

Abstract
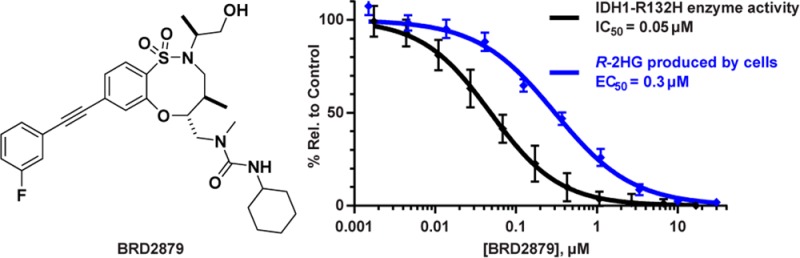
Evidence suggests that specific mutations of isocitrate dehydrogenases 1 and 2 (IDH1/2) are critical for the initiation and maintenance of certain tumor types and that inhibiting these mutant enzymes with small molecules may be therapeutically beneficial. In order to discover mutant allele-selective IDH1 inhibitors with chemical features distinct from existing probes, we screened a collection of small molecules derived from diversity-oriented synthesis. The assay identified compounds that inhibit the IDH1-R132H mutant allele commonly found in glioma. Here, we report the discovery of a potent (IC50 = 50 nM) series of IDH1-R132H inhibitors having 8-membered ring sulfonamides as exemplified by the compound BRD2879. The inhibitors suppress (R)-2-hydroxyglutarate production in cells without apparent toxicity. Although the solubility and pharmacokinetic properties of the specific inhibitor BRD2879 prevent its use in vivo, the scaffold presents a validated starting point for the synthesis of future IDH1-R132H inhibitors having improved pharmacological properties.
Keywords: BRD2879, isocitrate dehydrogenase, 2-hydroxyglutarate, glioma, AML, cancer, diversity-oriented synthesis, high-throughput screening, allele-selective probe, small-molecule probe
Systematic efforts to characterize the genomes of patient tumors are revealing the genomic alterations that cause and maintain different cancers. Somatic mutations in the genes encoding the isocitrate dehydrogenases IDH1 and IDH2 have been found in >70% of grade II–III gliomas and secondary glioblastomas,1,2 ∼17% of acute myeloid leukemias (AML),3,4 ∼56% of central and periosteal chondrosarcoma,5 and sporadically in other tumor types. Mutations are nearly always heterozygous and occur frequently at codons IDH1-R132, IDH2-R172, or IDH2-R140. IDH enzymes normally catalyze the interconversion of isocitrate and α-ketoglutarate (α-KG), but these mutations unmask an otherwise cryptic NADPH-dependent ketoreductase activity, allowing the enzyme to reduce α-KG to (R)-2-hydroxyglutarate (R-2HG).3,6 As a result, R-2HG levels are elevated >50-fold in samples from patients with IDH mutations.3
The pathogenesis of IDH mutant tumors is thought to center on the ability of R-2HG to act as an “oncometabolite”. Due to its structural similarity to α-KG, R-2HG competitively inhibits several α-KG-dependent dioxygenases when present at the high concentrations observed in IDH mutant tumors. In particular, R-2HG impairs DNA demethylation through inhibition of TET2,7 impairs histone demethylation through inhibition of various lysine demethylases,8,9 and modulates hypoxic stress response through activation of EGLN1.10 These molecular changes are thought to cause the enhanced proliferation and impaired differentiation observed in IDH mutant tumors. The mutual exclusivity of IDH and TET2 mutations in AML tumors7 and the ability of exogenous R-2HG to induce leukemogenesis in blood cells11 further implicate R-2HG as a critical mediator in how mutant IDH contributes to AML. In IDH1 mutant glioma models, hypermethylation of CTCF binding sites has been shown to cause genomic insulator dysfunction leading to aberrant activation of PDGFRA, providing an explanation for how R-2HG impacts cancer initiation in the brain.12
The discovery of mutant allele-selective small-molecule inhibitors of IDH1 and IDH2 has enabled the validation of these enzymes as therapeutic targets. In particular, studies of AGI-5198, AGI-6780, and GSK321 in disease models indicate that these inhibitors may shift cancer cells toward a more mature and less proliferative state, suggesting such compounds might form the basis of a future differentiation therapy.13−16 Early results from clinical trials of mutant-IDH inhibitors AG-12017 and AG-22118 in IDH mutant AML suggest that the compounds may be effective in this context.
While the clinical progress of existing IDH inhibitors is encouraging, the fact that cancers frequently develop resistant alleles to targeted therapy motivates the need to develop structurally and mechanistically diverse “next-in-class” compounds targeting IDH. The cocrystallization of mutant IDH1 with probes such as GSK321, VVS, and SYC-435 has already uncovered distinct binding modes for inhibitors of mutant IDH1 (Chart 1).16,19−21 To identify new scaffolds for inhibition of IDH1-R132H, we screened compounds derived from diversity-oriented synthesis (DOS). The DOS collection contains molecules with increased sp3 content and myriad stereochemical features compared with those found in standard commercial libraries,22,23 so we hypothesized that our screen could reveal a probe with unique binding characteristics. Here we report the discovery of BRD2879, a potent and cell-active inhibitor of IDH1-R132H with a markedly different structure from previously reported probes. While BRD2879 exemplifies a distinct class of IDH1 inhibitors, its physical properties need improvement to yield a suitable probe for in vivo applications.
Chart 1. Structures of Selected Mutant IDH Inhibitors.

a IDH1 inhibitor.
b IDH2 inhibitor.
c Clinical compound.
To identify small molecules that inhibit IDH1-R132H, we developed an in vitro assay measuring the enzyme’s ketoreductase activity in 1536-well plates. Enzyme activity was measured by detecting consumption of NADPH in a diaphorase-coupled reaction with a fluorescence readout. The screening buffer included 0.01% Tween 20 detergent to minimize false positives due to compound aggregation. IDH1 is reported to function with either Mn2+ or Mg2+ as a cofactor, and we performed screens under both conditions. Surprisingly, the screening results were markedly different depending on the cofactor used (Figure S1). Although enzyme turnover was substantially faster using the IDH1-R132H-Mn2+ complex (Figure S2), the potency of inhibitors in this model system proved to be a poor predictor of cellular activity (Table S1), and we prioritized the Mg2+ complex for further study. The IDH1-R132H-Mg2+ complex was screened in duplicate against 89,093 compounds from the Broad Institute’s DOS screening library. Primary screening at 15 μM yielded 551 positives with ≥60% inhibition in both replicates (hit rate 0.6%).
We retested positives in 8-point dose in the primary screening assay and in an orthogonal enzymatic assay detecting NADPH by absorbance to confirm compound activity and to mitigate detection-specific artifacts. We then tested compounds for selectivity with respect to wild-type IDH1. Wild-type IDH1 inhibition was measured using an assay analogous to that used for the primary screen, measuring the production of NADPH from NADP+ and isocitrate in a diaphorase-coupled reaction. Notably, only 15 of the positives from this screen inhibited wild-type IDH1 with an IC50 below 50 μM, and none of these showed an IC50 below 20 μM. This allele-selectivity is consistent with that seen for most previously published mutant IDH1 inhibitors and is likely due to the substantial differences in tertiary structure between wild-type and R132H mutant IDH1.24
To prioritize the 103 confirmed hits for follow-up investigation, we examined preliminary structure–activity relationships (SARs) present in the screening data as well as biological activity annotations in PubChem as a readout of compound selectivity. The DOS screening library consists of many groups of structural analogues for a given scaffold, including nearly all stereoisomers of each compound. This design enables the identification of series that display SARs suggestive of a specific molecular interaction with the protein target. Here, we identified BRD2879, an 8-membered sulfonamide containing three stereocenters (2S, 5R, 6R) that has an IC50 10- to 1000-fold lower than those of its stereoisomers (Figure 1A,B). This stereochemistry–activity pattern was consistent among structurally related compounds in the screening collection.
Figure 1.
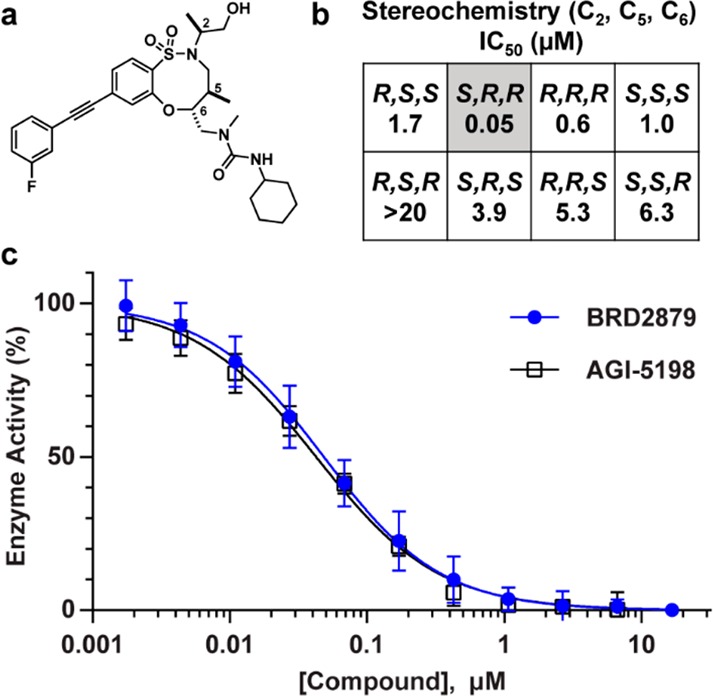
BRD2879, but not its stereoisomers, potently inhibits IDH1-R132H in vitro. (a) Structure of BRD2879. (b) The (2S, 5R, 6R) configuration shows >10× lower IC50 than its stereoisomers. Values are geometric means of three independent experiments; typical CV is 35%. (c) NADPH-dependent ketoreductase activity of IDH1-R132H after incubation with compounds. BRD2879 inhibits IDH1-R132H with comparable potency and Hill slope to previously disclosed AGI-5198. Values are mean ± SD of three independent experiments.
BRD2879 and other DOS-derived compounds have been tested in a variety of screens conducted through the NIH Molecular Libraries Program, and the data can be analyzed for evidence of indiscriminate activity.25 BRD2879 was inactive in all 40 screening assays for which results are available on PubChem, including a screen for inhibitors of KDM4C, which like IDH1 uses α-KG as a cofactor. Overall, the potency of BRD2879, its encouraging preliminary SAR, and its lack of activity in other screens led us to prioritize this scaffold for follow-up experiments.
In order to confirm the activity of BRD2879 and explore additional SAR of the scaffold, we resynthesized the probe along with 36 analogues and tested their activity in an enzymatic assay. The core structure 1 was synthesized according to the published procedure,26,27 and elaboration to the final compounds was accomplished by N-capping and Sonogashira reactions in either order, followed where appropriate by functionalization of the primary alcohol (exemplified in Scheme 1). A two-step Boc deprotection avoiding strong acid was used to avoid concurrent removal of the PMB group. All analogues were synthesized via this route with the exception of the “reverse amide” 36, which required a modified synthesis of the structural core (Scheme S1). The activity of resynthesized BRD2879 and analogues was confirmed in dose by monitoring the kinetic decrease in fluorescence of NADPH when incubated with IDH1-R132H and α-KG (Figure 1C). In comparison to the enzymatic assays used in screening, this direct fluorescence detection method allowed compound evaluation at lower enzyme concentrations. The assay modification was necessary to maintain [inhibitor] ≫ [enzyme] and to ensure that IC50 is a good approximation of binding affinity.
Scheme 1. Elaboration of the Sulfonamide Scaffold.

Reagents and conditions: (a) (1) TBSOTf, 2,6-lutidine, DCM, rt, 1 h. (2) HF-pyr, THF, rt, 0.5 h. (b) Cyclohexyl isocyanate, TEA, DCM, rt, 0.5 h, 86% (2 steps). (c) 1-Ethynyl-4-fluorobenzene, TEA, XPhos-Pd-G3, MeCN, 70 °C, 14 h, 94%. (d) DDQ, DCM, pH 7 buffer, rt, 5 h, 66%. (e) PPh3, DIAD, phthalimide, THF, 0–23 °C, 18 h. (f) Methylhydrazine, EtOH, 80 °C, 1 h, 36% (2 steps). (g) CH2O, Na(OAc)3BH, DCM, rt, 6 h, 51%.
Our SAR studies focused on the alkyne (R1) and urea (R2) side chains, as these are facile points of diversification designed into the DOS library (Table 1). Recalling the stereochemical SAR for BRD2879, we synthesized analogues retaining the (2S, 5R, 6R) configuration. Activity is maintained when the R1 side chain is any of a variety of large hydrophobic groups, with the 4-fluorophenyl substituent in compound 3 showing a slight improvement over the 3-fluorophenyl substituent in BRD2879. Compounds with smaller or more polar substituents in this position show reduced activity. Several analogues were synthesized with the 3-pyridyl substituent in this position in order to improve solubility, but this modification significantly reduced potency.
Table 1. Biochemical Potency of BRD2879 Analoguesa.

Values are geometric means of at least three independent experiments; typical CV is 35%.
Variations of the R2 substituent indicate a clear preference for the cyclohexyl urea group, and decreasing the size of the substituent leads to decreases in potency (5–8). Aromatic or hydrophilic residues are poorly tolerated. The urea linkage is essential, as replacement with an amide in either orientation (13, 36) reduces activity, as does N-methylation (16). These data suggest the urea may be a site for specific interaction of the compounds with mutant IDH1. Varying the primary alcohol did not significantly alter potency in the small set of analogues tested (4, 37), suggesting an opportunity for future modification or conjugation at this site.
To provide further evidence that BRD2879 inhibits IDH1-R132H through direct binding of the protein, we examined the effect of the compound on the melting temperature of recombinant IDH1-R132H as measured by differential scanning fluorimetry. The 3.7 °C melting point shift observed in the presence of BRD2879 is consistent with a direct binding interaction and is equivalent to the shift seen in the presence of AGI-5198. We then performed steady-state kinetics studies to determine the mechanism of action of BRD2879, finding that the compound is competitive with both the α-KG substrate and the Mg2+ cofactor (Figure S3), and that binding of BRD2879 and AGI-5198 is mutually exclusive (Figure S4).
In order to confirm the activity of our inhibitors in a more physiologically relevant context, we established a cell-based model system for mutant IDH1 activity. Since it has proved difficult to establish robust cell lines directly from IDH mutant tumors, we developed an engineered model based on overexpression of IDH1-R132H in HA1E-M cells (a HEK-293 derivative).28 These cells express high levels of R-2HG, which is released into the growth media. We determined the cellular activity of compounds by measuring R-2HG present in conditioned media by liquid chromatography–mass spectrometry (LC–MS) after 72 h of compound treatment. After harvesting media, we also determined viability of compound-treated cells by observing cell morphology and measuring ATP levels. In this model, AGI-5198 causes dose-dependent reduction in R-2HG levels with an EC50 of 0.06 μM, consistent with previous reports of its efficacy in cells.29 We found that BRD2879 also reduced R-2HG levels, though at lower potency (EC50 = 0.3 μM) as compared to its biochemical activity (Figure 2A). Furthermore, the potency of BRD2879’s stereoisomers in the cell-based assay correlated with their potency in the enzymatic assay, providing evidence for an IDH1-R132H-based mechanism-of-action in cells (Figure S5).
Figure 2.
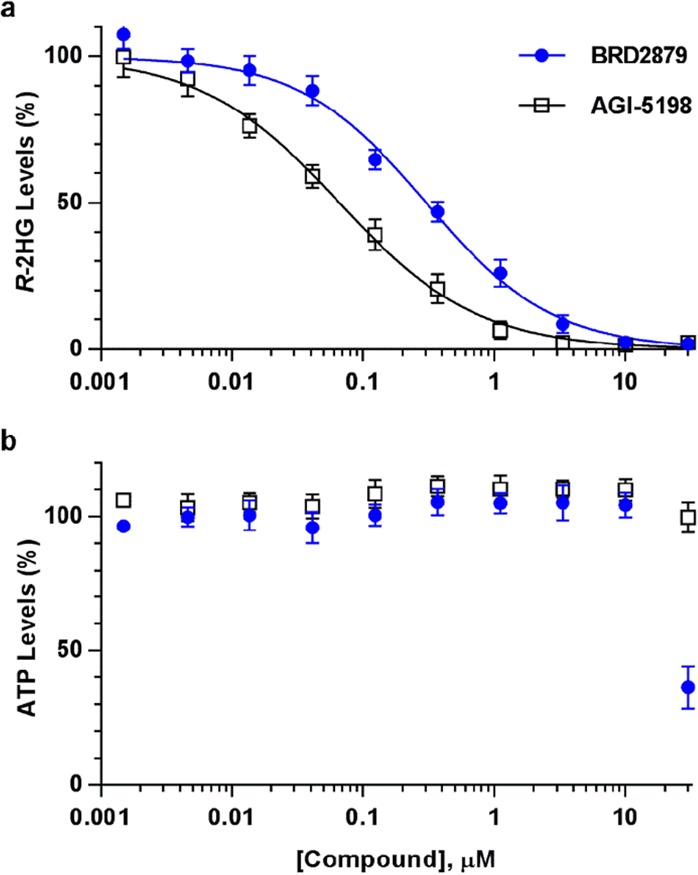
BRD2879 is effective in HA1E-M cells. (a) BRD2879 treatment causes dose-dependent reduction in R-2HG secreted by cells, though at lower potency than AGI-5198. (b) Cell viability, as measured by ATP levels, is maintained at doses up to 10 μM but drops off at higher doses. Measurements were taken after 72 h of treatment. Values represent percentages normalized to DMSO-treated control samples, mean ± SD from three independent experiments, each run in triplicate.
To determine the toxicity of BRD2879, we measured intracellular ATP levels and observed cell morphology of our engineered HA1E-M cells and the AML-derived cell lines U937 and THP1 after 3 days of compound treatment (Figure 2B, Figure S6). By these measures, BRD2879 began to show inhibitory effects on cell viability at the 10 μM concentration required for near-complete suppression of R-2HG production. U937 and THP1 are wild-type for IDH1, and we do not expect IDH1-R132H inhibition to affect the viability of engineered HA1E-M cells because the cell line was already capable of growth and proliferation before the IDH1-R132H enzyme was introduced. Thus, the observed toxicity is likely due to off-target effects and is possibly related to the compound’s poor solubility.
To assess further the suitability of BRD2879 for in vivo use, we measured several relevant physical properties of the probe (Table 2). The rapid degradation of the compound by mouse and human liver microsomes indicates optimization of the compound for metabolic stability will be required before in vivo use is possible. Additionally, the compound’s low solubility and high logD are liabilities even in cell-based model systems, as the solubility is barely sufficient to allow an efficacious dose in solution. We synthesized a small number of analogues in an attempt to improve solubility of the probe, but these modifications either reduced potency (25) or failed to improve solubility as expected (4), indicating the need for further effort in this area.
Table 2. Key Properties of BRD2879.
| in vitro enzyme inhibition, IC50a | |
|---|---|
| IDH1-R132H | 0.05 μM |
| IDH1-R132C | 2.5 μM |
| IDH1-WT | >20 μM |
| IDH2-R140Q | >20 μM |
| thermal stabilizationb | |
|---|---|
| IDH1-R132H | 3.7 °C |
| IDH1-R132C | 1.4 °C |
| cell-based R-2HG suppression, EC50c | |
|---|---|
| HA1E-M-IDH1-R132H | 0.3 μM |
| mechanism of inhibition | |
|---|---|
| w.r.t. α-KG | competitive |
| w.r.t. Mg2+ | competitive |
| microsomal stability, t1/2f | |
|---|---|
| mouse | 0.7 min |
| human | 0.7 min |
| plasma protein bindingd | |
|---|---|
| mouse | 96.3% |
| human | 99.5% |
Geometric mean of at least three independent experiments.
Mean of three independent experiments, each with seven replicates.
Mean of three independent experiments, each with three replicates.
Mean of three replicates in one experiment.
Single experiment, includes 1% DMSO.
Single experiment, calculated from a 6-point curve over 1 h.
Although the high molecular weight, lipophilicity, and low solubility of BRD2879 raise concerns that it may inhibit IDH1-R132H by nonspecific aggregation, we found that its activity in vitro is unaffected by increasing concentrations of Tween 20 detergent (Figure S7). Furthermore, the low activity of BRD2879’s enantiomer suggests that the compound’s activity may rely on specific interactions with the target rather than simply its physical properties. The thermal stabilization of purified enzyme, lack of activity against wild-type IDH1 and across many other assays, and ability to suppress R-2HG production in cells provide further evidence for this hypothesis. While BRD2879 is of limited utility in its present form, exploration of the SAR has revealed sites that are not critical for compound potency and that may be modified to improve solubility, selectivity, and susceptibility to metabolism. BRD2879 represents a new structural class of mutant IDH1 inhibitors that, with optimization, may prove useful in the study of this enzyme and its role in cancer.
Acknowledgments
We thank Dr. Jeremy R. Duvall, Dr. Ben Munoz, Dr. Zarko Boskovic, Micah Maetani, and Shawn D. Nelson, Jr. of the Broad Institute for providing advice regarding compound synthesis; and Dr. W. Frank An and Dr. Marshall Morningstar of the Broad Institute, Dr. Julie-Aurore Losman of DFCI, and Dr. Yongcheng Song of Baylor College of Medicine for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00264.
Supplementary figures, experimental details, compound characterization, and abbreviations (PDF)
This work was supported by the NCI’s Cancer Target Discovery and Development grant (U01CA176152, awarded to S.L.S.). S.L.S. is an Investigator at the Howard Hughes Medical Institute. M.M.H. was supported by a Howard Hughes Medical Institute Postdoctoral Fellowship. M.L. was supported by a Kekulé Fellowship (Fonds der Chemischen Industrie) and the German Academic Exchange Service (DAAD). O.V. was supported by the Wenner–Gren Foundations and the Swedish Chemical Society (Stiftelsen Bengt Lundqvists Minne).
The authors declare no competing financial interest.
Supplementary Material
References
- Parsons D. W.; Jones S.; Zhang X.; Lin J. C.; Leary R. J.; Angenendt P.; Mankoo P.; Carter H.; Siu I. M.; Gallia G. L.; Olivi A.; McLendon R.; Rasheed B. A.; Keir S.; Nikolskaya T.; Nikolsky Y.; Busam D. A.; Tekleab H.; Diaz L. A. Jr.; Hartigan J.; Smith D. R.; Strausberg R. L.; Marie S. K.; Shinjo S. M.; Yan H.; Riggins G. J.; Bigner D. D.; Karchin R.; Papadopoulos N.; Parmigiani G.; Vogelstein B.; Velculescu V. E.; Kinzler K. W. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.; Parsons D. W.; Jin G.; McLendon R.; Rasheed B. A.; Yuan W.; Kos I.; Batinic-Haberle I.; Jones S.; Riggins G. J.; Friedman H.; Friedman A.; Reardon D.; Herndon J.; Kinzler K. W.; Velculescu V. E.; Vogelstein B.; Bigner D. D. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P. S.; Patel J.; Wise D. R.; Abdel-Wahab O.; Bennett B. D.; Coller H. A.; Cross J. R.; Fantin V. R.; Hedvat C. V.; Perl A. E.; Rabinowitz J. D.; Carroll M.; Su S. M.; Sharp K. A.; Levine R. L.; Thompson C. B. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas S.; Lugthart S.; Kavelaars F. G.; Schelen A.; Koenders J. E.; Zeilemaker A.; van Putten W. J. L.; Rijneveld A. W.; Löwenberg B.; Valk P. J. M. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood 2010, 116, 2122–2126. 10.1182/blood-2009-11-250878. [DOI] [PubMed] [Google Scholar]
- Amary M. F.; Bacsi K.; Maggiani F.; Damato S.; Halai D.; Berisha F.; Pollock R.; O’Donnell P.; Grigoriadis A.; Diss T.; Eskandarpour M.; Presneau N.; Hogendoorn P. C.; Futreal A.; Tirabosco R.; Flanagan A. M. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa M. E.; Abdel-Wahab O.; Lu C.; Ward P. S.; Patel J.; Shih A.; Li Y.; Bhagwat N.; Vasanthakumar A.; Fernandez H. F.; Tallman M. S.; Sun Z.; Wolniak K.; Peeters J. K.; Liu W.; Choe S. E.; Fantin V. R.; Paietta E.; Lowenberg B.; Licht J. D.; Godley L. A.; Delwel R.; Valk P. J.; Thompson C. B.; Levine R. L.; Melnick A. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Yeoh K. K.; Tian Y. M.; Hillringhaus L.; Bagg E. A.; Rose N. R.; Leung I. K.; Li X. S.; Woon E. C.; Yang M.; McDonough M. A.; King O. N.; Clifton I. J.; Klose R. J.; Claridge T. D.; Ratcliffe P. J.; Schofield C. J.; Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Ward P. S.; Kapoor G. S.; Rohle D.; Turcan S.; Abdel-Wahab O.; Edwards C. R.; Khanin R.; Figueroa M. E.; Melnick A.; Wellen K. E.; O’Rourke D. M.; Berger S. L.; Chan T. A.; Levine R. L.; Mellinghoff I. K.; Thompson C. B. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P.; Lee S.; Duncan C. G.; Lopez G.; Lu G.; Ramkissoon S.; Losman J. A.; Joensuu P.; Bergmann U.; Gross S.; Travins J.; Weiss S.; Looper R.; Ligon K. L.; Verhaak R. G.; Yan H.; Kaelin W. G. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman J.-A.; Looper R. E.; Koivunen P.; Lee S.; Schneider R. K.; McMahon C.; Cowley G. S.; Root D. E.; Ebert B. L.; Kaelin W. G. (R)-2-Hydroxyglutarate Is Sufficient to Promote Leukemogenesis and Its Effects Are Reversible. Science 2013, 339, 1621–1625. 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan W. A.; Drier Y.; Liau B. B.; Gillespie S. M.; Venteicher A. S.; Stemmer-Rachamimov A. O.; Suvà M. L.; Bernstein B. E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. 10.1038/nature16490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D.; Popovici-Muller J.; Palaskas N.; Turcan S.; Grommes C.; Campos C.; Tsoi J.; Clark O.; Oldrini B.; Komisopoulou E.; Kunii K.; Pedraza A.; Schalm S.; Silverman L.; Miller A.; Wang F.; Yang H.; Chen Y.; Kernytsky A.; Rosenblum M. K.; Liu W.; Biller S. A.; Su S. M.; Brennan C. W.; Chan T. A.; Graeber T. G.; Yen K. E.; Mellinghoff I. K. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340, 626–630. 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F.; Travins J.; DeLaBarre B.; Penard-Lacronique V.; Schalm S.; Hansen E.; Straley K.; Kernytsky A.; Liu W.; Gliser C.; Yang H.; Gross S.; Artin E.; Saada V.; Mylonas E.; Quivoron C.; Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Yan S.; Murray S.; Wei W.; Gao Y.; Dang L.; Dorsch M.; Agresta S.; Schenkein D. P.; Biller S. A.; Su S. M.; de Botton S.; Yen K. E. Targeted Inhibition of Mutant IDH2 in Leukemia Cells Induces Cellular Differentiation. Science 2013, 340, 622–626. 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Kernytsky A.; Wang F.; Hansen E.; Schalm S.; Straley K.; Gliser C.; Yang H.; Travins J.; Murray S.; Dorsch M.; Agresta S.; Schenkein D. P.; Biller S. A.; Su S. M.; Liu W.; Yen K. E. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 2015, 125, 296–303. 10.1182/blood-2013-10-533604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye-Okafor U. C.; Bartholdy B.; Cartier J.; Gao E. N.; Pietrak B.; Rendina A. R.; Rominger C.; Quinn C.; Smallwood A.; Wiggall K. J.; Reif A. J.; Schmidt S. J.; Qi H.; Zhao H.; Joberty G.; Faelth-Savitski M.; Bantscheff M.; Drewes G.; Duraiswami C.; Brady P.; Groy A.; Narayanagari S.-R.; Antony-Debre I.; Mitchell K.; Wang H. R.; Kao Y.-R.; Christopeit M.; Carvajal L.; Barreyro L.; Paietta E.; Makishima H.; Will B.; Concha N.; Adams N. D.; Schwartz B.; McCabe M. T.; Maciejewski J.; Verma A.; Steidl U. New IDH1 mutant inhibitors for treatment of acute myeloid leukemia. Nat. Chem. Biol. 2015, 11, 878–886. 10.1038/nchembio.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Botton S.; Pollyea D. A.; Stein E. M.; DiNardo C.; Fathi A. T.; Roboz G. J.; Collins R.; Swords R. T.; Flinn I.; Altman J. K.; Tallman M. S.; Kantarjian H.; Colby K.; Fan B.; Goldwasser M.; Prahl M.; Agresta S.; Stone R. M. Clinical Safety and Activity of AG-120, a First-in-Class, Potent Inhibitor of the IDH1-Mutant Protein, in a Phase 1 Study of Patients with Advanced IDH1-Mutant Hematologic Malignancies. European Hematology Association Learning Center 2015, P563. [Google Scholar]
- Stein E. M.; Altman J. K.; Collins R.; DeAngelo D. J.. AG-221, an Oral, Selective, First-in-Class, Potent Inhibitor of the IDH2 Mutant Metabolic Enzyme, Induces Durable Remissions in a Phase I Study in Patients with IDH2 Mutation Positive Advanced Hematologic Malignancies. In 56th ASH Annual Meeting; American Society of Hematology: San Francisco, CA, 2014. [Google Scholar]
- Zheng B.; Yao Y.; Liu Z.; Deng L.; Anglin J. L.; Jiang H.; Prasad B. V. V; Song Y. Crystallographic Investigation and Selective Inhibition of Mutant Isocitrate Dehydrogenase. ACS Med. Chem. Lett. 2013, 4, 542–546. 10.1021/ml400036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G.; Shen J.; Yin M.; McManus J.; Mathieu M.; Gee P.; He T.; Shi C.; Bedel O.; McLean L. R.; Le-Strat F.; Zhang Y.; Marquette J.-P.; Gao Q.; Zhang B.; Rak A.; Hoffmann D.; Rooney E.; Vassort A.; Englaro W.; Li Y.; Patel V.; Adrian F.; Gross S.; Wiederschain D.; Cheng H.; Licht S. Selective Inhibition of Mutant Isocitrate Dehydrogenase 1 (IDH1) via Disruption of a Metal Binding Network by an Allosteric Small Molecule. J. Biol. Chem. 2015, 290, 762–774. 10.1074/jbc.M114.608497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.; Jiang H.; Zheng B.; Kogiso M.; Yao Y.; Zhou C.; Li X.-N.; Song Y. Inhibition of Cancer-Associated Mutant Isocitrate Dehydrogenases by 2-Thiohydantoin Compounds. J. Med. Chem. 2015, 58, 6899–6908. 10.1021/acs.jmedchem.5b00684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons P. A.; Bodycombe N. E.; Carrinski H. A.; Wilson J. A.; Shamji A. F.; Wagner B. K.; Koehler A. N.; Schreiber S. L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18787–18792. 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons P. A.; Wilson J. A.; Dancik V.; Muller S.; Carrinski H. A.; Wagner B. K.; Koehler A. N.; Schreiber S. L. Quantifying structure and performance diversity for sets of small molecules comprising small-molecule screening collections. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6817–6822. 10.1073/pnas.1015024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Zhong C.; Peng Y.; Lai Z.; Ding J. Molecular mechanisms of ″off-on switch″ of activities of human IDH1 by tumor-associated mutation R132H. Cell Res. 2010, 20, 1188–1200. 10.1038/cr.2010.145. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Xiao J.; Suzek T. O.; Zhang J.; Wang J.; Bryant S. H. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaurelle L. A.; Comer E.; Dandapani S.; Duvall J. R.; Gerard B.; Kesavan S.; Lee M. D. t.; Liu H.; Lowe J. T.; Marie J. C.; Mulrooney C. A.; Pandya B. A.; Rowley A.; Ryba T. D.; Suh B. C.; Wei J.; Young D. W.; Akella L. B.; Ross N. T.; Zhang Y. L.; Fass D. M.; Reis S. A.; Zhao W. N.; Haggarty S. J.; Palmer M.; Foley M. A. An aldol-based build/couple/pair strategy for the synthesis of medium- and large-sized rings: discovery of macrocyclic histone deacetylase inhibitors. J. Am. Chem. Soc. 2010, 132, 16962–16976. 10.1021/ja105119r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B.; Duvall J. R.; Lowe J. T.; Murillo T.; Wei J.; Akella L. B.; Marcaurelle L. A. Synthesis of a stereochemically diverse library of medium-sized lactams and sultams via S(N)Ar cycloetherification. ACS Comb. Sci. 2011, 13, 365–374. 10.1021/co2000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm J. S.; Zhao J. J.; Yao J.; Kim S. Y.; Firestein R.; Dunn I. F.; Sjostrom S. K.; Garraway L. A.; Weremowicz S.; Richardson A. L.; Greulich H.; Stewart C. J.; Mulvey L. A.; Shen R. R.; Ambrogio L.; Hirozane-Kishikawa T.; Hill D. E.; Vidal M.; Meyerson M.; Grenier J. K.; Hinkle G.; Root D. E.; Roberts T. M.; Lander E. S.; Polyak K.; Hahn W. C. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 2007, 129, 1065–1079. 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan Q.; Shi D.; Biller S. A.; Su S. M. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med. Chem. Lett. 2012, 3, 850–855. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.