

Abstract
Self-repair of nuclear DNA damage is the most known reason that leads to drug resistance of cancer tissue and limited therapeutic efficacy of anticancer drugs. Inhibition of protein phosphatase 2A (PP2A) would block DNA damage-induced defense of cancer cells to suppress DNA repair for enhanced cancer treatment. Here, we combined a PP2A inhibitor LB (4-(3-carboxy-7-oxa-bicyclo[2.2.1]heptane-2-carbonyl) piperazine-1-carboxylic acid tert-butyl ester) and the DNA damage chemotherapeutic drug cisplatin through a simple physical superposition. The two drugs administrated at a ratio of 1:1 exhibited an optional synergistic antitumor efficacy in vitro and in vivo. LB was demonstrated to specifically activate the protein kinase B (Akt) and mitogen-activated protein kinases (MAPK) signaling pathways by PP2A inhibition to overcome cell cycle arrest caused by cisplatin-induced DNA damage.
Keywords: Protein phosphatase 2A, cisplatin, DNA repair, combination treatment
Cancer remains one of the leading causes of morbidity and mortality around the world, which continues to increase with around 10 million new cases every year.1,2 Although traditional chemotherapy is quite beneficial in improving survival rate, it has also shown a lot of hindrances including severe toxicity, limited efficiency, and especially unexpected drug resistance.3−5 It is well-known that drug resistance is mediated by multiple defense mechanisms of cancer cells, such as DNA repair, inhibition of apoptosis, and cell cycle arrest.6 PP2A, a ubiquitous and multifunctional serine/threonine phosphatase, involves mitotic progression and cellular responses to DNA damage, playing a dynamic role in conquering DNA damage repair.7−9 PP2A inhibitors have been demonstrated to force cancer cells prematurely into mitosis and subsequently induce cell apoptosis. Hence, combination of PP2A inhibitors with chemotherapeutics should be an attractive strategy to improve therapeutic effect and combat drug resistance to tumor.
Recently, the derivatives of cantharidin without evident acute or chronic toxicity as PP2A inhibitors showed enhanced antitumor activity with DNA-damaging agents by specifically blocking PP2A.10,11 Zhuang studied the effect of a derivative LB combined with TMZ for the NB line SH-SY5Y.12 Results demonstrated that LB inhibited the cancer defense mechanisms in response to DNA damage induced by TMZ. Another study also showed that LB could improve the efficacy of DOX against stem cell-derived cancers.13 Platinum(II) complexes, cisplatin in particular, are employed in 50% of all cancer therapies.14,15 However, the clinic application of platinum(II) is limited by severe side effects and acquired resistance.16−18 There are certain known mechanisms of cisplatin resistance, including increased DNA adduct repair, perturbed cell cycle, and inhibition of apoptosis.19 Based on this, cantharidin derivatives and combination with cisplatin have been reported to overcome drug resistance and enhance antitumor efficacy of cisplatin. In our previous report, canthaplatin was obtained by appending a PP2A inhibitor LB to cisplatin in a platinum(IV) prodrug form for DNA repair blocking.20 Recently, we also demonstrated a polymer–(multifunctional single-drug) conjugate strategy, in which different drug components (platinum and PP2A inhibitor) were rationally integrated.21−24 Nevertheless, these systems were established on tedious chemical modification of platinum(II). Furthermore, the synergistic mechanism of platinum(II) and PP2A inhibitor was ambiguous and the optimal drug ratio was also vacant.
In this study, we demonstrated a combination treatment of cisplatin and LB through a simple physical superposition. This method would avoid fussy chemical modification and maintain the original valence state of cisplatin. The two drugs were typically internalized via passive diffusion through the cell membrane, and played a synergistic antitumor effect. Cisplatin would disrupt DNA structure through formation of intrastrand and interstrand cross-links.25,26 Nevertheless, in this process, cancer cells would trigger DNA damage repair mechanisms procedurally.27−29 We hypothesized that LB could block the replication checkpoints triggered by DNA repair and drive quiescent cancer cells into cycle through the significant inhibition effect of PP2A when combined with cisplatin (Scheme 1). Herein, we displayed the superior drug ratio of cisplatin and LB for synergistic effect in vitro and in vivo, and the possible synergetic mechanism was also investigated.
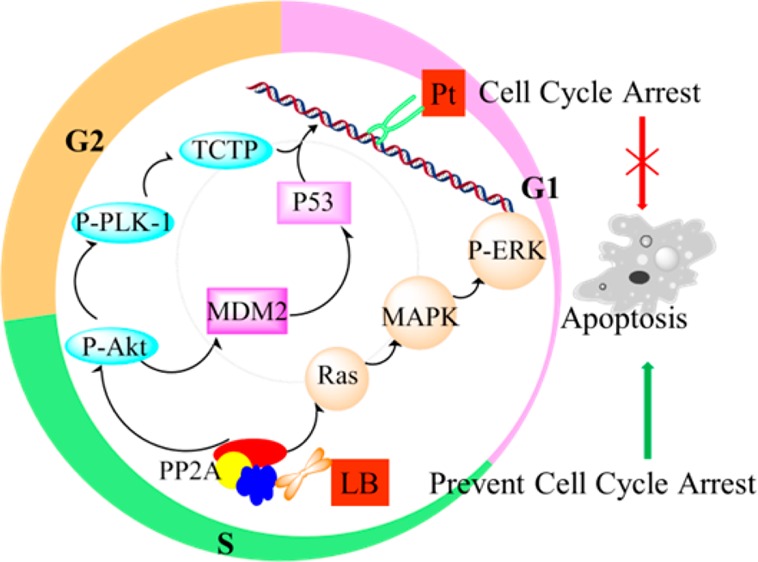
Scheme 1. Schematic Representation of the Synergistic Mechanism of Cisplatin and a PP2A Inhibitor, LB.
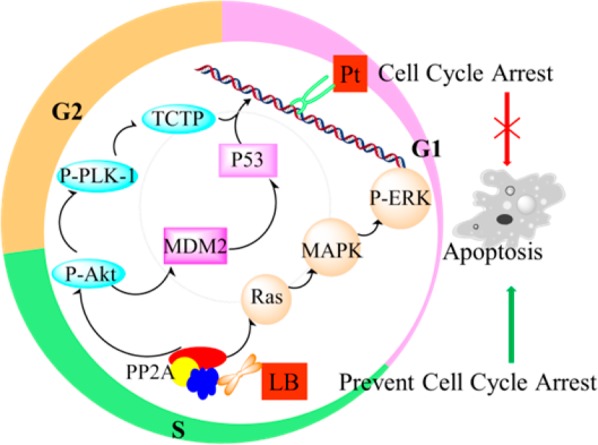
P-Akt, phosphorylated Akt; P-Plk-1, phosphorylated polo-like kinase 1; MDM2, murine double minute 2; TCTP, translationally controlled tumor protein; P-ERK, phosphorylated extracellular regulated protein kinases.
First, the potential for LB to specifically inhibit PP2A was investigated with a PP2A activity assay. As shown in Figure S2, it was found that the PP2A activity level in cervical cancer HeLa cells exposed to LB for 3 h decreased to 78.5% of the control level, while those exposed to combination treatment of LB and cisplatin decreased to 86.0%. The reduced PP2A activity level clearly proved that LB could possess the capability to inhibit PP2A function when combined with cisplatin.
The cytotoxicity of LB and cisplatin was evaluated against HeLa cells and lung carcinoma A549 cells through a MTT (3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide) assay. Both LB and cisplatin showed concentration- and time-dependent cytotoxicity (Figure S3). IC50 (the 50% inhibitory concentration) values of LB and cisplatin were shown in Table S1. Cisplatin showed high cytotoxicity to both cancer cells, while LB exhibited moderate cytotoxicity especially against A549 cells. The result indicated that the low-toxic LB would be a suitable sensitizer to promote the anticancer activity of cisplatin.
The cytotoxicity of combination treatment of cisplatin and LB with different mole ratios was further evaluated (Figure S4). The combination treatment with a cisplatin/LB ratio at 1:1 showed the highest anticancer activity to A549 cells. The combination index (CI) values lower than, equal to, and higher than 1, denote synergism, additivity, and antagonism, respectively.30,31 The CI50 values (CI at 50% inhibitory) against drug effect level were also carried out (Figure 1). The detail computational formula was shown in Supporting Information. For A549 cells after 24 h of incubation, all the CI50 values of combination treatment with different ratios are lower than 1, indicating the synergy effect of cisplatin and LB against A549 cells. The CI50 with a cisplatin/LB ratio of 1:1.5 was about 1.7-fold lower than that of 1:2.5. For HeLa cells after 24 h of incubation, combination treatment with a ratio of 1:0.5 achieved the best synergistic effect. However, when the ratio was 1:2 or 1:0.3, they did not display synergism efficiently, even exhibited antagonism after 48 and 72 h. These results demonstrated that balancing dosage of the two individual drugs was very important to achieve the optimal anticancer efficacy. It was also found that the CI values increased with time. Finally, the combination treatment with a cisplatin/LB molar ratio of 1:1 was chosen for further study because of the excellent synergism for both HeLa and A549 cells.
Figure 1.
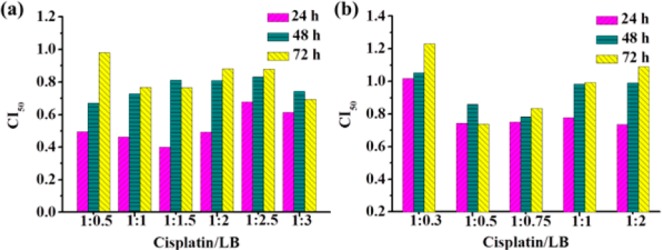
CI50 values of cisplatin and LB with different mole ratios against (a) A549 cells and (b) HeLa cells.
One of the hallmarks of cancer cells is the capability to evade apoptosis. The apoptosis rate of cancer cells after treatment is another important index to evaluate the synergistic effect. Annexin V-FITC and propidium iodide (PI) were used to evaluate the percentage of living cells (lower left), early apoptosis cells (lower right), and late apoptosis or necrosis cells (upper right) (Figure S5). After 24 h of treatment with drug concentration at IC50 values, combination treatment led to 41.5% of total apoptosis, while 36.9% for cisplatin and 35.5% for LB treatment against A549 cells (Figure 2). Similar results were also observed in HeLa cells. For instance, cisplatin, LB, and combination treatment induced 4.8%, 5.4%, and 6.4% of early apoptosis and 54%, 21.8%, and 64.1% of late apoptosis, respectively. In short, LB significantly increased the cell apoptosis when combined with cisplatin. The main known apoptosis mechanism induced by cisplatin is DNA cross-linking damage. However, cancer cells would establish defense mechanism in the fate of acute DNA damage. Hence, the enhanced anticancer activity is believed to be a result of combining cisplatin, which caused acute DNA damage, with LB, which forces cells with DNA damage to bypass cell cycle checkpoints and go through lethal mitosis by PP2A inhibition.
Figure 2.
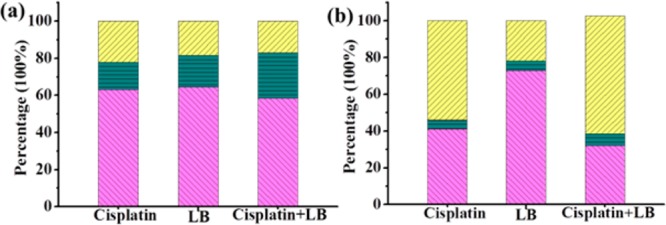
Apoptosis of (a) A549 cells and (b) HeLa cells after 24 h of drug treatments: (yellow) late apoptosis; (green) early apoptosis; (pink) living cells.
To determine the effect of PP2A inhibition on cell cycle arrest, in the fate of acute DNA damage, we analyzed cell cycle patterns of HeLa and A549 cells after exposure to cisplatin, LB, and combination treatment for 24 h. For HeLa cells, compared to control samples, 24 h exposure to LB did not alter the cell cycle percentages. Exposure to cisplatin alone increased the percentage of cells in S phase, indicating increased cancer cell senescence (Figure 3). As anticipated, combination treatment prominently increased percentage of cells in G2/M phases, corresponding to the loss of checkpoints ordinarily induced by acute DNA damage.7,13 Likewise, the DNA replication analysis of A549 cells revealed that 24 h exposure to LB decreased G1 phase, prominently increased G2/M phase, and little effect on S phase. Exposure to cisplatin alone at the same dose had little effect on cell cycle on the whole. Exposure to both drugs resulted in markedly decreased G1 and significantly increased S. It was evident that cisplatin arrested the cancer cells in S phase, whereas LB would abrogate cisplatin-induced cell cycle arrest and accelerate the malignant cell entry into G2/M phase. As mentioned above, the chemotherapeutic action of cisplatin was to cross-link with DNA, thereby interfering with transcription and/or DNA replication in tumor cells; LB could significantly inhibit the effect of PP2A, drive quiescent cancer cells with DNA damage into cycle and block some replication checkpoints, and in turn cause cell apoptosis.
Figure 3.
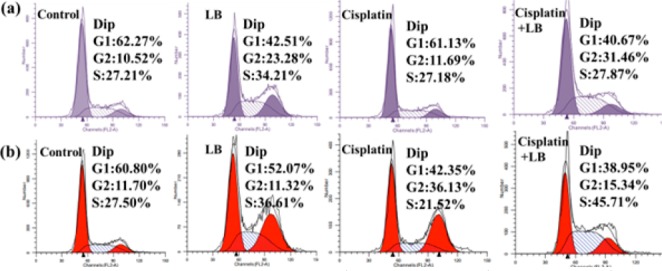
Cell cycle distribution of (a, purple color) HeLa and (b, red color) A549 cells after incubation of drugs for 24 h. The percentages of G1, S, and G2/M phases are indicated.
PP2A is a ubiquitous serine/threonine phosphatase expressed in eukaryotic cells.32,33 The inhibition of PP2A by LB would activate several important signaling pathways including Akt to accelerate inappropriate entry of cells into mitosis, bypass cell cycle arrest, and block some replication checkpoints triggered by DNA damage.7,9 Hence, to explore the mechanism of how LB abrogated cisplatin-induced cell cycle arrest on molecular level, we further performed immunofluorescence assay of HeLa cells after 24 h of drug treatment. As shown in Figure 4, all cells have the same strong expression of tubulin and total Akt. Compared to control and cisplatin treatment, both LB treatment and combination treatment induced higher levels of P-Akt. The increased expression of P-Akt would further activate the phosphorylation of MDM2 and suppress TCTP, which further induce apoptosis from continued cell cycle progression.
Figure 4.
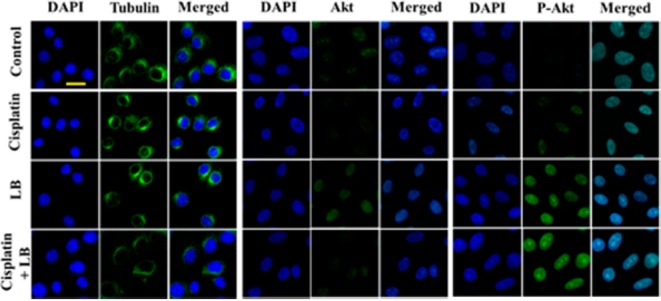
Immunofluorescence of tubulin, Akt, P-Akt (green), nuclear morphology (DAPI, blue), and merged in HeLa cells after 24 h exposure to DMEM only, 2.5 μM cisplatin, 2.5 μM LB, and both drugs at the same concentration (scale bar: 20 μm).
Previous studies have demonstrated that MAPK signaling pathway also played a critical role in PP2A action and regulating cell cycle progression.34−36 ERK is an important pathway in MAPK pathway, involved in the regulation of DNA damage response (DDR) and cell-cycle checkpoint pathways dominated by PP2A. Therefore, we next investigated whether the MAPK pathway was activated in HeLa cells using specific antibodies against the phosphorylated ERK. As shown in Figure 5a, cisplatin exhibited slight elevation on the expression of P-ERK, while enhanced expression of P-ERK were observed both in LB alone and in combination treatment. In contrast, no significant changes of total ERK were detected in response to different treatments. The results suggested that inhibition of PP2A by LB could also activate ERK in tumor cells and aberrantly accelerate cancer cell entry into mitosis, resulting in increased cell vulnerability to DNA damage, disordered replication, and death.
Figure 5.
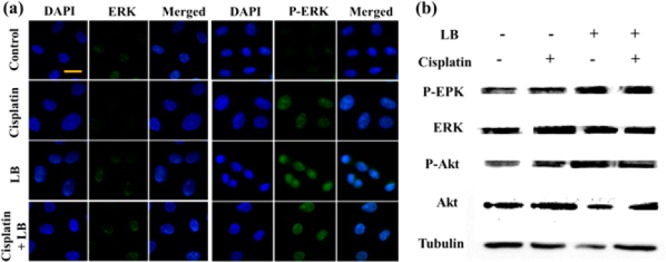
(a) Immunofluorescence and (b) Western blotting against HeLa cells after 24 h exposure to cisplatin, LB, and combination treatment (scale bar: 20 μm).
The expression of P-Akt and P-ERK was also evaluated by Western blotting. As shown in Figure 5b, it was clear to find that LB increased the expression of P-Akt and P-ERK compared to the control group. LB plus cisplatin assumed the similar phenomenon with LB alone, whereas cisplatin alone had little effect on the expression of P-Akt and P-ERK. All the results were in agreement with the above immunofluorescence studies. Taken together, the data demonstrated that PP2A inhibition by LB would activate the Akt and MAPK pathways, rendering cancer cells to overcome cisplatin-induced damaged DNA repair and undergo lethal apoptosis.
Finally, the synergistic antitumor efficacy of cisplatin and LB at a molar ratio of 1:1 was evaluated in vivo against KM mice with subcutaneous xenografted U14 (mouse uterine cervical cancer cell) tumor (Figure 6a). Tumor volume grew rapidly in control animals, and the average tumor volumes increased to about 1000 mm3 after 2 weeks. Cisplatin alone caused marked repression, and the tumor size decreased to about 270 mm3 after half a mouth. LB alone did not inhibit the growth of tumors for the first week while displayed a moderate effect in the second week. LB plus cisplatin at the same dose and schedule as administrated individually significantly decreased the growth of the xenografts. For instance, the average tumor volume for the combination group was about 97 ± 18 mm3, while cisplatin group was 178 ± 33 mm3 at day 10. It is reasonable that the improved antitumor efficacy of cisplatin combined with LB was attributed to PP2A inhibition, which rendered cancer cells less efficient in repairing DNA damage and more vulnerable to death.
Figure 6.
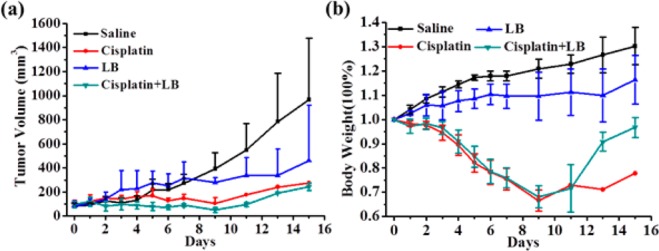
(a) Relative tumor volume and (b) body weight variation of KM mice treated with saline, cisplatin (5 mg/kg), LB (1.5 mg/kg), and cisplatin (5 mg/kg) + LB (1.5 mg/kg).
Body weight was also monitored to evaluate systemic toxicity (Figure 6b). LB alone had little effect on body weight, suggesting very low toxicity. Cisplatin alone caused a significant decrease in total body weight during the test period. Although combination treatment of cisplatin and LB also caused an obvious body weight loss in the first week, the body weight was finally comparable to the initial body weight at day 0 after 2 weeks. In short, these results demonstrated that the LB would not only improve the antitumor efficacy but also relieve the systemic toxicity of cisplatin at a certain degree in vivo.
In the present investigation, a PP2A inhibitor LB was used to combine with cisplatin for enhanced cancer therapy. The optimal drug ratio of cisplatin and LB for maximum synergistic effect in vitro was demonstrated. Meanwhile, the molecular mechanism assumed that LB could block cisplatin-induced DNA damage repair through activation of Akt and MAPK signaling pathways. As a result, the therapeutic efficacy of cisplatin was markedly sensitized by LB in vitro and in vivo. Hence, for future platinum-based drugs, or other DNA damage chemotherapeutic agents, combination with PP2A inhibitors that render cancer cells more susceptible may have more potential in the clinical application.
Acknowledgments
The authors would like to thank the financial support from the National Natural Science Foundation of China (No. 51403198 and 51573069) and Jilin Provincial Science and Technology Department (No. 20150520019JH).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00236.
Full experimental details for descriptions of biological assays (PDF)
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Palvai S.; More P.; Mapara N.; Basu S. Chimeric Nanoparticle: A Platform for Simultaneous Targeting of Phosphatidylinositol-3-Kinase Signaling and Damaging DNA in Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 18327–18335. 10.1021/acsami.5b04015. [DOI] [PubMed] [Google Scholar]
- Mallick A.; More P.; Ghosh S.; Chippalkatti R.; Chopade B. A.; Lahiri M.; Basu S. Dual Drug Conjugated Nanoparticle for Simultaneous Targeting of Mitochondria and Nucleus in Cancer Cells. ACS Appl. Mater. Interfaces 2015, 7, 7584–7598. 10.1021/am5090226. [DOI] [PubMed] [Google Scholar]
- He C.; Poon C.; Chan C.; Yamada S. D.; Lin W. Nanoscale Coordination Polymers Codeliver Chemotherapeutics and siRNAs to Eradicate Tumors of Cisplatin-Resistant Ovarian Cancer. J. Am. Chem. Soc. 2016, 138, 6010–6019. 10.1021/jacs.6b02486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z.-T.; Chen Z.-Y.; Sun C.-Y.; Li H.-J.; Wang H.-X.; Cheng Q.-Q.; Zuo Z.-Q.; Wang J.-L.; Liu Y.-Z.; Wang Y.-C.; Wang J. Overcoming tumor resistance to cisplatin by cationic lipid-assisted prodrug nanoparticles. Biomaterials 2016, 94, 9–19. 10.1016/j.biomaterials.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Shen J.; Su H.; Mu G.; Sun J. H.; Tan C. P.; Liang X. J.; Ji L. N.; Mao Z. W. Co-Delivery of Cisplatin Prodrug and Chlorin e6 by Mesoporous Silica Nanoparticles for Chemo-Photodynamic Combination Therapy to Combat Drug Resistance. ACS Appl. Mater. Interfaces 2016, 8, 13332. 10.1021/acsami.6b03881. [DOI] [PubMed] [Google Scholar]
- Zhuang Z.; Lu J.; Lonser R.; Kovach J. S. Enhancement of cancer chemotherapy by simultaneously altering cell cycle progression and DNA-damage defenses through global modification of the serine/threonine phospho-proteome. Cell Cycle 2009, 8, 3303–3306. 10.4161/cc.8.20.9689. [DOI] [PubMed] [Google Scholar]
- Chang K.-E.; Wei B.-R.; Madigan J. P.; Hall M. D.; Simpson R. M.; Zhuang Z.; Gottesman M. M. The Protein Phosphatase 2A Inhibitor LB100 Sensitizes Ovarian Carcinoma Cells to Cisplatin-Mediated Cytotoxicity. Mol. Cancer Ther. 2015, 14, 90–100. 10.1158/1535-7163.MCT-14-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C. S.; Ho W.; Zhang C.; Yang C.; Elder J. B.; Zhuang Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833. 10.1080/15384047.2015.1040961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Hong C. S.; Hu X.; Yang C.; Wang H.; Zhu D.; Moon S.; Dmitriev P.; Lu J.; Chiang J.; Zhuang Z.; Zhou Y. Inhibition of protein phosphatase 2A with the small molecule LB100 overcomes cell cycle arrest in osteosarcoma after cisplatin treatment. Cell Cycle 2015, 14, 2100–2108. 10.1080/15384101.2015.1041693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Chen Z. The Effects of Cantharidin and Cantharidin Derivates on Tumour Cells. Anti-Cancer Agents Med. Chem. 2009, 9, 392–396. 10.2174/1871520610909040392. [DOI] [PubMed] [Google Scholar]
- Hart M. E.; Chamberlin A. R.; Walkom C.; Sakoff J. A.; McCluskey A. Modified norcantharidins: synthesis, protein phosphatases 1 and 2A inhibition, and anticancer activity. Bioorg. Med. Chem. Lett. 2004, 14, 1969–1973. 10.1016/j.bmcl.2004.01.093. [DOI] [PubMed] [Google Scholar]
- Lu J.; Kovach J. S.; Johnson F.; Chiang J.; Hodes R.; Lonser R.; Zhuang Z. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 11697–11702. 10.1073/pnas.0905930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Peng Y.; Wang F.; Tan X.; Liu N.; Fan S.; Wang D.; Zhang L.; Liu D.; Wang T.; Wang S.; Zhou Y.; Su Y.; Cheng T.; Zhuang Z.; Shi C. A synthetic cantharidin analog for the enhancement of doxorubicin suppression of stem cell-derived aggressive sarcoma. Biomaterials 2010, 31, 9535–9543. 10.1016/j.biomaterials.2010.08.059. [DOI] [PubMed] [Google Scholar]
- Galanski M.; Jakupec M. A.; Keppler B. K. Update of the preclinical situation of anticancer platinum complexes: Novel design strategies and innovative analytical approaches. Curr. Med. Chem. 2005, 12, 2075–2094. 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- Wang D.; Lippard S. J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discovery 2005, 4, 307–320. 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- Dhar S.; Lippard S. J. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 22199–22204. 10.1073/pnas.0912276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.-J.; Meng H.; Wang Y.; He H.; Meng J.; Lu J.; Wang P. C.; Zhao Y.; Gao X.; Sun B.; Chen C.; Xing G.; Shen D.; Gottesman M. M.; Wu Y.; Yin J.-j.; Jia L. Metallofullerene nanoparticles circumvent tumor resistance to cisplatin by reactivating endocytosis. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7449–7454. 10.1073/pnas.0909707107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min Y.; Mao C.-Q.; Chen S.; Ma G.; Wang J.; Liu Y. Combating the Drug Resistance of Cisplatin Using a Platinum Prodrug Based Delivery System. Angew. Chem., Int. Ed. 2012, 51, 6742–6747. 10.1002/anie.201201562. [DOI] [PubMed] [Google Scholar]
- Xue X.; You S.; Zhang Q.; Wu Y.; Zou G. Z.; Wang P. C.; Zhao Y. L.; Xu Y.; Jia L.; Zhang X.; Liang X. J. Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction. Mol. Pharmaceutics 2012, 9, 634–644. 10.1021/mp200571k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D.; Cong Y.; Qi Y.; He S.; Xiong H.; Wu Y.; Xie Z.; Chen X.; Jing X.; Huang Y. Overcoming tumor resistance to cisplatin through micelle-mediated combination chemotherapy. Biomater. Sci. 2015, 3, 182–191. 10.1039/C4BM00305E. [DOI] [PubMed] [Google Scholar]
- Zhou D.; Xiao H.; Meng F.; Li X.; Li Y.; Jing X.; Huang Y. A polymer-(tandem drugs) conjugate for enhanced cancer treatment. Adv. Healthcare Mater. 2013, 2, 822–827. 10.1002/adhm.201200385. [DOI] [PubMed] [Google Scholar]
- Wang E.; Xiong H.; Zhou D.; Xie Z.; Huang Y.; Jing X.; Sun X. Co-delivery of oxaliplatin and demethylcantharidin via a polymer-drug conjugate. Macromol. Biosci. 2014, 14, 588–596. 10.1002/mabi.201300402. [DOI] [PubMed] [Google Scholar]
- Zhou D.; He S.; Cong Y.; Xie Z.; Chen X.; Jing X.; Huang Y. A polymer-(multifunctional single-drug) conjugate for combination therapy. J. Mater. Chem. B 2015, 3, 4913–4921. 10.1039/C5TB00576K. [DOI] [PubMed] [Google Scholar]
- Cong Y.; Wang Z.; He S.; Zhou D.; Li J.; Xie Z.; Chen X.; Jing X.; Huang Y. Multifunctional single-drug loaded nanoparticles for enhanced cancer treatment with low toxicity in vivo. RSC Adv. 2016, 6, 20366–20373. 10.1039/C5RA26372G. [DOI] [Google Scholar]
- Han X.; Sun J.; Wang Y.; He Z. Recent Advances in Platinum (IV) Complex-Based Delivery Systems to Improve Platinum (II) Anticancer Therapy. Med. Res. Rev. 2015, 35, 1268–1299. 10.1002/med.21360. [DOI] [PubMed] [Google Scholar]
- Wang X.; Wang X.; Guo Z. Functionalization of Platinum Complexes for Biomedical Applications. Acc. Chem. Res. 2015, 48, 2622–2631. 10.1021/acs.accounts.5b00203. [DOI] [PubMed] [Google Scholar]
- Peltomaki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J. Clin. Oncol. 2003, 21, 1174–1179. 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- Rabik C. A.; Dolan M. E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L. P.; Hamilton T. C.; Schilder R. J. Platinum resistance: The role of DNA repair pathways. Clin. Cancer Res. 2008, 14, 1291–1295. 10.1158/1078-0432.CCR-07-2238. [DOI] [PubMed] [Google Scholar]
- Chou T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Wientjes M. G.; An J. L. S. Evaluation of combination chemotherapy: Integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin. Cancer Res. 2004, 10, 7994–8004. 10.1158/1078-0432.CCR-04-1087. [DOI] [PubMed] [Google Scholar]
- Janssens V.; Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. 10.1042/bj3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K.-F.; Yu H.-C.; Liu T.-H.; Lee S.-S.; Chen P.-J.; Cheng A.-L. Synergistic interactions between sorafenib and bortezomib in hepatocellular carcinoma involve PP2A-dependent Akt inactivation. J. Hepatol. 2010, 52, 88–95. 10.1016/j.jhep.2009.10.011. [DOI] [PubMed] [Google Scholar]
- Chen T.; Wong Y.-S. Selenocystine Induces S-Phase Arrest and Apoptosis in Human Breast Adenocarcinoma MCF-7 Cells by Modulating ERK and Akt Phosphorylation. J. Agric. Food Chem. 2008, 56, 10574–10581. 10.1021/jf802125t. [DOI] [PubMed] [Google Scholar]
- Boldt S.; Weidle U. H.; Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis 2002, 23, 1831–1838. 10.1093/carcin/23.11.1831. [DOI] [PubMed] [Google Scholar]
- Chen J.; Li L.; Su J.; Li B.; Chen T.; Wong Y.-S. Synergistic Apoptosis-Inducing Effects on A375 Human Melanoma Cells of Natural Borneol and Curcumin. PLoS One 2014, 9, e101277–e101277. 10.1371/journal.pone.0101277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.