

Abstract

Expedited structure-based optimization of the initial fragment hit 1 led to the design of (R)-7 (AZD2716) a novel, potent secreted phospholipase A2 (sPLA2) inhibitor with excellent preclinical pharmacokinetic properties across species, clear in vivo efficacy, and minimized safety risk. Based on accumulated profiling data, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Keywords: Secreted phospholipase A2, sPLA2, inhibitor, fragment-based drug discovery, fragment screening, atherosclerosis, coronary artery disease
Secreted phospholipase A2 (sPLA2) are enzymes that hydrolyze the acyl ester at the sn-2 position of sn-3 glycerophospholipids,1 a process characterized by complex interfacial kinetics of substrate–enzyme binding and catalysis.2 Eleven sPLA2 enzymes (group Ib-XIIb) have so far been identified in mammals,3−5 several of which have been detected in human atherosclerotic lesions.6 Among these, group IIa, V, and X sPLA2 isoforms are present in human carotid atherosclerotic lesions and have been associated with disease progression. They have been implicated in several proatherogenic actions in the arterial wall.7−9 Due to their hydrolytic action on lipoprotein phospholipids, sPLA2s promote lipid accumulation, induce significant lipoprotein remodeling, macrophage activation, and foam cell formation.10,11 Furthermore, as the rate-limiting step in eicosanoid production, sPLA2-mediated release of arachidonic acid from the sn-2 position of phospholipids renders them highly pro-inflammatory enzymes.10,11 In addition, epidemiological data has shown that increased levels of sPLA2 protein and sPLA2 activity are independently associated with risk of cardiovascular events and prevalence of atherosclerosis.11 Owing to the pivotal role of sPLA2s in regulating lipoprotein function and inflammatory mechanisms, two crucial components of atherogenesis, sPLA2 inhibitors12 could be useful for the treatment of atherosclerosis. Interestingly, the archetypal sPLA2 inhibitor varespladib methyl (Figure 1) was evaluated in short duration clinical trials for the treatment of rheumatoid arthritis and acute coronary syndrome with negative results.13,14
Figure 1.

sPLA2 inhibitor varespladib methyl, its active metabolite varespladib, and initial fragment hit 1.
We thus set out to identify novel sPLA2 inhibitors that could be used in longer term coronary artery disease-based clinical studies to more properly assess the relevance of their lipoprotein-modifying effects15−17 on cardiovascular disease, alongside their anti-inflammatory properties.
Given the competitive landscape, medicinal chemistry precedents and available structural information for sPLA2 enzymes, we opted for a structure-based fragment approach to maximize the chances of novelty and developability.18 Analysis of potency data in combination with the available ligand-bound sPLA2 crystal structures19 indicated that primary amides are extremely effective sPLA2 warheads, as they establish three hydrogen bonds with sPLA2 and one coordination bond with the catalytic calcium ion. We assembled a selection of primary aromatic carboxamide-containing fragments (heavy atom count ≥10 and ≤18) by mining in house biochemical and biophysical assay data against the sPLA2-IIa and sPLA2-X isoforms. The selection was based on activity against sPLA2-IIa, which is the most widely expressed isoform in humans,20 but also on inspection of crystal structures and chemical evolution potential.
The triaging process identified compound 1 (LE: 0.39) originating from a legacy fragment/HTS campaign as the most promising fragment lead. As the translatability to a clinical setting was of special importance, we also triaged the activities for sPLA2 activity inhibition in human plasma (the mode-of-action biomarker to be used in clinical trials), as measured using a previously established protocol.21
Compound 1 inhibited sPLA2-IIa (IC50: 24 μM) and human plasma sPLA2 activity (ICu,50: 0.9 μM) in a concentration dependent manner (Table 1). Going forward, as plasma sPLA2 activity is the result of various sPLA2 isoforms and we were interested in identifying a broad spectrum sPLA2 inhibitor, we also monitored inhibition of sPLA2-V and sPLA2-X, given their potential role in lipoprotein modulation.16 Lastly, to avoid the need for a prodrug strategy (e.g., varespladib methyl) we carefully evaluated compound lipophilicity against passive permeability, solubility, and metabolic stability, prior to verifying the pharmacokinetic (PK) and pharmacodynamic (PD) profile in vivo.
Table 1. Initial Profile for the Fragment Hit 1.
| entry | sPLA2-IIa IC50 (μM) | plasma ICu,50 (μM)a | Fu (%) | LE/LLEb |
|---|---|---|---|---|
| varespladib | 0.028 | 0.008 | 12.5 | 0.37/7.2 |
| 1 | 24 | 0.9 | 1.8 | 0.39/2.2 |
Calculated as plasma sPLA2 IC50 (μM) × compound’s unbound fraction in human plasma (Fu)/100. Fu = 100 – human protein binding (%).
Ligand efficiency, LE (kcal/mol/HAC), calculated as −RT ln(sPLA2-IIa IC50)/heavy atom count. Ligand lipophilicity efficiency (LLE) calculated as pIC50 (sPLA2-IIa) – logD.
The crystal structure of 1 bound to sPLA2-X confirmed the binding mode of the primary amide, with hydrogen-bonding to sPLA2-X’s G28, H46, and D47 (corresponding to sPLA2-IIa G29, H47, and D48) and coordination to the calcium ion was observed, as shown in Figure 2. Additionally, the 4-benzyl substituent was located in a lipophilic pocket consisting of residues I2, L5, A6, V9, P17, I18, and M21 (corresponding to L2, F5, H6, I9, A17, A18, and G22 in sPLA2-IIa, Figure 2). While the two sPLA2 isoforms are identical in the amide coordinating residues, the lipophilic pocket that accommodates the benzyl group is slightly smaller in sPLA2-IIa. However, superposition of the sPLA2-IIa and sPLA2-X crystal structures suggested that the benzyl group of 1 could fit in the slightly smaller sPLA2-IIa pocket.
Figure 2.
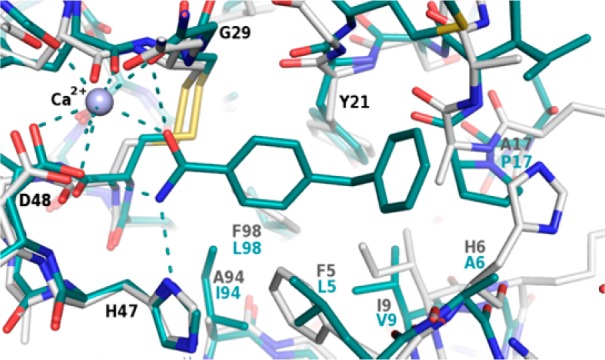
Superposition of the crystal structure of 1 bound to sPLA2-X (cyan) and a crystal structure of sPLA2-IIa (gray). Residues that differ between the two isoforms are labeled in cyan and gray, respectively. The calcium ion is depicted as a purple sphere and hydrogen bonds are displayed as dashed lines.
We therefore devised a chemical exploration strategy starting from the binding mode of 1. Here, special effort was placed upon establishing a second coordination bond to the catalytic calcium ion. The reasoning was 2-fold: (a) to increase affinity and functional inhibition of the enzyme as a result of a bidentate calcium chelate and additional van der Waals contact with the enzyme, and (b) to allow a more balanced lipophilicity profile of the final compounds as the additional calcium interacting moiety was anticipated to be a carboxylic acid. The ortho position of the benzamide ring was identified as a favorable substitution vector to deploy such a strategy. Based on iterative molecular modeling and careful consideration of theoretical affinity gain and ligand efficiency prediction, we synthesized compounds 2–6, according to Scheme 1 (for compound 4 refer to Scheme 2). Key steps involved the formation of the boronic acid by ortho lithiation22 of the 4-benzylbenzonitrile (step b) followed by a Suzuki–Miyaura coupling and a controlled hydrolysis (step d) to generate both the amide and carboxylic acid functions (Scheme 1).
Scheme 1. General Synthesis of Compounds 2, 3, 5, and 6.

Conditions: (a) benzylzinc bromide, Pd(Ph3)4, THF, 60 °C. (b) 1. nBuLi, tetramethylpiperidine; 2. B(OiPr)3, THF −78 °C. (c) Compound 2, 3, 5: 3-bromophenyl carboxylic acid A, PdCl2(dppf), Cs2CO3, DMF, 60–90 °C. Compound 6: Pd(P(Ph3)4, Cs2CO3, DMF 90. (d) Compound 2, 3, 5: n-propanol/H2O (10:1), KOH (10 equiv), 80–100 °C, or THF, H2SO4. Compound 6: KOH (10 equiv), MeOH/H2O, microwave 130 °C, 20 min.
Scheme 2. General Synthesis of sPLA2 Inhibitors 4 and 7–9.

Conditions: (a) appropriate malonate, Cs2CO3, DMF, 70 °C, 2 h. (b) NaOH (4 equiv), H2O/MeOH 3:1 80 °C 2 h. (c) HOAc (3 M), reflux. (d) HCl, MeOH 60 °C, 2.5 h. (e) (Bpin)2 (1.3 equiv), KOAc (2.5 equiv), PdCl2(dppf) (6.5 mol %), dioxane, 90 °C. (f) BnZnBr (1.5 equiv, 0.5 M in THF), Pd(PPh3)4 (5 mol %), THF, 60 °C, 2 h. (g) Compound 4: 3-(3-methoxy-3-oxopropyl)phenylboronic acid (commercial), Pd(Ph3)4. Compound 7: PdCl2(dbpf) (5 mol %), C. Cs2CO3, DMF, 90 °C, 1.5 h. (h) n-Propanol/H2O (10:1), KOH (10 equiv), 80–100 °C. (i) B, PdCl2(dppf), Cs2CO3, DMF, 90 °C.
Introduction of a 3-benzoic acid moiety at the 2-position of 4-benzylbenzamide 1, albeit nonoptimal, confirmed the potential for growth at that position (2, Table 2). Progressive elongation at the carboxylic acid position by introduction of methylene units (3–5) had a parabolic effect on potency, with the 3-phenylpropionic acid side chain yielding the most potent and ligand efficient derivative (4, Table 2). Replacement of the benzylic methylene by an ether oxygen or further elongating the hydrocarbon chain were not tolerated (cf. 5 and 6, Table 2), hinting at a specific conformational requirement for the carboxylic acid-containing side chain. The cocrystal structure of sPLA2-IIa and 4 confirmed the previously hypothesized ligand-mediated calcium chelation, as displayed in Figure 3.
Table 2. sPLA2 Potency and Ligand Efficiencies for Compounds 2–6a.

Results are mean of at least two experiments. Experimental errors within 20% of value.
Calculated as plasma sPLA2 IC50 (μM) × compound’s unbound fraction in human plasma (Fu)/100. Fu = 100 – human protein binding(%)
Ligand efficiency, LE (kcal/mol/HAC), calculated as −RT ln(sPLA2-IIa IC50)/heavy atom count. Ligand lipophilicity efficiency (LLE) calculated as pIC50 (sPLA2-IIa) – logD.
Not active at maximum tested concentration (25 μM).
Not determined.
Figure 3.
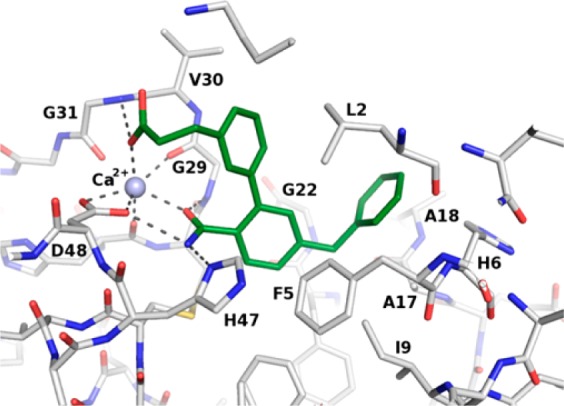
Cocrystal structure of 4 bound to sPLA2-IIa. The calcium ion is depicted as a purple sphere, and relevant hydrogen bonds are displayed as dashed lines.
The carbonyl oxygen atom of the amide group of 4 provided the first coordination bond to calcium, analogously to 1. The carboxylate moiety established the second coordination bond to calcium (d = 2.4 Å) and a hydrogen bond to the backbone amide group of G31, as shown in Figure 3, while the additional phenyl ring made significant van der Waals contacts with the side chains of L2, G29, and V30. The 4-benzylbenzamide component of 4 displayed a similar interaction pattern as in 1, except for the benzamide ring, which was rotated by ca. 50 degrees. This rotation is induced by the introduction of the substituent at the 2-position, and in the case of sPLA2-X, this comes with a penalty. This is exemplified by 2 and 3, where the affinity gain is very limited, despite the addition of more lipophilic interactions (Table 2). In type IIa, however, this conformational lock is further stabilized by an edge-to-face π interaction with F5, which is not available in sPLA2-X. This is reflected by the steep improvement in affinity when adding the substituents. For example, 3 despite having a linker too short to form the additional calcium interaction still gains more than 300-fold in affinity. This is further improved by the propanoic acid side chain of 4 where the second calcium coordination bond is properly established leading to a 2000-fold increase in potency compared to 1. The active site of sPLA2-IIa is smaller, F5 (L5 in sPLA2-X) affects the benzamide moiety, and I9 (V9 in sPLA2-X) is located close to the hinge between the two benzyl groups of 1 (d = 3.7 Å), thereby slightly altering the angle in which the 4-benzyl enters the pocket and potentially introducing some strain in the fragment, where the larger pocket of sPLA2-X offers a less restrained binding mode
The high ligand efficiency and potency of 4, coupled with its marked plasma sPLA2 inhibition ability (ICu,50 = 7 nM) triggered a broad characterization campaign to identify potential shortcomings.
As summarized in Table 3, compound 4 proved to be soluble, highly permeable, and metabolically stable; characteristics that translated well in vivo with high bioavailability and low systemic clearance recorded in rat and dog (Table 3). This provided a significant improvement over varespladib, which required a methyl ester prodrug approach (i.e., varespladib methyl) to afford moderate oral absorption (F = 40–55%) in the same species. Compound 4 did not show any significant inhibition of cytochrome P450 enzymes or ion channel activity relevant to cardiac function. Nevertheless, 4 inhibited the uptake of pivastatin in HEK293 cells transfected with the human organic anionic transporter polypeptide 1B1 (OATP1B1) at an estimated IC50 of 2.2 μM, as shown in Table 3. The OATP1B1 transporter is necessary for statin’s hypocholesterolemic action as it mediates their access to the liver compartment where they can then inhibit the function of HMG-CoA reductase.23−25
Table 3. Profilea of Compound 4.
| solubility (pH = 7.4) (μM) | 98 | |
| Papp (10–6 cm/s) | 40.1 | |
| HEP Clint (μL/min/10–6 cells) | 5.2 | |
| hERG, Nav1.5, IKs, Kv4.3, Cav3.2, Cav1.2 IC50 (μM) | >33.3 | |
| CYP450 IC50 (μM) | >20 | |
| OATP1B1 IC50 (μM) | 2.2 | |
| PK | Rat | Dog |
| dose i.v./p.o. (μmol/kg) | 2/4 | 1/2 |
| CL (mL/min/kg) | 1 | 0.3 |
| Vss (L/kg) | 0.22 | 0.26 |
| F (%) | 81 | 82 |
Please see the Supporting Information for experimental details.
Considering that an eventual sPLA2 inhibitor for the treatment of coronary artery disease will need to be coadministered with a statin, as an established standard of care, minimizing the risk for such drug–drug interaction was required. As OATP1B1 recognizes anionic compounds, we reasoned that modification of the molecular environment around the carboxylic acid of 4 might alleviate its interaction with OATP1B1. More specifically, substituting the carbon atom alpha to the carboxylic acid was of special interest as (a) it was postulated to provide a steric impediment to OTAP1B1, (b) it seemed compatible with the binding pocket of the sPLA2-IIa enzyme, and (c) it could enhance potency and/or selectivity through conformational “freezing” of the carboxylic acid side chain via gauche-like effects. Due to structure-based constraints and in order to minimize the impact on compound lipophilicity, we targeted small substituents and synthesized compounds 7–9, following Scheme 2.
According to Scheme 2, the appropriate dimethyl malonate was alkylated using 3-bromobenzyl bromide. The propionic acid derivative was then obtained by hydrolysis, decarboxylation, and re-esterification to yield the methyl ester. Boronylation was accomplished by standard protocols using (Bpin)2 and PdCl2(dppf) to yield the pinacol borane ester, which could be used in the subsequent Suzuki–Miyaura coupling using the benzylated chloro benzonitrile. Finally, racemic 7–9 could be obtained by hydrolysis using hydroxide in alcohol/water mixtures (careful monitoring of the reaction to avoid overhydrolysis to the corresponding diacid is needed).
Addition of a methyl group (7, Table 4) displayed an isoform-specific effect on potency: while it was neutral at sPLA2-IIa, it enhanced inhibition of sPLA2-V and reduced that of sPLA2-X. Overall, this yielded excellent plasma sPLA2 inhibition (ICu,50: 0.1 nM). Remarkably, structural manipulation of the carboxylic acid surroundings by an alpha-methyl group demonstrated the intended effect at a transporter level, and 7 was devoid of OATP1B1 inhibition at the maximum tested concentration (25 μM). Ethyl and cyclopropyl substitutions (8, 9) were not as efficient and had a deleterious effect on metabolic stability in human hepatocytes (cf. 8, 9, and 7, Table 4). We therefore proceeded to separate and characterize the two enantiomers of 7. In line with expectations, sPLA2 inhibition was affected by stereochemistry, and the R enantiomer proved to be the most active, half-maximally inhibiting sPLA2-IIa, -V, and -X at 10, 40, and 400 nM, respectively (Table 4). As a result, (R)-7 was the most potent plasma sPLA2 inhibitor in this study (ICu,50 = 0.1 nM) and, based on its minimized risk for drug–drug interactions with statins, was progressed to further profiling.
Table 4. sPLA2 Potency and Optimization Parameters for Compounds 4–9.
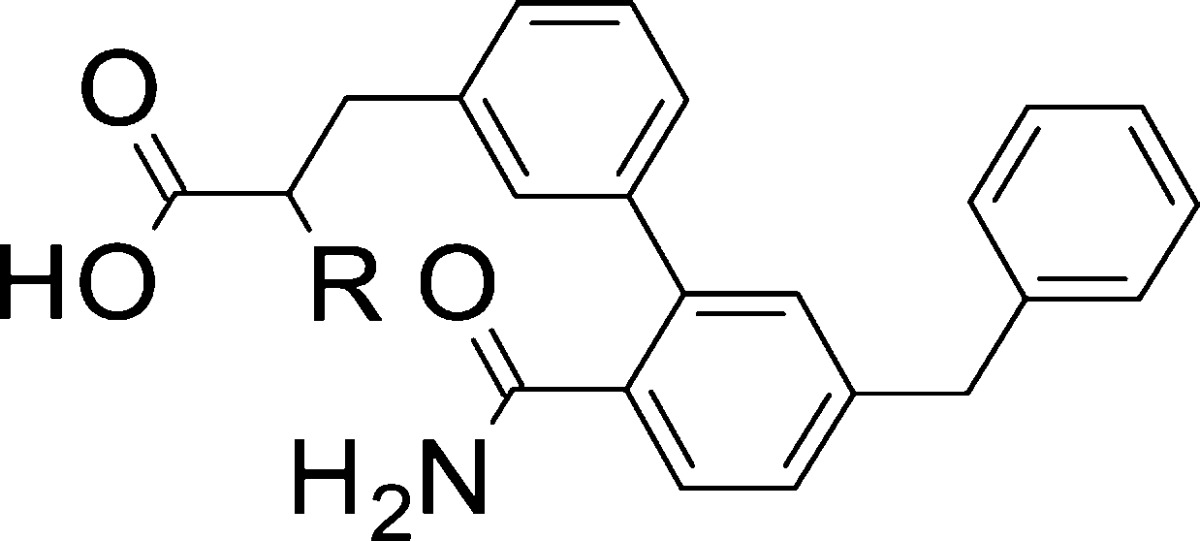
| entry | R | sPLA2-IIa IC50 (μM)a | sPLA2-V IC50 (μM)a | sPLA2-X IC50 (μM)a | plasma ICu,50 (nM)b | HEP Clint (μL/min/10–6 cells)c | OATP1B1 IC50 (μM)d |
|---|---|---|---|---|---|---|---|
| 4 | H | 0.012 | 0.36 | 0.28 | 7 | 5.2 | 2 |
| 7 | Me | 0.011 | 0.07 | 0.75 | 1 | 9.6 | NAe |
| 8 | Et | 0.021 | 0.07 | 0.43 | 0.8 | 25 | NDf |
| 9 | CyPr | 0.018 | 0.25 | 0.58 | 0.9 | 21 | NDf |
| (S)-7 | (S)-Me | 0.038 | 1.2 | 3.8 | ND | 9.3 | NDf |
| (R)-7 | (R)-Me | 0.010 | 0.04 | 0.4 | 0.1 | 12 | NAe |
Mean of at least two experiments. Experimental errors within 20% of value.
Calculated as Plasma sPLA2 IC50 (μM) × unbound fraction in human plasma (Fu)/100.
Intrinsic clearance of test compounds after incubation with human hepatocytes.
Inhibition of pivastatin uptake to HEK293 cells transfected with human OATP1B1.
Not active at maximum tested concentration (25 μM).
Not determined.
When incubated with HepG2 cells, (R)-7 effectively inhibited sPLA2 activity (IC50 < 14 nM) and suppressed production of sPLA2-IIa (IC50 176 ± 28 nM) via a mechanism not yet elucidated.7 Importantly, (R)-7 demonstrated significant sPLA2 activity inhibition (IC50 56 ± 10 nM) in atherosclerotic plaque homogenates, as obtained from carotid endarterectomy of coronary artery disease patients (N = 7).26
In vivo PK analysis of (R)-7 showed consistent high bioavailability and low clearance across different animal species, as summarized in Table 5.
Table 5. PK Parameters of Compound (R)-7.
| mouse | rat | dog | cynomolgus | |
|---|---|---|---|---|
| N i.v./p.o. | 2/2 | 2/2 | 2/2 | 2/2 |
| dose i.v./p.o. (μmol/kg) | 10/50 | 2/8 | 1/3 | 5/15.8 |
| oral AUC0-inf (μM × h) | 69.7 | 70 | 284.1 | 71.3 |
| Cl (mL/min/kg) | 9.2 | 1 | 0.2 | 2.8 |
| Vss (L/kg) | 2.8 | 0.21 | 0.15 | 1.1 |
| F (%) | 76.3 | 84 | 91 | 81 |
Building on the observed PK parameters and the in vitro sPLA2 inhibition measured in cynomolgus monkey plasma (ICu,50 1 nM), we resolved to evaluate the in vivo sPLA2 inhibitory effect of (R)-7.
A 30 mg dose of (R)-7 was orally administered to cynomolgus monkeys (N = 2), as shown in Figure 4. This generated a concentration-dependent inhibition of sPLA2 activity in plasma (ICu,80 = 13 ± 3 nM) that well reflected the time course of (R)-7’s exposure profile (Figure 4).
Figure 4.

Vehicle-corrected plasma sPLA2 activity inhibition: corresponding PK–PD relationship (a) and time course (b) following oral administration of 30 mg (R)-7 to cynomolgus monkeys (N = 2).
In summary, starting from the original fragment hit 1, two design cycles based on structural information, ligand efficiency reasoning, physicochemical property control, medicinal chemistry tactics, and readily available experimental data resulted in the discovery of (R)-7 (AZD2716), a novel, potent sPLA2 inhibitor with excellent PK properties, in vivo efficacy, and minimized risk for drug–drug interactions. Based on the available results and the favorable toxicological profile in rats, dogs, and cynomolgus monkeys, (R)-7 was selected as a clinical candidate for the treatment of coronary artery disease.
Acknowledgments
Tomas Åkerud, Cristian Bodin, Kenth Hallberg, Thomas Olsson, and Hans-Georg Beisel are acknowledged for early lead generation work and useful discussions. Åsa Månson and Fana Hunegnaw are acknowledged for synthetic chemistry contributions.
Biographies
Fabrizio Giordanetto graduated in Medicinal Chemistry (Genoa, Italy) followed by his Ph.D. (London, UK) while working for the chemistry unit of Pharmacia–Pfizer (Italy). After positions at AstraZeneca (Sweden) as Principal Scientist and Project Leader and at Taros (Germany) as Head of Medicinal Chemistry, he has recently joined DE Shaw Research LLC (New York, USA) where he leads medicinal chemistry activities and drug discovery projects. During his career, he worked on several drug discovery programs resulting in multiple clinical candidates spanning oncology, CNS, inflammation, and cardiovascular indications and >100 peer-reviewed publications, book chapters, and international patents.
Daniel Pettersen received his Ph.D. in Organic Chemistry from the University of Gothenburg, Sweden, under the supervision of Prof. Per Ahlberg. Following graduation, he spent 2 years as a Post Doctoral Fellow at the University of Bologna, Italy, in the area of organocatalysis in the laboratories of Prof. Alfredo Ricci. In 2009 he was employed at Astrazeneca, Gothenburg, Sweden, working as a medicinal chemist in the cardiovascular and metabolic areas. Since 2012 he holds a Team Leader position in the Medicinal Chemistry department working on projects in various drug discovery phases. Recent publications include fibrinolysis inhibitors and sPLA2 inhibitors.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00188.
Synthesis details for 2–9, assay protocols, and X-ray data (PDF)
Author Present Address
∇ (F.G.) D. E. Shaw Research, 120 West 45th Street, New York, New York 10036, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Project leader, lead author F.G.; Author D.P.; Medicinal chemists D.P., P.N., M.D., I.S., L.K., N.S.; Bioscientists E.H.-C., B.R.; Enzyme assays U.K.; DMPK L.-O.L.; X-ray J.S., N.D., M.C.
The authors declare no competing financial interest.
Supplementary Material
References
- Murakami M.; Sato H.; Miki Y.; Yamamoto K.; Taketomi Y. A new era of secreted phospholipase A2. J. Lipid Res. 2015, 56, 1248–1261. 10.1194/jlr.R058123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer A. G.; Ghomashchi F.; Le Calvez C.; Bollinger J.; Bezzine S.; Rouault M.; Sadilek M.; Nguyen E.; Lazdunski M.; Lambeau G.; Gelb M. H. Interfacial kinetic and binding properties of the complete set of human and mouse groups I, II, V, X, and XII secreted phospholipases A2. J. Biol. Chem. 2002, 277, 48535–48549. 10.1074/jbc.M205855200. [DOI] [PubMed] [Google Scholar]
- Kudo I.; Murakami M.. Phospholipase A2 enzymes. In Prostaglandins and Other Lipid Mediators; Elsevier, 2002; Vol. 68–69, pp 3–58. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Taketomi Y.; Miki Y.; Sato H.; Hirabayashi T.; Yamamoto K. Recent progress in phospholipase A2 research. In Cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Kimura-Matsumoto M.; Ishikawa Y.; Komiyama K.; Tsuruta T.; Murakami M.; Masuda S.; Akasaka Y.; Ito K.; Ishiguro S.; Ito K.; Ishiguro S.; Morita H.; Sato S.; Morita H.; Sato S.; Ishii T. Expression of secretory phospholipase A2s in human atherosclerosis development. Atherosclerosis 2008, 196, 81–91. 10.1016/j.atherosclerosis.2006.08.062. [DOI] [PubMed] [Google Scholar]
- Hurt-Camejo E.; Camejo G.; Peilot H.; Öörni K.; Kovanen P. Phospholipase A2 in Vascular Disease. Circ. Res. 2001, 89, 298–304. 10.1161/hh1601.095598. [DOI] [PubMed] [Google Scholar]
- Rosenson R. S.; Hurt-Camejo E. Phospholipase A2 enzymes and the risk of therosclerosis. Eur. Heart J. 2012, 33, 2899–2909. 10.1093/eurheartj/ehs148. [DOI] [PubMed] [Google Scholar]
- Mallat Z.; Lambeau G.; Tedgui A. Lipoprotein-Associated and Secreted Phospholipases A2 in Cardiovascular Disease. Circulation 2010, 122, 2183–2200. 10.1161/CIRCULATIONAHA.110.936393. [DOI] [PubMed] [Google Scholar]
- Rosenson R. S.; Gelb M. H. Secretory phospholipase A2: a multifaceted family of proatherogenic enzymes. Curr. Cardiol Rev. 2009, 11, 445–51. 10.1007/s11886-009-0064-2. [DOI] [PubMed] [Google Scholar]
- Shridas P.; Webb N. R. Diverse Functions of Secretory Phospholipases A2. Advances in Vascular Medicine 2014, 2014, 1–11. 10.1155/2014/689815. [DOI] [Google Scholar]
- Lind L.; Simon T.; Johansson L.; Kotti S.; Hansen T.; Machecourt J.; Ninio E.; Tedgui A.; Danchin N.; Ahlström H.; Mallat Z. Circulating levels of secretory- and lipoprotein-associated phospholipase A2 activities: relation to atherosclerotic plaques and future all-cause mortality. Eur. Heart J. 2012, 33, 2946–2954. 10.1093/eurheartj/ehs132. [DOI] [PubMed] [Google Scholar]
- For recent literature and references:Vasilakaki S.; Barbayianni E.; Leonis G.; Papadopoulos M. G.; Mavromoustakos T.; Gelb M. C.; Kokotos G. Development of a potent 2-oxoamide inhibitor of secreted phospholipase A2 guided by molecular docking calculations. Bioorg. Med. Chem. 2016, 24, 1683–1695. 10.1016/j.bmc.2016.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J. D.; Dmitrienko A. A.; Kivitz A. J.; Gluck O. S.; Weaver A. L.; Wiesenhutter C.; Myers S. L.; Sides G. D. A randomized, double-blinded, placebo-controlled clinical trial of LY333013, a selective inhibitor of group II secretory phospholipase A2, in the treatment of rheumatoid arthritis. J. Rheumatol. 2005, 32, 417–23. [PubMed] [Google Scholar]
- Nicholls S. J.; Kastelein J. J.; Schwartz G. G.; Bash D.; Rosenson R. S.; Cavender M. A.; Brennan D. M.; Koenig W.; Jukema J. W.; Nambi V.; Wright R. S.; Menon V.; Lincoff A. M.; Nissen S. E. Varespladib and cardiovascular events in patients with an acute coronary syndrome: the VISTA-16 randomized clinical trial. JAMA 2014, 3, 252–262. 10.1001/jama.2013.282836. [DOI] [PubMed] [Google Scholar]
- Ishimoto Y.; Yamada K.; Yamamoto S.; Ono T.; Notoya M.; Hanasaki K. Group V and X Secretory Phospholipase A(2)s-Induced Modification of High-Density Lipoprotein Linked to the Reduction of Its Antiatherogenic Functions. Biochim. Biophys. Acta, Mol. Cell Res. 2003, 1642, 129–138. 10.1016/S0167-4889(03)00120-4. [DOI] [PubMed] [Google Scholar]
- Hanasaki K.; Yamada K.; Yamamoto S.; Ishimoto Y.; Saiga A.; Ono T.; Ikeda M.; Notoya M.; Kamitani S.; Arita H. Potent Modification of Low Density Lipoprotein by Group X Secretory Phospholipase A2 Is Linked to Macrophage Foam Cell Formation. J. Biol. Chem. 2002, 277, 29116–29124. 10.1074/jbc.M202867200. [DOI] [PubMed] [Google Scholar]
- de Beer F. C.; Connell P. M.; Yu J.; de Beer M. C.; Webb N. R.; van der Westhuyzen D. R. HDL Modification by Secretory Phospholipase A2 Promotes Scavenger Receptor Class B Type I Interaction and Accelerates HDL Catabolism. J. Lipid Res. 2000, 41, 1849–1857. [PubMed] [Google Scholar]
- Erlanson D. A. Introduction to fragment-based drug discovery. Top. Curr. Chem. 2011, 317, 1–32. 10.1007/128_2011_180. [DOI] [PubMed] [Google Scholar]
- Schevitz R. W.; Bach N. J.; Carlson D. G.; Chirgadze N. Y.; Clawson D. K.; Dillard R. D.; Draheim S. E.; Hartley L. W.; Jones N. D.; Mihelich E. D.; Olkowski J. L.; Snyder D. W.; Sommers C.; Wery J.-P. Structure-based design of the first potent and selective inhibitor of human non-pancreatic secretory phospholipase A2. Nat. Struct. Biol. 1995, 2, 458–465. 10.1038/nsb0695-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeau G.; Gelb M. H. Biochemistry and physiology of mammalian secreted phospholipase A2. Annu. Rev. Biochem. 2008, 77, 495–520. 10.1146/annurev.biochem.76.062405.154007. [DOI] [PubMed] [Google Scholar]
- Pernas P.; Masliah J.; Olivier J. – L.; Salvat C.; Rybkine T.; Bereziat G. Type II Phospholipase A2 recombinant overexpression enhance stimulated arachidonic acid release. Biochem. Biophys. Res. Commun. 1991, 178, 1298–1305. 10.1016/0006-291X(91)91035-B. [DOI] [PubMed] [Google Scholar]
- Kristensen J.; Lysén M.; Vedsö P.; Begtrup M. Synthesis of ortho substituted arylboronic esters by in situ trapping of unstable lithio intermediates. Org. Lett. 2001, 10, 1435–1437. 10.1021/ol015598+. [DOI] [PubMed] [Google Scholar]
- Hsiang B.; Zhu Y.; Wang Z.; Wu Y.; Sasseville V.; Yang W. P.; Kirchgessner T. G. A novel human hepatic organic anion transporting polypeptide (OATP2). Identification of a liver-specific human organic anion transporting polypeptide and identification of rat and human hydroxymethylglutaryl-CoA reductase inhibitor transporters. J. Biol. Chem. 1999, 274, 37161–37168. 10.1074/jbc.274.52.37161. [DOI] [PubMed] [Google Scholar]
- Mizuno N.; Niwa T.; Yotsumoto Y.; Sugiyama Y. Impact of drug transporter studies on drug discovery and drug development. Pharmacol Rev. 2003, 55, 425–461. 10.1124/pr.55.3.1. [DOI] [PubMed] [Google Scholar]
- Shitara Y.; Horie T.; Sugiyama Y. Transporters as a determinant of drug clearance and tissue distribution. Eur. J. Pharm. Sci. 2006, 27, 425–446. 10.1016/j.ejps.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Hurt-Camejo E.; Andersen S.; Standal R.; Rosengren B.; Sartipy P.; Stadberg E.; Johansen B. Localization of Non Pancreatic Secretory Phospholipase A2 in Normal and Atherosclerotic Arteries: Activity of the Isolated Enzyme on Low Density Lipoproteins. Arterioscler., Thromb., Vasc. Biol. 1997, 17, 300–309. 10.1161/01.ATV.17.2.300. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.