with Trp271, Gly274, and Leu275 (see Supporting Information S36, Figure S1A). A similar stacking interaction to this was also observed in the ligand in the LPA1 X-ray crystal structure19 and represents a common conformational feature of our LPA1 antagonists. The results of the docking experiments also revealed that compounds 10 and 13 could adopt conformations in which their carboxylic acid moieties interacted with Lys39, His40, and Arg124 in LPA1. These two compounds would most likely show similar binding modes, which would explain why they exhibited similar levels of antagonist activity. In contrast, the docking experiments revealed that the carboxylic acid moiety of compound 14 could only interact with His40 in LPA1 (Figure 2C), explaining why this compound was less potent than compounds 10 and 13.
Figure 2.
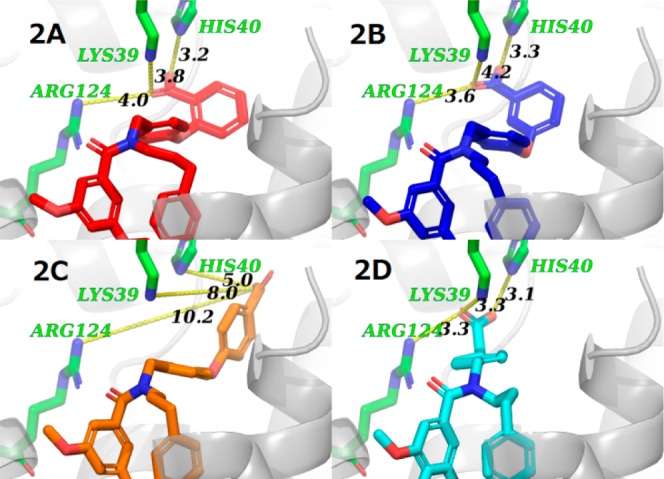
Docking results for some of our compounds using LPA1 crystal structure (PDB code 4z34).19 Docking poses of compounds (2A) 10 (red), (2B) 13 (blue) (2C) 14 (orange), and (2D) 17a (ONO-7300243). The hydrogen atoms have been omitted for clarity. The distances are highlighted by dashed lines (distances in Å).
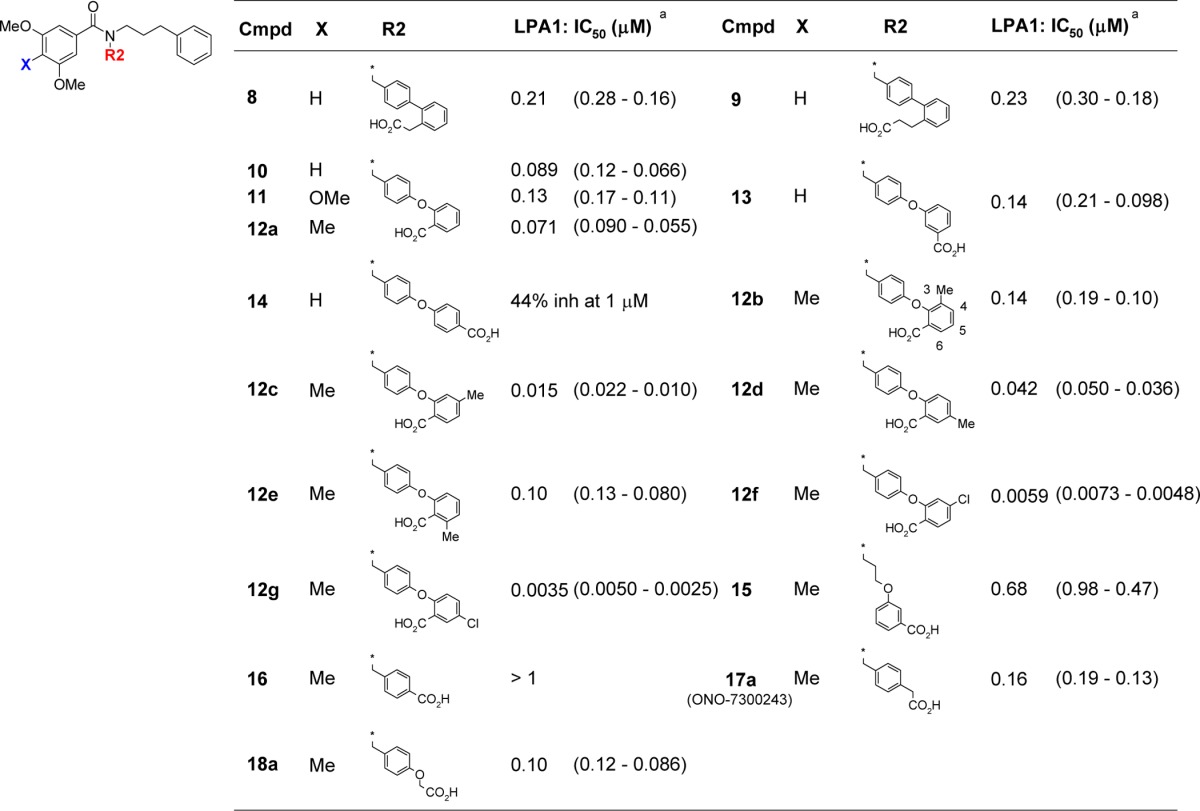
Further investigation of section B was conducted by fixing section A as a 3,5-dimethoxy 4-methyl substituent and varying the nature of section B moiety. The 3,5-dimethoxy-4-methyl substituent was selected as the optimum section A group based on the potency of compound derivative 12a being greater than that of the 3,4,5-trimethoxybenzamide derivative 11. Compounds 12c and 12d bearing a methyl substituent at the 4- or 5-position of their benzoic acid moiety in section B, respectively, showed improved antagonist activity compared with compound 12a. This result indicated that there was enough space around the benzoic acid group to accommodate additional substituents at the 4- and 5-positions of the benzene ring. In contrast, compounds 12b and 12e bearing a methyl group at the 3- or 6-position of the benzoic acid moiety did not improve the potency.
The introduction of an electron-withdrawing chloride at the 4- or 5-position of the benzene ring of the benzoic acid moiety afforded compounds 12f and 12g, respectively, with considerable increases in the activity compared with the methyl analogues. Based on these modifications, compound 12g bearing a 3,5-dimethoxy-4-methylphenyl substituent on section A and a 5-chloro substituent on section B was determined to be the most potent antagonist of all of the compounds synthesized so far. However, the effect of the chloride atom on the benzoic acid moiety in section B currently remains unclear because compounds 12f and 12g exhibited almost identical activity, despite their different substitution pattern. It is noteworthy that both of these compounds are highly lipophilic (clogP = 8.24) and that increasing lipophilicity can lead to an increase in the activity.20 The addition of a chloride substituent to give compound 12g led to an increase in the lipophilicity compared with compound 12d, as well as an increase in activity. In general, highly lipophilic compounds tend to behave in a promiscuous manner, hitting multiple targets, which can lead to unwanted side effects and poor pharmacokinetics.21 With this in mind, we decided to investigate an alternative strategy for increasing the antagonist activity without increasing the lipophilicity to discover a lead compound.
To address this problem, we investigated the possibility of removing one of the phenyl rings from the bis aryl portion of the molecule (section B) to decrease the molecular weight (Table 2, 15–18a). Although a benzylic phenyl ring was not essential for inducing antagonist activity in 15, a linker was required to maintain some distance between the carboxylic acid and amide functional groups in 16. The activity lost in 16 was recovered by the addition of a spacer, as shown in compounds 17a and 18a. The results of a docking experiment with compound 17a using the X-ray crystal structure of LPA1 suggested that basic residues in the active site of the protein could form interactions with acidic functional groups, as shown in compounds 10 and 13 (Lys39, His40, and Arg124, Figure 2D). This would explain why compounds 17a and 18a exhibited similar levels of antagonist activity against LPA1. Notably, compounds 17a and 18a allowed for a considerable reduction in the molecular weight, as well as the lipophilicity (clogP = 5.29 for 17a, 5.23 for 18a).
Table 2. Structure–Activity Relationship for Section B.
IC50 values were determined by nonlinear regression analysis of the dose–response curves (5 points) generated using GraphPad Prism ver.5.04 with 95% confidence intervals in parentheses.
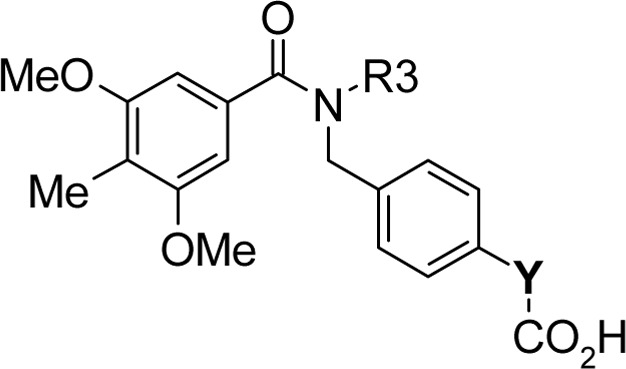
Regarding section C, the replacement of the phenylpropyl chain in 18a with a phenylethyl or phenylbutyl group gave compounds 18b and 18c, respectively, which both showed lower antagonist activities than 18a, indicating that the chain length is critical to the activity (Table 3). Compounds 18b and 18c could not form a π-stacking interaction between the phenyl rings of section A and C so that their activities were lost. The introduction of an sp2 carbon to this part of the molecule also led to a decrease in the potency, as demonstrated by 17b. Notably, the introduction of a fluoro group at the ortho (17c) or meta (17d) position of the phenyl ring was tolerated, whereas the para-fluoro analogue 17e ablated activity because of steric repulsion with Trp271 (see Supporting Information S36, Figure S1B). This phenyl propyl moiety is critical to the antagonist activity.
Table 3. Structure–Activity Relationship for Section Ca.
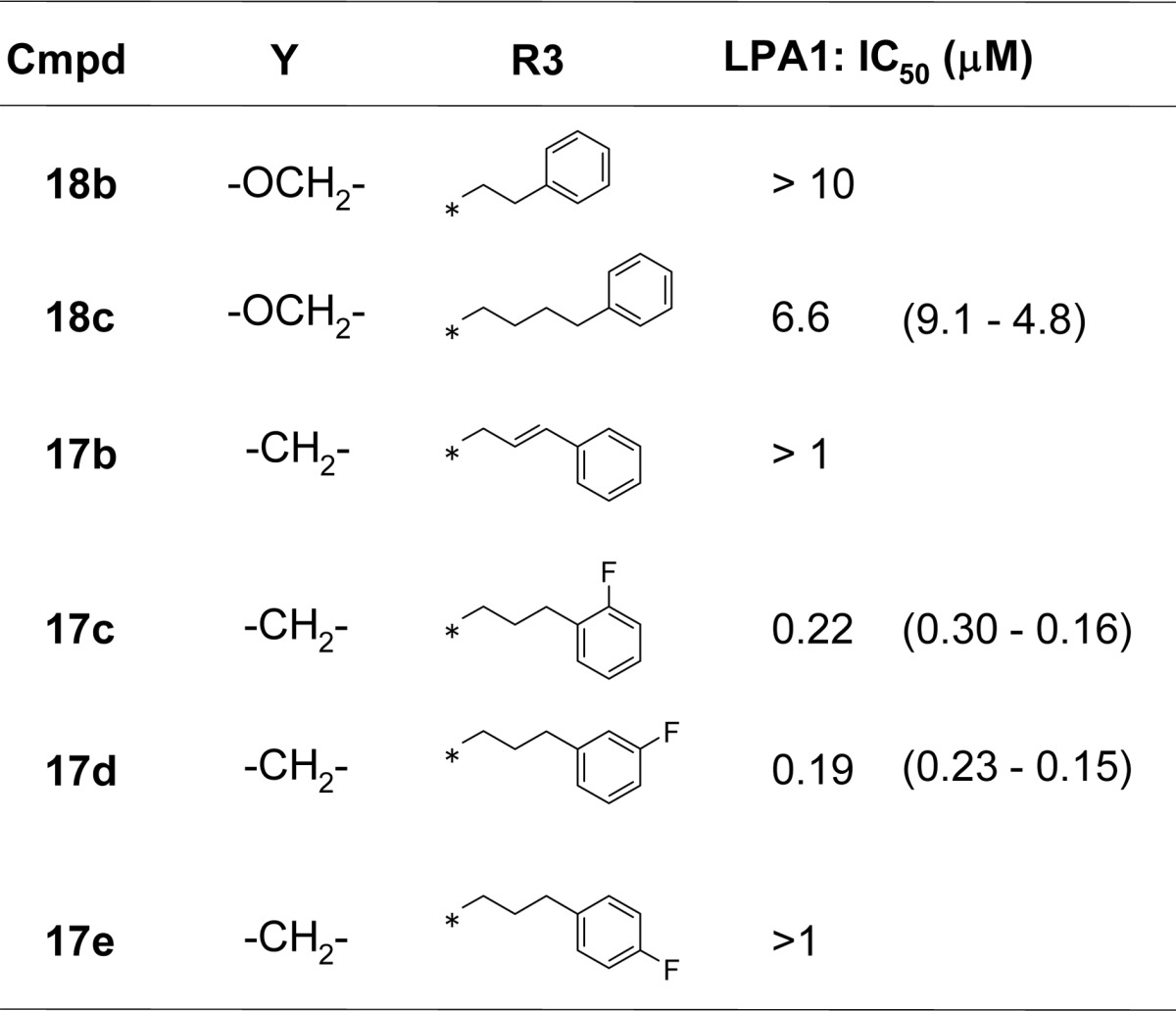
IC50 values were determined by nonlinear regression analysis of the dose–response curves (5 points) generated using GraphPad Prism ver.5.04 with 95% confidence intervals in parentheses.
Three representative compounds (12g, 17a, and 18a) were studied in an LPA-induced rat intraurethral pressure (IUP) model (Table 4). This IUP model was conducted as previously described by the intraduodenal (i.d.) administration of the compounds.22,23 Compound 12g, which showed the strongest in vitro LPAl antagonist activity (IC50 = 0.0035 μM), showed 53% inhibition of LPA-induced IUP increase at 10 mg/kg i.d. administration. Although ONO-7300243 (17a) showed only modest in vitro activity (IC50 = 0.16 μM), it showed much stronger effects in vivo (88% inhibition at 10 mg/kg i.d., 62% inhibition at 3 mg/kg i.d.) compared with compound 12g. To develop a deeper understanding of this difference, we compared the in vitro physicochemical properties of these two compounds. The results revealed that ONO-7300243 (17a) showed good membrane permeability and good metabolic stability against rat liver microsomes (MS). Furthermore, the molecular weight of ONO-7300243 (17a) was much lower than that of compound 12g (MW = 461 for 17a, 574 for 12g), leading to a lower lipophilicity (clog P = 5.29 for 17a, 8.24 for 12g). These data therefore indicate that the good physicochemical properties of compound 17a led to its good in vivo efficacy. It is noteworthy that ONO-7300243 (17a) and compound 18a exhibited good selectivity toward LPAl over LPA2, most likely because low molecular weight and low lipophilicity lead to reduced compound promiscuity and increased selectivity.20 Compounds 17a and 18a exhibited almost identical levels of antagonist activity in vitro. However, the in vivo potency of compound 18a was lower than that of compound 17a. These two compounds have showed similar physicochemical properties except for their Caco2 permeability. The main difference between compounds 17a and 18a is the acidity of their carboxylic acid group (i.e., the pKa value of phenyl acetic acid is 4.3, whereas that of phenoxyacetic acid is 3.1). This increased acidity could contribute to decreased membrane permeability and could explain the reduced in vivo activity (44% inhibition at 10 mg/kg i.d.).
Table 4. Representative Properties of ONO-7300243 (17a), 18a, and 12g.
| compd | 17a ONO-7300243 | 18a | 12g |
|---|---|---|---|
| In Vitro Efficacy (μM)a | |||
| LPA1 | 0.16 (0.19–0.13) | 0.10 (0.12–0.86) | 0.0035 (0.0050–0.0025) |
| LPA2 | 8.6 (10.3–7.2) | 23 (96–5.4) | 0.79 (1.9–0.33) |
| LPA3 | >10 | >10 | >10 |
| In Vivo Efficacy | |||
| Rat IUP (%)b | –88 | –44 | –53 |
| Physicochemical Properties | |||
| MW | 461 | 477 | 574 |
| clogP | 5.29 | 5.23 | 8.24 |
| solubility (μM) (pH 6.8 buffer) | 73 | 84 | 69 |
| Caco2 (Papp) | 15.9 × 10–6 | 3.2 × 10–6 | N.T.c |
| Metabolic Stabilityd | |||
| T1/2 (min) | |||
| Human MS | 28 | 22 | <15 |
| Rat MS | 29 | 24 | <15 |
IC50 values were determined by nonlinear regression analysis of the dose–response curves (5 points) generated using GraphPad Prism ver.5.04 with 95% confidence intervals.
Maximal inhibition rate of LPA-induced increase of intraurethral pressure (IUP) in anesthetized rats. Each compound (10 mg/kg) in 0.5% methylcellulose was administered intraduodenally at 0 min, and LPA (300 μg/kg) was injected intravenously at pre, 5, 15, 30, and 60 min.
N.T. = not tested.
0.5 mg/mL, NADPH; MS, microsomes.
We subsequently investigated the oral administration of ONO-7300243 (17a) to determine its effect on rat IUP. Compound 17a inhibited the LPA-induced IUP increase in a dose-dependent manner (ID50 = 11.6 mg/kg p.o.) up to 1 h after dosing (Figure 3A). Significant effects were observed at 10 and 30 mg/kg (p < 0.05 vs vehicle). We also assessed whether or not this LPA1 antagonist influenced in blood pressure in conscious rats (Figure 3B). ONO-7300243 (17a) (30 mg/kg, p.o.) led to a significant decrease in the IUP in conscious rats without LPA stimulation compared with the vehicle without affecting the mean blood pressure (MBP). These results therefore suggested that the LPA1 receptor was constantly activated to some extent in vivo, exhibiting LPA1 antagonist effects in normal rats without exogenous LPA stimulation. For comparison, tamsulosin (α1 adrenoceptor antagonist), which is used in clinical practice for the treatment of BPH, was tested in the same in vivo model (Figure 3C). Tamsulosin significantly reduced IUP at 1 mg/kg oral administration with the same potency for reducing IUP as ONO-7300243 (17a). This compound also led to a significant reduction in MBP at the same dose.22 This difference in the effect on blood pressure distinguishes LPA1 antagonists from α1 adrenoceptor antagonists. Tamsulosin has been optimized for once daily administration. In contrast, the results of a rat pharmacokinetic study of ONO-7300243 (17a) showed that this material had a rapid clearance (CLtot = 15.9 mL/min/kg at 3 mg/kg i.v.) and a short half-life (0.3 h) (see Supporting Information S35, Table S1). The main goal of the lead optimization stage for this compound would therefore involve making improvements to its pharmacokinetic profile to yield a promising compound.
Figure 3.
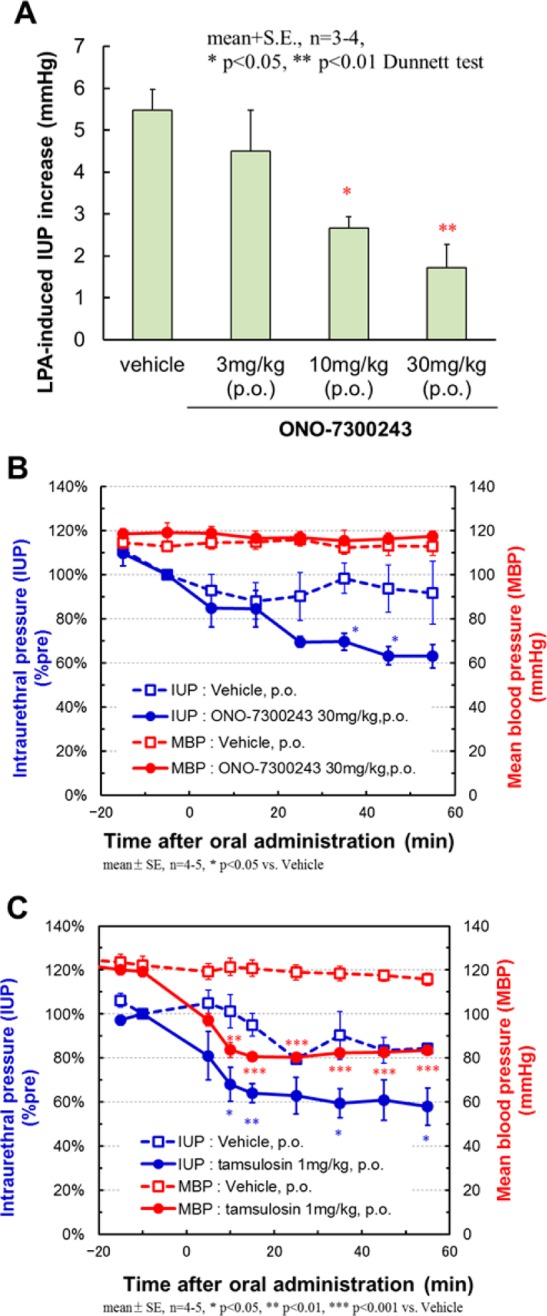
In vivo efficacy of ONO-7300243 (17a). (A) Dose responsibility of ONO-7300243 (17a). The compound was orally administered at each dose in conscious rats. After 60 min, LPA (300 μg/kg) was injected intravenously and IUP was measured in short-term anesthetized rats. (B,C) Change of IUP and mean blood pressure (MBP) without LPA stimulation in conscious rats. Vehicle, ONO-7300243 (17a), or tamsulosin (α1 adrenoceptor antagonist) was orally administered at 0 min, and IUP and MBP were measured continuously for 60 min. The mean pressure was calculated at 10 min intervals.
In conclusion, we have successfully obtained the orally effective lead compound ONO-7300243 (17a) as the result of modification of sections A and B of hit compound 7a. The oral administration of compound 17a in rats led to a significant reduction in IUP in a dose-dependent manner without affecting MBP. When dosed at 30 mg/kg p.o., the potency of ONO-7300243 was the same as tamsulosin dosed at 1 mg/kg p.o. The lead optimization of this compound will focus on improving its pharmacokinetic profile and should lead to a promising candidate for the treatment of BPH.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00225.
Experimental procedures, pharmacokinetic parameters of ONO-7300243, LPA1 receptor antagonist assay, and van der Waals contact of Section C (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tokumura A.; Majima E.; Kariya Y.; Tominaga K.; Kogure K.; Yasuda K.; Fukuzawa K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- Aoki J.; Taira A.; Takanezawa Y.; Kishi Y.; Hama K.; Kishimoto T.; Mizuno K.; Saku K.; Taguchi R.; Arai H. Serum Lysophosphatidic Acid Is Produced through Diverse Phospholipase Pathways. J. Biol. Chem. 2002, 277, 48737–48744. 10.1074/jbc.M206812200. [DOI] [PubMed] [Google Scholar]
- Umezu-Goto M.; Kishi Y.; Taira A.; Hama K.; Dohmae N.; Takio K.; Yamori T.; Mills G. B.; Inoue K.; Aoki J.; Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archbold J. K.; Martin J. L.; Sweet M. J. Towards selective lysophospholipid GPCR modulators. Trends Pharmacol. Sci. 2015, 35, 219–216. 10.1016/j.tips.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Mills G. B.; Moolenaar W. H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- Inoue M.; Rashid M. H.; Fujita R.; Contos J. J.; Chun J.; Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med. 2004, 10, 712–718. 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Tager A. M.; LaCamera P.; Shea B. S.; Campanella G. S.; Selman M.; Zhao Z.; Polosukhin V.; Wain J.; Karimi-Shah B. A.; Kim N. D.; Hart W. K.; Pardo A.; Blackwell T. S.; Xu Y.; Chun J.; Luster A. D. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. 10.1038/nm1685. [DOI] [PubMed] [Google Scholar]
- Kihara Y.; Maceyka M.; Spiegel S.; Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagida K.; Kurikawa Y.; Shimizu T.; Ishii S. Current progress in non-Edg family LPA receptor research. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 33–41. 10.1016/j.bbalip.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Kenny B.; Ballard S.; Blagg J.; Fox D. Pharmacological Options in the Treatment of Benign Prostatic Hyperplasia. J. Med. Chem. 1997, 40, 1293–1315. 10.1021/jm960697s. [DOI] [PubMed] [Google Scholar]
- American Urological Association Guideline: Management of Benign Prostatic Hyperplasia (BPH). http://www.auanet.org/education/guidelines/benign-prostatic-hyperplasia.cfm.
- Fukushima D.; Nakade S.. WO2002062389.
- Saga H.; Ohhata A.; Hayashi A.; Katoh M.; Maeda T.; Mizuno H.; Takada Y.; Komichi Y.; Ota H.; Matsumura N.; Shibaya M.; Sugiyama T.; Nakade S.; Kishikawa K. A novel highly potent Autotaxin/ENPP2 inhibitor produces prolonged decreases in plasma lysophosphatidic acid formation in vivo and regulates urethral tension. PLoS One 2014, 9, e93230. 10.1371/journal.pone.0093230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta H.; Sato K.; Murata N.; Damirin A.; Malchinkhuu E.; Kon J.; Kimura T.; Tobo M.; Yamazaki Y.; Watanabe T.; Yagi M.; Sato M.; Suzuki R.; Murooka H.; Sakai T.; Nishitoba T.; Im D. S.; Nochi H.; Tamoto K.; Tomura H.; Okajima F. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [DOI] [PubMed] [Google Scholar]
- Swaney J. S.; Chapman C.; Correa L. D.; Stebbins K. J.; Bundey R. A.; Prodanovich P. C.; Fagan P.; Baccei C. S.; Santini A. M.; Hutchinson J. H.; Seiders T. J.; Parr T. A.; Prasit P.; Evans J. F.; Lorrain D. S. A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. 10.1111/j.1476-5381.2010.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y.; Hamilton M.; Sidduri A.; Gabriel S.; Ren Y.; Peng R.; Kondru R.; Narayanan A.; Truitt T.; Hamid R.; Chen Y.; Zhang L.; Fretland A. J.; Sanchez R. A.; Chang K.-C.; Lucas M.; Schoenfeld R. C.; Laine D.; Fuentes M. E.; Stevenson C. S.; Budd D. C. Discovery of highly selective and orally active lysophosphatidic acid receptor-1 antagonists with potent activity on human lung fibroblasts. J. Med. Chem. 2012, 55, 7920–7939. 10.1021/jm301022v. [DOI] [PubMed] [Google Scholar]
- Schaffer M.; Pernerstorfer J.; Kadereit D.; Strobel H.; Czechtizky W.; Chen L. C.; Safarova A.; Weichsel A.; Patek M.. WO2009135590.
- Allanore Y.; Jagerschmidt A.; Jasson M.; Distler O.; Denton C.; Khanna D. OP0266 Lysophophatidic acid receptor 1 antagonist SAR100842 as a potential treatment for patients with systemic sclerosis: results from a phase 2A study. Ann. Rheum. Dis. 2015, 74, 172–173. 10.1136/annrheumdis-2015-eular.3472. [DOI] [Google Scholar]
- Chrencik J. E.; Roth C. B.; Terakado M.; Kurata H.; Omi R.; Kihara Y.; Warshaviak D.; Nakade S.; Asmar-Rovira G.; Mileni M.; Mizuno H.; Griffith M. T.; Rodgers C.; Han G. W.; Velasquez J.; Chun J.; Stevens R. C.; Hanson M. A. Crystal structure of antagonist bound human lysophosphatidic acid receptor 1. Cell 2015, 161, 1633–1643. 10.1016/j.cell.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manly C. J.; Chandrasekhar J.; Ochterski J. W.; Hammer J. D.; Warfield B. B. Strategies and tactics for optimizing the Hit-to-Lead process and beyond-A computational chemistry perspective. Drug Discovery Today 2008, 13, 99–109. 10.1016/j.drudis.2007.10.019. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Akiyama K.; Hora M.; Tatemichi S.; Masuda N.; Nakamura S.; Yamagishi R.; Kitazawa M. KMD-3213, a uroselective and long-acting α1a-adrenoceptor antagonist, tested in a novel rat model. J. Pharmacol Exp Ther 1999, 291, 81–91. [PubMed] [Google Scholar]
- Rat IUP was measured according to this report using LPA instead of norepinephrine. Our modified procedure is described in ref (13).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.