

In this study, Li et al. characterize the genomic distribution and transcriptional activity of C. elegans HSF-1 during larval development and show that the developmental HSF-1 transcriptional program is distinct from the heat-shock response. Overall, they show that dE2F and HSF-1 directly regulate a gene network, including a specific subset of chaperones, to promote protein biogenesis and anabolic metabolism, which are essential in development.
Keywords: heat-shock factor (HSF), E2F transcription factor, development, stress response, transcription regulation, molecular chaperones, Caenorhabditis elegans
Abstract
Heat-shock factor (HSF) is the master transcriptional regulator of the heat-shock response (HSR) and is essential for stress resilience. HSF is also required for metazoan development; however, its function and regulation in this process are poorly understood. Here, we characterize the genomic distribution and transcriptional activity of Caenorhabditis elegans HSF-1 during larval development and show that the developmental HSF-1 transcriptional program is distinct from the HSR. HSF-1 developmental activation requires binding of E2F/DP to a GC-rich motif that facilitates HSF-1 binding to a heat-shock element (HSE) that is degenerate from the consensus HSE sequence and adjacent to the E2F-binding site at promoters. In contrast, induction of the HSR is independent of these promoter elements or E2F/DP and instead requires a distinct set of tandem canonical HSEs. Together, E2F and HSF-1 directly regulate a gene network, including a specific subset of chaperones, to promote protein biogenesis and anabolic metabolism, which are essential in development.
The heat-shock response (HSR) is an evolutionarily conserved transcriptional response to environmental and physiological stress that is associated with protein mismanagement and proteotoxic damage (Labbadia and Morimoto 2015a). Acute and chronic exposure of cells to elevated temperature, organic solvents, and heavy metals can cause protein damage, which in turn activates heat-shock factor (HSF) that induces the expression of a group of molecular chaperones called heat-shock proteins (HSPs) and key components in protein degradation and processing pathways to restore protein homeostasis (proteostasis) (Labbadia and Morimoto 2015a). Yeast, Caenorhabditis elegans, and Drosophila express a single HSF, whereas vertebrates possess a family of HSFs (HSF1–4) (Akerfelt et al. 2010a). Among vertebrate HSFs, HSF1 is essential for the HSR and to survive exposure to diverse stress conditions (Akerfelt et al. 2010a).
Regulation of HSF1 in the HSR has been extensively studied at the molecular level. Upon heat shock and the appearance of misfolded proteins, HSF1 is converted from an inert monomer to a functional DNA-binding trimer that binds selectively to heat-shock elements (HSEs) located within target promoters (Perisic et al. 1989; Baler et al. 1993). HSF1 then interacts with general transcription factors and coactivators to robustly induce transcription at both initiation and elongation steps (Mason and Lis 1997; Yuan and Gurley 2000; Park et al. 2001; Duarte et al. 2016). The activity of HSF1 is strongly influenced by post-translational modifications and association with chaperones that regulate different steps of the HSR (Shi et al. 1998; Westerheide et al. 2009; Raychaudhuri et al. 2014; Kourtis et al. 2015). Together, these events ensure a rapid, robust, and transient transcriptional response to cellular stress.
In Drosophila, HSF is also essential for oogenesis and larval development (Jedlicka et al. 1997). The molecular basis of this requirement has not been addressed, except that HSF seems to be activated differently than in the HSR, since the expression of stress-inducible heat-shock genes does not change in an HSF mutant that displays larval lethality (Jedlicka et al. 1997). Likewise, in vertebrates, HSFs are vital for many developmental processes. HSF1 is a maternal factor required for gametogenesis in mice and cooperates with HSF2 and HSF4 for brain and lens development (Christians et al. 2000; Santos and Saraiva 2004; Chang et al. 2006; Takaki et al. 2006). During mammalian development, it has been proposed that HSF1 is required for chaperone gene expression and maintaining proteostasis. For example, HSF1 regulates Hsp90α transcription in oocytes to ensure meiotic maturation (Metchat et al. 2009), disruption of HSF1 in mice leads to decreased expression of small HSPs HSP25 and CRYAB and results in postnatal brain defects (Xiao et al. 1999; Santos and Saraiva 2004), and HSF1-regulated expression of several HSPs supports murine olfactory neurogenesis (Takaki et al. 2006). However, these observations have not addressed whether the activation of chaperone gene expression by HSF1 during development corresponds to a distinct regulatory process or a variant of the HSR. Furthermore, genomic analyses in mouse oocytes and testes have identified HSF1 target genes unrelated to HSPs as executors of HSF1 developmental function (Akerfelt et al. 2010b; Le Masson et al. 2011). Similarly, many of the HSF2 and HSF4 target genes that are crucial for development of cortex and sensory organs, respectively, do not correspond to classical HSP genes (Chang et al. 2006; Takaki et al. 2006). In these situations, it is not clear how HSFs are activated and directed to their genomic targets.
Here, we examine the essential role of HSF-1 in C. elegans larval development and show that the genomic occupancy and transcriptional activity of HSF-1 differs significantly from the HSR. HSF-1 transcriptional activation in development relies on a unique promoter architecture comprised of a GC-rich motif recognized by an E2F complex and an adjacent degenerate HSE for binding of HSF-1. We propose that the evolutionarily conserved E2F/DP complex serves as a transcriptional activator at HSF-1 developmental targets by recognizing the GC-rich motif and facilitating the recruitment of HSF-1 to the adjacent degenerate HSE. The functional interaction between E2F/DP and HSF-1 therefore provides the molecular basis for HSF-1 transcriptional control in C. elegans larval development and may have broader implications in HSF1 physiological function in metazoans.
Results
HSF-1 is essential for C. elegans larval development
The role of HSF-1 in C. elegans larval development was examined using a loss-of-function mutant allele, ok600, in which the C-terminal regulatory and transactivation domains of HSF-1 were eliminated by a frameshift deletion (Supplemental Fig. S1A). ok600 mutant animals raised at the ambient temperature of 20°C arrest in larval development at late L2 to early L3 based on molting status, size measurement, and hallmarks of gonadal development (Fig. 1A,B; Supplemental Fig. S1G). Larval arrest in ok600 animals was observed previously at all growth temperatures (Morton and Lamitina 2013). More than half of the HSF-1-deficient animals persisted for another 3 d after the arrest without further development, with some animals surviving for up to 8 d of life (Supplemental Fig. S1B). To establish that the larval arrest of ok600 animals is due to the loss of HSF-1 function, we introduced a single-copy HSF-1::GFP transgene into ok600 animals under the control of the hsf-1 promoter and 3′ untranslated region (UTR) (Supplemental Fig. S1C). HSF-1::GFP is expressed at levels similar to that of the endogenous HSF-1 gene (Supplemental Fig. S1D) and is localized to the nuclei of all major somatic tissues in L2 and young adult (YA) animals (Supplemental Fig. S1E,F). The HSF-1::GFP transgene fully restored larval development of the ok600 animals to that of N2 wild-type animals (Fig. 1B; Supplemental Fig. S1G), thus providing direct evidence that HSF-1 is essential for larval development.
Figure 1.

HSF-1 is essential for C. elegans larval development. (A) Histograms showing the percent of wild-type (N2) and hsf-1(ok600) animals that passed through each larval molt at 20°C. Error bars represent the SEM of four biological replicates. n = 30 N2; n = 55 hsf-1(ok600). (B) The size of wild-type (N2), hsf-1(ok600), and hsf-1(ok600); rmSi1[hsf-1::gfp] larvae. The rmSi1 transgene carries a single copy of hsf-1p::hsf-1(minigene)::gfp::hsf-1 3′ UTR. At least 30 animals of each genotype were measured. Error bars represent SD. The size of hsf-1(ok600) animals is unchanged from 34 h after egg lay (L2 stage; P = 0.68, one-way ANOVA analysis) and significantly differs from N2 and hsf-1(ok600); rmSi1[hsf-1::gfp] measured at 39 h after egg lay (L3 stage) and thereafter. t-test, P = 0.001. The timeline represents larval stages (L1–L4) at given time points. (C) The expression of hsp-16.41 and hsp-70(C12C8.1) in wild-type (N2), hsf-1(ok600), and hsf-1(ok600); rmSi1[hsf-1::gfp] animals exposed to heat shock (HS; 30 min at 34°C) at L2 stage. Error bars represent the SEM of biological triplicates. (D) Thermorecovery of wild-type (N2), hsf-1(ok600), and hsf-1(ok600); rmSi1[hsf-1::gfp] animals at L2 (30 h after egg lay) exposed to an extended heat shock (HS) for 4 h at 34°C followed by recovery (Rec) for 16 h at 20°C. Error bars represent the SEM of biological triplicates. n = 137 N2; n = 130 hsf-1(ok600); n = 136 hsf-1(ok600); rmSi1[hsf-1::gfp].
The expression of classical heat-shock genes hsp-16.41 and hsp-70 (C12C8.1) was severely compromised (>99% reduced) in hsf-1(ok600) animals exposed to heat shock compared with N2 animals and was significantly restored in the transgenic animals expressing the HSF-1::GFP transgene (Fig. 1C). Consistent with the loss of the HSR, hsf-1(ok600) animals also exhibited very poor thermorecovery following heat shock relative to N2 or the HSF-1::GFP-rescued animals (Fig. 1D). The correlation of larval arrest and deficiency of the HSR in hsf-1(ok600) animals raised the possibility that the requirement of HSF-1 in larval development might be linked to its function in the HSR.
HSF-1 associates with specific gene promoters during larval development
To investigate the molecular requirements for HSF-1 in development, we characterized the genome-wide occupancy of HSF-1 in L2 animals using chromatin immunoprecipitation (ChIP) coupled with massively parallel DNA sequencing (ChIP-seq) via the HSF-1::GFP fusion protein expressed in the hsf-1(ok600) background. This analysis provides a specific binding profile for HSF-1, since ChIP analysis of nuclear-localized GFP (NLS::GFP) (Supplemental Fig. S2A) failed to enrich any of the candidate HSF-1-bound promoters that show a wide range of occupancy detected with HSF-1::GFP (Supplemental Fig. S2B–D). In parallel, we also performed ChIP-seq to determine the genomic occupancy of RNA polymerase II (Pol II). In L2 animals grown at 20°C, HSF-1 associates with the promoters of 373 genes. HSF-1 binding is strongly enriched within 250 base pairs (bp) upstream of the transcription start sites (TSSs) (Fig. 2A) and is accompanied by Pol II binding at these promoters (Fig. 2A; Supplemental Fig. S2E), implicating a role for HSF-1 in transcriptional regulation during larval development.
Figure 2.
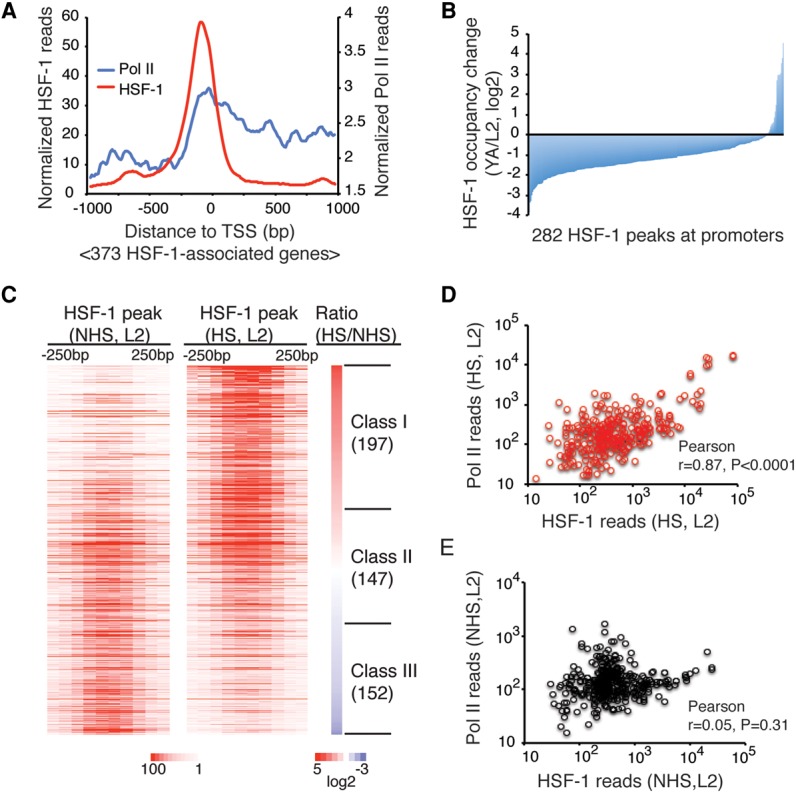
HSF-1 exhibits distinct genomic occupancy in development compared with the HSR. (A) Composite plots of HSF-1 and Pol II ChIP-seq reads within 1000 bp from the TSSs of 373 HSF-1-associated genes in L2 animals at 20°C. HSF-1-associated genes were defined as genes having HSF-1 ChIP-seq peak summits mapped within 1000 bp from the TSS. (B) Histogram of HSF-1 occupancy changes at promoter-associated HSF-1 peaks between L2 and YA animals at 20°C. The 282 peaks whose summits are within 1000 bp from the TSS detected in either L2 or YA animals were included. HSF-1 occupancy was calculated as normalized HSF-1 ChIP-seq reads within 250 bp from the peak summits. (C) Heat maps of normalized HSF-1 reads at ChIP-seq peaks (mapped to 50 bp bins, ±250 bp from peak summits) in L2 animals grown at 20°C without heat shock (NHS) or exposed for 30 min to 34°C heat shock (HS). Peaks were ranked by the ratio of HSF-1 reads in heat shock compared with without heat shock. The peaks with increased or decreased HSF-1 occupancy by 1.5-fold in heat shock were grouped into class I or class III, respectively, and the remaining peaks with similar HSF-1 occupancy were grouped into class II. The number of genes associated with each class of peaks is shown in parentheses. (D,E) Scatter plots of HSF-1 (−600 to +400 bp) and Pol II (−500 to +1000 bp) ChIP-seq reads at HSF-1-associated promoters from L2 animals in heat shock (D; 313 genes) and without heat shock (E; 373 genes).
The genomic distribution of HSF-1 is developmental stage-specific, which is made evident by comparison of promoter occupancy of HSF-1 in L2 and YA animals (Supplemental Fig. S2F). HSF-1 occupancy at most promoters decreases after the completion of larval development (Fig. 2B) despite the fact that HSF-1 protein levels are similar at these two stages (Supplemental Fig. S2G). Only 15% of the HSF-1-binding sites in L2 animals persist in YAs and remain significantly bound (Supplemental Fig. S2H). Eleven promoters are exceptions and show increased HSF-1 binding (at least 1.5-fold of L2) in YAs, including the hsb-1 gene, a negative regulator of HSF-1 in the stress response and longevity (Satyal et al. 1998; Chiang et al. 2012); four genes that function in oogenesis and embryogenesis; and three genes of the ubiquitin proteasome system (UPS) (Supplemental Table S1). These changes correlate with the onset of reproduction and increased UPS activity in adults (Liu et al. 2011). It is unlikely that the global impairment of HSF-1 binding is due to technical reasons of ChIP-seq in adult animals, as we did not observe a general decrease of Pol II occupancy at the same set of promoters (Supplemental Fig S2I, J). Rather, these data indicate that the stronger and broader binding of HSF-1 in L2 animals reflects the specific requirements for HSF-1 in larval development.
HSF-1 genomic occupancy in development is distinct from the HSR
We next tested whether the requirement for HSF-1 in larval development is the same as for the HSR by comparing the ChIP-seq profiles of HSF-1 in L2 animals with or without a 30-min heat shock. The results clearly indicate that heat shock causes a global change of HSF-1 binding (Fig. 2C). By using a cutoff of 1.5-fold change, three classes of HSF-1-binding sites corresponding to similar numbers of genes in each class were revealed, which acquire (class I), maintain (class II), or show reduced (class III) HSF-1 binding upon heat shock (Fig. 2C; Supplemental Fig. S3A–D). Class I (197 genes) is highly enriched with classical heat-shock-responsive genes represented by hsp-70, in which binding of HSF-1 and Pol II is at low levels during development and robustly induced by heat shock (Supplemental Fig. S3A). Class II (147 genes) shows readily detectable association of HSF-1 in development that does not change appreciably upon heat shock (Supplemental Fig. S3B,C). Class II genes, however, sort into two subgroups based on Pol II occupancy change upon heat shock. For example, Y55F3AR.2, a predicted disulfide oxidoreductase, which typifies the majority of the class II genes, is not induced by heat shock (Supplemental Fig. S3B), whereas a small subset of genes, including the cochaperone dnj-13, is induced by heat shock, albeit not as robustly as hsp-70 (Supplemental Fig. S3C). Class III (152 genes), in contrast, shows 1.5-fold or more reduced HSF-1 binding upon heat shock, and Pol II binding is typically not induced by heat shock (Supplemental Fig. S3D). Together, these results demonstrate that different DNA-binding preferences for HSF-1 between development and the HSR correspondingly direct distinct transcriptional programs.
In line with the genomic redistribution of HSF-1 in the HSR, we observed an overall positive correlation between acquired HSF-1 binding in heat shock and transcriptional induction measured by RNA sequencing (RNA-seq; Pearson r = 0.67) (Supplemental Fig. S3E,F). In contrast, there was an almost complete absence of correlation between HSF-1 binding in development and heat-shock induction (Pearson r = −0.024) (Supplemental Fig. S3G). Consistent with HSF-1 being the master regulator of the HSR, upon heat shock, the levels of Pol II at promoters correlate with the levels of HSF-1 (Fig. 2D). However, this is not the case during development, in which the occupancy of Pol II at promoters does not linearly correlate with the occupancy of HSF-1 (Fig. 2E). These genomic observations suggest that HSF-1 transcriptional regulation in development may be highly dependent on the promoter architecture and composition of other transcription factors, which is in sharp contrast to HSR that is primarily driven by the promoter occupancy of HSF-1.
HSF-1 directly regulates a gene network essential for C. elegans larval development
To determine the output of HSF-1 binding on transcription in larval development, we compared the transcriptomes of wild-type (N2), hsf-1(ok600), and hsf-1(ok600); rmSi1 animals that express the HSF-1::GFP transgene. In order to identify the primary expression changes resulting from the loss of HSF-1, we chose mid-L2 stage larvae (30 h after egg lay) to perform RNA-seq analysis. At this developmental stage, hsf-1(ok600) animals are indistinguishable in size and morphology from wild-type animals (Fig. 1B; Supplemental Fig. S1G). The expression of approximately 700 genes exhibited significant changes in hsf-1(ok600) animals (false discovery rate [FDR] 0.05), corresponding to equal numbers decreasing or increasing in mRNA levels. Of these, 44 genes appear to be directly activated by HSF-1, since their respective promoters are bound by HSF-1 in ChIP-seq analysis, and their expression is down-regulated in hsf-1(ok600) animals (Fig. 3A,B). A smaller but statistically significant group of HSF-1-associated genes (23 genes) is up-regulated in hsf-1(ok600) animals, and this could correspond to a class of genes whose expression is repressed by HSF-1 (Fig. 3A,C). The expression changes at HSF-1-associated genes in hsf-1(ok600) animals are largely corrected by the HSF-1::GFP transgene, establishing that these changes in expression are specifically dependent on HSF-1 (Fig. 3A; Supplemental Fig. S4A).
Figure 3.
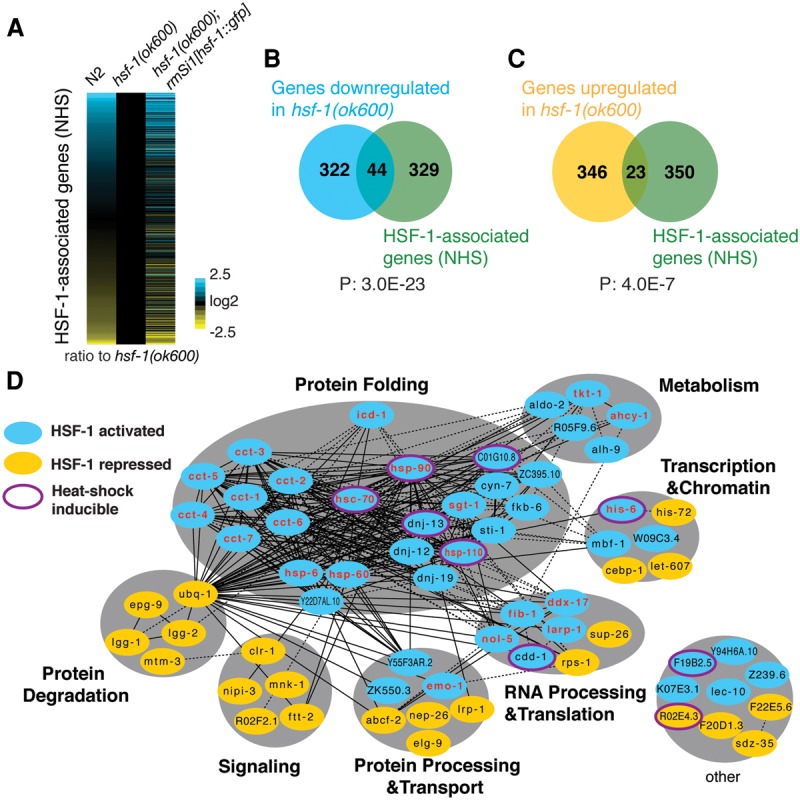
HSF-1 directly regulates genes essential for C. elegans larval development. (A) Relative expression of HSF-1-associated genes in wild-type (N2), hsf-1(ok600), and hsf-1(ok600); rmSi1[hsf-1::gfp] L2 animals (30 h after egg lay) at 20°C. Gene expression levels were determined by RNA-seq and normalized to that in hsf-1(ok600). (B,C) Venn diagrams showing the overlap of genes down-regulated (B) or up-regulated (C) in hsf-1(ok600) compared with N2 (FDR 0.05) and genes associated with HSF-1. P = Fisher's exact test. (D) The gene network directly regulated by HSF-1 in C. elegans larval development. HSF-1 directly activated genes corresponding to those genes directly associated with HSF-1 and down-regulated in hsf-1(ok600) (indicated in blue ovals); HSF-1 directly repressed genes corresponding to genes associated with HSF-1 and up-regulated in hsf-1(ok600) (indicated in yellow ovals). Solid lines represent physical protein interactions; dashed lines represent genetic interactions or coexpression. The 22 genes labeled in red are known to be required for larval development in genetic analyses. The eight genes induced upon heat shock are indicated with a purple outline around either blue or yellow ovals.
Of the HSF-1-associated genes, only a relatively small subset of genes exhibited significantly altered expression in hsf-1(ok600) animals. This could be explained by residual levels of maternal wild-type HSF-1 protein even though maternal hsf-1 mRNA is undetectable (Supplemental Fig. S1D). Another possibility is that loss of HSF-1 is compensated for by other transcription factors, as HSF-1-binding sites in development overlap extensively with the extreme highly occupied target (xHOT) regions that are bound by many transcription factors (P = 3.2 × 10−156) (Supplemental Fig. S4B) identified by modENCODE ChIP-seq analyses (Araya et al. 2014). It has been proposed that genes with essential cellular functions are located in xHOT regions and redundantly regulated by multiple transcription factors to ensure proper levels of expression (Gerstein et al. 2010; Van Nostrand and Kim 2013). Nevertheless, approximately half of the “HSF-1 directly regulated genes” that are associated with HSF-1 and differentially expressed in hsf-1(ok600) animals are in xHOT regions (Supplemental Fig. S4C), suggesting that HSF-1 is indispensible at these selective targets.
As the primary components of the HSF-1 developmental transcriptome, the set of “HSF-1 directly regulated genes” encodes proteins that physically and genetically interact to form a highly connected network important for metabolism, gene expression (transcription, RNA processing, and translation), and protein homeostasis (protein processing, transport, folding, and degradation) (Fig. 3D). Genes directly activated by HSF-1 are enriched for those that function in protein folding and anabolic metabolism. These include chaperones and cochaperones that serve as the core of the HSF-1 developmental network and promote cotranslational folding and protein maturation to ensure production of a functional proteome. Of these, only five of 23 HSF-1 developmentally activated chaperones and cochaperones are induced by heat shock (FDR 0.05). This result was further confirmed by testing the HSF-1-dependent HSR by RNA-seq analyses of the reduction-of-function mutant hsf-1(sy441), which lacks the transactivation domain and exhibits a severely compromised HSR (Supplemental Fig. S4D–G). HSF-1 also activates transcription of three key enzymes in the pentose phosphate pathway (tkt-1, aldo-2, and R05F9.6) that generates NADPH for reductive biosynthesis and ribose-5-phosphate for synthesis of nucleotides. HSF-1-dependent repression occurs at genes involved in protein degradation, including the UPS (ubq-1) and autophagy (lgg-1, lgg-2, and epg-9). Overall, via these direct targets, HSF-1 promotes biosynthesis that is essential for growth and development. Consistent with the requirement for HSF-1, half of the 44 HSF-1 directly activated genes correspond to previously identified “essential larval development genes” by RNAi or mutagenesis screens (P = 1.1 × 10−11) (Supplemental Fig. S4H), establishing the importance of HSF-1-mediated transcription in larval development.
HSF-1 activation in development is linked to unique promoter architecture
We next characterized the molecular mechanisms by which HSF-1 regulates its developmental targets by focusing on the 44 HSF-1 directly activated genes, since the larval arrest of hsf-1(ok600) is likely due to down-regulation of these essential larval development genes. Because the impact of HSF-1 binding in development is promoter context-dependent, we searched for DNA motifs overrepresented in the HSF-1 ChIP-seq peaks at these directly activated promoters. As expected, we identified a sequence that resembles the evolutionarily conserved HSE represented by three adjacent and inverted “NGAAN” pentamers (Fig. 4A, top; Akerfelt et al. 2010a). However, upon closer inspection, we noticed that the HSEs at HSF-1 developmentally activated genes are “degenerated” by more frequently containing one “relaxed pentamer” with mismatches at the GAA trinucleotides (Wilcoxon rank sum test, P = 0.042) (Supplemental Fig. S5A; Guertin et al. 2012) as compared with the HSEs at genes that are induced by heat shock (Fig. 4A, bottom). In addition, HSF-1 developmentally activated promoters typically have only a single HSE, while a significantly larger fraction of heat-shock-induced promoters has clusters of HSEs (Wilcoxon-rank sum test, P = 0.036) (Supplemental Fig. S5B). It is well established from biochemical and structural studies that the GAA sequence in the HSE is in direct contact with the DNA-binding domain of HSF-1 (Neudegger et al. 2016) and that promoters with tandem HSEs promote cooperative binding between multiple HSF-1 trimers to increase the affinity of HSF-1 to DNA (Xiao et al. 1991; Kroeger and Morimoto 1994).
Figure 4.

HSF-1 transcriptional activities in development require a unique promoter architecture. (A) The HSEs derived from HSF-1 ChIP-seq peaks that are either associated with HSF-1-activated genes in development and correspond to degenerate HSEs (top) or induced upon heat shock and correspond to canonical HSEs comprised of three inverted pentamer NGAAN sequences (bottom). Genes associated with HSF-1 ChIP-seq peaks within 1000 bp from the TSS and significantly increased expression (twofold or more; FDR 0.05) upon heat shock are defined as induced genes. (B) A GC-rich motif derived from HSF-1 ChIP-seq peaks associated with HSF-1-activated genes in development. (C) Histograms representing the position relationship of the GC-rich motif and HSE at HSF-1 ChIP-seq peaks in L2 animals at 20°C. HSF-1 peaks within 1000 bp of TSSs were included. HSF-1 peaks linked to activation are those at the promoters of HSF-1-activated genes in development. (D) Schematic representation of a transcriptional reporter system used to assay HSF-1 developmental targets. The unc-119p::unc-119::mCherry internal reference reporter is on the same construct of the GFP transcriptional reporter. (E–G) RT-qPCR analysis of transcriptional reporters of the cct-5 (E), sti-1 (F), and Y94H6A.10 (G) genes in L2 animals at 20°C. The mRNA levels of GFP were normalized to the mRNA levels of mCherry to calculate promoter activity. The relative activity of promoter variants carrying either mutations of the HSE (mHSE) or deletion of the GC-rich motif (ΔGC) is shown as the ratio to the wild-type (WT) promoters. (pHSE) Proximal HSE; (dHSE) distal HSE. Shown at the top of each panel is a schematic of the cct-5, sti-1, and Y94H6A.10 promoters, with the arrow indicating the TSS and direction of transcription. Error bars represent the SEM of biological triplicates. (H) Gbrowser view of HSF-1 and Pol II occupancy at the hsc70 (hsp-1) gene locus in L2 animals with or without heat shock. (I) Schematic representation of the hsc70 (hsp-1) promoter. The two arrows indicate the distal and proximal TSSs, respectively. (J,K) RT-qPCR analysis of transcription reporters of the hsc70 (hsp-1) gene in L2 animals at 20°C (J) or with a 30-min heat shock at 34°C (K). Because of the high abundance of hsc70 (hsp-1) mRNA at 20°C, the newly synthesized nascent transcript during heat shock was measured to calculate promoter activity at 34°C. (mdHSEs) Mutation of both distal HSEs; (ΔdHSEs) deletion of the region containing both distal HSEs.
How does HSF-1 bind to lower-affinity HSEs during development? One potential mechanism for HSF-1 developmental control is that HSF-1 binding is facilitated by cofactors at the same promoter. This prompted us to search for additional DNA motifs in close proximity to the HSE. Multiple correlation analyses suggest that a GC-rich motif (Fig. 4B) is an additional DNA signature for HSF-1 developmental activity. First, 70% of HSF-1 developmentally activated promoters contain this GC-rich motif, which is fourfold enriched relative to genes that are regulated only by heat shock (Supplemental Fig. S5C). Second, co-occurrence of the GC-rich motif and HSE at HSF-1-binding sites provides a much better prediction of HSF-1 developmental activity than either the HSE or GC-rich motif alone (Supplemental Fig. S5D–F). Finally, the GC-rich motif is typically located 20–40 bp from the HSE with a strong preference to be upstream of the HSE at HSF-1 developmentally activated genes (Fig. 4C). Taken together, these observations suggest that the regulation of developmental target genes by HSF-1 is linked to a unique promoter structure featuring the closely located GC-rich motif and the HSE.
HSF-1 function in development requires the GC-rich motif and the HSE
To test the functional requirements for the HSE and the GC-rich motif in developmental activation by HSF-1, we generated a transcriptional reporter system (Fig. 4D) comprised of a GFP reporter that is regulated by the test promoter together with a reference mCherry fusion reporter under the control of the HSF-1-independent unc-119 promoter on the same construct. This controls for expression variation of the reporter among transgenic lines by providing an internal normalization of GFP to mCherry expression (Supplemental Fig. S5G,H).
Five representative HSF-1 developmentally activated genes were examined using this reporter assay in transgenic L2 animals. All of these genes contain the GC-rich motif and the immediately downstream HSE in their respective promoters, with a subset of these promoters containing additional HSEs in other locations. All five genes require HSF-1 for developmental expression based on our RNA-seq analysis, and two of them are also heat-shock-responsive. The analysis of the wild-type and mutant promoter constructs would therefore provide a comparison of HSF-1 regulation in development and upon heat shock. For all promoter constructs, deletion of the GC-rich motif resulted in decreased reporter expression during development by ≥80%, thus providing evidence that the GC-rich motif is necessary for developmental activation (Fig. 4E–G,J; Supplemental Fig. S5L). Likewise, the proximal HSE immediately downstream from the GC-rich motif is also required for transcription in development. This is demonstrated for the promoters of the cct-5, sti-1, and hsc70 (hsp-1) genes that contain a single proximal HSE in which mutation of this HSE decreased reporter expression by at least 60% (Fig. 4E,F,J). For the Y94H6A.10 and hsp90 (daf-21) genes that contain two HSEs downstream from the GC-rich motif in their respective promoters, mutation of either HSE alone did not impair the reporter expression to the same level as mutation of both (Fig. 4G; Supplemental Fig. S5L). While this indicates that these HSEs are partially redundant, mutation of the proximal HSE adjacent to the GC-rich motif has a greater effect on the reporter than the HSE further downstream (Fig. 4G; Supplemental Fig. S5L). In the sti-1, hsc70, and hsp90 genes that also contain HSEs located upstream of the GC-rich motif, mutation of these upstream HSEs did not affect developmental expression of the reporter in L2 animals at 20°C (Fig. 4F,J; Supplemental Fig. S5L), in contrast to the effect of mutation at the proximal HSEs. From these results, we conclude that both the GC-rich motif and the adjacent downstream proximal HSE are essential for developmental expression by HSF-1. Furthermore, changes of these motifs have effects on the nascent transcripts similar to the mRNA levels at 20°C, indicating that this motif pair controls expression via transcription rather than post-transcriptional regulation (Supplemental Fig. S5I).
Among the genes tested in the reporter assay, hsc70 and hsp90 are also heat-shock-inducible. During development, HSF-1 binds to the region of the proximal HSEs and the GC-rich motif, whereas, upon heat shock, HSF-1 occupancy declines at the developmental sites and exhibits inducible binding at the upstream region of distal HSEs (Fig. 4H; Supplemental Fig. S5J). Accordingly, Pol II occupancy is induced upon heat shock and correlates with transcription initiation from the upstream region (Fig. 4H; Supplemental Fig. S5J). To demonstrate whether the GC-rich motif and proximal HSE are necessary for heat-shock induction of the hsc70 and hsp90 genes, we monitored nascent transcripts induced upon heat shock to obtain a more direct measure of promoter activity in the HSR. In contrast to the results for development at 20°C, activity of the hsc70 and hsp90 promoters at 34°C is independent of the GC-rich motif or the proximal HSEs but is regulated entirely by the tandem distal HSEs (Fig. 4K; Supplemental Fig. S5M). These results demonstrate that HSF-1-mediated transcription in development and the HSR can be uncoupled and rely on the differential use of HSF-1-binding sites.
An E2F complex binds to the GC-rich motif at HSF-1 developmental targets
The requirement for the GC-rich motif for HSF-1 activity in development suggests that the GC-rich motif corresponds to the binding site of a cofactor for HSF-1. The GC-rich motif shares extensive sequence similarity with an E2F-binding site (Fig. 5A) that was derived from ChIP-seq analysis of the endogenous C. elegans E2F ortholog EFL-1 (Latorre et al. 2015). This motif was also shown to be the “somatic E2F-binding site” based on ChIP-seq of an EFL-1::GFP fusion protein expressed in somatic tissues (Kudron et al. 2013). Similar to the mammalian counterparts, EFL-1 and the C. elegans DP ortholog DPL-1 form a heterodimer that binds DNA and regulates transcription with profound effects on cell proliferation, differentiation, and apoptosis (Ceol and Horvitz 2001; Page et al. 2001; Reddien et al. 2007; Schertel and Conradt 2007). EFL-1 and DPL-1 can associate with LIN-35, the C. elegans Rb protein, and the MuvB subcomplex to form the DRM complex (Harrison et al. 2006), which serves as a transcription repressor of cell cycle genes (Latorre et al. 2015). EFL-1 and DPL-1 can also activate transcription of genes that promote oogenesis and early embryogenesis in the gonad, which is likely independent of LIN-35/Rb (Chi and Reinke 2006).
Figure 5.

An E2F complex binds to the GC-rich motif at HSF-1 developmental genes. (A) The GC-rich motif from HSF-1 directly activated promoters in development (bottom) resembles an E2F-binding motif (top) derived from ChIP-seq peaks of the E2F-associated DRM complex (Latorre et al. 2015). (B) Box plots of EFL-1 ChIP-seq reads at HSF-1 peaks that are linked to either developmental activation (Dev.), heat-shock induction (HS), or all EFL-1 peaks at promoters (±250 bp around summits) (Kudron et al. 2013). Peaks linked to HSF-1 activation in both conditions were excluded. Boxes depict the 25th through 75th percentiles, and whiskers show the 10th through 90th percentiles. (C) Composite plots of EFL-1 ChIP-seq reads (Kudron et al. 2013) within 400 bp from the summits of the HSF-1 peaks that are linked to either developmental activation (Dev.) or heat-shock induction (HS). (D) Schematic representation of ChIP-qPCR analysis at the integrated sti-1 reporters. Paired arrows represent the primers used in qPCR analysis, blue lines and arrows indicate the sequences from the endogenous sti-1 promoter, and black lines and arrows indicate the sequences specific to the reporter transgene. (E) RT-qPCR analysis of the sti-1 reporter and the endogenous sti-1 gene in transgenic animals. Normalized expression levels of the reporter (GFP/mCherry) and the endogenous gene (sti-1/housekeeping genes) were measured in transgenic animals carrying the sti-1 reporter containing the wild-type (WT) promoter or promoter variants with mutations in the proximal HSE (mpHSE) or deletion of the GC-rich motif (ΔGC). Data are represented as ratios of the expression levels in animals carrying the wild-type reporter. Error bars represent the SEM of biological triplicates. (F) ChIP-qPCR analysis of HSF-1 and subunits of the DRM complex in transgenic animals carrying the wild-type sti-1 reporter. Occupancies of the transcription factors at three regions across the reporter were measured and are shown as fold enrichment over the upstream control region. Error bars represent the SEM of biological triplicates. (G) ChIP-qPCR analysis of HSF-1 and subunits of the DRM complex in transgenic animals carrying different sti-1 reporters. Transgenic animals with an integrated sti-1 reporter carrying the wild-type (WT) promoter, mutation of the proximal HSE (mpHSE), or deletion of the GC-rich motif (ΔGC) were analyzed. Occupancies of the transcription factors were measured at the promoter regions of the reporter as well as the endogenous sti-1 gene, and the normalized occupancy (reporter/endogenous gene) is shown in the histograms. Error bars represent the SEM of biological triplicates.
The similarity of E2F-binding sites to the GC-rich motif led us to ask whether EFL-1 co-occupies HSF-1 developmentally activated promoters. We observed strong EFL-1 binding at HSF-1-binding sites required for developmental activation but not at promoters that responded only to heat shock (Fig. 5B). Consistent with the close proximity of the GC-rich motif and HSE, EFL-1 is highly enriched at the HSF-1 peak summits (Fig. 5C). Furthermore, we observed enrichment for all other subunits of the DRM complex at promoters of HSF-1 developmental targets (Supplemental Fig. S6A) but not at genes regulated only upon heat shock (Supplemental Fig. S6B), supporting the idea that HSF-1 and the DRM complex are binding partners at HSF-1 developmentally regulated genes.
To demonstrate that the DRM complex binds to HSF-1 developmental targets via the GC-rich motif and establish how binding of the DRM complex affects HSF-1, the wild-type and mutant transcriptional reporters described previously were used to test for binding of the DRM complex and HSF-1. This was addressed by using the cochaperone gene sti-1 that contains the GC-rich motif and the proximal HSE close to the start codon, allowing for the design of primers to distinguish the promoters of the reporter construct from the corresponding endogenous gene (Fig. 5D). The sti-1 gene also exhibits significant levels of HSF-1 and DRM complex binding at this promoter (Supplemental Fig. S6C), providing sufficient dynamic range for ChIP-qPCR analysis. We first confirmed that expression of the stably integrated sti-1 reporter in transgenic animals declines when the GC-rich motif or the proximal HSE is altered, whereas expression of the endogenous sti-1 gene was unaffected (Fig. 5E). We then performed ChIP-qPCR analysis in animals expressing the wild-type reporter and showed that both HSF-1 and the DRM complex bound to the promoter of the sti-1 reporter. A fourfold enrichment of EFL-1, DPL-1, and MuvB subcomplex (LIN-54 and LIN-37 subunits) binding was observed at the sti-1 promoter compared with the control regions in the transgene, which corresponds to a similar level of HSF-1 binding (Fig. 5F). LIN-35 exhibits weaker but still significant binding at the promoter.
We next compared the binding of the DRM complex and HSF-1 on variant and wild-type promoters and normalized the occupancy at these reporters to the endogenous promoter of the sti-1 gene. Whereas mutation of the proximal HSE diminished HSF-1 binding, this had no effect on binding of the DRM complex (Fig. 5G), indicating that HSF-1 binding during development occurs through the proximal HSE and that binding of the DRM complex does not rely on HSF-1. Deletion of the GC-rich motif disrupted the binding of the DRM complex (Fig. 5G), providing evidence that the DRM complex binds to the promoter through the GC-rich motif. It is unlikely that the loss of DRM binding by deletion of the GC-rich motif is due to the secondary effect of transcriptional repression, since mutation of the proximal HSE decreased the activity of the reporter by 70% (Fig. 5E) without altering the binding of DRM to the promoter (Fig. 5G). Finally, deletion of the GC-rich motif resulted in the loss of HSF-1 binding even though the proximal HSE remains intact (Fig. 5G). From these results, we propose that the DRM complex binds to the GC-rich motif at HSF-1 developmental target promoters and likely facilitates the recruitment of HSF-1 to the proximal HSE.
EFL-1/DPL-1 functions as a coactivator for HSF-1 at its developmental targets independently of LIN-35
The DRM complex has been well studied as a transcriptional repressor (Kirienko and Fay 2007; Latorre et al. 2015); therefore, it is intriguing that DRM components appear to function as coactivators for HSF-1 in larval development. To study the contribution of DRM components on endogenous HSF-1-associated genes, we first tested whether LIN-35(Rb), the subunit of the DRM complex required for repression, regulates HSF-1 developmental target genes. As expected, we observed the derepression of polh-1 and dna-2, two canonical DRM-repressed genes (Fig. 6A), in lin-35(n745) mutant animals but did not see any significant change in expression of the HSF-1 developmentally regulated genes cct-5, sti-1, hsc70(hsp-1), or hsp90(daf-21) (Fig. 6B) that require the GC-rich motif for transcription. This is further supported by analysis of published RNA-seq data showing that the majority of HSF-1 developmental targets does not exhibit altered expression in lin-35(n745) animals (Supplemental Fig. S7A). We next tested whether the absence of LIN-35 has an effect on other DRM components at the promoters. As expected, we observed a pronounced decrease in LIN-35 binding by ChIP analysis on all promoters tested (Fig. 6C,D; Supplemental Fig. S7B,C). Loss of LIN-35 leads to dissociation of the entire DRM complex from the LIN-35-repressed polh-1 and dna-2 promoters (Fig. 6C; Supplemental Fig. S7B), since binding of EFL-1, DPL-1, and LIN-54, the three subunits in the DRM complex that bind DNA, was reduced by ∼80% (Tabuchi et al. 2011). In contrast, the binding of EFL-1, DPL-1, and LIN-54 at the hsc70(hsp-1) and sti-1 promoters does not change or decreases only slightly in the absence of LIN-35 (Fig. 6D ; Supplemental Fig. S7C), whereas HSF-1 binding is maintained. These results imply that EFL-1/DPL-1 and perhaps other DRM components may function as coactivators for HSF-1 independently of LIN-35.
Figure 6.

EFL-1/DPL-1 functions as a coactivator for HSF-1 at its developmental targets independently of LIN-35. (A,B) RT-qPCR analysis of the canonical DRM-repressed genes polh-1 and dna-2 (A) and the HSF-1 developmental target genes cct-5, sti-1, hsc70(hsp-1), and hsp90(daf-21) (B) in the wild-type N2 or lin-35(nn745) L2 larvae. Expression levels are plotted as the ratio to that of N2 animals. Error bars represent the SEM of biological triplicates. (C,D) ChIP-qPCR analysis of HSF-1 and subunits of the DRM complex at the promoters of the polh-1 (C) and hsc70(hsp-1) (D) genes. Occupancies of transcription factors were plotted as percent of input. Error bars represent the SEM of biological triplicates. (E) Western blot analysis of HSF-1 (HSF-1::GFP) and subunits of the DRM complex in hsf-1(ok600); rmSi1[hsf-1::gfp] L2 animals treated with dpl-1 RNAi, lin-54 RNAi, or the empty vector control L4440. α-Tubulin was probed as a loading control. (F,G) RT-qPCR analysis of polh-1 and dna-2 (F) and cct-5, sti-1, hsc70(hsp-1), and hsp90(daf-21) (G) in hsf-1(ok600); rmSi1[hsf-1::gfp] L2 animals treated with dpl-1 RNAi, lin-54 RNAi, or the empty vector control L4440. Expression levels are plotted as the ratio to that of the L4440 control. Error bars represent the SEM of biological triplicates.
To test this idea, we performed RNAi against dpl-1 and lin-54, respectively. We selected RNAi against dpl-1 over efl-1 as a strategy to impair EFL-1/DPL-1 function because DPL-1 is the sole ortholog of DP, whereas C. elegans expresses three E2F-like proteins. Animals were maintained on RNAi bacteria for two generations to eliminate both maternal and zygotic activities, since, in addition to zygotic DPL-1, maternal DPL-1 also contributes to larval development (Ceol and Horvitz 2001). In L2 animals treated with dpl-1 RNAi, DPL-1 and EFL-1 total protein levels decreased by ∼60% and ∼45%, whereas neither LIN-54 nor LIN-37 protein was reduced. In contrast, lin-54 RNAi treatment reduced LIN-54 protein by ∼80% and LIN-37 by ∼60% without affecting the levels of either EFL-1 or DPL-1 (Fig. 6E). These results are consistent with previous biochemical analysis of the DRM complex that implied the presence of subcomplexes (Harrison et al. 2006). As expected, RNAi to both dpl-1 and lin-54 led to derepression of polh-1 and dna-2, consistent with functional impairment of the DRM complex (Fig. 6F). However, only dpl-1 RNAi, but not lin-54 RNAi, decreased expression of the candidate HSF-1 developmental targets (Fig. 6G), indicating that EFL-1/DPL-1 can function as a coactivator for HSF-1 at these gene promoters. Because EFL-1/DPL-1 is required for embryogenesis (Ceol and Horvitz 2001; Page et al. 2001), a significant fraction of dpl-1 RNAi-treated animals was sterile (Supplemental Fig. S7D), thus making it impossible to collect sufficient synchronized L2 animals to monitor changes in HSF-1 occupancy upon dpl-1 RNAi treatment by ChIP. However, since neither dpl-1 nor lin-54 RNAi significantly altered HSF-1 protein levels (Fig. 6E) or affected the HSR, as shown by induction of the classical HSR genes hsp-70 (C12C8.1) and hsp-16.41 upon heat shock (Supplement Fig. S7E), the contribution of EFL-1/DPL-1 on HSF-1 target genes in development most likely results from its function on these selective promoters but not by regulating the general levels or activity of HSF-1.
Taken together, our data support a model in which EFL-1/DPL-1 recognizes the GC-rich motif at HSF-1 developmental target promoters, which serves as the coactivator for HSF-1 likely through promoting the recruitment of HSF-1 to the immediately downstream HSE to activate transcription during development (Fig. 7A).
Figure 7.
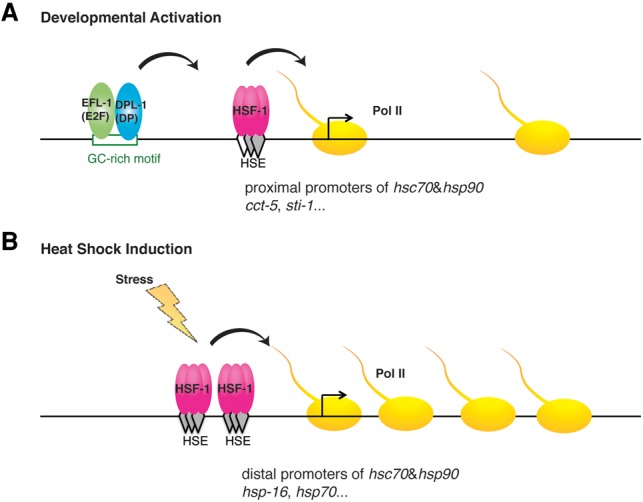
Model of HSF-1 transcription activation in larval development and the HSR. (A) Schematic of an HSF-1 developmentally activated promoter showing binding of EFL-1(E2F)/DPL-1(DP) to the GC-rich motif to function as an activator by enhancing the binding of HSF-1 to a degenerate HSE, resulting in transcriptional activation. (B) Schematic of classical heat-shock-responsive gene in which multiple HSF-1 trimers bind to tandem canonical HSEs, leading to inducible transcription.
Discussion
HSF1 has been studied extensively for its role in the HSR and involvement in diverse conditions of cell stress and disease. Numerous observations have established that HSF1 also functions in conditions that are not typically associated with acute or chronic stress, such as development, reproduction, innate immunity, and aging (Singh and Aballay 2006; Vihervaara and Sistonen 2014). Here, we show that the underlying molecular mechanisms by which HSF-1 activates transcription during C. elegans larval development require a unique promoter architecture comprised of a GC-rich motif for binding to EFL-1(E2F)/DPL-1(DP) that functions as a coactivator and likely aids the recruitment of HSF-1 to an adjacent “degenerate HSE” (Fig. 7A). Both the GC-rich motif and E2F/DP are required for HSF-1-dependent expression during development but are dispensable for the HSR. In contrast, HSF-1 uses a separate cluster of canonical HSEs that are necessary and sufficient for the HSR (Fig. 7B). Thus, HSF-1 uses distinct regulatory strategies during development and in response to heat shock and consequently regulates an overlapping but not identical set of genes.
Although many of the genes directly regulated by HSF-1 during development are components of the proteostasis network, there is a significant difference from those genes induced by heat shock. During C. elegans development, protein biogenesis likely accelerates rapidly to accommodate for the quick expansion in body size (Fig. 1B). Accordingly, HSF-1 in development activates a specific subset of chaperones that are essential for translation, folding, assembly, and transport to support rapid protein biogenesis and represses genes that function in protein degradation (Fig. 3D). In contrast, heat shock causes an overall repression of translation to prevent accumulation of misfolded proteins, and HSF-1 induces genes involved in both protein folding and degradation to restore proteome balance. The unexpected repression of genes in protein degradation by HSF-1 during larval development needs to be explored further; however, the regulation of HSF-1 on the proteostasis network is obviously different in development and the HSR. In support of the notion that development and stress signals have opposing effects on protein synthesis, chaperone genes that function intimately with nascent polypeptide folding are activated by HSF-1 in development but show reduced HSF-1 binding upon heat shock (Supplemental Table S2). These include β-NAC (icd-1) that functions at the ribosome exit tunnel in cotranslational folding (Preissler and Deuerling 2012; Kirstein-Miles et al. 2013) and subunits of the TRiC complex (cct genes) that assist folding of newly synthesized proteins (Lopez et al. 2015), which is consistent with a previous report that expression of these chaperones is repressed rather than activated by stress (Albanese et al. 2006). We show that the different promoter architectures and interaction with E2F underlie the distinct binding preference of HSF-1 in development and heat shock, allowing for differential regulation of HSF-1 targets in these conditions.
The E2F family of transcription factors has been extensively studied for its roles in the cell cycle-dependent gene expression and function in development by regulating cell proliferation, differentiation, and apoptosis (van den Heuvel and Dyson 2008; Sadasivam and DeCaprio 2013). The partnership described here between the evolutionarily conserved HSF-1 and E2F transcription factors supports a link between cell cycle control and proteostasis and could provide a mechanism to ensure the proper stoichiometry of a subset of chaperones and other HSF-1-regulated genes for rapid cell growth and proliferation. E2F is also highly responsive to the feeding and energy status of the cell and regulates expression of metabolic genes in different tissue types (Blanchet et al. 2011; Denechaud et al. 2016). The metabolic rate of C. elegans increases throughout development and reaches its highest level in L2 to L4 larvae followed by a steep decline through adulthood. In particular, there are global metabolic changes that occur in early larval development (late L1 and L2) when the animals switch from using stored lipids to consuming exogenous food sources (Braeckman et al. 2009). This could suggest that functional interaction between E2F and HSF-1 provides a way for the animal to coordinate energy production, protein synthesis, and protein quality control. We propose that the uncoupling of HSF-1 activity in development from the HSR enables HSF-1 to integrate these growth and environmental cues.
Previous studies on the promoters of heat shock genes have identified a number of cis elements and corresponding factors—including SP1 in mammals and GAGA factor in Drosophila—that alter chromatin structure and affect the inducibility of heat-shock gene transcription (Morgan 1989; Tsukiyama et al. 1994; Martinez-Balbas et al. 1995; Duarte et al. 2016). Binding of these factors at heat-shock gene promoters does not lead to the constitutive recruitment of HSF1 but rather directs nucleosome displacement to create DNA accessibility at HSEs for binding of HSF1 upon heat shock (Guertin and Lis 2010; Fuda et al. 2015). In C. elegans, a similar mechanism is likely used at some heat-shock promoters by DAF-16 (FOXO3) that uses the chromatin remodeler SWI/SNF to promote stress resistance (Hsu et al. 2003; Riedel et al. 2013). Additional levels of regulatory interaction for mammalian HSFs include the formation of mixed heterotrimers of HSF1 and HSF2 (Vihervaara et al. 2013) and bookmarking heat-shock promoters by HSF2 for rapid HSF1-mediated activation after mitosis (Xing et al. 2005). It is therefore possible that E2F/DP functions as the coactivator for HSF-1 in development by opening the chromatin structure to allow for HSF-1 binding. However, additional mechanisms must be used by E2F to enable constitutive binding of HSF-1 at the weak “degenerate HSEs” during larval development. Considering the proximity of the GC motif and “degenerate HSE”, it is tempting to hypothesize that EFL-1(E2F)/DPL-1(DP) physically interacts with HSF-1 and recruits it to selective promoters. Since EFL-1/DPL-1 is required for embryogenesis, a system that enables acute inhibition of EFL-1/DPL-1 during early larval development will be required in further studies for directly testing the hypothesis.
Our observations on E2F for HSF-1 developmental control support the speculation that HSF-1 cooperates with a number of other transcriptional regulators in the absence of classical cell stress. Because we used whole animals to perform RNA-seq analysis, our results are most sensitive for HSF-1 targets that are ubiquitously expressed. Consistent with this, the majority of the E2F and HSF-1 coregulated genes that we identified is broadly expressed. It is also possible that HSF-1 could partner with other transcriptional regulators in specific cell types to confer tissue-specific gene expression.
The HSF-1 developmental regulation that is uncoupled from the HSR shares common features with the observation in cancer cells that HSF1 is widely distributed on the genome to support carcinogenesis (Mendillo et al. 2012). Similar to our HSF-1 developmental program, the HSF1 cancer program is also tightly linked to protein production (Santagata et al. 2013) and regulates expression of chaperones and metabolic genes to support rapid growth (Mendillo et al. 2012). In addition, ChIP-seq data from the ENCODE project reveal the binding of E2Fs and HSF1 at the promoters of corresponding chaperone genes in cancer cells. As E2F can have pro-oncogenic roles, it will be of interest in future studies to explore how the HSF1 and E2F developmental program might become engaged in disease and its role in carcinogenesis.
Materials and methods
Worm maintenance and heat shock
C. elegans strains were maintained and handled using standard techniques (Brenner 1974). Animals were grown on NGM plates at 20°C unless stated. Age synchronization was achieved by egg lay in a 2-h period for all experiments that include the hsf-1(ok600) animals and by treatment of alkaline hypochlorite solution for the other experiments. hsf-1(ok600) animals were collected by picking GFP-negative young larvae hatched from eggs laid by OG576 hsf-1(ok600) I/hT2[bli-4(e937)] let-?(q782) qIs48 [pmyo-2::gfp; ppes-10::gfp; pges-1::gfp](I;III). A list of worm strains is in the Supplemental Material.
Heat shock was performed on solid NGM plates in a water bath preheated to 34°C. Plates were wrapped with parafilm and submerged for 30 min for RNA isolation and ChIP analysis or for 4 h for the thermorecovery assay.
RNA-seq and RT-qPCR
RNA was extracted using Trizol and a Qiagen RNeasy RNA extraction kit with on-column DNase I digestion according to the manufacturer's instructions. For RNA-seq analysis of N2, hsf-1(ok600), and hsf-1(ok600); rmSi1[hsf-1::gfp], total RNA from ∼500 L2 larvae (30 h after egg lay) pooled from multiple collections was used in library preparation following PrepX SPIA RNA-seq library protocol using a Nugen Ovation RNA-seq V2 kit. For RNA-seq analysis of the HSR, total RNA from ∼20,000 N2, hsf-1(sy441), and hsf-1(sy441); rmSi1[hsf-1::gfp] larvae were used for polyA selection and library construction following the Illumina TruSeq RNA sample preparation protocol. RNA-seq libraries were sequenced on an Illumina HiSeq 2000 instrument. All RNA-seq experiments were done in biological triplicates.
For candidate gene-based expression analysis, cDNA synthesis and qPCR were performed as described previously (Labbadia and Morimoto 2015b). Primers used in these experiments are in Supplemental Table S3.
ChIP
Chromatin preparation was essentially performed as described previously (Zhong et al. 2010) with slight modifications. Animals were collected from NGM plates, washed with M9, cross-linked with 2% formaldehyde at room temperature for 15 min, resuspended in FA buffer, lysed by douncing in a Kontes 2-mL glass dounce, and sonicated in a Bioruptor to yield 200- to 800-bp size DNA fragments.
Immunoprecipitation was performed as described previously (Boehm et al. 2003) except that magnetic Protein G Dynabeads were used. Extracts corresponding to 200 µg of protein from L2 larvae or 400 µg of protein from YAs was used for each pull-down. For ChIP-seq analysis of HSF-1 and Pol II, the hsf-1(ok600); rmSi1[hsf-1::gfp] animals were used. For each immunoprecipitation, 5 µL of antibody against GFP (polyclonal; Clontech) or 2.5 µL of antibody against Pol II (Novus) was used. DNA was pooled from six immunoprecipitations for each ChIP-seq experiment. Libraries were prepared following PrepX DNA library protocol with an Apollo 324 system and sequenced on an Illumina HiSeq 2000 instrument. All ChIP-seq experiments were performed with biological duplicates. ChIP analysis of HSF-1 and Pol II were also performed for candidate promoters in the hsf-1(ok600); rmSi1[hsf-1::gfp] animals, and AM1076 rmIs358[phsf-1(4 kb)::NLS-gfp::3′ UTRhsf-1]II in biological triplicates with normal rabbit IgG (2.5 µL per immunoprecipitation; Cell Signaling, #2729) was used as the negative control. For ChIP analysis of HSF-1 and the DRM complex subunits on the sti-1 promoter, L2 larvae carrying the integrated sti-1 transcriptional reporters and drSi41[hsf-1p::hsf-1::HA::unc-54utr; Cbr-unc-119(+)] (lines AM1199, AM1200, and AM1201) were used. For each immunoprecipitation reaction, 5 µL of anti-HA antibody (ChIP grade; Abcam) or 2.5 µL of antibody against EFL-1, DPL-1, LIN-35, LIN-54, or LIN-37 (Novus) was used. For ChIP analysis of HSF-1 and the DRM complex subunits on endogenous gene promoters, L2 animals expressing rmSi1[hsf-1::gfp] in the wild-type or lin-35(n745) background were used. For all candidate gene-based ChIP analyses, qPCR was used to determine percent input for each primer pair (Supplemental Table S4).
Transcriptional reporter assay
The control transcriptional reporter without promoter upstream of GFP was generated by inserting an unc-119(cDNA)::mCherry fusion gene (Ferguson and Fisher 2009) into the pPD95.75 vector (a gift from the Fire Lab) between the EagI and ApaI sites. Promoters of HSF-1 target genes were cloned by PCR amplification of N2 genomic DNA. Mutation and deletions of the promoters were introduced by asymmetric overlap extension PCR (Xiao and Pei 2011). Wild-type and mutant promoters were then inserted into the control transcription reporter between the BamHI and MscI sites. Primers used in cloning and mutagenesis are listed in Supplemental Table S5.
Transcriptional reporters were introduced into unc-119(ed3) animals by microinjection. Animals containing the transcriptional reporters were selected by following wild-type movement and expression of the UNC-119::mCherry transgene. Integrated lines expressing the sti-1 transcriptional reporters (lines AM1196, AM1197, and AM1198 corresponding to the wild-type promoter, mutation of HSE, and deletion of the GC motif, respectively) were generated by exposing lines expressing extrachromosomal arrays of the indicated transgenes to γ irradiation from caesium137 for 18 min. Animals were backcrossed at least five times to HT1593 unc-119(ed3)III before further use.
Immunoblotting
Animals were collected in M9 buffer and boiled in 2× SDS loading buffer as described previously (Morton and Lamitina 2013). Roche anti-GFP (7.1 and 13.1; 1:1000 dilution), Sigma anti-α-Tubulin (T5168; 1:4000 dilution), and Novus anti-EFL-1, anti-DPL-1, anti-LIN-54, and anti-LIN-37 (1:2000 dilution) primary antibodies and HRP-conjugated secondary antibodies (1:5000) were used for Western blot analysis to probe the levels of HSF-1, the loading control α-Tubulin, and subunits of the DRM complex.
RNAi
RNAi against dpl-1 and lin-54 was performed using clones from an ORFeome-based RNAi library (Rual et al. 2004). Bacterial cultures were grown overnight in LB with 12.5 µg/mL tetracycline and 100 µg/mL ampicillin and induced with 5 mM IPTG for 4 h. Egg laying was performed on RNAi bacteria, and animals were grown to gravid adults. At least 200 animals were then picked to new plates for the second generation of exposure to RNAi. Age-synchronized larvae were obtained by treatment of alkaline hypochlorite solution and hatched in M9 buffer. At least 1000 L2 larvae grown on RNAi bacteria (∼21 h at 20°C) were used in RNA isolation or Western blot analysis.
Phenotypic characterization of hsf-1(ok600) animals
The thermorecovery assay was performed as described previously (Labbadia and Morimoto 2015b) except that animals were heat-shocked for 4 h at 34°C and scored for motility defects after recovery for 16 h at 20°C. Methods for microscopy, scoring of molting status, survival analysis, and size measurement are described in the Supplemental Material.
Sequencing data processing
Detailed next-generation sequencing data processing and bioinformatic analyses are described in the Supplemental Material. The accession number for the ChIP-seq and RNA-seq raw and processed data reported here is Gene Expression Omnibus GSE81523.
Supplementary Material
Acknowledgments
We thank Erik Andersen for helpful discussions on the DRM complex, H. Robert Horvitz and David Fay for sharing strains, Jean-Louis Bessereau for generous advice and technical support on generating the Mos1 transposase-mediated single-copy gene insertion (MosSCI) transgenic line, Janay Terry for assisting with the NLS::GFP line, Yoko Shibata for confocal microscopy, and members of the Morimoto laboratory for their scientific advice and comments on the manuscript. We also thank the Biopolymers Facility at Harvard Medical School and the High-Throughput Genome Analysis Core at the Institute for Genomics and Systems Biology at the University of Chicago for sequencing support. J.L. was supported by post-doctoral awards from the National Ataxia Foundation, the BrightFocus Foundation for Alzheimer's Disease Research, and the Chicago Biomedical Consortium. R.I.M. was supported by grants from the National Institutes of Health (National Institute on Aging and National Institute of Mental Health), the Ellison Medical Foundation, the Glenn Foundation, the Chicago Biomedical Consortium, and the Daniel F. and Ada L. Rice Foundation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.283317.116.
References
- Akerfelt M, Morimoto RI, Sistonen L. 2010a. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol 11: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerfelt M, Vihervaara A, Laiho A, Conter A, Christians ES, Sistonen L, Henriksson E. 2010b. Heat shock transcription factor 1 localizes to sex chromatin during meiotic repression. J Biol Chem 285: 34469–34476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanese V, Yam AY, Baughman J, Parnot C, Frydman J. 2006. Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell 124: 75–88. [DOI] [PubMed] [Google Scholar]
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D, et al. 2014. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature 512: 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baler R, Dahl G, Voellmy R. 1993. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol 13: 2486–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchet E, Annicotte JS, Lagarrigue S, Aguilar V, Clape C, Chavey C, Fritz V, Casas F, Apparailly F, Auwerx J, et al. 2011. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol 13: 1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm AK, Saunders A, Werner J, Lis JT. 2003. Transcription factor and polymerase recruitment, modification, and movement on dhsp70 in vivo in the minutes following heat shock. Mol Cell Biol 23: 7628–7637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braeckman BP, Houthoofd K, Vanfleteren JR. 2009. Intermediary metabolism WormBook (ed. The C. elegans Research Community), WormBook, 10.1895/wormbook.1.146.1; http://www.wormbook.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol CJ, Horvitz HR. 2001. dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol Cell 7: 461–473. [DOI] [PubMed] [Google Scholar]
- Chang Y, Ostling P, Akerfelt M, Trouillet D, Rallu M, Gitton Y, El Fatimy R, Fardeau V, Le Crom S, Morange M, et al. 2006. Role of heat-shock factor 2 in cerebral cortex formation and as a regulator of p35 expression. Genes Dev 20: 836–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi W, Reinke V. 2006. Promotion of oogenesis and embryogenesis in the C. elegans gonad by EFL-1/DPL-1 (E2F) does not require LIN-35 (pRB). Development 133: 3147–3157. [DOI] [PubMed] [Google Scholar]
- Chiang WC, Ching TT, Lee HC, Mousigian C, Hsu AL. 2012. HSF-1 regulators DDL-1/2 link insulin-like signaling to heat-shock responses and modulation of longevity. Cell 148: 322–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christians E, Davis AA, Thomas SD, Benjamin IJ. 2000. Maternal effect of Hsf1 on reproductive success. Nature 407: 693–694. [DOI] [PubMed] [Google Scholar]
- Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, Nicolay BN, Dyson NJ, Bonner C, Pattou F, et al. 2016. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. J Clin Invest 126: 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte FM, Fuda NJ, Mahat DB, Core LJ, Guertin MJ, Lis JT. 2016. Transcription factors GAF and HSF act at distinct regulatory steps to modulate stress-induced gene activation. Genes Dev 30: 1731–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AA, Fisher AL. 2009. Retrofitting ampicillin resistant vectors by recombination for use in generating C. elegans transgenic animals by bombardment. Plasmid 62: 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuda NJ, Guertin MJ, Sharma S, Danko CG, Martins AL, Siepel A, Lis JT. 2015. GAGA factor maintains nucleosome-free regions and has a role in RNA polymerase II recruitment to promoters. PLoS Genet 11: e1005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Lis JT. 2010. Chromatin landscape dictates HSF binding to target DNA elements. PLoS Genet 6: e1001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Martins AL, Siepel A, Lis JT. 2012. Accurate prediction of inducible transcription factor binding intensities in vivo. PLoS Genet 8: e1002610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Ceol CJ, Lu X, Horvitz HR. 2006. Some C. elegans class B synthetic multivulva proteins encode a conserved LIN-35 Rb-containing complex distinct from a NuRD-like complex. Proc Natl Acad Sci 103: 16782–16787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. 2003. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science 300: 1142–1145. [DOI] [PubMed] [Google Scholar]
- Jedlicka P, Mortin MA, Wu C. 1997. Multiple functions of Drosophila heat shock transcription factor in vivo. EMBO J 16: 2452–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirienko NV, Fay DS. 2007. Transcriptome profiling of the C. elegans Rb ortholog reveals diverse developmental roles. Dev Biol 305: 674–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein-Miles J, Scior A, Deuerling E, Morimoto RI. 2013. The nascent polypeptide-associated complex is a key regulator of proteostasis. EMBO J 32: 1451–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourtis N, Moubarak RS, Aranda-Orgilles B, Lui K, Aydin IT, Trimarchi T, Darvishian F, Salvaggio C, Zhong J, Bhatt K, et al. 2015. FBXW7 modulates cellular stress response and metastatic potential through HSF1 post-translational modification. Nat Cell Biol 17: 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger PE, Morimoto RI. 1994. Selection of new HSF1 and HSF2 DNA-binding sites reveals difference in trimer cooperativity. Mol Cell Biol 14: 7592–7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudron M, Niu W, Lu Z, Wang G, Gerstein M, Snyder M, Reinke V. 2013. Tissue-specific direct targets of Caenorhabditis elegans Rb/E2F dictate distinct somatic and germline programs. Genome Biol 14: R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. 2015a. The biology of proteostasis in aging and disease. Annu Rev Biochem 84: 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J, Morimoto RI. 2015b. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol Cell 59: 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latorre I, Chesney MA, Garrigues JM, Stempor P, Appert A, Francesconi M, Strome S, Ahringer J. 2015. The DREAM complex promotes gene body H2A.Z for target repression. Genes Dev 29: 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Masson F, Razak Z, Kaigo M, Audouard C, Charry C, Cooke H, Westwood JT, Christians ES. 2011. Identification of heat shock factor 1 molecular and cellular targets during embryonic and adult female meiosis. Mol Cell Biol 31: 3410–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Rogers J, Murphy CT, Rongo C. 2011. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J 30: 2990–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez T, Dalton K, Frydman J. 2015. The mechanism and function of Group II chaperonins. J Mol Biol 427: 2919–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C. 1995. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83: 29–38. [DOI] [PubMed] [Google Scholar]
- Mason PB Jr, Lis JT. 1997. Cooperative and competitive protein interactions at the hsp70 promoter. J Biol Chem 272: 33227–33233. [DOI] [PubMed] [Google Scholar]
- Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, Fraenkel E, Ince TA, Whitesell L, Lindquist S. 2012. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 150: 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metchat A, Akerfelt M, Bierkamp C, Delsinne V, Sistonen L, Alexandre H, Christians ES. 2009. Mammalian heat shock factor 1 is essential for oocyte meiosis and directly regulates Hsp90α expression. J Biol Chem 284: 9521–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan WD. 1989. Transcription factor Sp1 binds to and activates a human hsp70 gene promoter. Mol Cell Biol 9: 4099–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton EA, Lamitina T. 2013. Caenorhabditis elegans HSF-1 is an essential nuclear protein that forms stress granule-like structures following heat shock. Aging Cell 12: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudegger T, Verghese J, Hayer-Hartl M, Hartl FU, Bracher A. 2016. Structure of human heat-shock transcription factor 1 in complex with DNA. Nat Struct Mol Biol 23: 140–146. [DOI] [PubMed] [Google Scholar]
- Page BD, Guedes S, Waring D, Priess JR. 2001. The C. elegans E2F- and DP-related proteins are required for embryonic asymmetry and negatively regulate Ras/MAPK signaling. Mol Cell 7: 451–460. [DOI] [PubMed] [Google Scholar]
- Park JM, Werner J, Kim JM, Lis JT, Kim YJ. 2001. Mediator, not holoenzyme, is directly recruited to the heat shock promoter by HSF upon heat shock. Mol Cell 8: 9–19. [DOI] [PubMed] [Google Scholar]
- Perisic O, Xiao H, Lis JT. 1989. Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell 59: 797–806. [DOI] [PubMed] [Google Scholar]
- Preissler S, Deuerling E. 2012. Ribosome-associated chaperones as key players in proteostasis. Trends Biochem Sci 37: 274–283. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Loew C, Korner R, Pinkert S, Theis M, Hayer-Hartl M, Buchholz F, Hartl FU. 2014. Interplay of acetyltransferase EP300 and the proteasome system in regulating heat shock transcription factor 1. Cell 156: 975–985. [DOI] [PubMed] [Google Scholar]
- Reddien PW, Andersen EC, Huang MC, Horvitz HR. 2007. DPL-1 DP, LIN-35 Rb and EFL-1 E2F act with the MCD-1 zinc-finger protein to promote programmed cell death in Caenorhabditis elegans. Genetics 175: 1719–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM, et al. 2013. DAF-16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, Vandenhaute J, Orkin SH, Hill DE, van den Heuvel S, et al. 2004. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14: 2162–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, DeCaprio JA. 2013. The DREAM complex: master coordinator of cell cycle-dependent gene expression. Nat Rev Cancer 13: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santagata S, Mendillo ML, Tang YC, Subramanian A, Perley CC, Roche SP, Wong B, Narayan R, Kwon H, Koeva M, et al. 2013. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 341: 1238303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Saraiva MJ. 2004. Enlarged ventricles, astrogliosis and neurodegeneration in heat shock factor 1 null mouse brain. Neuroscience 126: 657–663. [DOI] [PubMed] [Google Scholar]
- Satyal SH, Chen D, Fox SG, Kramer JM, Morimoto RI. 1998. Negative regulation of the heat shock transcriptional response by HSBP1. Genes Dev 12: 1962–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schertel C, Conradt B. 2007. C. elegans orthologs of components of the RB tumor suppressor complex have distinct pro-apoptotic functions. Development 134: 3691–3701. [DOI] [PubMed] [Google Scholar]
- Shi Y, Mosser DD, Morimoto RI. 1998. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 12: 654–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V, Aballay A. 2006. Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci 103: 13092–13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi TM, Deplancke B, Osato N, Zhu LJ, Barrasa MI, Harrison MM, Horvitz HR, Walhout AJ, Hagstrom KA. 2011. Chromosome-biased binding and gene regulation by the Caenorhabditis elegans DRM complex. PLoS Genet 7: e1002074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki E, Fujimoto M, Sugahara K, Nakahari T, Yonemura S, Tanaka Y, Hayashida N, Inouye S, Takemoto T, Yamashita H, et al. 2006. Maintenance of olfactory neurogenesis requires HSF1, a major heat shock transcription factor in mice. J Biol Chem 281: 4931–4937. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, Becker PB, Wu C. 1994. ATP-dependent nucleosome disruption at a heat-shock promoter mediated by binding of GAGA transcription factor. Nature 367: 525–532. [DOI] [PubMed] [Google Scholar]
- van den Heuvel S, Dyson NJ. 2008. Conserved functions of the pRB and E2F families. Nat Rev Mol Cell Biol 9: 713–724. [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Kim SK. 2013. Integrative analysis of C. elegans modENCODE ChIP-seq data sets to infer gene regulatory interactions. Genome Res 23: 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihervaara A, Sistonen L. 2014. HSF1 at a glance. J Cell Sci 127: 261–266. [DOI] [PubMed] [Google Scholar]
- Vihervaara A, Sergelius C, Vasara J, Blom MA, Elsing AN, Roos-Mattjus P, Sistonen L. 2013. Transcriptional response to stress in the dynamic chromatin environment of cycling and mitotic cells. Proc Natl Acad Sci 110: E3388–E3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM Jr, Sistonen L, Morimoto RI. 2009. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science 323: 1063–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao YH, Pei Y. 2011. Asymmetric overlap extension PCR method for site-directed mutagenesis. Methods Mol Biol 687: 277–282. [DOI] [PubMed] [Google Scholar]
- Xiao H, Perisic O, Lis JT. 1991. Cooperative binding of Drosophila heat shock factor to arrays of a conserved 5 bp unit. Cell 64: 585–593. [DOI] [PubMed] [Google Scholar]
- Xiao X, Zuo X, Davis AA, McMillan DR, Curry BB, Richardson JA, Benjamin IJ. 1999. HSF1 is required for extra-embryonic development, postnatal growth and protection during inflammatory responses in mice. EMBO J 18: 5943–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing H, Wilkerson DC, Mayhew CN, Lubert EJ, Skaggs HS, Goodson ML, Hong Y, Park-Sarge OK, Sarge KD. 2005. Mechanism of hsp70i gene bookmarking. Science 307: 421–423. [DOI] [PubMed] [Google Scholar]
- Yuan CX, Gurley WB. 2000. Potential targets for HSF1 within the preinitiation complex. Cell Stress Chaperones 5: 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.