

Abstract
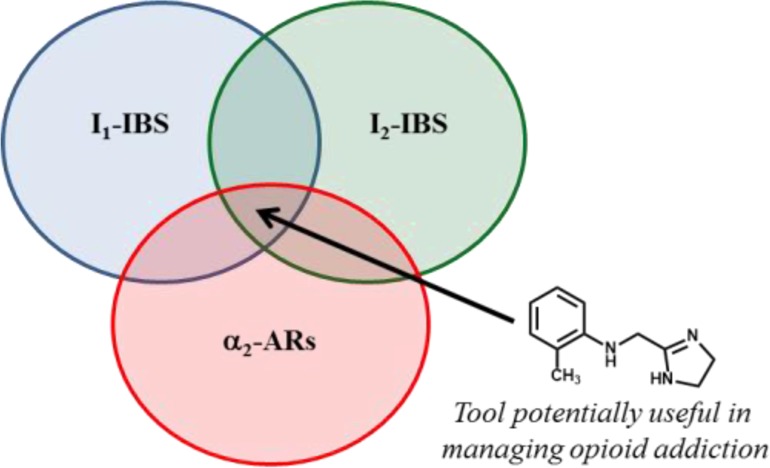
Tolerance and dependence associated with chronic opioid exposure result from molecular, cellular, and neural network adaptations. Such adaptations concern opioid and nonopioid systems, including α2-adrenoceptors (α2-ARs) and I1- and I2-imidazoline binding sites (IBS). Agmatine, one of the hypothesized endogenous ligands of IBS, targeting several systems including α2-ARs and IBS, proved to be able to regulate opioid-induced analgesia and to attenuate the development of tolerance and dependence. Interested in the complex pharmacological profile of agmatine and considering the nature of its targets, we evaluated two series of imidazolines, rationally designed to simultaneously interact with I1-/I2-IBS or I1-/I2-IBS/α2-ARs. The compounds showing the highest affinities for I1-/I2-IBS or I1-/I2-IBS/α2-ARs have been selected for their in vivo evaluation on opiate withdrawal syndrome. Interestingly, 9, displaying I1-/I2-IBS/α2-ARs interaction profile, appears more effective in reducing expression and acquisition of morphine dependence and, therefore, might be considered a promising tool in managing opioid addiction.
Keywords: opioid addiction, imidazolines, α2-adrenoceptors, combined interactions, agmatine biological profile
Opioid addiction, involving tolerance, drug-seeking, and physical dependence, is defined as a chronic relapsing condition and represents a major health and social problem in most societies. Tolerance and dependence associated with chronic opioid exposure result from molecular, cellular, and neural network adaptations. Such adaptations concern opioid and nonopioid systems, including α2-adrenoceptors (α2-ARs) and imidazoline binding sites (IBS). Therefore, agents directed to these targets can also affect the pharmacological actions of opioids and represent useful tools in managing opioid addiction.1
α2-ARs have been demonstrated to be extremely sensitive to opioid exposure and to play a key role in opiate withdrawal symptoms.2 They are widely distributed both in the central nervous system (CNS) and in peripheral tissues.3 By pharmacological and molecular cloning techniques, α2-ARs have been classified into three subtypes, namely, α2A-AR, mediating hypotension, sedation and analgesia, α2B-AR, mediating vasoconstriction, and α2C-AR, modulating many CNS processes and contributing to spinal α2-agonist mediated analgesia and adrenergic-opioid synergy.2−4
IBS, discovered about 30 years ago by Bousquet et al.,5 are distributed in both central and peripheral nervous systems.6 At present they appear to be divided into three subtypes: I1-IBS, preferentially recognized by [3H]-clonidine and related compounds, I2-IBS, recognized by [3H]-idazoxan, and I3-IBS, atypical site constituting a potential target for the treatment of diabetes.6,7 I1-IBS are involved in the regulation of cardiovascular function, in the modulation of ocular pressure, and in the secretion of renal sodium. I2-IBS are involved in CNS pathologies such as Parkinson’s disease, depression, tolerance and addiction to opioids, and food intake in rats.6,8
For many years, the nonsubtype selective α2-AR agonist clonidine (Chart 1) has been the mainstay of nonopioid treatment for relief of withdrawal symptoms during detoxification.2
Chart 1. Chemical Structures of Clonidine, Moxonidine, 2-BFI, and Agmatine.

Since clonidine also interacts with I1-IBS and is endowed with preferential I1-IBS/α2-AR selectivity (selectivity ratio = 4), a potential role of I1-IBS signaling in addiction is hypothesized. It has also been reported that, as observed with moxonidine (I1-IBS/α2-AR selectivity ratio = 40), the coincident stimulation of α2-ARs and I1-IBS prevents relapse of cocaine-seeking, indicating novel pharmacological approaches for addiction treatment.9
Moreover, a significant number of studies suggested that some substances such as 2-BFI (Chart 1), displaying high affinity for I2-IBS over α2-ARs, were able to regulate the opioid-induced analgesia,10 and to attenuate the development of tolerance and dependence.11
Positive effects in managing opioid addiction are also provided by agmatine (Chart 1), which has been considered one of the endogenous ligands for IBS.12 Several studies demonstrated that agmatine potentiates opioid analgesia and attenuates opioid tolerance and dependence mainly by interaction with IBS.13 Moreover, it attenuates ethanol withdrawal symptoms14 and displays anxiolytic, antidepressant, anticonvulsive, antiproliferative, and neuroprotective effects.15 Nevertheless, the efficacy of agmatine is weakened by the fact that, similarly to the other monoamine transmitter molecules, its fast peripheral metabolization reduces its penetration into the brain, limiting its use as a therapeutic agent. Moreover, only high and frequent doses of exogenous agmatine induced significant effects in animal models.16 Agmatine is one of the few neurotransmitters with multireceptorial affinities. Indeed, it interacts with serotonin 5-HT receptors, antagonizes NMDA receptors, inhibits nitric oxide synthase, and binds to α2-ARs and I1-, I2-IBS.12,15,16 The multiplicity of systems recognized by agmatine makes it difficult to identify its certain targets modulating opioid functions as well as other interesting aforementioned effects.
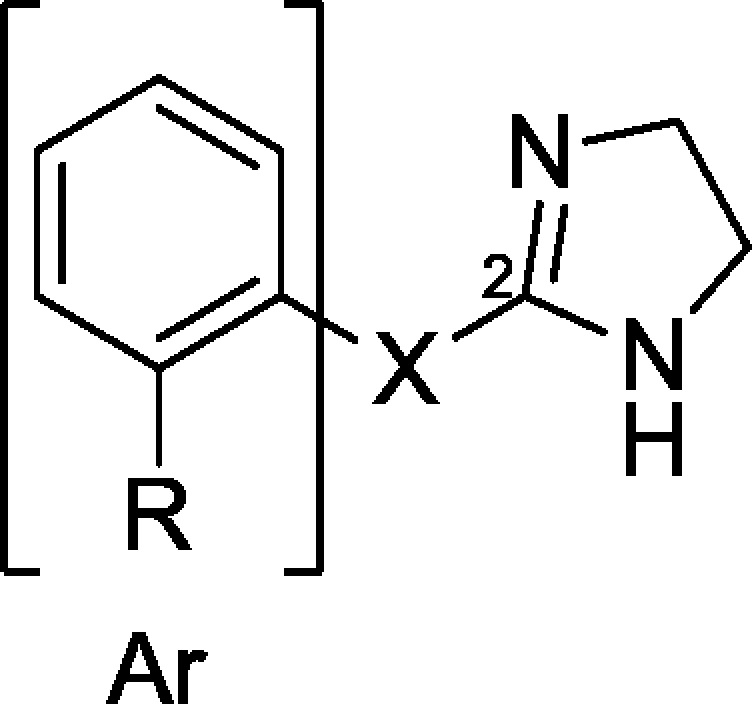
Some advantageous combined interactions also emerged by our studies regarding imidazolines able to interact with different systems, including IBS and α2-ARs, and built on the common scaffold I characterized by an aromatic area (Ar) linked to the position 2 of the imidazoline nucleus by a biatomic bridge (X) (Chart 2). Interestingly, our results highlighted that the peculiar chemical nature of X addressed the ligand to specific biological targets, whereas the ortho-phenyl substituents were responsible for its functional behavior.
Chart 2. Chemical Structures of Imidazolines 1–13 Sharing the Common Scaffold I.

In particular, the -OCH(CH3)- bridge proved to be suitable for α2-AR and 5-HT1A receptor (5-HT1A-R) interactions,17 whereas the −O–CH2– linker promoted the simultaneous α2-AR/I2-IBS recognition (Chart 2).18 Indeed, we demonstrated that ligands such as allyphenyline (1), displaying effective α2C-AR agonism/α2A-AR antagonism/5-HT1A-R agonism, or compound 2, producing significant α2C-AR agonism/α2A-AR antagonism/I2-IBS interaction, behaved as multifunctional agents and were able, at the same dose, to reduce opioid withdrawal syndrome and associated depression.17−19 At low dose 1 also reduced hyperanxiety-like behavior after alcohol intoxication.20
In contrast, the −CH=CH– bridge induced efficacious I1- and I2-IBS recognition, as demonstrated by tracizoline (3) (pKi: I1-IBS 7.72, I2-IBS 8.72, α2-ARs < 6),21 whereas the −NH–CH2– chain might be tolerated by ligands addressed to I1-, I2-IBS and α2-ARs, as reported for 4 (pKi: I1-IBS 9.30, I2-IBS 7.48, α2-ARs 7.14).22,23
Our studies also indicated that the introduction of suitable decorations in the ortho position of the phenyl ring conferred to the ligand an interesting profile modulation from antagonism to agonism. Such a modulation was demonstrated both for selective α2-AR24 and I1-IBS ligands,25 suggesting some analogies in the nature of critical binding sites for both systems, as hypothesized by Hieble and Ruffolo.26 As aforementioned, the simultaneous stimulation of α2-ARs and I1-IBS might be advantageous for addiction treatment.9
On the basis of these observations, interested in the complex pharmacological profile of agmatine, we focused our attention on some of its targets, namely, α2-ARs and I1-, I2-IBS. Therefore, we evaluated the in vitro IBS and α2-ARs profiles of two series of imidazolines rationally designed to simultaneously interact with I1-/I2-IBS or I1-/I2-IBS/α2-ARs. Indeed, compounds 5–8, bearing the −CH=CH– bridge and being structurally related to 3, might interact with I1- and I2-IBS, whereas simultaneous I1-/I2-IBS/α2-AR interactions might be expected for 9–13, structurally related to 4 and characterized by the −NH–CH2– linker (Chart 2). Our aim was to discover compounds useful for exploring the biological effects modulated by these target interaction combinations and novel tools potentially useful in managing opioid addiction. Therefore, in vivo assays on the expression and acquisition of morphine dependence were performed with the most interesting compounds.
Imidazolines 7, 8, 11, and 12 have been synthesized for the first time in this study. To make relevant structure–activity relationships, compounds 5, 6(21) and 9, 10, 13(27) have also been evaluated.
Compounds 7, 8, 11, and 12 were prepared according to the synthetic procedures reported in Scheme 1. In particular, the reaction of the aldehydes 14(28) and 15(29) with ethylenediamine in the presence of N-bromosuccinimide gave the imidazolines 7 and 8, respectively. The condensation of the commercially available anilines 16 and 17 with 2-(chloromethyl)-4,5-dihydro-1H-imidazole at 170 °C in diglime afforded the imidazolines 11 and 12, respectively.
Scheme 1. Synthesis of Imidazolines 7, 8, 11, and 12.

Reagents and conditions: (a) ethylenediamine, dichloromethane, 1 h, then N-bromosuccinimide, overnight at room temperature; (b) diglime, 170 °C, 20 h.
The biological profiles of 5–13 were evaluated by radioligand competition binding assays at I1-IBS, I2-IBS, and α2-ARs using [125I]-p-iodoclonidine (on rat kidney membranes), [3H]-2-BFI (on rat brain membranes), and [3H]-RX821002 (on rat brain membranes), respectively, as radioligands, according to previous reported procedures.30 Moreover, the functional profiles of ligands interacting with α2-ARs (9–13) at α2A-, α2B-, and α2C-AR subtypes have been assessed with Cytosensor microphysiometer on CHO cells expressing each individual human α2-AR subtype.24
The data reported in Table 1 are in agreement with our expectations. Indeed, compounds 5–8, bearing the −CH=CH– bridge, prove to be able to effectively produce simultaneous I1-/I2-IBS interactions. Similarly to compound 3,21 they are inactive at α2-ARs. ortho-Phenyl substituents endowed with low steric bulk, such as −CH3 or −Cl, appear to be more suitable for the binding at I1- and I2-IBS than bulkier substituents, such as allyl or phenyl groups, as demonstrated by the higher affinities showed by 5 and 6 with respect to 7 and 8. A preferential binding affinity for I2- over I1-IBS is also observed for all the compounds of this series.
Table 1. Binding Affinity (pKia) at I1- and I2-IBS on Rat Brain Membranes; Binding Affinity (pKia), Antagonist Potency (pKbb), Agonist Potency (pEC50b), and Intrinsic Activity (iab) at Human α2-Adrenergic Receptors Subtypes on CHO Cells.

pKi affinity values for I1-IBS, I2-IBS, and α2-ARs were assessed by measuring the ability of the tested compounds to displace [125I]-p-iodoclonidine (rat kidney membranes), [3H]-2-BFI (rat brain membranes), and [3H]-RX 821002 (rat brain membranes), respectively.
pKb, pEC50, and ia values were determined by applying the Cytosensor microphysiometry system to the CHO cells expressing individual each human α2-AR subtype. Intrinsic activity of the tested compounds is expressed as the fraction of that of the full agonist (−)-noradrenaline taken as equal to 1. The data are expressed as mean ± SEM of 3–5 separate experiments.
Reference (21).
Re-examined in the experimental conditions of the present study.
On the contrary, compounds 9–13, bearing the −NH–CH2– linker, show I1-/I2-IBS/α2-AR affinities. In particular, in the case of compound 13, a significant I1-IBS selectivity vs I2-IBS and α2-AR is displayed. The ortho-methyl group proved to be particularly suitable for the combined I1-/I2-IBS/α2-AR interaction, as demonstrated by the affinity values of compound 9. In comparison to the first series of compounds (5–8), the imidazolines 9–13 display higher affinity for I1-IBS and lower for I2-IBS. The functional profile of the compounds bearing the −NH–CH2– linker (9–13) at α2-AR subtypes highlights the crucial role of the ortho-phenyl pendant. Indeed, while compound 13 significantly activates both α2C-AR and α2A-AR subtypes, compounds 9–12 show interesting α2C-AR agonism/α2A-AR antagonism. The behavior of 9–13 might be ascribed to the different conformations adopted by the ligands, similarly to what was previously observed for analogues bearing the −OCH(CH3)– bridge.31
Because compounds 5 and 9 displayed the best biological profiles at I1-/I2-IBS and I1-/I2-IBS/α2-ARs, respectively, they were selected for their in vivo evaluation on opiate withdrawal syndrome. Both of them are characterized by the presence of the ortho-methyl substituent in the phenyl ring that, in ligands selectively interacting with I2-IBS, was demonstrated to be compatible with a significant reduction of the morphine withdrawal signs.30
Figures 1 and 2 show the effects of 5 and 9, respectively, on naloxone-precipitated withdrawal syndrome behavior. Repeated administration of morphine produced physical dependence, as assessed by a summary of characteristic set of behavioral responses, including jumping, rearing, forepaw tremor and teeth chattering following the naloxone challenge, as compared to vehicle. Acute administration of 5 or 9, 15 min prior to the naloxone injection, significantly decreased both the number of jumping, a sign featuring the morphine withdrawal syndrome (Figures 1A and 2A), and the frequencies of other withdrawal manifestations, reported as the sum of the frequency of rearing, forepaw tremor, and teeth chattering (Figures 1B and 2B) in morphine-dependent mice.
Figure 1.

Effects of acute (A,B) or repeated (C,D) intraperitoneal (i.p.) administration of 5 (5, 10, mg/kg) on expression and acquisition of morphine dependence, respectively. Naloxone-precipitated withdrawal symptoms, both in control and morphine treated mice, are given as a measure of the frequency of jumping (A,C) and somatic signs (B,D) expressed as summary of rearing, forepaw tremors, and teeth chatter. Data represent mean (±SEM) of 6–10 animals. Significant differences: ##p < 0.01, compared to vehicle; *p < 0.05, **p < 0.01, compared to morphine group.
Figure 2.

Effects of acute (A,B) or repeated (C,D) i.p. administration of 9 (5, 10, mg/kg) on expression and acquisition of morphine dependence, respectively. Naloxone-precipitated withdrawal symptoms, both in control and morphine treated mice, are given as a measure of the frequency of jumping (A,C) and somatic signs (B,D) expressed as summary of rearing, forepaw tremors, and teeth chatter. Data represent mean (±SEM) of 6–9 animals. Significant differences: ##p < 0.01, compared to vehicle; **p < 0.01, compared to morphine group.
In particular, treatment with 9 at the doses of 5 and 10 mg/kg almost completely nullifies the number of jumping (96% and 98%, respectively). Similarly, the reduction of the frequency of the other signs of withdrawal is 86% and 93% at 5 and 10 mg/kg, respectively.
The combined I1- and I2-IBS interactions produced by 5 appear less effective in reducing the expression of morphine dependence. Indeed, the reduction of the number of jumping is comparable to that produced by 9 only at 10 mg/kg (90%). Moreover, only half reduction of the other withdrawal signs is observed.
Figures 1 and 2 also show the effects of repeated coadministration of 5 or 9 with morphine on the naloxone-precipitated withdrawal symptoms. In particular, pretreatment of the mice with 5 or 9, 15 min before each morphine injection, significantly attenuates both the development of jumping (Figures 1C and 2C) and the total signs of withdrawal (Figures 1D and 2D).
Also in this case 9 is the most effective compound, showing a reduction of about 80% of jumping and other withdrawal signs at 5 and 10 mg/kg. Analogous reduction of jumping was produced by 5 at the dose of 10 mg/kg, whereas no effect is observed at 5 mg/kg (Figure 1C). Moreover, significantly lower effects are also observed in the attenuation of the other signs of morphine withdrawal (34% and 46% at 5 and 10 mg/kg, respectively).
In conclusion, according to what was previously observed for compounds 3 and 4, the −CH=CH– and −NH–CH2– bridges of the imidazoline scaffold I reported in Chart 2 confirm to be suitable for ligands able to produce I1-/I2-IBS or I1-/I2-IBS/α2-ARs interactions, respectively. Such combined interactions, rationally selected on the basis of some of the numerous targets of agmatine, were demonstrated to produce advantageous pharmacological responses. Indeed, 5 and 9, showing the best biological profiles at I1-/I2-IBS and I1-/I2-IBS/α2-ARs, respectively, significantly affected both expression and acquisition of morphine dependence. In particular, the combined I1-/I2-IBS/α2-ARs interaction appeared more favorable, as demonstrated by the higher efficacy displayed by 9 both in contrasting and preventing morphine withdrawal. Moreover, such a combination also appeared more favorable than that produced by 2 (I2-IBS/α2-ARs), already evaluated in the same assays18 (see Table S2). Therefore, 9 might be considered a promising tool useful in managing opioid addiction, potentially lacking in sedative and hypotensive side effects, due to its α2A-AR antagonist profile.
Acknowledgments
This work was supported by grants from the University of Camerino (Fondo di Ateneo per la Ricerca 2014–2015) and from Natural Sciences and Engineering Research Council of Canada.
Glossary
ABBREVIATIONS
- 5-HT1A-R
5-HT1A receptor
- α2-ARs
α2-adrenoceptors
- CNS
central nervous system
- IBS
imidazoline binding sites
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00290.
Synthetic procedures, experimental details of in vivo assays, and elemental analysis of the final compounds (PDF)
The authors declare no competing financial interest.
Dedication
This article is dedicated to Prof. Maria Pigini, who died on February 8, 2016.
Supplementary Material
References
- Veilleux J. C.; Colvin P. J.; Anderson J.; York C.; Heinz A. J. A rewiew of opioid dependence treatment: pharmacological and psychological interventions to treat opioid addiction. Clin. Psychol. Rev. 2010, 30, 155–166. and references therein 10.1016/j.cpr.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Gowing L.; Farrell M. F.; Ali R.; White J. M. Alpha2-adrenergic agonists for the management of opioid withdrawal. Cochrane Database Syst. Rev. 2014, 3, CD002024. 10.1002/14651858.CD002024.pub2. [DOI] [PubMed] [Google Scholar]
- Tan C. M.; Limbird L. E.. The α2-adrenergic receptors. In The Receptors: The Adrenergic Receptors: In the 21st Century; Perez D. M., Ed.; Humana Press Inc.: Totowa, NJ, 2006; pp 241–265. [Google Scholar]
- Fairbanks C. A.; Stone L. S.; Wilcox G. L. Pharmacological profile of alpha 2 adrenergic receptor agonists identified using genetically altered mice and isobolographic analysis. Pharmacol. Ther. 2009, 123, 224–238. and references therein 10.1016/j.pharmthera.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet P.; Feldman J.; Schwartz J. Central cardiovascular effects of alpha adrenergic drugs: differences between catecholamines and imidazolines. J. Pharmacol. Exp. Ther. 1984, 230, 232–236. [PubMed] [Google Scholar]
- Dardonville C.; Rozas I. Imidazoline binding sites and their ligands: an overview of the different chemical structures. Med. Res. Rev. 2004, 24, 639–661. and references therein 10.1002/med.20007. [DOI] [PubMed] [Google Scholar]
- Nikolic K.; Agbaba D. Pharmacophore development and SAR studies of imidazoline receptor ligands. Mini-Rev. Med. Chem. 2012, 12, 1542–1555. and references therein 10.2174/138955712803832636. [DOI] [PubMed] [Google Scholar]
- Head G. A.; Mayorov D. N. Imidazoline receptors, novel agents and therapeutic potential. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 17–32. 10.2174/187152506775268758. [DOI] [PubMed] [Google Scholar]
- Smith R. J.; Aston-Jones G. α2-Adrenergic and imidazoline receptor agonists prevent cue-induced cocaine seeking. Biol. Psychiatry 2011, 70, 712–719. and references therein 10.1016/j.biopsych.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Blázquez P.; Boronat M. A.; Olmos G.; Garcìa-Sevilla J. A.; Garzón J. Activation of I2-imidazoline receptors enhances supraspinal morphine analgesia in mice: a model to detect agonist and antagonist activities at these receptors. Br. J. Pharmacol. 2000, 130, 146–152. 10.1038/sj.bjp.0703294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boronat M. A.; Olmos G.; García-Sevilla J. A. Attenuation of tolerance to opioid-induced antinociception by idazoxan and other I2-ligands. Ann. N. Y. Acad. Sci. 1999, 881, 359–363. 10.1111/j.1749-6632.1999.tb09380.x. [DOI] [PubMed] [Google Scholar]
- Raasch W.; Schäfer U.; Chun J.; Dominiak P. Biological significance of agmatine, an endogenous ligand at imidazoline binding sites. Br. J. Pharmacol. 2001, 133, 755–780. 10.1038/sj.bjp.0704153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.; Su R.-B.; Li J. Agmatine and imidazoline receptors: their role in opioid analgesia, tolerance and dependence. Cell. Mol. Neurobiol. 2008, 28, 629–641. 10.1007/s10571-007-9164-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taksande B. G.; Kotagale N. R.; Patel M. R.; Shelkar G. P.; Ugale R. R.; Chopde C. T. Agmatine, an endogenous imidazoline receptor ligand modulates ethanol anxiolysis and withdrawal anxiety in rats. Eur. J. Pharmacol. 2010, 637, 89–101. and references therein 10.1016/j.ejphar.2010.03.058. [DOI] [PubMed] [Google Scholar]
- Halaris A.; Plietz J. Agmatine. Metabolic pathway and spectrum of activity in brain. CNS Drugs 2007, 21, 885–900. 10.2165/00023210-200721110-00002. [DOI] [PubMed] [Google Scholar]
- Regunathan S. Agmatine: biological role and therapeutic potentials in morphine analgesia and dependence. AAPS J. 2006, 8, E479–E484. and references therein 10.1208/aapsj080356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bello F.; Diamanti E.; Giannella M.; Mammoli V.; Marchioro C.; Mattioli L.; Titomanlio F.; Piergentili A.; Quaglia W.; Benedetti G.; Varrone M.; Pigini M. Low doses of allyphenyline and cyclomethyline, effective against morphine dependence, elicit an antidepressant-like effect. ACS Med. Chem. Lett. 2012, 3, 535–539. 10.1021/ml300064v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bello F.; Diamanti E.; Giannella M.; Mammoli V.; Mattioli L.; Titomanlio F.; Piergentili A.; Quaglia W.; Lanza M.; Sabatini C.; Caselli G.; Poggesi E.; Pigini M. Exploring multitarget interactions to reduce opiate withdrawal syndrome and psychiatric comorbidity. ACS Med. Chem. Lett. 2013, 4, 875–879. 10.1021/ml400232p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bello F.; Mattioli L.; Ghelfi F.; Giannella M.; Piergentili A.; Quaglia W.; Cardinaletti C.; Perfumi M.; Thomas R. J.; Zanelli U.; Marchioro C.; Dal Cin M.; Pigini M. Fruitful adrenergic α2C-agonism/α2A-antagonism combination to prevent and contrast morphine tolerance and dependence’. J. Med. Chem. 2010, 53, 7825–7835. 10.1021/jm100977d. [DOI] [PubMed] [Google Scholar]
- Ubaldi M.; Del Bello F.; Domi E.; Pigini M.; Nasuti C. Investigation of allyphenyline efficacy in the treatment of alcohol withdrawal symptoms. Eur. J. Pharmacol. 2015, 760, 122–128. 10.1016/j.ejphar.2015.04.005. [DOI] [PubMed] [Google Scholar]
- Gentili F.; Bousquet P.; Brasili L.; Dontenwill M.; Feldman J.; Ghelfi F.; Giannella M.; Piergentili A.; Quaglia W.; Pigini M. Imidazoline binding sites (IBS) profile modulation: key role of the bridge in determining I1-IBS or I2-IBS selectivity within a series of 2-phenoxymethylimidazoline analogues. J. Med. Chem. 2003, 46, 2169–2176. and references therein 10.1021/jm021113r. [DOI] [PubMed] [Google Scholar]
- Nicolotti O.; Carotti A.; Carrieri A.; Pigini M.; Gentili F.; Brasili L.; Giannella M.; Quaglia W.; Piergentili A.; Bousquet P.; Dontenwill M. Pharmacophore development and 3D-QSAR study of I1-imidazoline binding site ligands. Med. Chem. Res. 2004, 13, 170–189. 10.1007/s00044-004-0023-9. [DOI] [Google Scholar]
- Gentili F.; Bousquet P.; Brasili L.; Caretto M.; Carrieri A.; Dontenwill M.; Giannella M.; Marucci G.; Perfumi M.; Piergentili A.; Quaglia W.; Rascente C.; Pigini M. a2-Adrenoreceptors profile modulation and high antinociceptive activity of (S)-(−)-2-[1-(biphenyl-2-yloxy)ethyl]4,5-dihydro-1H-imidazole. J. Med. Chem. 2002, 45, 32–40. 10.1021/jm0110082. [DOI] [PubMed] [Google Scholar]
- Gentili F.; Cardinaletti C.; Vesprini C.; Carrieri A.; Ghelfi F.; Farande A.; Giannella M.; Piergentili A.; Quaglia W.; Laurila J. M.; Huhtinen A.; Scheinin M.; Pigini M. α2-Adrenoreceptors profile modulation. 4. From antagonist to agonist behavior. J. Med. Chem. 2008, 51, 4289–4299. 10.1021/jm800250z. [DOI] [PubMed] [Google Scholar]
- Del Bello F.; Bargelli V.; Cifani C.; Gratteri P.; Bazzicalupi C.; Diamanti E.; Giannella M.; Mammoli V.; Matucci R.; Micioni Di Bonaventura M. V.; Piergentili A.; Quaglia W.; Pigini M. Antagonism/agonism modulation to build novel antihypertensives selectively triggering I1-imidazoline receptor activation. ACS Med. Chem. Lett. 2015, 6, 496–501. 10.1021/acsmedchemlett.5b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieble J. P.; Ruffolo R. R. Jr. Possible structural and functional relationships between imidazoline receptors and alpha 2-adrenoceptors. Ann. N. Y. Acad. Sci. 1995, 753, 8–21. 10.1111/j.1749-6632.1995.tb32387.x. [DOI] [PubMed] [Google Scholar]
- Urech E.; Marxer A.; Miescher K. 2-Aminoalkyl-imidazoline. Helv. Chim. Acta 1950, 33, 1386–1407. 10.1002/hlca.19500330539. [DOI] [Google Scholar]
- Padwa A.; Hornbuckle S. F.; Fryxell G. E.; Zhang Z. J. Tandem cyclization-cycloaddition reaction of rhodium carbenoids. Studies dealing with intramolecular cycloadditions. J. Org. Chem. 1992, 57, 5747–5757. 10.1021/jo00047a032. [DOI] [Google Scholar]
- Hoffman W. F.; Alberts A. W.; Cragoe E. J. Jr.; Deana A. A.; Evans B. E.; Gilfillan J. L.; Gould N. P.; Huff J. W.; Novello F. C.; Prugh J. D.; Rittle K. E.; Smith R. L.; Stokker G. E.; Willard A. K. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 2. Structural modification of 7-(substituted aryl)-3,5-dihydroxy-6-heptenoic acids and their lactone derivatives. J. Med. Chem. 1986, 29, 159–169. 10.1021/jm00152a001. [DOI] [PubMed] [Google Scholar]
- Gentili F.; Cardinaletti C.; Vesprini C.; Ghelfi F.; Farande A.; Giannella M.; Piergentili A.; Quaglia W.; Mattioli L.; Perfumi M.; Hudson A.; Pigini M. Novel ligands rationally designed for characterizing I2-imidazoline binding sites nature and functions. J. Med. Chem. 2008, 51, 5130–5134. 10.1021/jm800400k. [DOI] [PubMed] [Google Scholar]
- Diamanti E.; Del Bello F.; Carbonara G.; Carrieri A.; Fracchiolla G.; Giannella M.; Mammoli V.; Piergentili A.; Pohjanoksa K.; Quaglia W.; Scheinin M.; Pigini M. Might the observed α2A-adrenoreceptor agonism or antagonism of allyphenyline analogues be ascribed to different molecular conformations?. Bioorg. Med. Chem. 2012, 20, 2082–2090. 10.1016/j.bmc.2012.01.035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.