

Abstract
Early detection is critical in prevention and treatment of kidney disease. However currently clinical laboratory and histopathological tests do not provide region-specific and accurate biomarkers for early detection of kidney disease. The present study was conducted to identify sensitive biomarkers for early detection and progression of tubulo-interstitial nephropathy in aristolochic acid I-induced rats at weeks 4, 8 and 12. Biomarkers were validated using aristolochic acid nephropathy (AAN) rats at week 24, adenine-induced chronic kidney disease (CKD) rats and CKD patients. Compared with control rats, AAN rats showed anemia, increased serum urea and creatinine, progressive renal interstitial fibrosis, activation of nuclear factor-kappa B, and up-regulation of pro-inflammatory, pro-oxidant, and pro-fibrotic proteins at weeks 8 and 12. However, no significant difference was found at week 4. Metabolomics identified 12-ketodeoxycholic acid, taurochenodesoxycholic acid, LPC(15:0) and docosahexaenoic acid as biomarkers for early detection of tubulo-interstitial nephropathy. With prolonging aristolochic acid I exposure, LPE(20:2), cholic acid, chenodeoxycholic acid and LPC(17:0) were identified as biomarkers for progression from early to advanced AAN and lysoPE(22:5), indoxyl sulfate, uric acid and creatinine as biomarkers of advanced AAN. These biomarkers were reversed by treatment of irbesartan and ergone in AAN rats at week 24 and adenine-induced CKD rats. In addition, these biomarkers were also reversed by irbesartan treatment in CKD patients.
Keywords: Chronic kidney disease, Metabolomics, Inflammation, Renal fibrosis, Lipid metabolism, Biomarker
Graphical abstract
Highlights
-
•
Tubulo-interstitial nephropathy (TIN) is a common cause of chronic kidney disease.
-
•
Plasma biomarker discovery and validation was performed by UPLC-based metabolomics.
-
•
CKD progression associated with activated redox signaling and lipid dysfunction.
-
•
TIN was associated with abnormal amino acids, purine and phospholipid metabolisms.
1. Introduction
Kidney is a common target of environmental and drug toxicity which is a major clinical problem. Early detection of drug-associated kidney injury is essential in prevention of kidney disease. Currently, evaluations of kidney injury are primarily based on measurements of serum creatinine (Scr) and urea, and routine urinalysis which are not region-specific and are significantly altered only after substantial kidney injury has occurred. Serum cystatin C has been considered to be more reliable than Scr and urinary protein in the early stage of chronic kidney disease (CKD) [1]. However cystatin C is affected by obesity, hyperthyroidism and steroid therapy. Therefore, region-specific and accurate biomarkers are needed for earlier detection of CKD to avoid iatrogenic morbidity and mortality.
Metabolomics is the latest system biology technology for understanding complex disease processes. Metabolomics focuses on identification of biochemical signatures related to pathogenesis of disease that could be used for the diagnosis and monitoring of the disease progression, and response to therapeutic interventions. A number of metabolomic studies have reported that metabolite changes of serum, urine and kidney tissues in CKD [2], [3], [4], [5], [6]. There has been a lack of systematic studies simultaneously determining changes in kidney tissue and serum to establish mechanistic links. Tubulo-interstitial nephropathy is a common cause of CKD. Aristolochic acid nephropathy (AAN) is a rapidly progressive interstitial nephritis that can lead to end stage renal disease (ESRD) [7]. Pharmacological and metabolomic investigations have revealed that histological lesions and metabolic signatures of AAN in experimental animals are similar to those of human tubulo-interstitial nephropathy [8], [9]. Long-term feeding of adenine to rats causes metabolic abnormalities similar to human CKD [10], [11]. Ultra performance liquid chromatography-quadrupole time-of-flight high-definition mass spectrometry (UPLC-QTOF/HDMS) is regarded as one of the best analytical techniques in sensitivity, selectivity and reproducibility [12], [13], [14]. The present study was undertaken to examine the effect of AAN on inflammatory, oxidative, and fibrotic cascades in the kidney tissue and to explore the longitudinal changes in the serum metabolome and kidney histology with long-term exposure to AAI in rats. Potential biomarkers were validated using AAN rats at week 24. Biomarkers were further validated using adenine-induced CKD rats and advanced CKD patients (Study flowchart in Fig. 1).
Fig. 1.

Flowchart of the study strategy in this study.
2. Materials and methods
2.1. AAN model
Male Sprague-Dawley rats were randomized to the AAN and control groups. The preparation of the AAN model and blood collections in the study animals were carried out as described in detail previously [15]. The AAN group was orally administered 20 mg/kg body weight/day of AAI by oral gavage for 12 weeks and used as the discovery phase. Serum samples were collected at weeks 4, 8 and 12.
2.2. Validation analysis by AAN rats, adenine-induced CKD rats and patients with CKD
We validated the biomarkers using the additional independent groups of control, AAN and irbesartan (IRB)-treated AAN and ergone (ERG)-treated AAN (ergone, a natural product from Cordyceps sinensis or Polyporus umbellatus) rats at week 24 [16]. In addition, we also validated the biomarkers using the adenine-induced CKD rats including control, CKD and IRB-treated CKD and ERG-treated CKD rats at week 6.
Blood samples from 25 CKD patients and 25 age and gender-matched healthy controls were obtained from the Affiliated Hospital of Shaanxi Institute of Traditional Chinese Medicine. The patients were also treated by irbesartan. The detailed clinical characteristics of the CKD patient included in this study are summarized in supplementary Table S1.
All the serum samples were used to test and validate potential biomarkers identified in the discovery phase. The biomarkers were quantitative analysis by using UPLC-QTOF/HDMS. All the study was approved by the Ethical Committee of Northwest University and all procedures were conducted in accordance with the Helsinki Declaration.
2.3. Chemical reagents and antibodies
Aristolochic acid I, creatinine, chenodeoxycholic acid, cholic acid, taurochenodesoxycholic acid, eicosapentaenoic acid, arachidonic acid (AA), docosahexaenoic acid, 5-hydroxyeicosatetraenoate (5-HETE), indoxyl sulfate and uric acid and p-Cresol sulfate were obtained from the National Institutes for Food and Drug Control. L-tryptophan was purchased from Amresco Company. Antibodies against nuclear factor-kappa B (NF-κB), cyclooxygenase-1 (COX-1), 12-lipoxygenase (12-LPO), NADPH oxidase 4 (NOX4), gp91phox, p67phox, ras-related C3 botulinum toxin substrate 1 (Rac1), plasminogen activator inhibitor-1 (PAI-1), alpha smooth muscle actin (α-SMA), transforming growth factor-β receptor I (TGF-β RI), transforming growth factor-β receptor II (TGF-β RII), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and histone H3 were purchased from Abcam Company. Antibodies against inhibitor kappa B alpha (IκBα), phosphorylated inhibitor kappa B alpha (p-IκBα), transforming growth factor-β 1 (TGF-β1), connective tissue growth factor (CTGF) and tissue inhibitor of metalloproteinase-1 (TIMP-1) were purchased from Santa Cruz Biotechnology.
2.4. Biochemical, histological and Western blot analyses
Plasma biochemistry was analyzed as described in detail previously [17]. White blood cell count (WBC), red blood cell count (RBC) or hemoglobin (HGB) was analyzed by HF-3800 analyzer. Kidney histology and fibrotic degree were assessed as described in detail previously [18]. All the Western blot analyses were performed as described previously [9].
2.5. Sample preparation and UPLC-MS analysis
Serum samples were prepared as described previously. The UPLC analysis was performed on a 2.1 mm×100 mm ACQUITY 1.8 µm HSS T3 using a Waters Acquity™ UPLC system equipped with a Waters Xevo™ G2 QTof MS. A gradient of 0.1% formic acid in acetonitrile (A) and 0.1% formic acid in water (B) used as follows: a linear gradient of 0–10 min, 1–99% A; 11.0–12.0 min, 99.0–1.0% A and 12.0–15.0 min, 1% A. The flow rate was 0.45 ml/min. The autosampler was maintained at 4 °C. The lyophilized serum samples were dissolved in 100 µL of acetonitrile/water (4:1). Every 2 µL sample solution was injected for each run.
Mass spectrometry of the optimal conditions were as follows: capillary voltage of 2.5 kV for negative ion mode, cone voltage of 35 V, desolvation gas temperature of 450 °C, source temperature of 120 °C, desolvation gas flow of 800 L/h, cone gas flow of 30 L/h, The scan range was from 50 to 1200 m/z. Leucine–enkephalin was used for accurate mass acquisition. Waters MassLynx v4.1 was used for all the acquisition and analysis of data.
2.6. Data analysis
The acquired UPLC-HDMS raw data in negative ion mode were first pre-processed by the Progenesis QI and Markerlynx XS. Orthogonal partial least square-discriminant analysis (OPLS-DA) and principal component analysis (PCA) was performed to discriminate between ANN and control groups. The potential metabolites between AAN and control groups were identified on the basis of variable importance in the projection (VIP) on the normalized peak intensity, where metabolites with VIP >1.0 were selected. A panel of potential metabolites responsible for the difference in the AAN group and control group was obtained.
Fold change (FC) were calculated based on mean ratios for AAN/control groups. Differential metabolites between AAN and control groups were identified by use of two-tailed Student's t-test (normal distribution) and two-tailed Mann-Whitney U test (abnormal distribution) with a threshold of P<0.05 in the SPSS 19.0 software. The resultant P values from two-tailed Student's t-test were further adjusted using the Hochberg and Benjamini false discovery rate (FDR) method. Potential metabolites with both multivariate and univariate statistical significance (VIP >1 and P<0.05) were considered to be potential biomarkers. Class-specific metabolomic pattern was visualized using heat map and z-score plots analyses. In addition, PLS-DA-based receiver operating characteristics (ROC) analysis was performed for the selection of candidate biomarkers, and ROC curves were plotted using MedCalc or SPSS.
2.7. Metabolic network analysis
The related metabolic pathways in AAN rats were performed by means of the quantitative enrichment analysis algorithm described in metabolite set enrichment analysis (MSEA) method [19]. Visualization of the remarkably disturbed metabolic pathways in AAN rats was performed by MetScape software running on Cytoscape [20].
3. Results
3.1. General data of AAN rats
Compared with the control group, the AAN rats showed a greatly increasing trend in plasma urea, creatinine and uric acid concentrations as well as WBC at week 4, but did not arrive at statistical significance. However, the AAN group exhibited significantly higher WBC, plasma urea, creatinine, uric acid and phosphorus concentrations, and a significantly lower RBC, HGB and plasma calcium concentrations at weeks 8 and 12 (Fig. 2A)..
Fig. 2.

Clinical biochemical parameters and histological analyses. (A) Serum creatinine, urea, uric acid, calcium and phosphorus of basic serum biochemical parameter and WBC, RBC, HGB and HCT of basic blood routine parameters comparisons between control and AAN groups. (B) Kidney tissues were stained using PAS method in control group and AAN group at weeks 4, 8 and 12. (C) Kidney tissues were stained using Masson staining in control group and AAN group at weeks 4, 8 and 12. All values presented as mean±SD. All statistical significances were calculated by two-tailed Student’s t-test with 95% confidence interval. *P<0.05, **P<0.01 compared to control group.
Representative photomicrographs of periodic acid-schiff (PAS) and Masson stained rats’ kidney tissues are shown in Fig. 2B and C. No significant abnormalities were found in the kidneys of the AAN rats at week 4. In contrast, kidney tissues from the AAN rats exhibited progressive tubulo-interstitial lesions at weeks 8 and 12. At week 8, PAS staining showed tubular cell swelling, vacuolization, slight granular degeneration, and modest monocyte and lymphocyte infiltration. At week 12, PAS staining showed inflammatory cell infiltration, tubular necrosis and atrophy, tubular brush border loss and interstitial fibrosis. Masson staining of the kidney tissue showed renal interstitial fibrosis, accumulation of myofibroblasts, and tubular damage at weeks 8 and 12. However, none of these changes were observed in AAN rats at week 4. The morphometric analysis of the kidney tissues revealed significant increase in renal interstitial fibrosis at weeks 8 and 12, but not week 4 in AAN rats.
3.2. Inflammatory, oxidative and fibrotic pathways
Expressions of inflammatory, oxidative and fibrotic proteins in the kidney tissues are shown in Fig. 3. Compared to control group, kidney tissues obtained from the AAN group at weeks 8 and 12 showed a marked increase in p-IκB and decrease in IκBα as well as elevated nuclear translocation of NF-κB subunit, indicating activation of NF-κB, the master regulator of pro-inflammatory and pro-fibrotic cytokine and chemokines. This was accompanied by significant up-regulation of pro-inflammatory and ROS-generating molecules including COX-1, 12-LPO, NOX4, gp91phox, p67phox and Rac1. Activation of inflammatory and oxidative pathways in the kidneys of AAN rats was accompanied by marked up-regulation of TGF-β RI, TGF-β RII, CTGF, PAI-1, TIMP-1 and α-SMA at weeks 8 and 12, pointing to activation of fibrotic pathway. However, no significant difference was found in expression of the measured proteins between AAN and control rats at week 4.
Fig. 3.

Western blot analyses of pro-inflammatory, pro-oxidant and fibrotic proteins. Western blots analysis between control and AAN groups and protein abundance of p-IκBα, IκBα and nuclear contents of active subunit of NF-κB, COX-1, 12-LPO and protein abundance of NAD(P)H oxidase subunits (NOX4, Rac1, gp91phox and p67phox) from the renal tissues of control and AAN groups at weeks 4, 8 and 12. Representative western blots of fibrotic protein including TGF-β RI, TGF-β RII, CTGF, PAI-1, TIMP-1 and α-SMA in the renal tissues of control and AAN groups at weeks 4, 8 and 12. Box plots for relative optical density of protein expression in control and AAN groups. All statistical significances were calculated by two-tailed Student’s t-test with 95% confidence interval. *P<0.05, **P<0.01 compared with the control group.
3.3. Metabolic profile and multivariate analysis
We applied UPLC to obtain the metabolic profiles in positive and negative ion modes. We observed that higher noise and matrix effect in positive ion mode resulted in a higher baseline, so negative ion mode was applied for final analysis (Fig. S1).
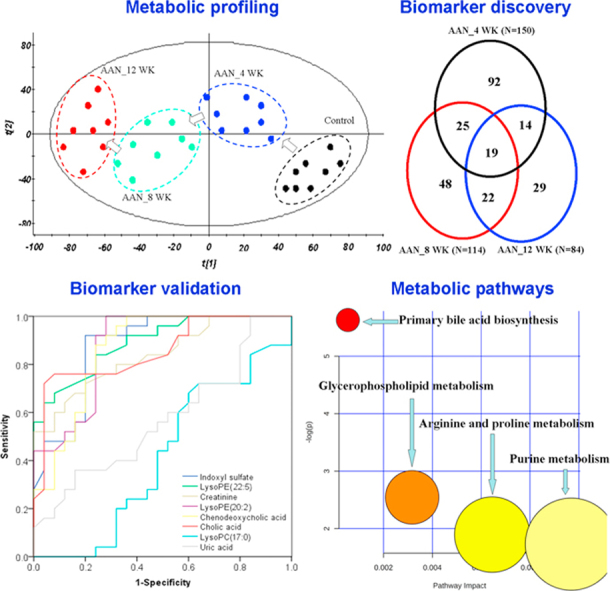
To evaluate the systemic changes of metabolome in AAN rats and find biomarkers, a two-predictive component OPLS-DA was performed on the serum metabolites from control and AAN. The OPLS-DA score plots could be readily divided into two clusters indicating that serum metabolic pattern was significantly altered in AAN group (Fig. S2A–C). The metabolic patterns of AAN group at weeks 4, 8 and 12 were plotted by PCA, in which arrows express the variable trend in metabolic patterns at weeks 0, 4, 8, and 12 (Fig. 4A). This indicated that AAI has caused different metabolic disturbance and pathobiological changes in different stages.
Fig. 4.

Metabolomic profiling of AAN. (A) The PCA score scatter plot of the different AAN at weeks 4, 8 and 12. (B) Vann diagram of the total remarkable variables when the AAN and control groups were compared (VIP>1.0, P<0.01, AUC>0.85). (C) Z-score plots for the data of 150 (week 4), 114 (week 8) and 84 (week 12) variables normalized to the mean of the control group. (D) Heat map of five metabolites at weeks 4, 8 and 12. The metabolites occur in AAN rats at weeks 4, 8 and 12. The color of each section is proportional to the significance of change of metabolites (red, upregulated; green, downregulated). Rows: metabolites; Columns: samples. (E) Comparison of the relative intensity of five metabolites including lysoPE(20:2), CA, CDCA, cystathionine sulfoxide and lysoPC(17:0). These metabolites were significantly increased or decreased on AAN progression from early stage to advanced stage. These metabolites could potentially serve as biomarkers for progressive AAN. (F) PCA score plot based on the five metabolites of the control group and AAN group. All statistical significances were calculated by two-tailed Student’s t-test with 95% confidence interval. *P<0.05, **P<0.01 compared to the control group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Selection and identification of important differential metabolites
Initially, variables were selected according to the VIP values from S-plots, which reflect the influence of each variable in AAN and control groups. Fig. S2E–G show the S-plots based on serum metabolic profiling at weeks 4, 8, and 12. It was found that 321, 306, and 336 variables have VIP values of greater than 1.0 at weeks 4, 8 and 12, respectively. Subsequently, using a combination of the Student's t-test and ROC curve, these variables were selected. Variables that had P<0.05 and the area under the curve (AUC) value> 0.75 were selected to further identify potential biomarkers. 150, 114, and 84 variables were selected at weeks 4, 8 and 12, respectively. PCA and heatmap showed that these variables could separate AAN from control groups (Fig. S3). A total of 249 variables with statistically significant differences were further studied (Fig. 4B). Fig. 4C presents the z-score plots of 150, 114, and 84 significantly changed variables at weeks 4, 8 and 12, respectively. These variables could better reflect the metabolic changes of the kidney injury. To further select the potential biomarkers, two-tailed Mann-Whitney U test and FDR were also applied to calculate the significance of each identified metabolite. In addition, xenobiotics, different fragment ions from the same metabolites and variables >3-fold different between the control groups were excluded. Potential metabolites were identified according to the previously reported method [21]. 13, 17 and 18 differential metabolites were identified as potential biomarkers at weeks 4, 8 and 12, respectively (Table 1).
Table 1.
Identification biomarkers of AAN detected by UPLC-QTOF/HDMS in negative ion mode in the 4th, 8th and 12th week.
| No. | Metabolite | VIPa | FCb | Pc | Pd | FDRe | Metabolic pathway |
|---|---|---|---|---|---|---|---|
| 4th week | |||||||
| 1 | LysoPC(15:0) | 4.15 | 0.74 | 1.21E−04 | 5.88E−04 | 1.75E−04 | Lipid metabolism |
| 2 | LysoPC(17:0) | 3.71 | 0.82 | 4.78E−03 | 3.57E−02 | 4.78E−03 | Lipid metabolism |
| 3 | Chenodeoxycholic acid | 3.51 | 1.99 | 3.58E−04 | 7.71E−04 | 3.88E−04 | Bile acid metabolism |
| 4 | LysoPE(20:2) | 3.22 | 1.37 | 4.76E−05 | 7.78E−04 | 8.84E−05 | Lipid metabolism |
| 5 | Cholic acid | 2.93 | 1.61 | 1.12E−04 | 1.60E−03 | 1.82E−04 | Bile acid metabolism |
| 6 | Taurochenodesoxycholic acid | 2.74 | 2.06 | 2.90E−05 | 4.75E−04 | 6.28E−05 | Bile acid metabolism |
| 7 | Eicosapentaenoic acid | 2.56 | 0.71 | 6.54E−11 | 4.49E−06 | 4.25E−10 | fatty acid metabolism |
| 8 | Arachidonic acid | 2.47 | 3.48 | 6.69E−07 | 9.48E−06 | 2.17E−06 | Prostaglandin and leukotriene metabolism |
| 9 | 7-Ketodeoxycholic acid | 2.24 | 0.67 | 1.90E−04 | 3.39E−05 | 2.47E−04 | Bile acid metabolism |
| 10 | Cystathionine sulfoxide | 2.06 | 0.54 | 2.56E−05 | 7.78E−04 | 6.66E−05 | Progestational hormone metabolism |
| 11 | 16-Dehydroprogesterone | 1.77 | 1.95 | 6.24E−16 | 4.13E−06 | 8.11E−15 | Progestational hormone metabolism |
| 12 | Docosahexaenoic acid | 1.65 | 5.43 | 8.42E−08 | 4.73E−06 | 3.65E−07 | fatty acid metabolism |
| 13 | 12-Ketodeoxycholic acid | 1.26 | 0.74 | 2.75E−04 | 3.64E−06 | 3.25E−04 | Bile acid metabolism |
| 8th week | |||||||
| 1 | 5-HETE | 3.91 | 0.58 | 1.30E−07 | 4.87E−07 | 1.11E−06 | Arachidonic acid metabolism |
| 2 | LysoPE(20:1) | 3.37 | 1.46 | 7.17E−06 | 2.95E−07 | 1.74E−05 | Lipid metabolism |
| 3 | Chenodeoxycholic acid | 3.18 | 2.27 | 5.48E−04 | 6.25E−03 | 9.32E−04 | Bile acid metabolism |
| 4 | 7-Ketodeoxycholic acid | 3.15 | 4.54 | 2.85E−06 | 1.64E−06 | 1.21E−05 | Bile acid metabolism |
| 5 | Tryptophan | 2.96 | 0.55 | 1.09E−09 | 7.23E−07 | 1.85E−08 | Tryptophan metabolism |
| 6 | 2-Methylacetophenone | 2.72 | 4.15 | 1.91E−07 | 1.34E−04 | 1.08E−06 | Unknown |
| 7 | Cholic acid | 2.52 | 1.69 | 1.55E−02 | 1.17E−02 | 1.55E−02 | Bile acid metabolism |
| 8 | LysoPE(24:6) | 2.42 | 1.77 | 1.17E−02 | 6.35E−03 | 1.33E−02 | Lipid metabolism |
| 9 | LysoPE(20:2) | 2.39 | 1.36 | 6.35E−03 | 3.28E−03 | 7.71E−03 | Lipid metabolism |
| 10 | LysoPC(17:0) | 2.17 | 0.74 | 3.28E−03 | 7.78E−04 | 4.29E−03 | Lipid metabolism |
| 11 | LysoPE(18:0) | 1.87 | 1.41 | 7.78E−04 | 2.76E−03 | 1.20E−03 | Lipid metabolism |
| 12 | LysoPE(22:4) | 1.76 | 1.41 | 1.64E−03 | 2.64E−03 | 2.32E−03 | Lipid metabolis |
| 13 | Leukotriene A4 | 1.54 | 0.49 | 2.42E−05 | 9.36E−06 | 5.14E−05 | Prostaglandin and leukotriene metabolism |
| 14 | LysoPE(18:1) | 1.44 | 1.84 | 5.69E−06 | 7.08E−07 | 1.61E−05 | Lipid metabolism |
| 15 | Glycocholic acid | 1.32 | 0.52 | 3.89E−06 | 3.00E−07 | 1.32E−05 | Bile acid metabolism |
| 16 | Dihydroxy-3-oxo-4-cholenoic acid | 1.29 | 5.15 | 8.97E−05 | 3.31E−05 | 1.69E−04 | Bile acid metabolism |
| 17 | Cystathionine sulfoxide | 1.27 | 0.79 | 1.17E−02 | 3.56E−02 | 1.24E−02 | Progestational hormone metabolism |
| 12th week | |||||||
| 1 | LysoPE(18:0) | 4.67 | 1.99 | 8.66E−07 | 2.14E−05 | 5.20E−06 | Lipid metabolism |
| 2 | 5-HETE | 4.49 | 0.82 | 6.80E−06 | 2.68E−06 | 1.11E−05 | Arachidonic acid metabolism |
| 3 | LysoPC(17:0) | 3.97 | 0.67 | 1.04E−06 | 7.71E−04 | 4.68E−06 | Lipid metabolism |
| 4 | LysoPE(20:2) | 3.75 | 1.81 | 3.30E−09 | 7.78E−04 | 5.94E−08 | Lipid metabolism |
| 5 | Indoxyl sulfate | 3.57 | 4.37 | 1.06E−06 | 2.40E−06 | 3.82E−06 | Tryptophan metabolism |
| 6 | LysoPE(20:1) | 3.26 | 1.54 | 1.17E−06 | 2.50E−05 | 3.51E−06 | Lipid metabolism |
| 7 | LysoPC(15:0) | 2.98 | 0.91 | 5.93E−04 | 4.97E−03 | 7.12E−04 | Lipid metabolism |
| 8 | Leukotriene A4 | 2.93 | 0.57 | 1.92E−06 | 2.54E−05 | 4.94E−06 | Prostaglandin and leukotriene metabolism |
| 9 | Uric acid | 2.81 | 2.20 | 1.46E−03 | 5.28E−03 | 1.64E−03 | Adenine metabolism |
| 10 | LysoPE(24:6) | 2.76 | 1.62 | 5.14E−05 | 3.78E−05 | 7.71E−05 | Lipid metabolism |
| 11 | Creatinine | 2.53 | 1.52 | 7.13E−03 | 6.24E−03 | 7.55E−03 | Arginie and proline metabolism |
| 12 | LysoPE(18:1) | 2.31 | 1.84 | 4.35E−06 | 1.05E−05 | 8.70E−06 | Lipid metabolism |
| 13 | Cholic acid | 2.2 | 2.16 | 2.21E−04 | 3.23E−03 | 2.84E−04 | Bile acid metabolism |
| 14 | Chenodeoxycholic acid | 1.86 | 3.06 | 1.55E−07 | 7.78E−04 | 1.40E−06 | Bile acid metabolism |
| 15 | Cystathionine sulfoxide | 1.53 | 0.84 | 2.20E−02 | 3.54E−02 | 2.20E−02 | Progestational hormone metabolism |
| 16 | LysoPE(16:0) | 1.47 | 1.74 | 4.80E−06 | 2.50E−04 | 8.64E−06 | Lipid metabolism |
| 17 | LysoPE(22:5) | 1.34 | 2.13 | 5.90E−05 | 1.84E−04 | 8.17E−05 | Lipid metabolism |
| 18 | Taurochenodesoxycholic acid | 1.21 | 0.4 | 3.42E−06 | 5.81E−07 | 7.70E−06 | Bile acid metabolism |
VIP was obtained from OPLS-DA model with a threshold of 1.0.
FC was obtained by comparing those identified metabolites in AAN rats with control rats. FC with a value >1 indicated a relatively higher intensity present in AAN rats, whereas a value <1 indicated a relatively lower intensity compared with control rats.
P values from Student’s t-test.
P values from Mann-Whitney U-test.
FDR value was obtained from the adjusted P value of FDR correction by Benjamini-Hochberg method.
3.5. Biomarkers of progressive AAN
LysoPE(20:2), cholic acid (CA), chenodeoxycholic acid (CDCA), cystathionine sulfoxide and lysoPC(17:0) were significantly altered in AAN rats at weeks 4, 8 and 12. To identify biomarkers indicative of progressive AAN from early stage to advanced stage during the course of the AAN, a criterion was applied for selecting identified metabolites that were unidirectionally changed across the different AAN stages. Metabolites that were increased in one stage and decreased in another stage were not selected. By applying this selection criterion, a subset of three metabolites including lysoPE(20:2), CA and CDCA were consistently increased with AAN progression, whereas lysoPC(17:0) tended to consistently decrease with AAN progression from control to early stage to advanced stage (Fig. 4D and E). PCA shows that AAN subgroups and control group can be clearly separated (Fig. 4F). Therefore these metabolites could potentially serve as biomarkers for progressive AAN.
3.6. Early biomarkers of AAN
Metabolites showed early response in AAN rats (4th week). Interestingly, the rise in taurochenodesoxycholic acid (TCDCA), AA and 12-ketodeoxycholic acid (12-KDCA) as well as the decline in lysoPC(15:0), eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) were observed in AAN rats prior to detectable changes in conventional chemical markers and histological evidence of kidney injury (Fig. 5A). The alterations of these metabolites were most dramatic at the early time point, and less pronounced at the advanced AAN and as such represent potential biomarkers for the early detection of AAN.
Fig. 5.

Box-and-whisker plot analysis and ROC analysis based on PLS-DA models of early kidney injury biomarkers. (A) Combined box-and-whisker and dot plot of normalized intensity of six metabolites (lysoPC(15:0), TCDCA, AA, EPA, DHA, 12-KDCA) of control and AAN groups. Box-and-whisker and dot plot illustrated significant changes in the levels of identified early biomarkers between control and AAN groups at week 4. Significant increases TCDCA, AA and 12-KDCA were observed in the AAN group, accompanied by concomitant decrease lysoPC(15:0), EPA and DHA displayed the opposite trends in association with early kidney injury. (B) ROC curve of the diagnostic performance of the six identified biomarkers. ROC analysis was employed to quantify the diagnostic performance of these biomarkers, yielding an AUC and sensitivity of each identified metabolite from control and AAN groups at week 4 (C). *P<0.05, **P<0.01. Hierarchical cluster analysis and ROC curves of five differential biomarkers in advanced AAN. (D) PCA of four differential biomarkers including uric acid, lysoPE(22:5), indoxyl sulfate and creatinine only appeared at week 12. The AAN and control could be separated completely based on these four differential biomarkers. (E) Clustered heatmap of four differential biomarkers only appeared at week 12. These biomarkers showed a response on advanced AAN. (F) ROC curves analysis. ROC curves for the individual metabolites with high sensitivity and specificity from four biomarkers. The AUC, 95% confidence interval, sensitivity and specificity for four biomarkers were mentioned.
The suitability of the identified metabolites for use as biomarkers of early kidney injury was examined by ROC analysis (Fig. 5B and C). Six metabolites had an AUC of more than 0.75 and were superior to standard biomarker creatinine for detecting early renal injury. Among these identified potential candidates, 12-KDCA, lysoPC(15:0), TCDCA and DHA had high predictive performance and had high sensitivity and specificity. Although AA and EPA had high AUC value, their sensitivity or specificity was low and as such they were excluded. Therefore, four metabolites could be used as effective serum biomarkers for the early detection of renal injury.
3.7. Biomarkers of advanced AAN
LysoPE(22:5), indoxyl sulfate, uric acid and creatinine were found at week 12 only. PCA and heatmap showed that the AAN and control groups could be separated completely based on these biomarkers (Fig. 5D and E). The suitability of the four metabolites for use as biomarkers of advanced AAN was examined by ROC analysis (Fig. 5F). In addition to the current standard biomarker, Scr and uric acid, lysoPE(22:5) and indoxyl sulfate were identified as the top-ranked candidates, with an AUC of more than 0.94. Thus indoxyl sulfate and lysoPE(22:5) could be regarded as biomarkers of advanced AAN.
3.8. Biomarkers validation by advanced AAN rats at 24 weeks
To confirm the usefulness of candidate biomarkers of advanced AAN, serum profile of control, AAN and drug-treated AAN rats was analyzed at week 24. Similar to AAN rats at week 12 subgroup of AAN rats at week 24 showed significantly increased lysoPE(22:5), CDCA, lysoPE(20:2), CA, creatinine, indoxyl sulfate and uric acid as well as significantly increased lysoPC(17:0) with high sensitivity and specificity (Table S1). AAN rats had improved renal function (Fig. 6A) and reversed 8 biomarkers (Fig. 6B) by oral administration each day continuously for 24 weeks. Biomarker validation showed that these biomarkers were associated with only the disease state and had very few interference from the non-disease factors.
Fig. 6.

Validation of differential biomarkers by animal models and CKD patients. (A) Levels of serum creatinine and urea in control, AAN and drug-treated rats at week 24. (B) Comparison of the relative intensity of 8 metabolites in control, AAN and drug-treated rats at week 24. These metabolites were also significantly increased or decreased in AAN rats at week 24. These metabolites were reversed by treatment with irbesartan and ergone. (C) Levels of serum creatinine and urea in control, CKD and drug-treated rats at week 6. (D) Irbesartan ameliorated fibrosis development in adenine-induced CKD rats. Representative images of H&E, masson, TGF-β1, and α-SMA staining of kidney section from control, CKD and drug-treated rats. (E) Protein expression of NF-κB and α-SMA in the renal tissues of control, CKD and drug-treated rats. (F) Comparison of the relative intensity of 8 metabolites in control, CKD and drug-treated rats. (G) Representative images of masson staining of kidney section from CKD patients. (H) Comparison of the relative intensity of 8 metabolites in control, CKD and drug-treated patients. (I) ROC curves for the diagnosis of chronic renal injury with the individual biomarkers and the combination of the eight (all eight) in CKD patients. (J) ROC curves for the diagnosis of chronic renal injury with the combination of the eight biomarkers. (K) Diagnostic performance of the potential biomarkers based on the PLS-DA model. Black dots represent CKD patients, and black circles represent healthy controls. The black dots with red squares and black circles with red squares are for the incorrectly predicted samples in CKD patient and controls, respectively. (L) PCA score plot based on the eight potential biomarkers in CKD patients and controls. (M) Summary of ingenuity pathway analysis with MetPA including purine metabolism, arginine and proline metabolism, glycerophospholipid metabolism and primary bile acid biosynthesis. The size and color of each circle was based on pathway impact value and p-value, respectively. (N) Metabolic pathways were visualized by means of cytoscape software. Metabolic pathways included purine metabolism and primary bile acid biosynthesis of CKD patients. The biomarkers in our study were represented by red hexagons. Hexagons with green lines mean that the alteration of the biomarkers in CKD had statistical significance. The size of hexagons indicated the FC of the corresponding metabolite in CKD relative to controls. In addition, pink hexagons indicated metabolites participating in the metabolic pathway but not been detected in our study. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.9. Biomarkers validation by adenine-induced CKD rats
In order to determine whether the biomarkers found in AAN rats are shared with another model of chronic tubulo-interstitial nephropathy, serum biochemistry, renal histology, protein expression and serum metabolomics were obtained in rats with CKD at week 8. Adenine-induced CKD rats had improved renal function (Fig. 6C), renal histology (Fig. 6D), lower expression of NF-κB and α-SMA (Fig. 6E) by drug treatment compared to CKD rats. Similar to AAN rats, CKD rats showed elevation of lysoPE(22:5), indoxyl sulfate, CA, creatinine, lysoPE(20:2) and CDCA (Fig. 6F). In addition uric acid level did not reach statistical significance (P=0.503). Interestingly unlike the AAN rats, rats with adenine-induced CKD showed a significant increase in serum lysoPC(17:0) (Fig. 6F). In addition to the current standard biomarkers creatinine, AUC values of five biomarkers were equal to or greater than 0.87 and had high sensitivity and specificity (Table S2). Eight biomarkers in CKD rats were reversed by drug treatment (Fig. 6F). Nevertheless, the identified metabolic signature demonstrated a concerted dysregulation of the metabolic pathways similar to AAN rats. The results showed that such abnormal metabolic changes were a common feature of chronic interstitial nephropathy of diverse etiologies.
3.10. Biomarkers validation by patients with CKD
In order to examine whether eight potential biomarkers found in AAN rats are relevant to human CKD, serum samples from healthy controls, CKD patients and IBR-treated CKD patients were analyzed using UPLC-QTOF/HDMS. Irbesartan had improved renal histology of CKD patients (Fig. 6G). Results of the metabolic profiles showed that like AAN rats, indoxyl sulfate, lysoPE(22:5), creatinine, lysoPE(20:2), CDCA and CA were elevated in CKD patients. Although not statistically significant, lysoPC(17:0) and uric acid showed a trend of elevation (Table 2 and Fig. 6H). These biomarkers were reversed by treatment of irbesartan. Six biomarkers including indoxyl sulfate, lysoPE(22:5), creatinine, lysoPE(20:2), CDCA and CA had high AUC values (>0.84) as well as high sensitivity and specificity (Fig. 6I and Table 2). The AUC value of the combination of the eight biomarkers was larger than each individual biomarker (Fig. 6J). To predict class probabilities for each sample, validation was performed on eight potential biomarkers (Fig. 6K). 22 out of the 25 patients with CKD were correctly grouped (88% sensitivity). 24 out of the 25 healthy controls were located in the control area (96% specificity). PCA showed CKD patients could be separated from controls (Fig. 6L). These results indicated that these metabolites were important metabolic biomarkers which can distinguish the CKD patients from healthy subjects and as such could serve as suitable serum biomarkers of chronic tubulo-interstitial nephropathy. Metabolic pathway analysis revealed that these biomarkers were associated with purine metabolism, arginine and proline metabolism, glycerophospholipid metabolism and primary bile acid biosynthesis (Fig. 6M and N).
Table 2.
Analyses of statistically significant differences and ROC curve of biomarkers validation in patients with advanced CKD.
| Metabolite | Pa | Pb | FDRc | AUC | Sensitivity (%) | Specificity (%) | 95%Cl |
|---|---|---|---|---|---|---|---|
| Indoxyl sulfate | 3.90E−07 | 1.98E−06 | 7.80E−07 | 0.89 | 86.5 | 89.7 | 0.80−0.97 |
| LysoPE(0:0/22:5) | 2.39E−07 | 4.88E−06 | 6.37E−07 | 0.88 | 82.5 | 91.4 | 0.78−0.97 |
| Creatinine | 2.02E−07 | 4.88E−06 | 8.08E−07 | 0.88 | 92.4 | 83.6 | 0.78−0.97 |
| LysoPE(20:2/0:0) | 5.71E−06 | 1.01E−05 | 7.61E−06 | 0.86 | 0.91 | 0.85 | 0.76−0.96 |
| Chenodeoxycholic acid | 1.15E−06 | 1.61E−05 | 1.84E−06 | 0.86 | 0.85 | 0.89 | 0.75−0.96 |
| Cholic acid | 1.99E−07 | 4.05E−05 | 1.59E−06 | 0.84 | 0.78 | 0.87 | 0.73−0.94 |
| LysoPC(17:0) | 1.11E−01 | 2.99E−01 | 1.27E−01 | 0.58 | 0.72 | 0.54 | 0.43−0.73 |
| Uric acid | 2.11E−01 | 3.99E−01 | 2.11E−01 | 0.56 | 0.64 | 0.52 | 0.39−0.72 |
P values from Student's t-test.
P values from Mann-Whitney U test.
FDR value was obtained from the adjusted P value of FDR correction by Benjamini-Hochberg method.
4. Discussion
AAN is a rapidly progressive tubulo-interstitial nephropathy. Metabolomics approach was employed in rats with AAN and differential metabolites were identified. MSEA revealed altered metabolic pathways in AAN rats (Figs. S4, S5 and S6). A total of 27 metabolites were associated with metabolisms of lipids, amino acids and purine metabolism. Some of these metabolites were identified as potential biomarkers to discriminate the early and advanced stages of CKD (Fig. S7).
Using a zebra fish model, a recent study revealed that AAI-induced renal injury was associated with oxidative stress and inflammation as evidenced by increased gene expression of pro-inflammatory molecules such as cyclooxygenase, myeloperoxidase and tumor necrosis factor alpha mRNA expression [22]. Using cultured glomerular mesangial cells, another study demonstrated that AAI up-regulated CTGF expression which could be suppressed by increased inducible nitric oxide synthase-derived nitric oxide [23]. Using cultured human HK-2 cells, the study showed that exposure to AAI led to a dose-dependent increased reactive oxidative species production, glutathione depletion and cell death [24]. Another study showed that administration of a synthetic nuclear factor-erythroid-2-related factor 2 activator ameliorated aristolochic acid-induced renal injury [25]. These findings illustrated that AAN is closely associated with oxidative stress and inflammation.
Earlier studies have demonstrated the importance of plasma phospholipid abnormalities in renal injury [26]. Phospholipids have been reported to be biomarkers for chronic glomerulonephritis, chronic renal failure, ESRD and diabetic nephropathy [26]. Plasma metabolomics have been employed in pathophysiology of AAI-induced renal injury and lysoPC(20:5) and lysoPC(20:4) have been identified as potential biomarkers in AAN rats [27]. We also found phospholipids to be the most important metabolites. Lipids accounted for 85%, 76% and 72% of all identified metabolites at weeks 4, 8, and 12, respectively. These findings indicated that lipids were affected in different stages of AAN. The underlying mechanism of the observed disturbances of lipids is not completely clear. As noted above, oxidative stress and inflammation play a major role in the pathogenesis of AAN by promoting generation of reactive oxygen, nitrogen, and halogen species which attack and denature lipids, proteins and nucleic acid and deplete anti-oxidant defense system [28]. In fact nephrotoxic drugs have been shown to increase the lipid peroxidation product, malondialdehyde and reduced catalase, glutathione, glutathione peroxidase and superoxide dismutase levels in the kidney tissue [26]. In addition, patients with renal injury exhibit oxidative stress which is marked by increased pro-oxidants and impairment of the antioxidant capacity, factors which contribute to tissue damage and upregulation of pro-fibrotic pathways [29]. In presence of oxidative stress, free radicals activate phospholipase A2, which hydrolyses phosphatidylcholine to produce LysoPC. This phenomenon may explain the observed increase in LysoPE and LysoPC in our AAN rats.
Bile acids are produced from conjugation of cholesterol and its incorporation in the bile by the liver for secretion in the intestine. Within the intestine, particularly colon, bile acids are deconjugated, oxidized and dehydroxylated by bacterial flora forming secondary bile acids which are transported back to the liver where they can be further modified to form tertiary bile acids. Plasma concentration of bile acids rises and kidney became the sole route of their excretion in patients and animals with obstructive jaundice in whom bile acids may have nephrotoxic effects [30]. CDCA is the primary bile acid, and 7-ketolithocholic acid is the major intermediate byproduct of the intestinal bacterial conversion of CDCA to ursodeoxycholic acid. Compared with control group, serum bile acids including CDCA, CA, TCDCA, 7-ketodeoxycholic acid (7-KDCA) and 12-KDCA were significantly increased in our AAN rats. It was reported that increased 12-KDCA concentration was observed in 5/6 nephrectomized CKD rats [31]. Recent studies have revealed dramatic changes in the composition and function of the gut microbiome in humans and experimental animals with renal failure [32], [33], events that can significantly alter bile acid metabolism in presence of renal disease. In fact elevation of plasma bile acids is mainly due to the expansion of small-intestinal bacteria capable of deconjugating bile acids and thereby facilitating their absorption. It has been reported that plasma 3-oxocholic acid and 7-KDCA are significantly increased in rats with AA-induced renal injury [27] and elevated plasma bile acids work in concert with other risk factors in exacerbating kidney disease [30]. The bile acid metabolites including CDCA, CA, deoxycholic acid and ursodeoxycholic acid have been identified in the serum and dialysate effluent of hemodialysis patients. The pre-dialysis and post-dialysis serum concentrations of secondary bile acids have been shown to be significantly higher in ESRD patients than in healthy subjects [34], [35]. Patients with CKD have been reported to exhibit alteration in the bile acid balance. Total serum bile acids are consistently elevated in patients with different stages of CKD or after kidney transplantation [36]. The observed changes in plasma bile acids in AAN may be due to alteration of synthesis and secretion of bile acids, and confirm the effect of kidney disease on bile acid metabolism [37].
Administration of the farnesoid X receptor agonist, CDCA, attenuated proteinuria, mesangial expansion, and tubulo-interstitial fibrosis and diminished up-regulation of collagen IV, TGF-β, PAI-I, α-SMA, tumor necrosis factor alpha. interleukin 6, and NAD(P)H oxidase in the renal cortex of high-fructose-fed Wistar rats [38]. Renal interstitial fibrosis and inflammation and oxidative stress observed in the AAN rats employed in the present study were accompanied by marked changes in bile acid metabolites. Given the well demonstrated action of bile acid metabolites in regulation of oxidative, inflammatory and fibrotic pathways, the observed changes in bile acid metabolism may contribute to the renal injury in this model.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Nos. 81673578, 81603271), the Program for New Century Excellent Talents in University of Ministry of Education of China (No. NCET-13-0954) and the project As a Major New Drug to Create a Major National Science and Technology Special (No. 2014ZX09304307-002).
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2016.09.014.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Weiss R.H., Kim K. Metabolomics in the study of kidney diseases. Nat. Rev. Nephrol. 2011;8(1):22–33. doi: 10.1038/nrneph.2011.152. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Y.Y. Effect of ergosta-4,6,8(14),22-tetraen-3-one (ergone) on adenine-induced chronic renal failure rat: a serum metabonomic study based on ultra performance liquid chromatography/high-sensitivity mass spectrometry coupled with MassLynx i-FIT algorithm. Clin. Chim. Acta. 2012;413(19–20):1438–1445. doi: 10.1016/j.cca.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 3.Posada-Ayala M. Identification of a urine metabolomic signature in patients with advanced-stage chronic kidney disease. Kidney Int. 2014;85(1):103–111. doi: 10.1038/ki.2013.328. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z.H. Metabolomics insights into chronic kidney disease and modulatory effect of rhubarb against tubulointerstitial fibrosis. Sci. Rep. 2015;5:14472. doi: 10.1038/srep14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y.Y. Intrarenal metabolomic investigation of chronic kidney disease and its TGF-β1 mechanism in induced-adenine rats using UPLC Q-TOF/HSMS/MSE. J. Proteome Res. 2013;12(2):692–703. doi: 10.1021/pr3007792. [DOI] [PubMed] [Google Scholar]
- 6.Zhao Y.Y. Urinary metabonomics study on biochemical changes in an experimental model of chronic renal failure by adenine based on UPLC Q-TOF/MS. Clin. Chim. Acta. 2012;413(5–6):642–649. doi: 10.1016/j.cca.2011.12.014. [DOI] [PubMed] [Google Scholar]
- 7.Debelle F.D., Vanherweghem J.L., Nortier J.L. Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 2008;74(2):158–169. doi: 10.1038/ki.2008.129. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y.Y., Lin R.C. Metabolomics in nephrotoxicity. Adv. Clin. Chem. 2014;65:69–89. [PubMed] [Google Scholar]
- 9.Zhao Y.Y. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci. Rep. 2015;5:12936. doi: 10.1038/srep12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kieffer D.A. Resistant starch alters gut microbiome and metabolomic profiles concurrent with amelioration of chronic kidney disease in rats. Am. J. Physiol. Ren. Physiol. 2016;310(9):F857–F871. doi: 10.1152/ajprenal.00513.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y.Y. Serum metabonomics study of adenine-induced chronic renal failure rat by ultra performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Biomarkers. 2012;17(1):48–55. doi: 10.3109/1354750X.2011.637180. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y.Y. UPLC based metabolomics applications for discovering biomarkers of diseases in clinical chemistry. Clin. Biochem. 2014;47(15):16–26. doi: 10.1016/j.clinbiochem.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 13.Zhao Y.Y. Renal metabolic profiling of early renal injury and renoprotective effects of Poria cocos epidermis using UPLC Q-TOF/HSMS/MSE. J. Pharm. Biomed. Anal. 2013;81–82:202–209. doi: 10.1016/j.jpba.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y.Y. Ultra-performance liquid chromatography-mass spectrometry as a sensitive and powerful technology in lipidomic applications. Chem. Biol. Interact. 2014;220:181–192. doi: 10.1016/j.cbi.2014.06.029. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y.Y. Urinary metabolomics and biomarkers of aristolochic acid nephrotoxicity by UPLC-QTOF/HDMS. Bioanalysis. 2015;7(6):685–700. doi: 10.4155/bio.14.309. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y.Y. A pharmaco-metabonomic study on chronic kidney disease and therapeutic effect of ergone by UPLC-QTOF/HDMS. PloS One. 2014;9(12):e115467. doi: 10.1371/journal.pone.0115467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y.Y. Ultra performance liquid chromatography-based metabonomic study of therapeutic effect of the surface layer of Poria cocos on adenine-induced chronic kidney disease provides new insight into anti-fibrosis mechanism. PloS One. 2013;8(3):e59617. doi: 10.1371/journal.pone.0059617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Z.H. An integrated lipidomics and metabolomics reveal nephroprotective effect and biochemical mechanism of Rheum officinale in chronic renal failure. Sci. Rep. 2016;6:22151. doi: 10.1038/srep22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia J., Wishart D.S. MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 2010;38:W71–W77. doi: 10.1093/nar/gkq329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karnovsky A. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics. 2012;28(3):373–380. doi: 10.1093/bioinformatics/btr661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y.Y. Application of faecal metabonomics on an experimental model of tubulointerstitial fibrosis by ultra performance liquid chromatography/high-sensitivity mass spectrometry with MSE data collection technique. Biomarkers. 2012;17(8):721–729. doi: 10.3109/1354750X.2012.724450. [DOI] [PubMed] [Google Scholar]
- 22.Ding Y.J., Chen Y.H. Developmental nephrotoxicity of aristolochic acid in a zebrafish model. Toxicol. Appl. Pharmacol. 2012;261(1):59–65. doi: 10.1016/j.taap.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 23.Tsai K.D. Downregulation of connective tissue growth factor by LPS/IFN-γ-induced nitric oxide is reversed by aristolochic acid treatment in glomerular mesangial cells via STAT-1α and NF-κB signaling. Chem. Biol. Interact. 2014;210:86–95. doi: 10.1016/j.cbi.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 24.Yu F.Y. Aristolochic acid I induced oxidative DNA damage associated with glutathione depletion and ERK1/2 activation in human cells. Toxicol. Vitro. 2011;25(4):810–816. doi: 10.1016/j.tiv.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 25.Wu J. Bardoxolone methyl (BARD) ameliorates aristolochic acid (AA)-induced acute kidney injury through Nrf2 pathway. Toxicology. 2014;318:22–31. doi: 10.1016/j.tox.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Y.Y., Vaziri N.D., Lin R.C. Lipidomics: new insight into kidney disease. Adv. Clin. Chem. 2015;68:149–171. doi: 10.1016/bs.acc.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Zhao Y.Y., Lin R.C. UPLC–MSE application in disease biomarker discovery: the discoveries in proteomics to metabolomics. Chem. Biol. Interact. 2014;215:7–16. doi: 10.1016/j.cbi.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 28.El-Beshbishy H.A. Abrogation of cisplatin-induced nephrotoxicity in mice by alpha lipoic acid through ameliorating oxidative stress and enhancing gene expression of antioxidant enzymes. Eur. J. Pharmacol. 2011;668(1–2):278–284. doi: 10.1016/j.ejphar.2011.06.051. [DOI] [PubMed] [Google Scholar]
- 29.Shidfar F. Effects of omega-3 fatty acid supplements on serum lipids, apolipoproteins and ma-londialdehyde in type 2 diabetes patients. East Mediterr. Health J. 2008;14(2):305–315. [PubMed] [Google Scholar]
- 30.Kaler B. Are bile acids involved in the renal dysfunction of obstructive jaundice? An experimental study in bile duct ligated rats. Ren. Fail. 2004;26(5):507–516. doi: 10.1081/jdi-200031753. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Z.H. Metabolomic signatures of chronic kidney disease of diverse etiologies in the rats and humans. J. Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00583. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri N.D. Chronic kidney disease alters the composition of intestinal microbial flora. Kidney Int. 2013;83(2):308–315. doi: 10.1038/ki.2012.345. [DOI] [PubMed] [Google Scholar]
- 33.Wong J. Expansion of urease and uricase-containing, indole- and p-cresol-forming and contraction of short chain fatty acid-producing intestinal bacteria in ESRD. Am. J. Nephrol. 2014;39(3):230–237. doi: 10.1159/000360010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hood C.J. Feasibility study of colestipol as an oral phosphate binder in hemodialysis patients. Nephrology. 2015;20(4):250–256. doi: 10.1111/nep.12388. [DOI] [PubMed] [Google Scholar]
- 35.Chu L. Mechanism underlying an elevated serum bile acid level in chronic renal failure patients. Int. Urol. Nephrol. 2015;7(2):345–351. doi: 10.1007/s11255-014-0901-0. [DOI] [PubMed] [Google Scholar]
- 36.Levi M. Role of bile acid-regulated nuclear receptor FXR and G protein-coupled receptor TGR5 in regulation of cardiorenal syndrome (cardiovascular disease and chronic kidney disease) Hypertension. 2016;67(6):1080–1084. doi: 10.1161/HYPERTENSIONAHA.115.06417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosińska P. The role of AST-120 and protein-bound uremic toxins in irritable bowel syndrome: a therapeutic perspective. Ther. Adv. Gastroenterol. 2015;8(5):278–284. doi: 10.1177/1756283X15587866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Z. Effect of chenodeoxycholic acid on fibrosis, inflammation and oxidative stress in kidney in high-fructose-fed Wistar rats. Kidney Blood Press. Res. 2012;36(1):85–97. doi: 10.1159/000341485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material