

In this study, Takahashi et al. investigated the role of chromokinesin KIF4A in condensin complexes. They found that KIF4A associates with condensin I but not with condensin II during mitosis, providing new insight into the regulation of condensin I and mitotic chromsome organization.
Keywords: chromosome condensation, centromere, mitosis, SMC
Abstract
The chromokinesin KIF4A has been implicated in shaping mitotic chromosomes, but its functional relationship to condensin complexes remains controversial. Here, we found that, in mitosis, KIF4A associates with condensin I but not with condensin II. Mutational analyses indicated that the enrichment of condensin I to chromosomal axes depends on its association with KIF4A in a way that likely involves its motor activity. Remarkably, this interaction is required for condensin I to confer physiological properties to chromosomes. These observations provide an insight into how condensin I is enriched at chromosomal axes and underscore the significance of axial structure in organizing mitotic chromosomes.
Chromosome segregation in eukaryotic cells requires the reorganization of chromatin into mitotic chromosomes. Formation of mitotic chromosomes depends on the disentangling of replicated sister DNA molecules and packaging of them into a highly compacted state, which confers sufficient levels of rigidity to prevent DNA breakage and unraveling of chromatids during mitosis. This disentanglement and compaction are universally mediated by the activity of a protein complex called condensin (for review, see Hirano 2012; Houlard et al. 2015).
In many eukaryotes, two condensin complexes, called condensins I and II, have crucial roles in mitotic chromosome assembly (for review, see Hirano 2012). These complexes are composed of two SMC family proteins, Smc2 and Smc4, both of which contain the ATPase domain, plus three non-SMC proteins that provide the distinct features of condensins I and II (Ono et al. 2003; Yeong et al. 2003). The nuclear condensin II facilitates chromosome formation in prophase, whereas the cytoplasmic condensin I loads onto chromosomes only after the nuclear envelope disintegrates in prometaphase (Hirota et al. 2004; Ono et al. 2004). These condensins associate with chromatin with different dynamics: Condensin II associates stably with chromatin after DNA replication (Ono et al. 2013), in contrast to the dynamic (unstable) association of condensin I with chromatin (Gerlich et al. 2006). Because the functions of both condensins I and II are essential in ensuring faithful chromosome segregation, their distinct behavior implies different layers of mechanisms in the organization of the higher-order structure of mitotic chromosomes. One prevailing hypothesis is that condensins execute these events as they are being arranged along the chromosomal axes (Hirano 2012; Thadani et al. 2012), but what promotes condensin association to the axial structures and what the significance of its axial enrichment is remain unknown.
The kinesin KIF4A shows characteristic localization to chromosome axes throughout mitosis; hence, the name chromokinesins (Wang and Adler 1995). Perturbation of KIF4A by knockdown or by turning off the transcription revealed defects in chromosomal structure, and thus KIF4A has been implicated in controlling mitotic chromosome assembly (Mazumdar et al. 2004; Samejima et al. 2012). Notably, KIF4A has been suggested to physically interact with condensins I and II (Mazumdar et al. 2004), and a close inspection of chromosome morphology indicated a functional link between KIF4A and condensins (Samejima et al. 2012). However, the observation that depletion of KIF4A aggravated the structural defects caused by depletion of condensins raised the idea that KIF4A and condensins function in parallel to achieve proper assembly of chromosomes. How KIF4A functions in chromosome assembly and how it might relate to condensins remain unclear.
In this study, we show that KIF4A associates specifically with condensin I in mitosis. This interaction is mediated through the carboxyl tail of KIF4A and the CAP-G subunit of condensin I. Identification of a binding site in KIF4A tail allowed us to create condensin I nonbinding mutants of KIF4A to address the significance of this interaction. We found that enrichment of condensin I to chromosomal axes requires its association with KIF4A and the motor function of KIF4A. Importantly, without binding to KIF4A, condensin I fails to confer rigidity to centromeres, indicating that this interaction is essential for condensin I function. Thus, chromosome assembly in prometaphase and metaphase seems to be dynamically mediated by a cooperative action of these two distantly related ATPases.
Results and Discussion
KIF4A binds to condensin I, but not to condensin II, in mitosis
To investigate the function of KIF4A in chromosome assembly, we first sought to clarify whether KIF4A interacts with either or both condensin I and condensin II (Mazumdar et al. 2004), particularly as condensins I and II are known to have distinct roles in mitotic chromosome assembly (Hirota et al. 2004; Ono et al. 2004). In binding assays, KIF4A and condensin I or II were immunoprecipitated using antibodies specific for KIF4A, CAP-G, or CAP-D3, respectively, from DNase-treated chromatin-enriched cell extracts (Fig. 1A). We consistently found that condensin I subunits (CAP-G and CAP-H) coimmunoprecipitate with KIF4A, whereas condesin II subunits (CAP-D3 and CAP-G2) do not. Conversely, KIF4A was found in immunoprecipitates of condensin I but not of condensin II.
Figure 1.
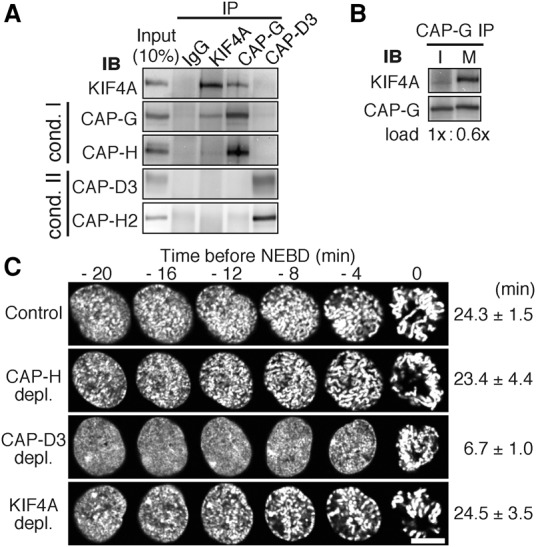
KIF4A binds to condensin I, but not to condensin II, in mitosis. (A) Coimmunoprecipitation of KIF4A with condensin I. Chromosome-enriched fractions prepared from mitotic HeLa cells were subjected to immunoprecipitation (IP) analysis with antibodies to KIF4A, CAP-G, and CAP-D3 or with nonimmune IgG and were immunoblotted (IB) with the antibodies indicated. (B) KIF4A interacts with condensin I in mitosis. Interphase and mitotic cell extracts were subjected to immunoprecipitation analysis with CAP-G antibodies (Supplemental Fig. S1). Based on these results, loading volumes of CAP-G immunoprecipitation samples were adjusted (1.0× interphase and 0.6× mitotic samples were loaded) such that CAP-G signals become equivalent between interphase (I) and mitosis (M). (C) Chromosome condensation in prophase occurs without a delay in KIF4A-depleted cells. Prophase image sequences of H2B-EGFP-expressing RPE1 cells were aligned on the time axis according to time before NEBD. Bar, 10 µm. The time when chromosome condensation first became recognizable is depicted. Mean ± SD.
Colocalization of condensin I with KIF4A is found only from prometaphase to anaphase (Mazumdar et al. 2004; Takahashi et al. 2016), suggesting that the interaction occurs during this period. Indeed, KIF4A was coimmunoprecipitated with condensin I in mitotic extracts but undetectably in interphase extracts (Fig. 1B). These results indicate that KIF4A binds to condensin I in mitosis. The fact that KIF4A does not detectably coimmunoprecipitate with condensin II discounts the possibility that binding to condensin I occurs indirectly through DNA.
If the function of condensin II relies on KIF4A, then the initial phase of chromosome condensation in prophase must be affected in cells that had been depleted of KIF4A (Hirota et al. 2004; Ono et al. 2004; Abe et al. 2011). However, live-cell imaging analysis of cells expressing GFP-tagged histone H2B revealed no significant delays in chromosome condensation in prophase after depleting KIF4A (Fig. 1C). These observations are consistent with the results of immunoprecipitation assays that indicated that KIF4A specifically interacts with condensin I but not with condensin II.
KIF4A tethers CAP-G through its C-terminal tail
KIF4A is known to have a motor domain at the N terminus, a tail domain at the C terminus, and the intervening coiled-coil domain facilitating dimerization of the protein (Fig. 2A; for review, see Seeger and Rice 2013). To determine which of the KIF4A domains mediates the interaction, we performed pull-down assays using purified recombinant protein fragments fused with GST. Only the C-terminal tail domain efficiently precipitated all five elements of the condensin I complex (Fig. 2B). It precipitated only condensin I, but not condensin II, from mitotic extracts, consistent with the results in the immunoprecipitation assay (Fig. 1A).
Figure 2.
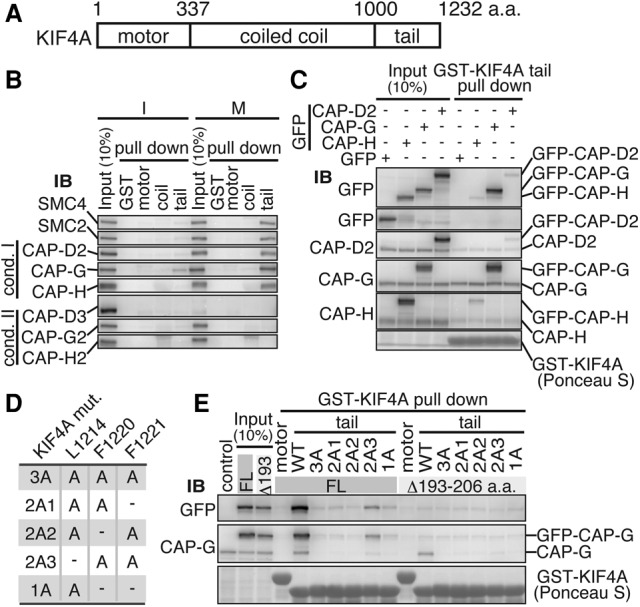
KIF4A tethers CAP-G through its C-terminal tail. (A) Schematic domain structure of human KIF4A. (B) The tail domain of KIF4A mediates binding to condensin I in mitosis. GST-tagged recombinant proteins of motor, coiled-coil, and tail domains of KIF4A were incubated with interphase (I) or mitotic (M) HeLa cell extracts, and proteins precipitated with glutathione-beads were immunobloted with antibodies to condensin subunits. (C) KIF4A binds to the CAP-G subunit of condensin I. Mitotic extracts from HEK293T cells transiently expressing GFP-tagged full-length CAP-H, CAP-G, or CAP-D2 were used in the pull-down assay with the KIF4A tail domain. Precipitated proteins were analyzed by immunoblotting with antibodies to GFP and condensin I-specific subunits. Note that the abundance of GFP-tagged CAP-G was significantly higher than that of CAP-H or CAP-D2 in the precipitates. (D) KIF4A mutants bearing various combinations of alanine substitutions at the conserved hydrophobic amino acids in the tail domain. (E) The KIF4A L1214 residue is essential for binding with CAP-G. HEK293T cell mitotic extracts expressing either GFP-tagged full-length CAP-G (FL) or Δ193–206, a deletion mutant that does not bind to KIF4A (Supplemental Fig. S2H), were used in the pull-down assay with the indicated KIF4A tail domain mutants or the motor domain as a negative control. Precipitated proteins were analyzed by immunoblotting with antibodies to GFP and CAP-G. All KIF4A tail mutants containing L1214A cannot bind to CAP-G efficiently.
We then asked which subunit of condensin I binds to KIF4A. To do this, we exogenously expressed GFP-tagged versions of condensin I-specific subunits CAP-D2, CAP-G, or CAP-H in 293T cells, and their mitotic extracts were incubated with recombinant KIF4A tail. A considerable amount of GFP-CAP-G was found in the KIF4A tail precipitate, whereas much smaller amounts of GFP-CAP-D2 and GFP-CAP-H were detected (Fig. 2C). It seemed reasonable to conclude that KIF4A interacts with condensin I through the CAP-G subunit and that the small amounts of exogenous CAP-D2 and CAP-H were precipitated indirectly as components of the condensin I complex.
Creation of KIF4A mutants that lack the ability to bind to CAP-G
To delineate the binding site in CAP-G, we examined a number of truncated mutants of CAP-G and found that an N-terminal portion of CAP-G mediates the binding to KIF4A. However, we were discouraged to define the binding site within this part, as it was also required for CAP-G to assemble into the condensin complex (see Supplemental Fig. S2A–I). Therefore, we instead dissected the tail domain of KIF4A into smaller parts and investigated their ability to bind to GFP-tagged full-length CAP-G. This indicated that the binding site resides within 49 amino acids from the carboxyl end of KIF4A (Supplemental Fig. S3A,B). Within this fragment, we focused on three hydrophobic residues: L1214, F1220, and F1221, which are highly conserved among species (Supplemental Fig. S3C). To test whether these amino acids mediate the interaction, we mutated these residues to alanines individually or in combination (Fig. 2D). The pull-down assay using these mutants revealed that mutation of the KIF4A tail at all three residues (3A mutant) almost completely abolished the precipitation of CAP-G. A combination of F1220A and F1221A mutations showed a minor effect; thus, mutating only the L1214 (1A mutant) expectedly abolished the binding to CAP-G to an extent similar to that of the 3A mutant (Fig. 2E).
Based on these analyses in vitro, we tested whether KIF4A bearing mutations at these residues fails to interact with condensin in mitotic cells. Cells were transfected with GFP-tagged constructs of KIF4A (either wild type, 3A, or 1A mutants), and these KIF4A proteins were precipitated from mitotic extracts by the GFP tag. All subunits of condensin I were detected in the wild-type KIF4A precipitate but were undetectable in both 3A-KIF4A and 1A-KIF4A precipitates, verifying that these KIF4A mutants no longer associate with condensin I in mitotic cells (Fig. 3A).
Figure 3.
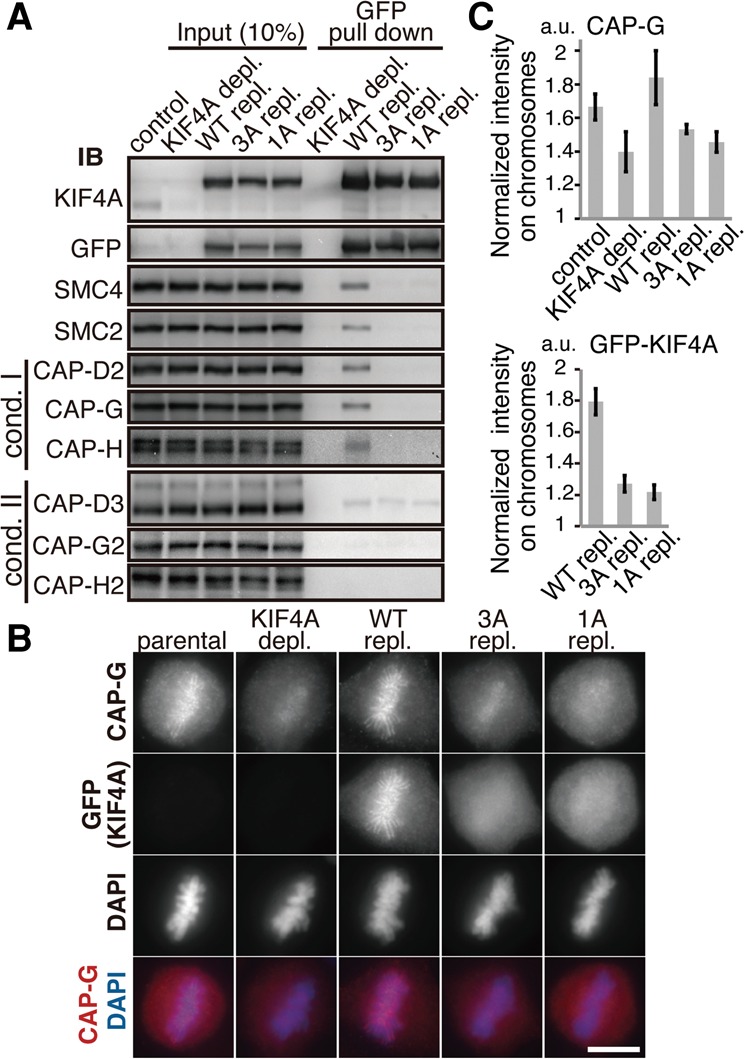
KIF4A is required for chromosomal axis enrichment of condensin I. (A) Creation of condensin I-binding-deficient mutants of KIF4A. Mitotic extracts from HEK293T cells transiently expressing GFP-tagged KIF4A wild type, 3A, or 1A were subjected to pull-down assay by GFP, and the resulting precipitates were immunoblotted for condensin subunits. The endogenous protein was depleted (depl.) and replaced (repl.) with GFP-tagged versions to prevent heterodimer formation between endogenous, wild-type, and mutant KIF4A. (B) Enrichment of condensin I to the chromosomal axis requires binding to KIF4A. HeLa cell lines expressing GFP-KIF4A either wild type or the 3A or 1A mutant were fixed and processed for immunofluorescence microscopy for GFP and CAP-G. DNA was counterstained with DAPI. Bar, 10 µm. Endogenous KIF4A was depleted and replaced with a GFP-tagged version as in A. (C) Quantitative analysis of fluorescence signal on chromosomes. DAPI-defined chromosome regions were measured for fluorescence intensities of GFP-KIF4A or CAP-G, and the ratios to their cytoplasmic intensities of the same area were calculated.
KIF4A is required to regulate the behavior of condensin I
Having condensin I-binding-deficient mutants in hand, we wished to address the significance of the KIF4A–condensin I interaction. To do this, we generated HeLa cell lines that stably express GFP-tagged wild-type, 3A, or 1A versions of KIF4A and analyzed the consequences of depleting endogenous KIF4A in these cells (Fig. 3B). The wild-type KIF4A was found at chromosomal axes from prophase to anaphase, recapitulating endogenous KIF4A localization (Supplemental Fig. S4A). In contrast, 3A-KIF4A and 1A-KIF4A mutants showed reduced levels of chromosomal association and failed to focus into the chromosome axis (Supplemental Fig. S4A). The kinesin function on microtubule dynamics seemed unaffected in these KIF4A mutants, as the microtubule association at the midbody was intact. These results indicated that the chromosomal localization of KIF4A and its accumulation to the chromosome core depend on the binding to condensin I.
We conversely asked the requirement of KIF4A for the axial localization of condensin I. Depletion of KIF4A caused substantial delocalization of condensin I from chromosomal axes, suggesting that KIF4A is essential for condensin I's axial localization (Fig. 3B,C). Remarkably, expression of the wild-type KIF4A completely restored the axial localization of condensin I, whereas expression of 3A-KIF4A and 1A-KIF4A failed to rescue (Fig. 3B,C), indicating that the ability of KIF4A to support condensin I localization depends on the physical interaction of KIF4A with CAP-G. These data together indicate that enrichment of condensin I and KIF4A at the chromosomal axis reciprocally requires the interaction between the two.
It was nevertheless possible that condensin I is loaded onto chromosome without KIF4A. We analyzed chromosome-enriched fractions biochemically to address this. In wild-type KIF4A-expressing cells, both KIF4A and CAP-G were equivalently distributed into cytoplasmic and chromosomal fractions. In contrast, in 3A mutant-expressing cells, both of them revealed a pronounced distribution to the cytoplasm. Noticeably, CAP-G was also found in the chromosome fraction when KIF4A was barely detectable (Supplemental Fig. S4B). Thus, it appeared that condensin I can be loaded onto chromatin without the aid of KIF4A.
A part of cellular KIF4A and condensin I is involved in their interaction
The finding that the vast majority of condensin I was delocalized from the chromosomal axis when KIF4A failed to associate with condensin I prompted us to address how much of the cellular populations of condensin I and KIF4A are bound to each other. Fractionation of total cell extracts by sucrose density gradient centrifugation indicated that there are no apparent overlapping populations between KIF4A and condensin I in interphase. In mitosis, however, KIF4A emerged in heavier-density fractions and thereby cofractionated at least in part with condensin I subunits (Supplemental Fig. S5A). Interestingly, when the fractionation assay was carried out in the presence of ATP-γS, a hardly hydrolysable analog of ATP, KIF4A migration to the heavier fractions became pronounced (Fig. 4A). It was reasonable to interpret that these shifts for both KIF4A and condensin I are consequences of their association because they were abolished by depleting either condensin I or KIF4A (Supplemental Fig. S5B). We could suggest that such effect of ATP-γS is causally related to ATP hydrolysis of KIF4A, as the KA-KIF4A mutant that lacks its ATP-hydrolyzing activity (see below) was found to migrate in the heavier-density fractions, unlike its wild-type counterpart (Supplemental Fig. S5C).
Figure 4.
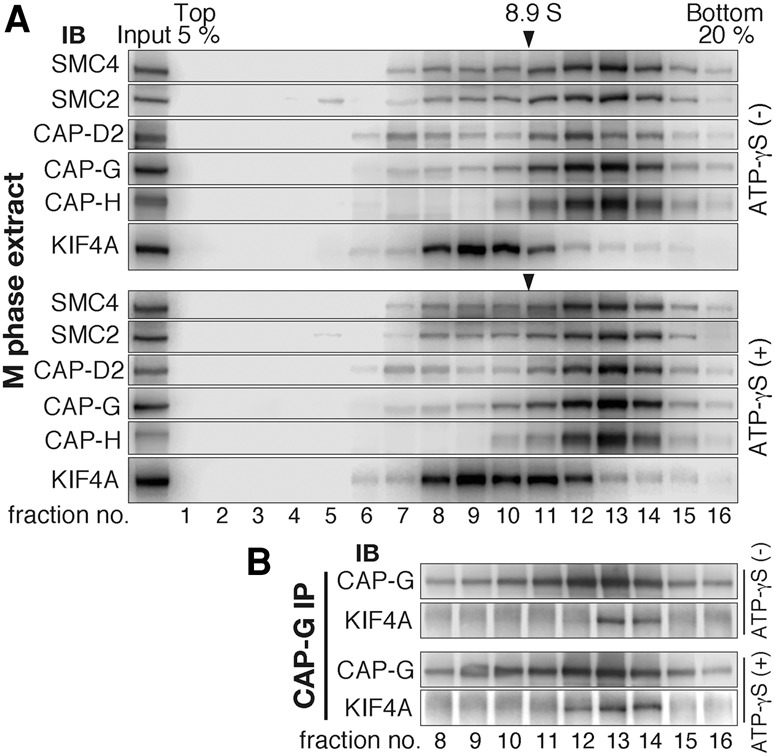
The proportion of KIF4A-bound condensin I in the cellular pool of KIF4A and condensin I. (A) Total extracts of mitotic HeLa cells were prepared in the presence or absence of ATP-γS, fractionated by 5%–20% sucrose density gradient centrifugation, and analyzed by immunoblotting. Peaks of the 8.9S marker (arrowhead) verify the repeatability of experiments, and the efficiency of mitotic synchronization was confirmed by immunoblotting of Cdc27 (Supplemental Fig. S5D). Note that KIF4A-bound condensin I shows a detectable shift for KIF4A, but the shift for condensin I was less clear because the weight change was sufficiently large for KIF4A but not for condensin I. (B) Immunoprecipitation analysis to examine where KIF4A-bound condensin I migrates. Fractions from 8 to 16 in A were each subjected to immunoprecipitation with CAP-G antibodies and immunoblotted for CAP-G and KIF4A.
To address which of the density fractions indeed contains the KIF4A-bound condensin I, we immunoprecipitated condensin I with CAP-G antibodies from all of the fractions containing condensin I. Reasonably, KIF4A was found to coimmunoprecipitate in heavier-density fractions for KIF4A (Fig. 4B). Of note, these fractions did not overlap with fractions in which the majority of KIF4A migrated (Fig. 4A). Also, fractionation of S-phase extracts in which KIF4A-bound condensin I is absent (Supplemental Fig. S5A) suggested that the majority of condensin I-positive fractions in mitotic extracts is composed of KIF4A-unbound condensin I. These results indicate that only a part of cellular KIF4A and condensin I is involved in their interaction. This would be a reason why the interaction has been difficult to detect in immunoprecipitation assays.
How, then, can KIF4A have a profound impact on condensin I when their interacting populations are limited? A plausible possibility would be that KIF4A transiently associates with condensin I and regulates localization of condensin I through a catalysis-like mechanism. In line with this, ATP-γS treatment caused a moderate increase of the KIF4A-bound condensin I complex, presumably because deceleration of ATP hydrolysis stabilized the interaction between KIF4A and condensin I (Fig. 4B). The fluorescence recovery after photobleaching experiment indicating a different kinetics of KIF4A and condensin I on chromosomes (Gerlich et al. 2006; Samejima et al. 2012) also implies that their interaction occurs transiently.
The motor function of KIF4A is involved in the axial enrichment of condensin I
The motor activity of KIF4A has been primarily studied in the context of central spindle regulation (Zhu and Jiang 2005; Bastos et al. 2013). To address whether this motor function is involved in acting with condensin I, we generated cell lines that express a Myc-tagged motor-inactive KA mutant of KIF4A (Lys94 in the ATP-binding Walker A consensus site was mutated to alanine) (Wu and Chen 2008). When endogenous KIF4A was depleted, the KA mutant of KIF4A hardly converged into the midbody during cytokinesis, as characterized (Supplemental Fig. S6A; Zhu and Jiang 2005). Unlike the wild-type KIF4A, we noticed that the KA mutant failed to assemble into a fine axial structure of chromosomes (Fig. 5A). Concurrently, under these conditions, the axial localization of condensin I became unfocused or poorly defined. As the motor-inactive KIF4A could still associate with condensin I (Supplemental Fig. S6B), these results are consistent with the idea that enrichment of condensin I to chromosomal axes relies on the motor activity of KIF4A.
Figure 5.
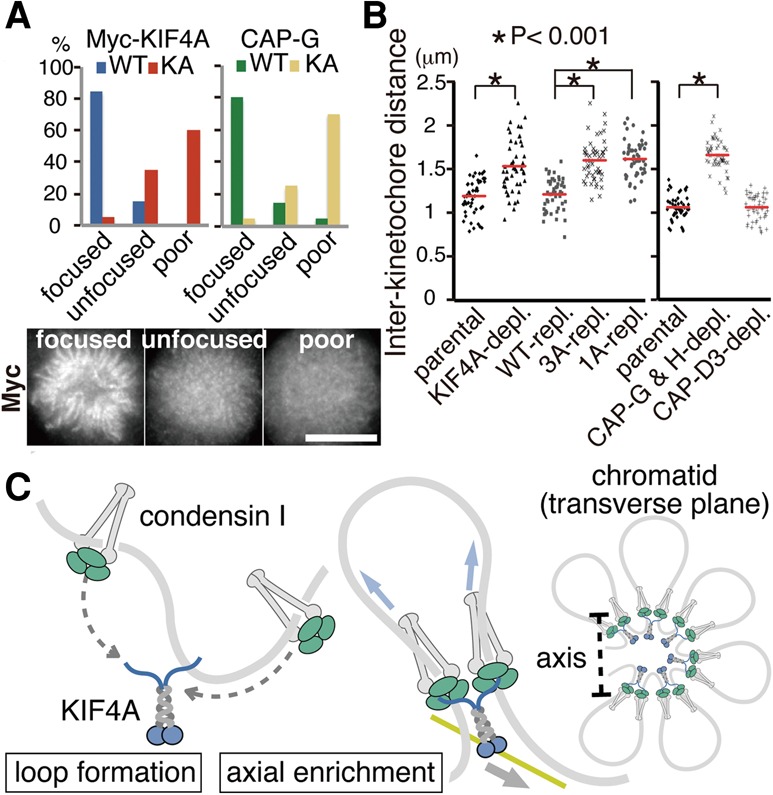
The functional relevance of KIF4A on condensin's function in organizing mitotic chromosomes. (A) The motor activity of KIF4A is involved in the axial enrichment of condensin I. HeLa cell lines expressing Myc-tagged KIF4A wild type or KA from transgenes were first depleted of endogenous KIF4A and then fixed and stained with antibodies to Myc and CAP-G. The staining patterns of KIF4A and CAP-G were classified into three categories, as indicated. Representative images are shown. Bar, 10 µm. (B) Interaction with KIF4A is required for condensin I to confer rigidity at centromeres. Quantification of interkinetochore distances on metaphase chromosomes in parental HeLa cells and KIF4A mutant-expressing cells in place of endogenous protein are shown. Interkinetochore distances in condensin I (CAP-G and CAP-H)-depeleted, or condensin II (CAP-D3)-depleted cells were measured in parallel (each n = 50 cells). (C) A hypothetical model predicting how condensin I and KIF4A might contribute to the organization of mitotic chromosomes. See the text for details.
The function of condensin I depends on binding to KIF4A
Condensin I is known to confer a rigidity to centromeric chromatin, and thus enlargement of the distance between sister kinetochores by microtubule-derived tension would be a readout for the activity of condensin I (Oliveira et al. 2005; Gerlich et al. 2006). Depletion of KIF4A caused a marked increase of the interkinetochore distance, as previously reported (Samejima et al. 2012). We found that the degree of the increase in the absence of KIF4A was largely equivalent to condensin I depletion. Importantly, this loss of centromere rigidity could be restored by expressing wild-type KIF4A but not the 3A-KIF4A or 1A-KIF4A mutants, indicating that the interaction is essential for the function of condensin I (Fig. 5B).
To further verify this idea, we asked whether condensin I-mediated resolution of chromosomal arms is affected when condensin I lacks the interaction with KIF4A. We found that cells depleted of KIF4A failed to efficiently release cohesion between chromosome arms and thus kept closed during the prolonged period in mitosis, as seen in cells depleted of condensin I (Hirota et al. 2004). Remarkably, this phenotype in KIF4A-depleted cells was again restored by expressing wild-type KIF4A but not the 3A-KIF4A or 1A-KIF4A mutant (Supplemental Fig. S7).
These results demonstrate the functional relevance of KIF4A on condensin I's function in shaping mitotic chromosomes. Our recent study suggested that the interaction between KIF4A and condensin I requires the activity of mitotic kinases Cdk1 and Aurora B (Takahashi et al. 2016). Intriguingly, these phenotypes of centromeres and chromosome arms that emerged in nonbinding mutants resemble chromosomal defects caused by Aurora B inactivation (Gimenez-Abian et al. 2004; Ono et al. 2004; Lipp et al. 2007). Thus, an appealing prediction would be that Aurora B supports the function of condensin I by promoting its binding to KIF4A.
How might KIF4A and condensin I act on chromatin to promote chromosome assembly?
Given that KIF4A forms a dimer, we speculate that two complexes of condensin I can be tethered by a KIF4A dimer. Our findings allow a speculative model in which two layers of processes underlie condensin I-mediated chromatid compaction (Fig. 5C). First, when condensin topologically holds the chromatin fiber (Cuylen et al. 2011), tethering two condesin I complexes would bring two distant DNA segments into proximity and create a loop in the chromatids. Second, the finding that accumulation of the KIF4A–condensin I complex to the chromosomal axis requires the motor function of KIF4A opens the possibility that the motor drags condensin I-mediated loops toward the chromatid core along putative cytoskeletal filaments, such as microtubules. The motor might also act to reel in and enlarge chromatin loops, and, as a result, the KIF4A–condensin I complex becomes enriched at the axis. Discovering which filament system KIF4A associates with and travels along is a challenging question for the future.
Is the KIF4A–condensin I interaction conserved throughout evolution?
Bioinformatic analyses have not identified a KIF4A homolog in yeasts. This may account for the fact that chromosome condensation occurs only mildly in these organisms. KIF4A is a conserved kinesin in metazoans, with a high level of amino acid conservation in the motor and coiled-coil domains. In contrast, the tail domain is poorly conserved, except for the very last 25 amino acids. As a secondary structure prediction suggests that the L1214 is a part of an α helix, it might comprise a hydrophobic bond to the HEAT repeats of CAP-G. Our binding assays imply that the adjacent two phenylalanines—at 1220 and 1221—support this interaction. Considering the conservation of these key amino acids mediating the interaction, it is reasonable to predict that the interaction and also the binding module between KIF4A and condensin I are assumed throughout evolution. Compacted chromosomes have been successfully assembled recently in vitro by condensin I with the minimum set of purified factors (Shintomi et al. 2015). As the above amino acids in KIF4A are also conserved in frogs, it is interesting to test whether addition of Xenopus KIF4A in this system might generate chromosomes with a higher degree of compaction.
Conclusions
In summary, although KIF4A has not been characterized as a condensin component, it was found enriched in chromosomes assembled in mitotic egg extracts and thus was initially called CAP-D, in a manner similar to canonical subunits of condensin I such as CAP-D2, CAP-G, and CAP-H (Hirano et al. 1997). However, unlike five subunits of condensin I that form a stable pentameric complex, KIF4A/CAP-D cannot be detected in association with condensin I, possibly due to the dynamic, unstable nature of the interaction. Nevertheless, KIF4A has an essential role in ensuring the function of condensin I. Experiments with condensin I-binding-deficient mutants highlight the significance of the interaction: Neither KIF4A nor condensin I alone can sufficiently accumulate on the chromosomal axis and confer physiological properties to chromosomes, thus establishing that the functions of KIF4A and condensin I are inseparable.
Materials and methods
RNAi
Cells were transfected with 50 or 100 nM siRNA duplex using RNAiMAX reagent (Life Technologies), which was performed during the thymidine synchronization in some experiments. Cells were used for the experiment at 48–72 h after siRNA transfection. Depletion efficiencies were monitored by Western blotting analysis. The siRNA sense sequences used for targeting KIF4A, CAP-G, CAP-H, and CAP-D3 were 5′-CCUCCAGGUCCAGACUACUACUCUA-3′, 5′-AAUACAUGAUAACACUGCCCGUCUA-3′, 5′-UACACAACCUAACUCUGGCAACUCG-3′, and 5′-UGAAUUGGGAUUCUGCUGACUGCUG-3′, respectively.
Sucrose density gradient centrifugation
To prepare cell extracts for sucrose density gradient centrifugation, cells synchronized in M or S phase were suspended in five packed cell volumes of a hypotonic buffer (10 mM K-HEPES at pH 7.7, 10 mM KCl, 1.5 mM MgCl2, 1 mM dithiothreitol [DTT], 0.1 µM okadaic acid, 50 ng/mL nocodazole). Swollen cells were then homogenized for 15 strokes, made into isotonic condition by adding 0.11× cell lysate volumes of a hypertonic buffer (110 mM K-HEPES at pH 7.7, 920 mM KCl, 7 mM MgCl2, 51 mM EGTA, 200 mM β–glycerophosphate, 101 mM NaF, 20 mM Na3VO4, 0.1 µM okadaic acid, 1 mM DTT, protease inhibitor coctail, 50 ng/mL nocodazole) with or without 1 mM ATP-γS (A060-05, Biolog), and incubated for 1 h at 30°C followed by 2 U/µL OmniCleave endonuclease treatment for 20 min on ice. After centrifugation at 15,000 rpm, cell lysates were overlaid to 5%–20% linear sucrose density gradients, prepared with a lysis buffer (20 mM K-HEPES at pH 7.7, 100 mM KCl, 2 mM MgCl2, 5 mM EGTA, 20 mM β-glycerophosphate, 10 mM NaF, 2 mM Na3VO4, 0.1 µM okadaic acid, 1 mM DTT, protease inhibitor) using Gradient Master (BioComp), and centrifuged at 34,000 rpm (Beckman SW40Ti swing rotor) for 30 h at 4°C. Sixteen fractions were reproducibly collected using a Pistone fractionator (BioComp). For calibration, a standard protein, β-amylase 8.9S, was used (Sigma–Aldrich, MWGF1000).
Detailed information for experimental procedures—including antibodies, cells, transgenes, immunoblotting, immunoprecipitation, pull-down assays, immunofluorescence microscopy, and live-cell imaging—are in the Supplemental Material.
Supplementary Material
Acknowledgments
We are grateful to Kenji Sugase, Ayako Furukawa (Suntory Foundation for Life Sciences, Osaka), and Alexander Schleiffer (Research Institute of Molecular Pathology, Vienna) for structural information; Keiji Kimura (Tsukuba University, Tsukuba) and Ai Takemoto (Japanese Foundation for Cancer Research [JFCR], Tokyo) for advice on biochemical experiments; and Utako Kato (JFCR, Tokyo) for generating cell clones. Research in the laboratory of T.H. is supported by grants from the Japan Society for the Promotion of Science (JSPS) and the Ministry of Education, Culture, Sports and Technology of Japan (MEXT).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.282855.116.
References
- Abe S, Nagasaka K, Hirayama Y, Kozuka-Hata H, Oyama M, Aoyagi Y, Obuse C, Hirota T. 2011. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev 25: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastos RN, Gandhi SR, Baron RD, Gruneberg U, Nigg EA, Barr FA. 2013. Aurora B suppresses microtubule dynamics and limits central spindle size by locally activationg KIF4A. J Cell Biol 202: 605–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuylen S, Metz J, Haering CH. 2011. Condensin structures chromosomal DNA through topological links. Nat Struct Mol Biol 18: 894–901. [DOI] [PubMed] [Google Scholar]
- Gerlich D, Hirota T, Koch D, Peters JM, Ellenberg J. 2006. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr Biol 16: 333–344. [DOI] [PubMed] [Google Scholar]
- Gimenez-Abian JF, Sumara I, Hirota T, Hauf S, Gerlich D, de la Torre C, Ellenberg J, Peters JM. 2004. Regulation of sister chromatid cohesion between chromosome arms. Curr Biol 14: 1187–1193. [DOI] [PubMed] [Google Scholar]
- Hirano T. 2012. Condensins: universal organizers of chromosomes with diverse functions. Genes Dev 26: 1659–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano T, Kobayashi R, Hirano M. 1997. Condensins, chromosome condensation protein complexes containing XCAP-C, XCAP-E and a Xenopus homolog of the Drosophila Barren protein. Cell 89: 511–521. [DOI] [PubMed] [Google Scholar]
- Hirota T, Gerich D, Koch B, Ellenberg J, Peters JM. 2004. Distinct functions of condensin I and II in mitotic chromosome assembly. J Cell Sci 117: 6435–6445. [DOI] [PubMed] [Google Scholar]
- Houlard M, Godwin J, Metson J, Lee J, Hirano T, Nasmyth K. 2015. Condensin confers the longitudinal rigidity of chromosomes. Nat Cell Biol 17: 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipp JJ, Hirota T, Poser I, Peters JM. 2007. Aurora B controls the association of condensin I but not condensin II with mitotic chromosomes. J Cell Sci 120: 1245–1255. [DOI] [PubMed] [Google Scholar]
- Mazumdar M, Sundareshan S, Misteli T. 2004. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J Cell Biol 166: 613–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RA, Coelho PA, Sunkel CE. 2005. The condensin I subunit Barres/CAP-H is essential for the structural integrity of centromeric heterochromatin during mitosis. Mol Cell Biol 25: 8971–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Losada A, Hirano M, Myers MP, Neuwald AF, Hirano T. 2003. Differential contributions of condensin I and condensin II to mitotic chromosome architecture in vertebrate cells. Cell 115: 109–121. [DOI] [PubMed] [Google Scholar]
- Ono T, Fang Y, Spector DL, Hirano T. 2004. Spatial and temporal regulation of Condensin I and II in mitotic chromosome assembly in human cells. Mol Biol Cell 15: 3296–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Yamashita D, Hirano T. 2013. Codensin II initiates sister chromatid resolution during S phase. J Cell Biol 200: 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samejima K, Samejima I, Vagnarelli P, Ogawa H, Vargiu G, Kelly DA, de Lima Alves F, Kerr A, Green LC, Hudson DF, et al. 2012. Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. J Cell Biol 199: 755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Rice SE. 2013. Intrinsic disorder in the kinesin superfamily. Biophys Rev 10.1007/s12551-012-0096-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintomi K, Takahashi TS, Hirano T. 2015. Reconstitution of mitotic chromatids with a minimum set of purified factors. Nat Cell Biol 17: 1014–1023. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Tanaka K, Wakai T, Hirota T. 2016. Phosphoproteomic analysis of human mitotic chromosomes identified a chromokinesin KIF4A. Biomed Res 37: 161–165. [DOI] [PubMed] [Google Scholar]
- Thadani R, Uhlmann F, Heeger S. 2012. Condensin, chromatin crossbarring and chromosome condensation. Curr Biol 22: 1012–1021. [DOI] [PubMed] [Google Scholar]
- Wang SZ, Adler R. 1995. Chromokinesin: a DNA-binding, kinesin-like nuclear protein. J Cell Biol 128: 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Chen PL. 2008. Structural requirements of chromokinesin Kif4A for its proper function in mitosis. Biochem Biophys Res Commun 372: 454–458. [DOI] [PubMed] [Google Scholar]
- Yeong FM, Hombauer H, Wendt KS, Hirota T, Mudrak I, Mechtler K, Loregger T, Marchler-Bauer A, Tanaka K, Peters JM, et al. 2003. Identification of a subunit of a novel Kleisin-β/SMC complex as a potential substrate of protein phosphatase 2A. Curr Biol 13: 2058–2064. [DOI] [PubMed] [Google Scholar]
- Zhu C, Jiang W. 2005. Cell cycle-dependent translocation of PRC1 on the spindle by Kif4 is essential for midzone formation and cytokinesis. Proc Natl Acad Sci 102: 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.