

Abstract
The introduction of the elite pineapple variety, MD-2, has caused a significant market shift in the pineapple industry. Better productivity, overall increased in fruit quality and taste, resilience to chilled storage and resistance to internal browning are among the key advantages of the MD-2 as compared with its previous predecessor, the Smooth Cayenne. Here, we present the genome sequence of the MD-2 pineapple (Ananas comosus (L.) Merr.) by using the hybrid sequencing technology from two highly reputable platforms, i.e. the PacBio long sequencing reads and the accurate Illumina short reads. Our draft genome achieved 99.6% genome coverage with 27,017 predicted protein-coding genes while 45.21% of the genome was identified as repetitive elements. Furthermore, differential expression of ripening RNASeq library of pineapple fruits revealed ethylene-related transcripts, believed to be involved in regulating the process of non-climacteric pineapple fruit ripening. The MD-2 pineapple draft genome serves as an example of how a complex heterozygous genome is amenable to whole genome sequencing by using a hybrid technology that is both economical and accurate. The genome will make genomic applications more feasible as a medium to understand complex biological processes specific to pineapple.
Keywords: pineapple, plant genome, fruit ripening, hybrid assembly
1. Introduction
The MD-2 is one of the best tasting pineapple varieties, that is, not only good in terms of its flesh flavour, but also in its overall fruit appearance resulting in an increase in its marketing value. Taxonomically, the pineapple is derived from the Bromeliaceae family, under the order of Poales, which includes the grass, sedges and cattail family. The family hosts 56 genera with approximately 2,600 species and many of them are ornamental. In the family, pineapple is the only species with an active global trade for its flavourful fruits. The species is usually diploid with 2n = =501 and with an estimated haploid genome size of 526 Mb.2 Since the plant is self-incompatible and usually parthenocarpic, the plant is commonly propagated vegetatively. This method of cultural system is known to increase its heterozygosity and development of an inbred pure line is difficult if not impossible. The high level of heterozygosity in pineapple is a challenge with regards to decoding its whole genome. Nevertheless, with the advances in sequencing technology and bioinformatics software, sequencing a complex heterozygous genome such as the pineapple is now feasible.
The fruit is known to be non-climacteric, as it does not have the accompanied burst of ethylene production and respiration spike during ripening, as normally observed in climacteric fruits such as banana and tomato. More importantly, the fruit will not improve its flavour once harvested and post-harvest exposure to external ethylene application will only assist in degreening its skin but will not improve its palate. Current understanding of fruit ripening regulation is limited mainly to the climacteric fruits. By the virtue of the genomic tool available for the model plant (i.e. tomato), many of the key regulator to achieve ripening has been elucidated.3 In parallel, increasing number of studies are now devoted to understand the non-climacteric fruit, and various fruits have been proposed as the model plant to decipher non-climacteric fruit ripening. Several different hormones had been recognized in regulating fruit ripening.4 Nonetheless, the focus in the study of ripening has always been the ethylene hormone, as it is known to be produced abundantly during the process and its suppression deems to inhibit ripening of many fruit species.5,6 Altogether, these results have been inconclusive as to what control the climacteric switch in plant and no single model has been proposed to represent the regulation of non-climacteric fruit ripening.7 Many hypotheses have been proposed as to why the non-climacteric fruit can still ripen without the ethylene burst.
In pineapple, general pathways and transcripts involved in its ripening process have been recognized through microarray analysis8 and comparative expressed-sequence-tag (EST) analysis,9 respectively. Moreover, assembly of pineapple fruit transcriptome RNASeq data has enriched transcriptomic database significantly.10 But, thus far, the role of ethylene or other hormones in the ripening process of pineapple fruit is still vague. This information is highly crucial as it leads to the revelation of the intricate regulation of ripening process in pineapple fruits, which ultimately enables us to better control its quality based on its economic preferences. With the advances in genomic sequencing technology, decoding a genome has become a routine procedure. However, the challenge lies in the assembly and in making sense of the big data being produced. Recently, the genome of pineapple has been published but the sample used to assemble the genome was derived from pineapple variety F153. This sequencing project, we have embarked on, is independent of the recently published genome assembly by Ming et al.11 of the F153 pineapple variety. In this paper, we report the sequence of the commercially important MD-2 pineapple which was assembled using predominantly error corrected long PacBio reads. With the challenges of repetitive and complex multi-allelic region of plant genomes, we believe that the long sequencing read technology is able to tackle the problem and to provide a complete genome assembly as it was shown with the human genome.12 It is our motivation, to solve the complex heterozygous genome of pineapple using the long sequencing read technology, but due to its low input and high error rate, the technology still requires error correction before it can be used directly in genome assembly, especially at medium coverage (i.e. less than ×30 coverage). In order to achieve this, we borrowed the accuracy from Illumina short reads to improve the long PacBio reads using novoLR package13 from Novocraft to perform the error correction. The genome serves as an example of how hybrid sequencing technology is feasible to assemble complex plant genome such as the pineapple.
2. Materials and methods
2.1. Plant materials
The cultivated MD-2 pineapple was provided by the Malaysian Pineapple Industry Board. The variety was originally developed through a series of pineapple breeding programme and was initially released to Del Monte Fresh Produce Hawaii Inc for evaluation and commercialization. The variety was derived from a cross of another two distinct Pineapple Research Institute hybrids, namely 58-1184 and 59-44314 which contribute to its high Cayenne genetic background in MD-2.
2.2. Sample preparation and sequencing
Sample preparations for sequencing using Illumina and PacBio platforms were as in Redwan et al.15 DNA sample extracted from pineapple leaves were processed for sequencing library preparation using TruSeq PCR-Free kit (Illumina) with 350 and 550 bp average insert sizes according to manufacturer protocol. The sequencing service and library preparation were provided by Macrogen, Korea. The two libraries were sequenced using HiSeq platform (Illumina), each on a single lane using 100 bp paired-end sequencing format. In addition, 750 bp average insert size sequencing library was also prepared in-house with minor modification to the manufacturer protocol. The library was then sequenced on the MiSeq platform (Illumina) using the 300 bp paired-end sequencing format. All the sequence reads obtained from Illumina platforms were quality trimmed and length filtered using fqtrim software (available at: http://ccb.jhu.edu/software/fqtrim/ (9 June 2016 date last accessed)) to a minimum quality of Q20 and 50 bp in length. In conjunction, all of the Illumina adaptors were included in the parameter for fqtrim to perform adaptor trimming. Altogether, the three libraries provided 81.28 Gb of trimmed and filtered sequencing data which translated to ×154.7 coverage of the pineapple genome (Supplementary Table 1a).
Library preparation for PacBio sequencing was performed on high-quality genomic DNA using P4-C2 and P5-C3 sequencing chemistry and library preparation according to PacBio Sample Net-Shared Protocol available online at http://www.pacb.com/ (9 June 2016 date last accessed). The 20 kb library was sequenced using PacBio RSII platform with 32 SMRT® cells. Final data of 14.85 Gb sequencing reads was pre-processed by SMRTBellCleaner™ software from Novocraft to align and trim SMRTbell™ adaptor from the filtered subreads of PacBio. This reduced the total long reads to 11.78 Gb in total size which was error corrected using the 350 and 750 bp sequencing library of Illumina short reads (Supplementary Table 1b).
2.3. Contamination detection
Preliminarily, short reads assembly were attempted using ABySS software,16 but the assembly obtained were collapsed compared with the estimated genome size with N50 of only 5,564 bp. The contigs from the assembly were used to inspect for possible contamination of the sequence reads by using Blobology software.17 Ten thousand contigs with a size larger than 1,000 bp were randomly selected and were BLASTN against the nt database of NCBI in order to find the taxonomy classification of the best hits with E-value 10e-6. The trimmed, filtered reads were then mapped back to the contigs using novoAlign™ at default parameter to produce alignment BAM file which were then used to collate the gc coverage of the reads. Based on the taxid file of the contigs identity obtained from the similarity search and the gc distribution, the final results obtained were plotted using R-script makeblobplot.R from the Blobology software. Based on the taxon-annotated GC-coverage plots (Supplementary Fig. 1), the majority of contigs sampled formed only a single ‘blob’ represented mostly by Poales, the order taxonomical classification of pineapple. The majority of the sampled contigs had GC-content in between 20% and 60%.
2.4. Error correction of long PacBio reads
Advancement in long-read sequencing technology such as from PacBio has proven to alleviate many of the difficulties in assembly plant genomes. However, direct use of PacBio in genome assembly is not possible at low to medium sequencing coverage, due to the innate high error rate of single-pass sequence reads. Thus, in order to improve the accuracy of the PacBio, high accuracy short reads library from 350 to 750 bp average insert size were used by using novoLR™ package13 developed by Novocraft. The programme is divided into two parts; pre-processing of the PacBio reads by novoLRcleaver™ and aligning and variant calling by novoAlign™ and novoLRcorrector™, respectively. Error correction began by mapping the short reads onto the long reads using the novoAlign™ programme and variant calling was performed using novoLRcorrector™ to produce error corrected PacBio reads. After error correction, the number of error-corrected read base was further reduced to 56% (8.34 Gb) of the initial total subreads produced (Supplementary Table 1b). Much of the data was lost because any subreads that were a replicate of each other (i.e. derived from the same DNA template) were removed by novoLRcleaver™ and only the longest replicates were chosen to represent the template. This is because reads at the same start and end will not help in improving the contiguity of the genome assembly; rather it would further complicate assembly process with the minor variants it may carry due to random error rate innate to PacBio sequencing profile. As the sequencing library preparation improved in terms of the template DNA fragment size (by increasing the BP start to 9,000 bp), more read base survived the novoLRcleaver™ process, as there were less shorter templates (Supplementary Table 1b).
2.5. Genome assembly
The genome of MD-2 pineapple was assembled using only the ×15.9 (8.34 Gb) of error corrected PacBio sequence reads, with highest read length of 27,913 bp and average read length of 4,684 bp. Assembly was performed using Celera Assembler software version 8.3rc1 (http://wgs-assembler.sourceforge.net/) (9 June 2016, date last accessed) with parameters as summarized in Supplementary Table 2. The first assembly produced a draft with a total size that was 48% larger than the haploid genome size of pineapple and N50 of 25,277 bp. The expanded genome size from the assembly was due to the failure of the software to resolve the double haplotype that existed in the genome due to its high heterozygosity rate. An overlap-based assembly as adopted in Celera Assembler should be able to resolve low heterozygosity rate as it allows tolerant mismatch in the discovery of overlaps among the sequence reads. However, with higher allele differences between the haplotype, assembler may produce different composites of the polymorphic paths into the assembly,18 leading to the construction of different unitigs representing the variants observed and thus causing an inflated assembly.
In order to reduce the redundancy that may be present in the assembly, contigs were binned into two based on length cut-off of 25,000 bp. The bin with contigs smaller than 25,000 bp were then mapped to another bin containing contigs larger than 25,000 bp using GMAP19 with default parameter and any shorter contigs with hits and target coverage of more than 80% were removed from the assembly. In addition, the error corrected PacBio reads were also mapped to the assembly to remove contigs with an average coverage of less than one, as it may represent a spurious combination of the polymorphic block not present in the genome.18 Mapping with the error-corrected PacBio reads onto the genome was performed using Blasr20 with parameter ‘-bestn 5 –minPctIdentity 90 –placeRepeatsRandomly’. At this point, the assembly did not improve much, with less than 16% reduction of the total size assembly compared with the original assembly. Furthermore, all of the Illumina short reads from the three libraries were mapped to the genome using novoAlign™ with parameter ‘-t 20,3 –hlimit 6 -H 20 -p 5,20 -r All 50’ producing a BAM alignment file. The mapping quality of the short reads was then accessed from the alignment file and the assembly was then disjointed at low-quality mapping site (Q-score of less than 10). Subsequently, the same method to reduce redundancy by a similar search as above was carried out again for the second time to the fragmented contigs but with length cut-off of 1,000 bp. Once again, the short reads were mapped to the draft using the same parameter for scaffolding purpose via BESST software21 after the previous fragmentation. Thereupon, the draft assembly achieved the total size of 508 Mb, which was 96.6% of the estimated genome size of a pineapple and improved N50 of 34,762 bp.
The contiguity of the draft was further improved using multiple scaffold software for different sequence data. First, SSPACE-LongRead22 software was used to scaffold the draft using error corrected PacBio reads, then Quiver tool (default parameter) via SMRT analysis was used to perform consensus calling using uncorrected PacBio reads, followed by another round of scaffolding using in-house PacBio long transcripts sequence data of pineapple using GMAP as aligner and L_RNA_Scaffolder23 for scaffolding. The draft was then further improved using SSPACE-LongRead software but in this round, all filtered subreads uncorrected PacBio reads were used. Finally, consensus calling was performed on the final draft using novoLRpolish™, utilizing all the trimmed and filtered Illumina short reads, and uncorrected novoLRcleaver™ processed long PacBio reads. After multiple rounds of scaffolding, the draft assembly improved significantly with N50 of 153,084 bp, maximum scaffold length of 1,287,057 bp and a total assembly size of 524.070 Mb.
Genome quality assessment was evaluated by remapping short reads, and the long reads error corrected PacBio to the final draft using novoAlign™ and Blasr, respectively. More than 91% of the short reads mapped to the genome in proper pairs and more than 96% of long PacBio reads mapped to the genome. The high percentage of reads mapped back to the draft suggests that most of the reads were incorporated into the genome and thus most of the genome were assembled. In addition, the genome was also evaluated using Core Eukaryotic Genes Mapping Approach (CEGMA)24 in order to identify the correct exon–intron structure of 248 Core Eukaryotic Genes (CEGs) in the assembly. The analysis found 99% of the CEGs and 90% of the match was complete. This number of CEG retrieved is higher than other genomes assembled using next generation sequencing technology; pear25 (98.4%), adzuki bean26 (86%) and date palm27 (94%). Mapping of MD-2 reference genome to the F153 pineapple assembly was performed using lastal alignment tool28 and the comparison metrics was produced using the COMPASS tool.29 Variant analysis was performed as in Wit et al.30 using minimum mapping quality of Q30.
2.6. Repeat and gene annotation
Repeat annotation was performed based on the advanced tutorial to construct repeat library from MAKER31 software. De novo identification of miniature inverted-repeat transposable elements (MITE) and Long Terminal Repeat (LTR) retrotransposons were performed using MITE-Hunter32 and LTRharvest33 software, respectively. Consensus sequences from de novo repeat library were then combined with LTR identified using RepeatModeler, to constitute the final repeat library. Gene prediction and annotation were conducted using MAKER pipeline31 with gene predictor tools including SNAP,34 AUGUSTUS35 and GENEMARK-ES.36 mRNA sequencing of a tissue sample from the mature yellow fruit of local pineapple variety from Babagon, Sabah were sequenced using PacBio. In addition to our previous RNA-Seq assembly data10 on the mature yellow pineapple fruit, another RNA-Seq assembly was performed on the mature green pineapple fruit following the same method. However, de novo assembly of the mature green RNA-Seq sequences was performed using Oases-MK (http://www.ebi.ac.uk/~zerbino/oases/ 9 June 2016, date last accessed), using the combination of kmer range from 23 to 65 in a step of two. The assembled transcripts were then clustered using TGI Clustering Tool37 software by default. All of these transcriptomic sequencing data in addition to the available pineapple EST data downloaded from Genbank NCBI were used as EST evidence to the predicted gene in the MAKER pipeline. Due to the limited transcriptome data to represent other tissues of pineapple, RefSeq protein sequences from Poales order were downloaded from NCBI to be recruited as the protein homology evidence. The final gene set contained 27,087 genes. In addition, tRNAscan-SE38 in MAKER pipeline was also enabled for identification of tRNA in the genome.
Gene function was assigned according to best hit in a similar search against SwissProt and TrEMBL database39 using BLASTP at E-value cut-off of 1e-5. The gene ids were then modified to add the gene function according to the best hit search and genes with no identity were indicated as ‘Protein of unknown function’. Motifs and domain of the genes were determined using InterProScan40 v5.15-54.0 against multiple databases including Pfam, PROSITE, PRINTS, ProDom, SMART, Panther, TMHMM and SignalP_EUK with pathway and GO lookup. Gene features comparison across other sequenced genomes in subclass Commelinidae and Arabidopsis thaliana were performed using GenomeTool41 and protein homology search were carried out using OrthoMCL.42
Non-coding RNA (ncRNA) including microRNA (miRNA), small nuclear RNAs (snRNA), small nucleolar RNA (snoRNAs) and other ncRNAs were identified using INFERNAL-v1.1 software43 using RFAM covariance database.44 In the analysis, for the case of overlap prediction, hit with higher E-value was selected.
2.7. Phylogeny construction
Single-copy gene among the nine taxa were obtained via orthology analysis using OrthoMCL.42 Oryza sativa, Sorghum bicolor, Brachypodium distachyon and Aegilops tauschii were selected to represent Poacea family, and Musa acuminata and Elaeis guineensis to represent the non-Polaes order in subclass commelinid. A. thaliana was chosen to represent the dicotyledonous group for comparison and Amborella trichopoda were included as the most recent common ancestor among the angiosperm. Four hundred and nine single-copy genes identified by OrthoMCL were concatenated into a single super long sequence for each taxon. The sequences were then aligned by using the amino acid sequence as the guide via MAFFT.45 The aligned amino acids were then back translated using EMBOSS Backtranseq tool prior to subsequent phylogenetic analysis. The phylogenetic tree was constructed using the same matrix via GTR + GAMMA model implemented in RAxML.46 Gene family expansion and contraction was analysed using CAFÉ47 on the 1,000 largest core gene family shared across all taxa in the tree. Divergence times in the phylogenetic tree were estimated using RelTime method in MEGA6 calibrated using divergence time between Brachypodium and Oryza (40–45 million years ago)48 and Arabidopsis and Oryza (130–200 million years ago).49
2.8. Transcriptome analysis
Differential expression analysis in this study is part of an extension to the previously published de novo transcriptome of mature yellow pineapple fruit.10 All of the tissue samples and RNA extraction were performed concurrently as in Ong et al.10 The fruits were collected from a commercial pineapple field located at Babagon, Sabah. The fruit was a local variety but a variant of the Smooth Cayenne. Nevertheless, its average fruit size is smaller than Smooth Cayenne but with higher Brix value (ranges from 8 to 20°) and the pH value of the fruit ranges from 3.7 to 3.2. The green mature fruit and yellow mature fruit were harvested randomly at 12 and 16 weeks after flowering, respectively. The green mature fruit was fruit that had all of its eyes fully expanded but the skin was still green and the yellow mature fruit was a fruit harvested during the time when more than 80% of its eyes turned yellow. Total RNA was extracted by using the modified method of Li et al.50 Total RNA was sequenced using Genome Analyzer IIx and was sequenced in 75 bp paired-end format with average insert size of 200 bp. All sequence reads were filtered and trimmed using Perl script Condetri51 with a minimal length of 50 bp and average Q-score of 25. RNASeq reads were then mapped onto the draft genome and analysed for differential expression by using the Tuxedo suite pipeline.52
3. Results and discussion
3.1. Sequencing and assembly
Sequencing of the MD-2 pineapple was carried out using the two forefront sequencing platforms, Illumina and PacBio, to produce short and long sequencing reads, respectively. Genomic DNA was obtained from leaf tissues and was sequenced using the Illumina platform in three libraries, each with different average insert sizes; 350, 550 and 750 bp. The first two libraries each produced 42 Gbp and 46 Gbp of 100 paired-end reads, respectively, and the latter produced 9 Gbp of 300 bp paired-end reads. All of the reads were trimmed to yield a final coverage of ×154. Sequencing of 20 kb template size library on the PacBio platform produced 14.85 Gb sequence data. After error correction, the data was reduced to 8.34 Gb, which translated to a ×15.9 of high accuracy long sequencing reads of the pineapple genome. Maximum read length before the assembly was 37,591 bp which was reduced to 27,913 bp after error correction. Two approaches were taken to find the most optimum assembly: de novo assembly of only error corrected long PacBio reads using the well-known, Celera Assembler53 and the recent strategy of long reads assembly-based mapping on the de-bruijn assembled short reads contigs, implemented in DBG2OLC.54 After comprehensive comparison between the two assemblies, the Celera assembly was selected as it contained more CEGs as assessed with CEGMA and better mapability of the pineapple transcripts obtained from the NCBI database and in-house database (Table 1). Although DBG2OLC strategy produced larger N50, Celera assembly is superior in terms of its accuracy which is what we considered to be more important. After contiguity improvement using multiple software for scaffolding, the final Celera assembly contained 8,448 scaffolds covering 99.6% of the genome with 901 scaffolds (i.e. 50% of the assembly) at a length of at least 153,084 bp (i.e. N50). The CEGMA24 assessment found 245 out of 248 (98.8%) CEGs with 93.2% of the matches were with alignment spanning more than 70% (i.e. complete). In addition, more than 95% of the short reads mapped to the genome in a proper pair and 96% of the long PacBio reads mapped uniquely to the genome. Earlier before the advancement of next-generation sequencing, sequencing a heterozygous sample was thought to be impossible. Nowadays, with massive parallel sequencing technology of the second-generation sequencing such as Illumina and the long sequencing technology such as the PacBio platform, de novo assembly of the heterozygous diploid sample is feasible. Hybrid assembly of the heterozygous diploid sample of MD-2 pineapple is yet another evidence of its practicability. To our knowledge, this is the second genome published utilizing the hybrid sequence technology in plant genome assembly following the Chinese orchid herb55 but the first in showcasing its feasibility with diploid heterozygous plant sample. The N50 of the MD-2 pineapple draft assembly is lower than many of the inbred fruit tree genome assembly utilizing NGS but the total genome coverage in relative to their estimated genome size exceeds many other plant genomes. It is important to note that the N50 is higher in comparison to other draft assembly of complex samples such as the 20 Gbp white spruce,56 the 2.57 Gb hop57 and the weed horseweed.58 The integrity of the scaffold is good enough for gene annotation as shown by the number of CEGs found which was comparable to the Setaria italica draft genome59 but better than the draft genome of pear25 and the Chinese orchid.55 The number of the CEG found in full length and partial were also exceeded the recently published pineapple genome.11 Furthermore, more than 99% of 114,077 complementary DNAs (cDNAs) of pineapple could be aligned to the genome with 87% of the matches had over 90% coverage and identity (Table 1). The cDNAs used for validation were derived from the previous fruit transcriptomic studies,10 pineapple EST sequences from Genbank and the new long RNA sequences derived from Iso-Seq sequencing using PacBio RSII. Transcripts mapped partially onto the genome could be used to further improve the gene annotation and assembly accuracy. Variant analysis of the MD-2 pineapple draft revealed one heterozygosity per 448 bp, which included 1,009,925 of SNPs and 183,133 of indels.
Table 1.
Assembly statistics of CELERA and DBG2OLC assemblies
| DBG2OLC | Celera | |
|---|---|---|
| Total number | 3,325 | 8,448 |
| Total size | 4.44E + 08 | 5.24E + 08 |
| Longest scaffold | 2208934 | 287057 |
| N50 | 326628 | 153084 |
| L50 | 360 | 901 |
| N75 | 144,165 | 67,283 |
| N90 | 58,670 | 27,416 |
| N95 | 32,808 | 16,741 |
| Percentage of assembly in scaffolded contigs | 69.40% | 82.40% |
| Percentage of assembly in unscaffolded contigs | 30.60% | 17.60% |
| Average number of contigs per scaffold | 1.7 | 2.1 |
| Average length of break (>25 Ns) between contigs in scaffold | 3,522 | 1,695 |
| Number of transcripts mapped (114,077) | 113,273 | 113,729 |
| Number of transcripts mapped more than or equal to 80% | 100,154 | 104,297 |
| Number of transcripts mapped more than or equal to 90% | 93,628 | 99,532 |
| CEGMA Completea | 231 | 231 |
| CEGMA Partialb | 244 | 245 |
aNumber of CEGs found with more than 70% identity.
bNumber of CEGs found with less than 70% identity.
3.2. Pineapple F135 assembly
Just recently, a pineapple genome from variety F135 was published.11 Initially, the genome had suffered low assembly contiguity due to its heterozygosity and the problem was alleviated by using haplotype phasing. This method is made possible by phasing out the haplotype that was not present in the sequencing data of an F1 progeny which was derived from a cross between the sequenced sample and another variety. With ×400 Illumina reads, ×2 Moleculo synthetic long reads, ×1 454 reads, ×5 PacBio single-molecule long reads and 9,400 BACs, the assembly achieved scaffold N50 of 11.8 Mb and genome coverage of 72.6%. Similar to other short-reads-based assemblies, the assembly also falls short in assembling the repeats, leading to the reduced genome coverage. Nevertheless, the assembly provides as an intact reference for numerous genome studies in pineapple. In comparison to our genome assembly, our draft which was assembled using the only ×15.4 of error-corrected long PacBio reads and ×154 of Illumina reads (only for error-correction and scaffolding) is inferior in term of contiguity but is able to achieve higher genome coverage and contained a higher number of CEGs. Mapping of the assembly scaffold onto the F153 assembly using lastal28 confirmed our assembly validity, as the assembly could cover most of the F153 assembly (Supplementary Fig. 2). Moreover, genome validation using the COMPASS tool showed that more than 89.7% of the F153 assembly was assembled in our genome. In addition, yet again the DBG2OLC assembly was not any superior to the CELERA assembly in term of coverage from the COMPASS metrics (Table 2). Despite of our larger assembly size, most of our assembled scaffolds were validated by the F153 assembly as shown by the validity metrics and parsimony. Due to our larger assembly size in comparison to F153 assembly, the validity metric scored higher than one, which denoted that there were more alignments of the assembly onto the reference than the total length of the reference itself. In addition, the parsimony metrics of 1.8 inferred that there was a slightly more of the assembled length than the total alignment length that can form the continuous coverage on the reference (i.e. coverage island).
Table 2.
COMPASS metrics for CELERA and DBG2OLC assembly using the F153 assembly as reference
| COMPASS metrics | CELERA | DBG2OLC |
|---|---|---|
| Coverage (fraction of the reference was assembled) | 0.897 | 0.864 |
| Validity (fraction of the assembly can be validated by the reference) | 1.036 | 1.007 |
| Multiplicity (replicated or collapsed repeat during assembly) | 1.916 | 1.640 |
| Parsimony (assembled bp versus validated bp) | 1.850 | 1.629 |
Interestingly, both assemblies using the long error corrected reads showed replicated repeats in comparison to the F153 assembly and the multiplicity is larger in CELERA assembly as compared with the DBG2OLC, which contained smaller assembly total size. The problem of multiplicity can be highlighted from the high coverage peak observed upon the mapping of our assembly scaffolds onto the F153 assembly (Supplementary Fig. 2). In order to confirm the collapsed repeat in the region, the 350 bp Illumina library was mapped to the F153 assembly and similar plot was produced. Many of the high coverage peaks observed in the mapping of the scaffold to the F153 assembly were also observed at the same genome coordinate in the mapping of the Illumina reads (Fig. 1). In many genome evaluations, higher coverage at a certain region in genome indicates the problem of collapse repeats that had been assembled in fewer copies than in the real genome.60 The fact that there were more of our scaffolds mapped at the same region where there were extremely high short reads coverage, inferred that in our assembly this collapse repeats had been expanded and assembled in few scaffolds. However, due to a limited number of long reads to resolve the repeats, the scaffolds were not been able to be merged longer. Interestingly, at the regions where there high coverage mapping of our assembly onto the F153 assembly, there was no mapping found from the DBG2OLC scaffold. This showed that the region had been eliminated during assembly in DBG2OLC which altogether may reduce its complexity so as to enable the better contiguity as shown by its longer N50.
Figure 1.

The plot showed the distribution of coverage of Illumina short reads and the MD-2 scaffolds mapping to the F153 pineapple genome assembly. These were the ‘linkage’ constructed that have high mapping coverage of short reads that match the same region where multiple scaffolds from MD-2 assembly mapped. The regions highlighted the high ‘multiplicity’ as shown in the COMPASS metrics. Different rows are for the different linkage of the F153 assembly and on the left are the mapping from the Illumina short reads and on the right are the respective mapping of the MD-2 scaffolds on the same linkage. For all of the linkages, the mapping covered throughout the genome, but may not be visible in the plot as the mapping value was undersized by the high coverage value.
3.3. Gene prediction
Protein-coding genes were annotated using MAKER automated annotation software by recruiting pineapple transcriptome and RefSeq protein from Poales as evidence to the ab initio gene predictions. The prediction yielded 27,017 putative gene models and 90.6% of the proteins were classified into 4,396 unique protein family based on the PANTHER database. Putative gene function was assigned based on best homology match via Blastp to SwissProt and TrEMBL database and 94.8% of the genes were with putative function. In addition, protein domain of the gene models were assigned via InterProScan40 which identified 3,911 protein domain (based on Pfam database) in 20,937 genes (77.4% of all genes) and 15,293 of the predicted genes were annotated with at least one GO term (Supplementary Table 3). INFERNAL analysis by using RFAM covariance model to detect ncRNA families identified 215 miRNA, 9 rRNA, 347 snoRNA, 63 snRNA and 1250 other ncRNA. Four hundred and seventy-five tRNA were identified using tRNAscan-SE38 via MAKER pipeline. Overall, the pineapple genome had fewer predicted gene models than other species in the same subclass Commelinidae, however, it had a similar number when compared with A. thaliana (TAIR 10). As well as this, Coding DNA Sequence (CDS) length, gene length, exon length and the number of individual exons remained similar between Commelinidae and A. thaliana (Fig. 2).
Figure 2.

Comparison of the gene features among eight sequenced plant genomes including the pineapple. From top left is length distribution of (a) CDS, (b) exon and (c) gene, followed by (d) the exon number. There was no obvious difference observed for all features, except for gene length of S. bicolor and A. thaliana.
3.4. Repeat analysis
A total of 236.9 Mb (45.21%) of repeated elements was identified in the unmasked Ananas comosus genome based on the advanced repeat library construction protocol by Maker (Fig. 3). The most abundant repeat elements was class I (LTR)/Gypsy elements, constituting about 18% (96 Mb) of the genome. Other class I LTR elements identified was LTR/Copia, representing 4.68% (20 Mb) of the genome and unclassified LTR element, consisting of 4.7% (24 Mb) of the genome. The occurrence of non-LTR class I only constituted of 2.5% (5 kb) of the genome for LINE elements, and 0.27% (1.2 kb) for SINE elements. Altogether, 10.4% (43 Mb) of the genome was identified to be class II DNA transposons, with the highest element being the unclassified DNA elements. In comparison to other Poales sequenced genome, the number of the repetitive elements were higher than rice (35%),61 date palm (21.9%),62 banana (26.9%)63 but lower than sorghum (61%)64 and comparable with the foxtail millet genome (46%).59
Figure 3.

Pie charts show the percentage of different repeated elements identified in MD-2 pineapple genome. The most abundant components identified were LTR/Gypsys, DNA Class II transposons and followed by unidentified LTR and LTR/Copia.
3.5. Comparative analysis of orthologous genes and phylogenetic analysis
Orthologs analysis with other Commelinidae subclass sequenced genomes, unveiled 9,701 gene families in common to A. comosus and four other sequenced genomes in its same subclass, namely O. sativa, B. distachyon, M. acuminata and E. guineensis (Fig. 4). The largest cluster from the common orthologous group among the six species consisted of 471 proteins, with 21% of the members derived from M. accuminata and 19% from E. guineensis. These proteins were with similarity hit to Leucine Rich Repeat receptor-like serine-threonine-protein kinase which was known to be highly duplicated in plant genome as the gene family underwent several rounds of recombination, resulting in gene death and birth within the family.65 In addition, the orthologous analysis also showed 902 clusters unique only to A. comosus. These clusters may contain the in-paralog specific to the gene families of A. comosus or genes that have underwent sufficient structural rearrangement, causing enough variation to be unique only to the genome.
Figure 4.
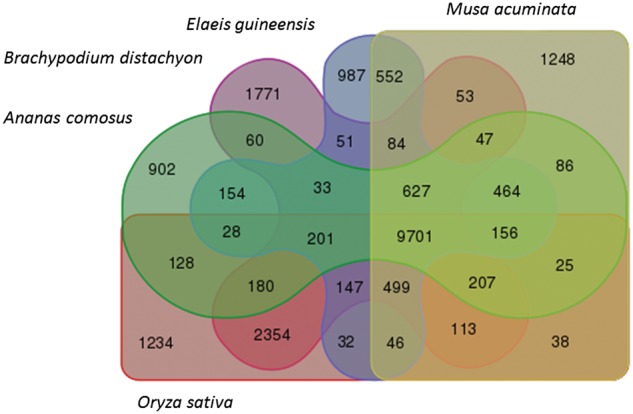
Venn diagram illustrates the shared orthologous gene cluster among pineapple and four other sequenced grass genomes, namely M. acuminata, E. guineensis, B. distachyon and O. sativa. Orthology analysis was performed using OrthoMCL.
A phylogenetic tree was constructed using 409 single-copy genes shared by pineapple and nine other angiosperm species (Fig. 5). The topology of the tree followed that of the current angiosperm classification,66 placing the pineapple at the base after the divergent of the Poales from other member of its subclass. The grass family formed the largest evolutionary distance (depicted by the branch length) as compared with other members of the commelinid, suggesting significant genome variations across the family than other commelinids. This is in agreement with the recent chloroplast study in subclass Commelinidae, where major modifications to the chloroplast genome was observed only among the grass family.15 On the other hand, pineapple maintained relatively similar genetic distance with M. acuminata, which inferred substantial genetic conservation in pineapple as compared with other Poales after the divergent from commelinid. Comparison of GC content across transcripts (CDS) from all nine taxa also supported genetic similarity between pineapples to the banana than to other Poales members (Supplementary Fig. 3).
Figure 5.

Phylogenetic tree, gene family contraction and expansion and time line divergence of pineapple among the subclass Commelinidae. The phylogenetic tree was constructed using 409 single-copy-genes shared by pineapple and six other Commelinidae and A. thaliana and Am. trichopoda were included as the out-group. Pie charts at each node depict the gene family expansion and contraction and underlined number at each node represents the divergence of pineapple from other commelinids in millions of years ago (MYA). Divergent time was calculated using RelTime using the same matrix used to construct the phylogenetic tree.
In addition, common gene family clusters across all nine taxon which were inferred to be the conserved gene family across the angiosperm were inspected for contraction and expansion. The analysis revealed significant contraction of the gene member had occurred since divergent of Poales from the commelinid and the largest expansion of the orthologous clusters gene members occurred in Zingiberales and Arecales, represented by M. acuminate and E. guineensis, respectively. Many of the green plant sequenced thus far showed large-scale duplication events which may lead to speciation but most importantly to drive evolution forward. The orthologus cluster gene expansion analysed in this study showed the largest expansion was observed in Zingiberales which is known to have at least three duplication events that were not shared by the Poales.67 However, the contraction event observed among Poales in this analysis does not eliminate the occurrence of expansion event among the groups. Strong evidence in previous studies have been shown to support whole genome duplication among the Poales in at least a single event after divergent from commelinid.68 It is interesting to note that across taxa, the largest gene reduction was observed in pineapple. In parallel of pineapple’s low gene density (33% reduction as compared with 430 Mb rice genome), genome-wide gene reduction unique to the species may have occurred. This hypothesis can be investigated through comparative genomic by synteny analysis but with the level of contiguity presented in this draft, this sort of analysis is computer-intensive and difficult for inference as the scaffolds are not presented at the chromosome size. In addition, the divergent time between A. comosus and the grass family was estimated to be circa 110.38 Myr and the split of the commelinid was estimated to be around 128 Myr, in accordance with the commelinid divergent time estimated in previous study.69
3.6. Role of ethylene in pineapple fruit ripening
In the study of fruit ripening, ethylene has been the focal point in dissecting this complex process, as the hormone is emitted abundantly during ripening. But this is only true with the climacteric fruits and the role of the hormone has only been clearly deciphered in fruits that release and respond to the hormone to induce their ripening process (i.e. climacteric fruits). In non-climacteric fruits, there are still large gaps of knowledge and arguments on the role of ethylene in this group of fruits which do not produce and response to ethylene during their ripening process. In bridging this gap, we performed differential expression analysis of the mature pineapple fruits RNASeq libraries differing in their level of ripening as determined by their skin colour. Overall, 99 genes were differentially expressed (>2-fold) at a significant level of P < 0.0001 (Supplementary Table 4, Supplementary Fig. 4), but for the sake of brevity, the focus is given only to ethylene-related transcripts.
Among the differentially regulated transcripts identified, four of the seven transcripts categorized under transcription regulator were related to ethylene and these were the ERF109, ERF3, ERF008 and TEM1 AP2/ERF and B3 domain transcription repression. The presence of differentially regulated ethylene-related transcription factors in pineapple fruit supports the current notion of the involvement of ethylene in non-climacteric fruit ripening, despite the absence of ethylene burst during its ripening process.70 In addition, a transcript, ACMD2_01443, with homology to ACO1 (1-aminocyclopropane-1-carboxylate oxidase 1) gene was also down-regulated. The product of this gene is known to be the rate-limiting enzyme in the synthesis of ethylene together with the ACS (1-aminocyclopropane-1-carboxylic acid) gene. The ACS gene were present in the genome in seven copies, but none were differentially regulated (Fig. 6). Similar observation of ACO gene down-regulation was made in other non-climacteric fruits, namely strawberry70 and grape.71 In climacteric fruits, ACO expression can be varied, with a different copy of the ACO genes up-regulated and down-regulated significantly through ripening. The various expression pattern of ACO copies during ripening of climacteric fruits were denoted to different copies of the ACO gene required to maintain system I and to induce system II.7
Figure 6.

Biosynthesis of ethylene and expression of its two rate-limiting enzymes, ACC Synthase (ACS) and ACC Oxydase (ACO). This figure depicts the biosynthetic pathway of ethylene, which is integral to the YANG cycle. Seven and 13 transcripts were identified in the genome with a putative function to ACS and ACO, respectively, and only one ACO transcript (i.e. ACMD2_01443) was differentially regulated during ripening of pineapple fruit. Asterisk marks the significantly differentially regulated transcript.
Recent studies showed that young non-climacteric fruit of strawberry and citrus produced ethylene burst prior to ripening and then it gradually subsided as ripening continues.70 The ethylene burst occurred was in concomitant with the high ACO expression and thus, the gene may be responsible for the transient peak in non-climacteric fruit ripening. It is interesting to investigate whether the down-regulation of ACO gene observed in pineapple and the peak observed in green mature fruit followed the same ethylene production pattern. Further physiological study of pineapple fruit ripening is required to support the hypothesis. It is important to note that not all fruits in the same climacteric pattern carry the same mechanism to achieve ripening, as some differences may occur. For example, although most of the climacteric fruits have varied expression pattern of the ACO copies during ripening, differential expression analysis of pear fruit revealed that all four copies of the ACO gene were up-regulated.72 But the key to their similarity is that the high expression of the ACO gene occurred in concomitant with the ethylene burst and is maintained through until the fruit ripens. The similar key for the non-climacteric fruit to achieve ripening is hoped to be deciphered in the near future.
There are two hypotheses, as to why the non-climacteric fruits do not undergo auto-induced system II ethylene. First, an investigation using two melon varieties with differing climacteric pattern suggests blockage in the auto-induced synthesis of ethylene.73 This hypothesis cannot be inferred in other genomes as the loci causing the blockage is not yet elucidated but it is known that it does not relate to ethylene controlling enzyme (i.e. ACS, ACO) and ethylene receptor gene (ERS) based on genetic mapping.73 Second, the study of the strawberry fruit ripening proposes that the non-climacteric fruits function with type-II ethylene receptor, and thus do not require abundant ethylene because the ethylene negative regulators, CTR is loosely bound by the type-II ethylene receptor (i.e. as compared with type I in climacteric fruit). Thus, only minimal amount of ethylene is required to release the CTR from negatively regulating the ethylene response pathway.70 In the pineapple genome, there were five copies of the type-II ethylene receptor (ETR2) and six copies of type-I ethylene receptor (a single ETR1 copy and five of ERS1) (Fig. 7). However, unlike in strawberry, none of the ethylene receptors mentioned above were differentially regulated. Nonetheless, most of them were expressed during the ripening process and one copy of ETR2 gene had constantly high Fragments Per Kilobase of transcript per Million value in comparison to all other receptors. Similarly, no significant changes of the ETR2 expression was observed during ripening of capsicum fruit but another variant of type-II ethylene receptor ETR4 were found constantly abundant throughout ripening of the fruit and upon exposure to ethylene inhibitor the expression decreased significantly and the ripening was delayed.74 In grape ETR2 expression increased as ripening progressing in concomitant with the type-I ethylene receptor, ETR1.71 Although it is inconclusive to determine the contribution of ETR2 in inducing ethylene response pathway in pineapple, its high expression in both green and yellow mature fruit over the type-1 receptor suggested its importance to achieve ripening. Non-climacteric ripening may not require the ethylene burst to be maintained throughout the ripening process, but the initial burst as observed in strawberry and citrus may probably be sufficient to utilize similar ethylene response pathway and to promote ethylene-dependent genes to achieve ripening.
Figure 7.

Regulation of ethylene production, from left is class I, auto-inhibition and class II auto-catalytic and level of expression of ethylene receptors during ripening of pineapple fruit. In class I, during the basal level of ethylene production, ethylene response pathway does not occur as the negative regulator (CTR) is bound and activated by the ethylene receptors, which lead to subsequent degradation of EIN2, EIN3 and EIL1 through ubiquitination by SCP complex and 26 proteosome. In class II, the presence of the hormone will further induce its production, leading to ethylene spike observed during floral senescence and fruit ripening process. Most importantly, the binding of ethylene at its receptors will release and inactivate the CTR, promoting cleavage of carboxyl end of EIN2 to the nucleus and activate the nuclear transcription factor EIN3/EIL1 and ERF1, which induce the ethylene responsive genes. Two types of ethylene receptors have been identified at the ER lumen. It is hypothesized that non-climacteric fruits rely on type-II ethylene receptor, which binds to CTR more loosely compared with type-I. Thus, only minimal amount of ethylene required to release the negative regulator, CTR. On the left is the heatmap of the transcripts with homology to ethylene receptors. None of the ethylene receptors were differentially expressed, but one transcript with homology to ETR2, type II ethylene receptors was the highest expression in green mature fruit.
In conclusion, the MD-2 pineapple draft genome presented here serves as another milestone in the sequencing technology. Sequencing a heterozygous genome is proven feasible by combining the long PacBio reads with the highly accurate Illumina short reads as they complement each other. The short reads even though they are highly accurate, they are not been able to resolve large tandem repeats that may exist in the genome. Similarly, the long reads even though they are long and may extend through the large repeats and complex region, at low sequencing outputs its accuracy hinders its use independently. Transcriptomic study of ripening pineapple fruit with the assistance of the draft genome as a reference suggests a similar role of ethylene in the regulation of ripening in non-climacteric tropical pineapple fruit. The availability of pineapple draft genome will revamp pineapple research as more molecular applications are now feasible to achieve greater understanding in the biology of pineapple.
Data availability
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LSRQ00000000. The version described in this paper is version LSRQ01000000.
Supplementary Material
Acknowledgements
We thank Noor Hydayaty MD. Yusuf and Malaysian Pineapple Industry Board for the pineapple sample, Caroline Chan from Pacific Biosciences (Asia Pacific) and Dana Chow from TreeCode Sdn Bhd for assistance with the Pacific Biosciences RSII, Science Vision Sdn Bhd for assistance in Illumina sequencing, and Novocraft Sdn Bhd and Malaysia Genome Institute for the computing facility used in this project.
Conflict of interest
None declared.
Supplementary data
Supplementary data are available at www.dnaresearch.oxfordjournals.org.
Funding
This project is funded by the Ministry of Education and the Ministry of Science, Technology and Innovation, Malaysia, through the Fundamental Research Grant Scheme (FRG0319-SG-2013) and Science Fund (SCF0087-BIO-2013), respectively.
Authorship
S.V.K. initiated and conceived the study and participated in its coordination. R.M.R. designed and performed the experiment and drafted the manuscript. R.M.R. and A.S. performed and analysed the data, and helped to draft and revise the manuscript. S.V.K., R.M.R. and A.S. revised the manuscript. All authors read and approved the final manuscript.
References
- 1.Zanella C.M., Janke A., Palma-Silva C., et al. 2012, Genetics, evolution and conservation of Bromeliaceae, Genet. Mol. Biol., 35, 1020–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arumuganathan K., Earle E.D. 1991, Nuclear DNA content of some important plant species, Plant Mol. Biol. Report., 9, 415. [Google Scholar]
- 3.Moore S. 2002, Use of genomics tools to isolate key ripening genes and analyse fruit maturation in tomato, J. Exp. Bot., 53, 2023–30. [DOI] [PubMed] [Google Scholar]
- 4.Cherian S., Figueroa C.R., Nair H. 2014, ‘ Movers and shakers’ in the regulation of fruit ripening: a cross-dissection of climacteric versus non-climacteric fruit, J. Exp. Bot., 65, 4705–22. [DOI] [PubMed] [Google Scholar]
- 5.Golding J.B., Shearer D., Wyllie S.G., McGlasson W. 1998, Application of 1-MCP and propylene to identify ethylene-dependent ripening processes in mature banana fruit, Postharvest Biol. Technol., 14, 87–98. [Google Scholar]
- 6.Pech J.C., Bouzayen M., Latché A. 2008, Climacteric fruit ripening: ethylene-dependent and independent regulation of ripening pathways in melon fruit, Plant Sci., 175, 114–20. [Google Scholar]
- 7.Paul V., Pandey R., Srivastava G.C. 2012, The fading distinctions between classical patterns of ripening in climacteric and non-climacteric fruit and the ubiquity of ethylene – an overview, J. Food Sci. Technol., 49, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koia J.H., Moyle R.L., Botella J.R. 2012, Microarray analysis of gene expression profiles in ripening pineapple fruits, BMC Plant Biol., 12, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moyle R., Fairbairn D.J., Ripi J., Crowe M., Botella J.R. 2005, Developing pineapple fruit has a small transcriptome dominated by metallothionein, J. Exp. Bot., 56, 101–12. [DOI] [PubMed] [Google Scholar]
- 10.Ong W.D., Voo L.Y., Kumar V.S. 2012, De novo assembly, characterization and functional annotation of pineapple fruit transcriptome through massively parallel sequencing, PLoS One, 7, e46937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ming R., VanBuren R., Wai C.M., et al. 2015, The pineapple genome and the evolution of CAM photosynthesis, Nat. Genet., 47, 1435–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaisson M.J., Huddleston J., Dennis M.Y., et al. 2015, Resolving the complexity of the human genome using single-molecule sequencing, Nature, 517, 608–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hercus C. 2015, novoLR package. Novocrarft Technology Sdn. Bhd. Kuala Lumpur, Malaysia, http://www.novocraft.com/support/download/.
- 14.Chan Y.K., D’Eeckenbrugge G.C., Sanewski G.M. 2003, Breeding and variety improvement. In: Bartholomew D.P., Paull R.E., Rohrbach K.G., (eds), The pineapple: botany, production and uses. CAB International, Wallingford, UK, pp. 33–55. [Google Scholar]
- 15.Redwan R.M., Saidin A., Kumar S.V. 2015, Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae, BMC Plant Biol., 15, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simpson J.T., Wong K., Jackman S.D., Schein J.E., Jones S.J., Birol I. 2009, ABySS: a parallel assembler for short read sequence data, Genome Res., 19, 1117–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar S., Jones M., Koutsovoulos G., Clarke M., Blaxter M. 2013, Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon-annotated GC-coverage plots, Front. Genet., 4, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Price J.C., Udall J.A., Bodily P.M., et al. 2012, De novo identification of ‘heterotigs’ towards accurate and in-phase assembly of complex plant genomes. In: Proceedings of the 2012 International Conference on Bioinformatics & Computational Biology. WORLDCOMP, pp. 144–50. Available: http://schatzlab.cshl.edu/publications/2012.Heterotig.pdf. [Google Scholar]
- 19.Wu T.D., Watanabe C.K. 2005, GMAP: a genomic mapping and alignment program for mRNA and EST sequences, Bioinformatics, 21, 1859–75. [DOI] [PubMed] [Google Scholar]
- 20.Chaisson M.J., Tesler G. 2012, Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory, BMC Bioinformatics, 13, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahlin K., Vezzi F., Nystedt B., Lundeberg J., Arvestad L. 2014, BESST – efficient scaffolding of large fragmented assemblies, BMC Bioinformatics, 15, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boetzer M., Pirovano W. 2014, SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information, BMC Bioinformatics, 15, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xue W., Li J.T., Zhu Y.P., et al. 2013, L_RNA_scaffolder: scaffolding genomes with transcripts, BMC Genomics, 14, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parra G., Bradnam K., Korf I. 2007, CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes, Bioinformatics, 23, 1061–7. [DOI] [PubMed] [Google Scholar]
- 25.Chagné D., Crowhurst R.N., Pindo M., et al. 2014, The draft genome sequence of European pear (Pyrus communis L. ‘Bartlett’), PLoS One, 9, e92644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang Y.J., Satyawan D., Shim S., et al. 2015, Draft genome sequence of adzuki bean, Vigna angularis, Sci. Rep., 5, 8069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Al-Dous E.K., George B., Al-Mahmoud M.E., et al. 2011, De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera), Nat. Biotechnol., 29, 521–7. [DOI] [PubMed] [Google Scholar]
- 28.Kielbasa S.M., Wan R., Sato K., Kiebasa S.M., Horton P., Frith M.C. 2011, Adaptive seeds tame genomic sequence comparison. Genome Res., 21, 487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fass J.2014, COMPASS-scripts to COMpare a DNA sequence ASSembly to a trusted reference sequence, p. https://github.com/jfass/compass, last accessed in 9th June 2016.
- 30.De Wit P., Pespeni M.H., Ladner J.T., et al. 2012, The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis, Mol. Ecol. Resour., 12, 1058–67. [DOI] [PubMed] [Google Scholar]
- 31.Campbell M.S., Law M., Holt C., et al. 2014, MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations, Plant Physiol., 164, 513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han Y., Wessler S.R. 2010, MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences, Nucleic Acids Res., 38, e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellinghaus D., Kurtz S., Willhoeft U. 2008, LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons, BMC Bioinformatics, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korf I. 2004, Gene finding in novel genomes, BMC Bioinformatics, 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stanke M., Steinkamp R., Waack S., Morgenstern B. 2004, AUGUSTUS: a web server for gene finding in eukaryotes, Nucleic Acids Res., 32, W309–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ter-Hovhannisyan V., Lomsadze A., Chernoff Y.O., Borodovsky M. 2008, Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training, Genome Res., 18, 1979–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pertea G., Huang X., Liang F., et al. 2003, TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets, Bioinformatics, 19, 651–2. [DOI] [PubMed] [Google Scholar]
- 38.Schattner P., Brooks A.N., Lowe T.M. 2005, The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs, Nucleic Acids Res., 33, W686–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bairoch A., Apweiler R. 2000, The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000, Nucleic Acids Res., 28, 45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zdobnov E. M., Apweiler R. 2001, InterProScan – an integration platform for the signature-recognition methods in InterPro, Bioinformatics, 17, 847–8. [DOI] [PubMed] [Google Scholar]
- 41.Gremme G., Steinbiss S., Kurtz S. 2013, GenomeTools: a comprehensive software library for efficient processing of structured genome annotations, IEEE/ACM Trans. Comput. Biol. Bioinform., 10, 645–56. [DOI] [PubMed] [Google Scholar]
- 42.Li L., Stoeckert C.J., Jr., Roos D.S. 2003, OrthoMCL : identification of ortholog groups for eukaryotic genomes, Genome Res., 13, 2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nawrocki E.P., Kolbe D.L., Eddy S.R. 2009, Infernal 1.0: inference of RNA alignments, Bioinformatics, 25, 1335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nawrocki E.P., Burge S.W., Bateman A., et al. 2015, Rfam 12.0: updates to the RNA families database, Nucleic Acids Res., 43, D130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh K., Standley D.M. 2013, MAFFT multiple sequence alignment software version 7: improvements in performance and usability, Mol. Biol. Evol., 30, 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ptitsyn A., Moroz L.L. 2012, Computational workflow for analysis of gain and loss of genes in distantly related genomes. In: Proceedings of the Ninth Annual MCBIOS Conference. BioMed Central Ltd., London, UK, pp. 1–6 [DOI] [PMC free article] [PubMed]
- 48.The International Brachypodium Initiative. 2010, Genome sequencing and analysis of the model grass Brachypodium distachyon, Nature, 463, 763–8. [DOI] [PubMed] [Google Scholar]
- 49.Vandepoele K., Saeys Y., Simillion C., Raes J., Van De Peer Y. 2002, The automatic detection of homologous regions (ADHoRe) and its application to microcolinearity between Arabidopsis and rice, Genome Res., 12, 1792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J.H., Tang C.H., Song C.Y., Chen M.J., Feng Z.Y., Pan Y.J. 2006, A simple, rapid and effective method for total RNA extraction from Lentinula edodes, Biotechnol. Lett., 28, 1193–7. [DOI] [PubMed] [Google Scholar]
- 51.Smeds L., Künstner A. 2011, CONDETRI – a content dependent read trimmer for Illumina data, PLoS One, 6, e26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trapnell C., Roberts A., Goff L., et al. 2014, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks, Nat. Protoc., 7, 562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myers E.W., Sutton G.G., Delcher A.L., et al. 2000, A whole-genome assembly of Drosophila, Science, 287, 2196–204. [DOI] [PubMed] [Google Scholar]
- 54.Ye C., Hill C., Ruan J., Ma Z.S. 2015, DBG2OLC : Efficient Assembly of Large Genomes Using the Compressed Overlap Graph, arXiv, 1410.2801, 1–20. [Google Scholar]
- 55.Yan L., Wang X., Liu H., et al. 2015, The genome of Dendrobium officinale illuminates the biology of the important traditional Chinese orchid herb, Mol. Plant, 8, 922–34. [DOI] [PubMed] [Google Scholar]
- 56.Birol I., Raymond A., Jackman S.D., et al. 2013, Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data, Bioinformatics, 29, 1492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Natsume S., Takagi H., Shiraishi A., et al. 2015, The draft genome of hop (Humulus lupulus), an essence for brewing, Plant Cell Physiol., 56, 428–41. [DOI] [PubMed] [Google Scholar]
- 58.Peng Y., Lai Z., Lane T., et al. 2014, De novo genome assembly of the economically important weed horseweed using integrated data from multiple sequencing platforms, Plant Physiol., 166, 1241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang G., Liu X., Quan Z., et al. 2012, Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential, Nat. Biotechnol., 30, 549–54. [DOI] [PubMed] [Google Scholar]
- 60.Hunt M., Kikuchi T., Sanders M., Newbold C., Berriman M., Otto T.D. 2013, REAPR: a universal tool for genome assembly evaluation, Genome Biol., 14, R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.International Rice Genome Sequencing Project. 2005, The map-based sequence of the rice genome, Nature, 436, 793–800. [DOI] [PubMed] [Google Scholar]
- 62.Al-Mssallem I.S., Hu S., Zhang X., et al. 2013, Genome sequence of the date palm Phoenix dactylifera L., Nat. Commun., 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davey M.W., Gudimella R., Harikrishna J.A., Sin L.W., Khalid N., Keulemans J. 2013, A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter- and intra-specific Musa hybrids, BMC Genomics, 14, 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Paterson A.H., Bowers J.E., Bruggmann R., et al. 2009, The Sorghum bicolor genome and the diversification of grasses, Nature, 457, 551–6. [DOI] [PubMed] [Google Scholar]
- 65.Baumgarten A., Cannon S., Spangler R., May G. 2003, Genome-level evolution of resistance genes in Arabidopsis thaliana, Genetics, 165, 309–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bremer B., Bremer K., Chase M.W., et al. 2009, An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III, Bot. J. Linn. Soc., 161, 105–21. [Google Scholar]
- 67.D’Hont A., Denoeud F., Aury J.M., et al. 2012, The banana (Musa acuminata) genome and the evolution of monocotyledonous plants, Nature, 488, 213–7. [DOI] [PubMed] [Google Scholar]
- 68.Tang H., Bowers J.E., Wang X., Paterson A.H. 2010, Angiosperm genome comparisons reveal early polyploidy in the monocot lineage, Proc. Natl. Acad. Sci. U. S. A., 107, 472–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hertweck K.L., Kinney M.S., Stuart S.A., et al. 2015, Phylogenetics, divergence times and diversification from three genomic partitions in monocots, Bot. J. Linn. Soc., 178, 375–93. [Google Scholar]
- 70.Trainotti L., Pavanello A., Casadoro G., Colombo V.G., Padova I. 2005, Different ethylene receptors show an increased expression during the ripening of strawberries : does such an increment imply a role for ethylene in the ripening of these non-climacteric fruits? J. Exp. Bot., 56, 2037–46. [DOI] [PubMed] [Google Scholar]
- 71.Fortes A.M., Agudelo-Romero P., Silva M.S., Ali K., Sousa L., Maltese F. 2011, Transcript and metabolite analysis in Trincadeira cultivar reveals novel information regarding the dynamics of grape ripening, BMC Plant Biol., 11, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang G., Li T., Li X., et al. 2014, Comparative transcriptome analysis of climacteric fruit of Chinese pear (Pyrus ussuriensis) reveals new insights into fruit ripening, PLoS One, 9, e107562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Périn C., Gomez-Jimenez M., Hagen L., et al. 2002, Molecular and genetic characterization of a non-climacteric phenotype in melon reveals two loci conferring altered ethylene response in fruit, Plant Physiol., 129, 300–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aizat W.M., Able J.A., Stangoulis J.C.R., Able A.J. 2013, Characterisation of ethylene pathway components in non-climacteric capsicum, BMC Plant Biol., 13, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession LSRQ00000000. The version described in this paper is version LSRQ01000000.