

Abstract
Analysis of transcription regulatory networks has revealed many principal features that govern gene expression regulation. MicroRNAs (miRNAs) have emerged as another major class of gene regulators that influence gene expression post-transcriptionally, but there remains a need to assess quantitatively their global roles in gene regulation. Here, we have constructed an integrated gene regulatory network comprised of transcription factors (TFs), miRNAs, and their target genes and analyzed the effect of regulation on target mRNA expression, target protein expression, protein–protein interaction, and disease association. We found that while target genes regulated by the same TFs tend to be co-expressed, co-regulation by miRNAs does not lead to co-expression assessed at either mRNA or protein levels. Analysis of interacting protein pairs in the regulatory network revealed that compared to genes co-regulated by miRNAs, a higher fraction of genes co-regulated by TFs encode proteins in the same complex. Although these results suggest that genes co-regulated by TFs are more functionally related than those co-regulated by miRNAs, genes that share either TF or miRNA regulators are more likely to cause the same disease. Further analysis on the interplay between TFs and miRNAs suggests that TFs tend to regulate intramodule/pathway clusters, while miRNAs tend to regulate intermodule/pathway clusters. These results demonstrate that although TFs and miRNAs both regulate gene expression, they occupy distinct niches in the overall regulatory network within the cell.
Keywords: gene regulation, biological networks
INTRODUCTION
Gene expression is controlled and fine-tuned at multiple levels within a hierarchical gene regulatory network. Transcription factors (TFs) activate or repress gene expression by binding transcription factor binding sites (TFBS) in gene promoters or cis-regulatory modules (Harbison et al. 2004; Maston et al. 2006). TFs were believed to be the primary regulators of gene expression until research in the past decade revealed microRNAs (miRNAs) as a second major class of gene expression regulator (Bartel 2004). miRNAs are small, noncoding RNAs that fine-tune gene expression post-transcriptionally. Mature miRNAs bind complementary sequences of target mRNAs, causing mRNA degradation, and/or translation repression (Bartel 2004). miRNAs regulate many biological processes and have been implicated in the development of human diseases including cancer (Brennecke et al. 2003; Bartel 2004; Flynt and Lai 2008). Recent research suggested that the majority of human genes might be targets of miRNAs (Friedman et al. 2009). Many miRNA targets are TFs, which in turn regulate miRNA expression, forming an intricate regulatory network (O'Donnell et al. 2005; Tsang et al. 2007). For example, proto-oncoprotein c-MYC simultaneously regulates the expression of transcription factor E2F1 and a cluster of six miRNAs that represses E2F1 expression. This positive co-regulation of E2F1 and its miRNA regulators allows the proliferative signal in the cell to be tightly controlled (O'Donnell et al. 2005). In addition, miRNAs often regulate their own expression through auto-regulatory feedback loops with specific TFs (Krol et al. 2010).
As the canonical gene expression regulator, TFs have been well characterized, and system-wide properties of the transcription regulatory network have been explored (Lee et al. 2002; Shen-Orr et al. 2002; Yu and Gerstein 2006). In recent years, researchers are increasingly interested in the combinatorial interactions between TFs and miRNAs. An integrated regulatory network that includes both transcriptional and post-transcriptional regulation is necessary to provide a more complete picture of gene expression regulation and may reveal basic regulatory principles underlying disease phenotypes. Recent studies have found recurring co-regulatory motifs involving both TFs and miRNAs, such as TF-miRNA co-regulating pairs and feed-forward loops, indicating prevalent crosstalk and cooperation between these two modes of gene regulation (Shalgi et al. 2007; Tsang et al. 2007; Su et al. 2010).
As genome-wide transcription-factor-binding data were not readily available, previous studies on the integrated regulatory network inferred TF-gene regulatory relationships from computationally predicted TFBS. Recently, large-scale ChIP-seq experiments from the ENCODE project generated system-wide data on transcription factor binding patterns (ENCODE Project Consortium 2012). Making use of the transcription factor binding data from the ENCODE project, Gerstein et al. (2012) created a human transcriptional regulatory network (Gerstein et al. 2012). They integrated miRNA regulation data with this transcription regulation network and found that top-level TFs have more regulatory relationships with miRNAs, and also identified enriched miRNA-TF co-regulation motifs in the network. However, the differences between the architectures of miRNA and TF regulation in the context of the overall gene regulation network are still unclear. In this study, we constructed a human integrated regulatory network by combining the ENCODE transcriptional regulation network and high-confidence miRNA-target predictions to investigate the possible differences in the roles of TFs and miRNAs in gene regulation and the synergistic actions of these two types of regulators. Previous studies of TF-miRNA co-regulation in the regulatory network suggested relationships between regulation and gene expression (Shalgi et al. 2007; Tsang et al. 2007; Su et al. 2010), but downstream effects of regulation, especially at the organismal level, remain unclear. Here we studied the effects of TF and miRNA regulation at three levels: mRNA and protein expression, protein–protein interaction, and organism-level disease phenotypes, and found that TFs and miRNAs exhibit distinct roles in the regulation of gene expression.
RESULTS
Effects of TF and miRNA regulation on target gene expression, protein–protein interaction, and disease association
By combining TF-gene regulatory relationships from the ENCODE project (ENCODE Project Consortium 2012; Gerstein et al. 2012) and high-confidence miRNA-target predictions from TargetScan (Lewis et al. 2005; Grimson et al. 2007; Agarwal et al. 2015), we constructed a human integrated gene regulatory network with a total of 35,304 regulatory relationships among 83 TFs, 77 miRNA families, and 11,407 target genes. Previous studies have established that both TFs and miRNAs work cooperatively to regulate their gene targets (Hobert 2008; Martinez and Walhout 2009). Here we investigated the additive effects of TF regulation and miRNA regulation on their target genes.
First, we examined the expression relationships among genes regulated by the same TF(s) or miRNA(s) at both mRNA and protein levels. Previous studies have shown in multiple organisms that genes regulated by the same TF tend to be coexpressed (Yu et al. 2003; Kim et al. 2006; Marco et al. 2009; Gu et al. 2011). Here, we found that the degree of expression correlation depends on the number of shared regulators. We calculated the log odds ratio of mRNA coexpression of gene pairs co-regulated by one or more TFs compared to random expectation (see Methods). We found that the likelihood of two genes being coexpressed at the mRNA level increases with the number of common TFs that regulate them (Fig. 1A). On the other hand, genes regulated by the same miRNAs do not tend to be coexpressed at the mRNA level compared to random gene pairs (Fig. 1B). Even gene pairs co-regulated by four or more miRNAs are not more likely to coexpress (LOD = 0.21, P = 0.28). Although mRNA expression levels affect the amount of protein produced in the cell, studies found that there is only a modest correlation between mRNA abundance and protein abundance (Ghaemmaghami et al. 2003; Vogel and Marcotte 2012). MicroRNAs not only regulate target mRNA levels but also act by repressing the translation of target mRNAs into proteins. Therefore, the effects of miRNAs on their targets could be better reflected at the level of protein expression. To investigate the effects of TF and miRNA regulation on protein expression of their target genes, we computed the pair-wise protein coexpression using protein expression profiles from the Human Proteome Map (Kim et al. 2014) and calculated the log odds ratio of protein coexpression of target pairs co-regulated by one or more common regulators compared to random expectations. Interestingly, we found that even at the protein level, genes co-regulated by the same miRNAs are not more likely to be coexpressed (Fig. 1D). These observations are the opposite of the commonly accepted view that co-regulation leads to coexpression. However, the observation is consistent with our current understanding of miRNAs: They only have a moderate repressive effect on target gene expression and do not control the on/off state of the target genes (Bartel 2004). Furthermore, the overall effect of miRNA regulation on gene expression is much more subtle compared to TF regulation (Bartel 2004).
FIGURE 1.

Synergistic effects of TFs and miRNAs on target gene co-expression. LOD values are calculated for the enrichment of mRNA co-expression between gene pairs co-regulated by multiple TFs (A) or miRNAs (B). LOD values are calculated for the enrichment of protein coexpression between gene pairs co-regulated by multiple TFs (C) or miRNAs (D). (*) P < 0.05. P-values are calculated by the Z-test. Error bars indicate ± SE. Difference in the fractions of coexpressed gene pairs between the first and last bins is calculated by the χ2 test.
Proteins are the functional units of the cell and carry out their diverse activities through interactions with other proteins. To investigate the effects of regulation on protein function, we studied the interactions between protein products of genes regulated by the same TF(s) or miRNA(s). We found that the protein products of genes co-regulated by either TFs or miRNAs are significantly more likely to physically interact with each other than randomly expected, and the likelihood of physical interaction increases with the number of shared regulators (Fig. 2A,B). For example, protein products of genes co-regulated by three or more common regulators are almost twice as likely to physically interact compared to random expectation (OD = 1.88, P < 10−6 for TF regulation; OD = 1.99, P < 10−4 for miRNA regulation). Furthermore, protein products of genes co-regulated by more TFs or miRNAs tend to be in closer proximity in the protein interaction network (Supplemental Fig. 1). Within the cell, proteins can form stable complexes or dynamically interact with each other to carry out subcellular functions. The stable interactions bring proteins into tightly regulated functional modules, while transient interactions connect and coordinate these modules (Das et al. 2012). To investigate the relationship between regulation and protein interaction dynamics, we identified stable complexes in the protein–protein interaction network using the ClusterONE algorithm (Nepusz et al. 2012). We found that genes co-regulated by the same miRNAs are less likely to encode proteins in the same complex compared to genes co-regulated by TFs (Fig. 2C, P < 10−5). Previous studies found that genes encoding subunits in the same protein complex are globally coexpressed (Jansen et al. 2002; Yu et al. 2008a). On the other hand, genes encoding proteins involved in transient interactions only coexpress under specific conditions and do not have highly correlated expression profiles (Das et al. 2012). This explains our observation that although genes co-regulated by the same miRNAs are more likely to encode for interacting proteins, co-regulation by miRNAs has little effect on the global coexpression of target genes.
FIGURE 2.
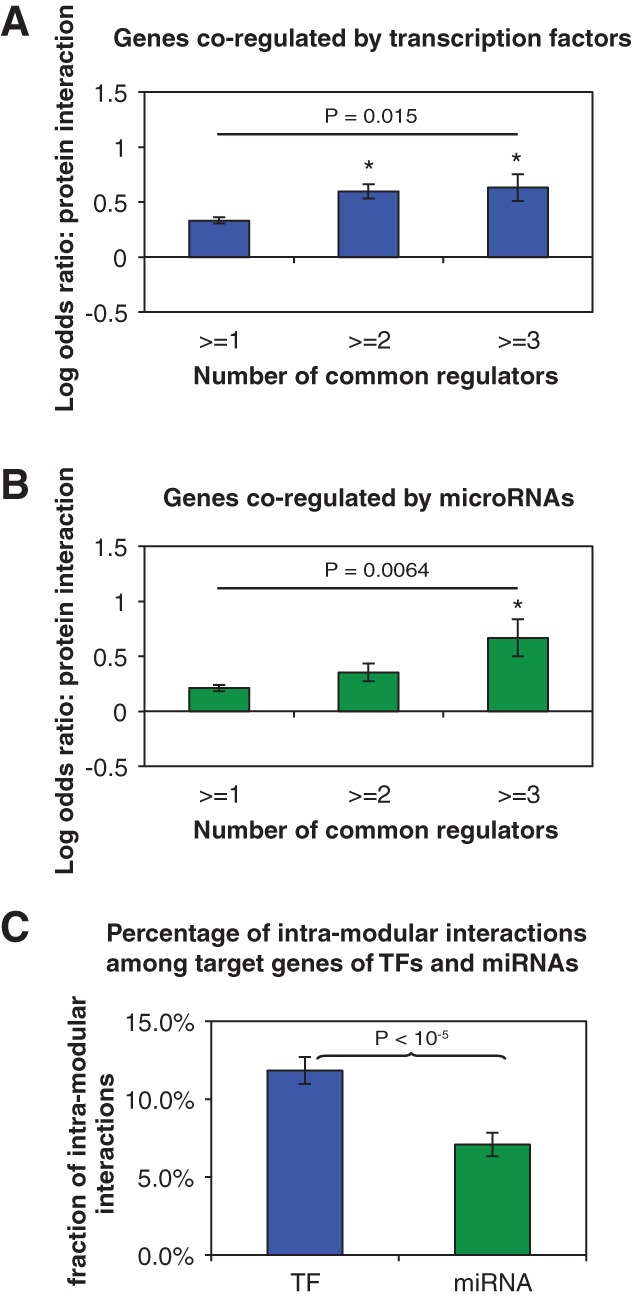
Synergistic effects of TFs and miRNAs on the protein–protein interaction of targets. LOD values are calculated for the enrichment of protein–protein interactions between gene pairs co-regulated by multiple TFs through TFs (A) or miRNAs (B). (*) P < 0.05. P-values are calculated by the Z-test. Error bars indicate ± SE. Difference in the fractions of genes pairs encoding interacting proteins between the first and last bins is calculated by the χ2 test. (C) The percentage of intramodular interactions among interacting protein pairs encoded by co-regulated genes.
Misregulation of gene expression often leads to disease phenotypes. Both TFs and miRNAs have been found to be associated with a wide range of human diseases, including the development and progression of cancers (Croce 2009; Esteller 2011). However, the relationship between regulatory network architecture and the involvement of genes in different human diseases is not well studied at a genome-wide scale. To this end, we compiled a comprehensive list of disease genes and their associated diseases from HGMD and OMIM databases and studied the relationship between co-regulation and coassociation of genes to diseases. Specifically, we calculated the enrichment of co-regulated disease genes that are associated with the same disease with respect to random expectations. Genes jointly regulated by more TFs are more likely to be associated with the same diseases (LOD = 0.32, 0.75, 0.96, and 1.25 for gene pairs jointly regulated by more than 1, 2, 3, and 4 TFs, respectively; Fig. 3A). Similarly, genes co-regulated by multiple miRNAs are also more likely to cause the same diseases (LOD = 0.22, 0.57, and 1.24 for genes jointly regulated by more than 1, 2, and 3 miRNAs, respectively; Fig. 3B). Together with the expression and interaction analyses above, our results demonstrate that genes co-regulated by multiple TFs tend to be more related on all three functional levels we examined. This shows that genes sharing more regulating TFs tend to be more functionally similar and tend to form tightly regulated modules that function together in the same biological processes/pathways. In contrast, miRNAs do not regulate the coordinated expression of genes in the same functional module and tend to regulate intercomplex protein–protein interactions, but target genes co-regulated by the same miRNAs are still significantly more likely to be associated with the same disease. This suggests that miRNAs play an important role in intermodular regulation, where they coordinate target genes in related biological processes/pathways.
FIGURE 3.
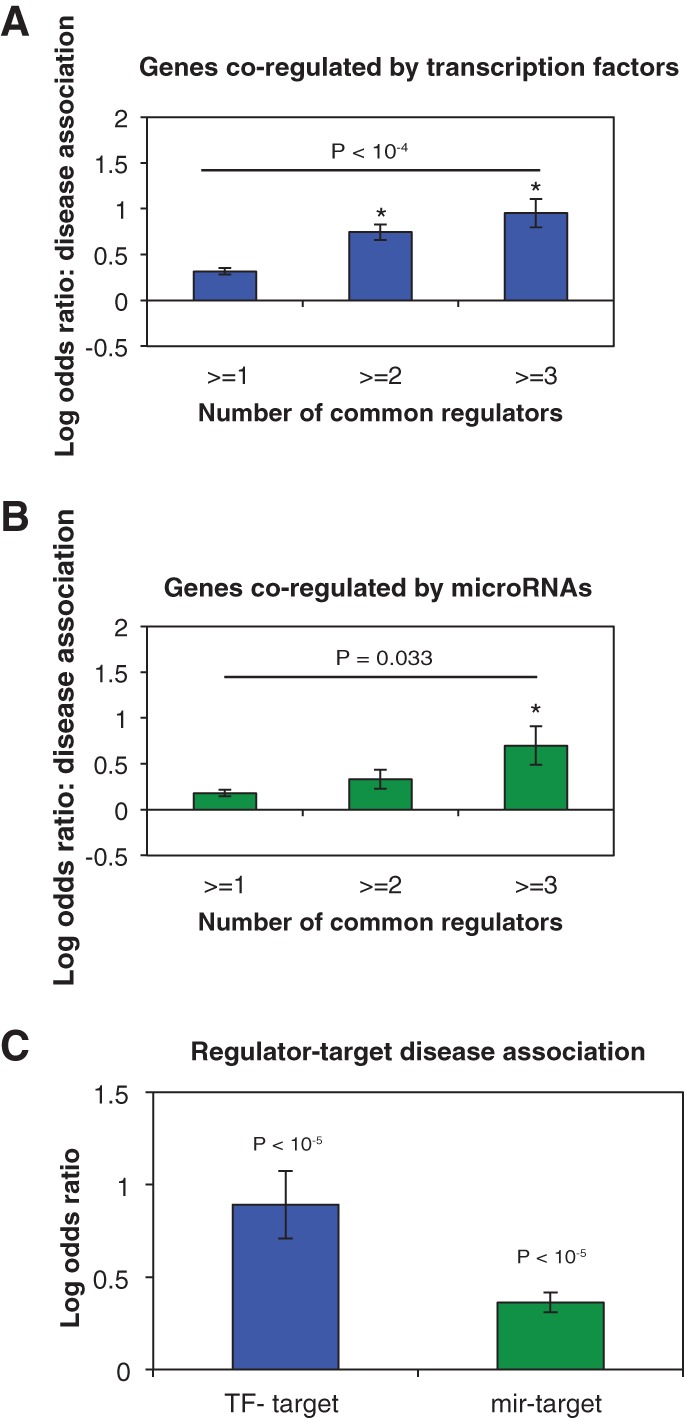
Effects of TF and miRNA regulation on target gene disease association. LOD values of the enrichment for co-regulated genes to cause the same disease are calculated for gene pairs co-regulated by multiple TFs through TFs (A) or miRNAs (B). Difference in the fractions of gene pairs associated with the same disease between the first and last bins is calculated by the χ2 test. (C) LOD values of the enrichment for regulator-gene pairs to be associated with the same disease compared to random expectations. (*) P < 0.05. P-values are calculated by the Z-test. Error bars indicate ±SE.
Although TargetScan provides a genome-wide, unbiased prediction of miRNA targets, most of the targets predicted are not verified experimentally. On the other hand, the transcription regulation network is derived from ChIP-seq experiments. The difference in the data reliability of each data source could affect the interpretation of our results. Unfortunately, it is difficult to benchmark the fractions of false-positive associations across the different data sources in a systematic way. To further validate our results, we generated a high-quality set of experimentally verified miRNA-gene interactions using manually curated data from Tarbase (Vergoulis et al. 2012) and miRTarBase (Hsu et al. 2014). To avoid study bias arising from small-scale, gene-specific experiments, we only included data generated by high-throughput experimental methods such as CLIP-seq and Degradome-seq. Since these miRNA targets were derived from high-throughput experiments, the quality of this miRNA-target set is likely more comparable to the TF target set derived from ENCODE. We repeated all of our calculations with this high-quality experimentally verified miRNA-target set and found the results to be consistent with those described above (Supplemental Fig. 2). Validating our results from two independent data sources confirms that our results are robust across a range of false-positive rates in the input. As TargetScan identifies miRNA targets on the genome scale for all known miRNA families, while studies that experimentally identify miRNA targets usually focus on a small number of miRNAs, the miRNA-target network predicted by TargetScan is significantly larger. Since analyses with the predicted miRNA-target set and with the observed miRNA-target set yield similar results, we use the predicted miRNA-target set for all subsequent analyses to ensure sufficient statistical power.
Functional relationship between regulators and their target genes
Genes associated with the same disease tend to have correlated gene expression and their protein products tend to physically interact, forming functional modules in the cellular network (Goh et al. 2007; Feldman et al. 2008). As TFs and miRNAs control the timing and level of expression of their target genes, we postulate that disruption of the regulator functions and disruption of the target protein functions are likely to result in the same diseased state. By comparing the diseases associated with TFs and the diseases associated with their gene targets, we found that overall, TFs are significantly more likely to cause the same disease as their gene targets compared to random TF-target pairs (LOD = 0.89, P < 10−5; Fig. 3C). To perform the same analysis on miRNAs, we compiled a list of literature-curated miRNA-disease associations from the HMDD (Lu et al. 2008) and miR2Disease (Jiang et al. 2009) databases. Similarly, we found that miRNAs are also significantly more likely to be associated with the same disease as the genes they regulate (LOD = 0.36, P < 10−10; Fig. 3C). This confirms our hypothesis that disruptions of TF or miRNA functions tend to have similar effects as disruptions of their target gene functions.
Different roles of TF and miRNA regulation: intramodular vs. intermodular
Recent studies found that crosstalk and cooperation between TFs and miRNAs are highly prevalent and could be an integral part of the gene regulatory network (Shalgi et al. 2007; Yu et al. 2008b; Chen et al. 2011; Lin et al. 2012). To further understand the crosstalk between TFs and miRNAs, we investigated the regulatory relationships between TFs and miRNAs. From our gene regulatory network, we found 1004 miRNA pairs co-regulated by at least one common TF, and 262 TF pairs co-regulated by at least one common miRNA.
First we examined the functional similarity of TFs that are targeted by the same miRNA(s). We computed the fraction of shared targets for each TF pair (number of shared targets/total number of targets of the two TFs) and compared the distribution of the fraction of shared targets of TF pairs regulated by the same miRNA and that of random TF pairs. We found that on average, two TFs co-regulated by the same miRNA do not share more targets than two random TFs (P = 0.96 by Wilcoxon rank-sum test; Fig. 4A), suggesting that TFs regulated by the same miRNA may regulate different biological processes. However, TFs do tend to share targets with the miRNA that regulates them, implying functional overlap between an miRNA and the TFs it regulates (Supplemental Fig. 3). In contrast, ∼81% of miRNA pairs regulated by the same TF share gene targets, which is significantly higher than random expectation (P < 10−3; Fig. 4C). This shows that miRNAs regulated by the same TFs tend to have higher functional overlap. As a comparison, we also calculated the distribution of the fraction of shared targets of TF pairs regulated by the same TF. We found that TFs that are regulated by the same TF tend to share more gene targets compared to random TF pairs (P < 10−6; Fig. 4B), suggesting that TFs regulated by the same TF also tend to have related functions. In summary, TF and miRNA pairs that are co-regulated by an upstream TF tend to target the same genes, whereas TF pairs co-regulated by the same miRNAs do not tend to share more targets compared to random TF pairs.
FIGURE 4.

Interregulation of TFs and miRNAs. Distribution of the fraction of shared targets of TF pairs regulated by the same miRNA (A), TF pairs regulated by the same TF (B), and miRNA pairs regulated by the same TF (C). P-values are calculated using the Wilcoxon rank-sum test.
Furthermore, we found that second degree targets co-regulated by an upstream TF are more likely to be coexpressed and to physically interact, whereas second degree targets co-regulated by the same miRNAs are not more likely to coexpress or physically interact compared to random expectations (Supplemental Figs. 4, 5). To further verify the above observations, we formulated a functional similarity metric for TFs and miRNAs based on the overlap of their target gene sets, the proximity of their target genes in the protein–protein interaction network and the similarity of their target genes according to the Gene Ontology biological processes terms (see Methods). Consistent with our previous observations, we found that TFs regulated by the same miRNAs are not more functionally similar than random TF pairs, while miRNA pairs (P < 10−15) and TF pairs (P < 10−12) regulated by the same TF are significantly more similar than random miRNA pairs or TF pairs without regulation (Wilcoxon rank-sum test; Supplemental Fig. 6). This suggests that TFs regulate groups of functionally similar downstream regulators that tend to target the same genes or functionally related genes. In contrast, miRNAs regulate functionally disparate TFs that are likely to be involved in different biological processes.
Thus far, we found that genes co-regulated by TFs form tightly regulated functional modules and that miRNAs/TFs regulated by the same TF are functionally similar. These results suggest that TFs tend to regulate genes within the same functional module, where genes regulated by the same TF participate in the same biological process/pathway (Fig. 5A). Examples of intramodular multilevel regulation are shown in Figure 5A. The oncogenic transcription factor MYC is involved in many different types of human cancers (Dang 2012). For example, overexpression of MYC is associated with human prostate cancer (Gurel et al. 2008; Koh et al. 2011). It has been found that MYC regulates the expression of many tumor-suppressing miRNAs in lymphoma and prostate cancer cells (Chang et al. 2008; Koh et al. 2011). In our integrated regulatory network, MYC regulates hsa-miR-19b and hsa-miR-92a, which are themselves also associated with prostate cancer. These miRNAs in turn regulate PTEN, a tumor suppressor gene that was found to be inactivated in somatic prostate cancers (Cairns et al. 1997). Here, the transcription factor MYC regulates a group of miRNAs, which in turn regulate the downstream gene PTEN in the same disease module. In this disease module, we also found another miRNA, hsa-miR-19a, that has not been previously associated with prostate cancer. As hsa-miR-19a is in the same miRNA family as hsa-miR-19b, it is likely that it also plays a role in prostate cancer.
FIGURE 5.

Intramodular regulation and intermodular regulation. (A) Schematic and an example of intramodular regulation by TF. Transcription factor MYC and downstream targets are associated with prostate cancer. (B) Schematic and an example of intermodular regulation by miRNA. miR-27ab targets three different TFs that each regulates different biological processes.
On the other hand, genes regulated by the same miRNA(s) do not tend to coexpress globally; their protein products tend to interact intermodularly, but they are still more likely to be associated with the same disease. In addition, a single miRNA may regulate multiple TFs that carry out different functions. Taken together, our results suggest that miRNAs are involved in intermodular regulation, where genes regulated by the same miRNA may not necessarily be in the same protein complex or pathway but are involved in related cellular processes (Fig. 5B). An example of intermodular multilevel regulation is shown in Figure 5B.
DISCUSSION
In this study, we have constructed an integrated gene regulatory network comprising both transcriptional regulation and post-transcriptional regulation, and investigated on a global scale the differences between the two layers of regulation on three functional levels. Our results revealed that TFs are involved in intramodular regulation, where multiple TFs act cooperatively to regulate a set of genes that tend to coexpress, interact physically, and associate with the same diseases. On the other hand, miRNAs coordinate related cellular pathways/processes through intermodular regulation. Gene targets regulated by the same miRNA(s) show higher expression variability, they do not tend to encode proteins in the same complex, although their protein products are in closer proximity in the protein–protein interactome network, and they are more likely to be associated with the same diseases. A previous study by Liang and Li on the correlation between miRNA regulation and protein–protein interactions found that miRNAs have a higher propensity to target intermodular protein hubs compared to intramodular protein hubs (Liang and Li 2007), which further supports our observations. miRNA regulation of genes across different functional modules/pathways is biologically important, as different functional modules have diverse expression profiles and regulation is essential for the coordination among different functional modules in the cell. Indeed, clustering of miRNAs by functional similarity revealed that disease-associated miRNAs tend to be at the interface between adjacent functional modules compared to non-disease-associated miRNAs (Xu et al. 2011).
In conclusion, we found that although TFs and miRNAs share similar regulatory logic, such as combinatorial regulation of gene targets in recurring network motifs (Shen-Orr et al. 2002; Shalgi et al. 2007; Gerstein et al. 2012), they appear to occupy distinct niches in the gene regulatory network, and that these differences impact the role that TFs and miRNAs play in mediating disease risk. Our findings provide new insights into the global architecture and organization principles of the gene regulatory network.
MATERIALS AND METHODS
Integrated gene regulatory network
The TF-gene regulatory relationships were derived from ENCODE data generated by ChIP-seq experiments. To ensure the quality of the network, we downloaded the filtered, high-confidence set of TF-gene associations from the supplementary website of Gerstein et al. (2012). The high-confidence TF regulatory network was filtered based on a probabilistic model called target identification from profiles (TIP) (Cheng et al. 2011). TIP learns a characteristic binding profile for each TF and predicts a regulatory score between the TF and each of its potential target genes based on the binding regions of the gene. TF-gene interaction was then ranked using the regulatory score, and those with FDR < 0.01 were included in the high-confidence TF-gene association provided by Gerstein and colleagues. Among the 119 transcription-related factors studied in the ENCODE project, we only considered sequence specific transcription factors that recognize and bind to specific DNA sequence motifs. The resulting transcriptional regulatory network comprises 83 sequence specific transcription factors and 8243 target genes.
Currently, several major miRNA-target prediction algorithms, such as TargetScan (Lewis et al. 2005), miRanda-mirSVR (John et al. 2004; Betel et al. 2010), PicTar (Krek et al. 2005), and PITA (Kertesz et al. 2007), make genome-wide predictions of miRNA targets based on target site conservation and other target site features. Studies of protein levels after miRNA knockdown and transfection demonstrated that TargetScan and miRanda-mirSVR outperform other miRNA prediction algorithms in terms of prediction accuracy (Baek et al. 2008; Selbach et al. 2008; Betel et al. 2010). In this study, we obtained miRNA identities and their predicted targets from TargetScan. To ensure that only high-confidence miRNA-gene regulatory relationships are included, we filtered TargetScan predictions based on both conservation criteria (Lewis et al. 2005; Friedman et al. 2009) and estimates of site performance, referred to as Context Score (Grimson et al. 2007; Agarwal et al. 2015). We used only gene targets with a Pct ≥ 0.5, which indicates that there is at least a 50% probability that a sequence is selectively maintained as a miRNA-target site, and a Context Score ≤ −0.2. Grimson et al. (2007) demonstrated by siRNA transfection experiments that predicted miRNA targets with a lower Context Score are more down-regulated in response to siRNA expression. Context Scores of −0.2 or lower were chosen as the cutoff because such targets were measurably down-regulated (∼25% change in expression on average for conserved sites) in the siRNA transfection experiments (Grimson et al. 2007). To avoid redundancies in the network and subsequent analyses, mature miRNAs with identical seed regions were grouped into miRNA families based on the miRNA family information from TargetScan. In total, we obtained regulatory relationships between 77 conserved miRNA families and 5858 predicted targets from TargetScan.
In addition, we compiled an experimentally verified miRNA-target set from Tarbase (Vergoulis et al. 2012) and miRTarBase (Hsu et al. 2014). This data set includes 6470 interactions among 59 miRNA families and 3660 target genes that are verified by high-throughput experiments.
Gene expression data and PCC calculation
We used the mRNA expression data generated by the Genotype-Tissue Expression (GTEx) project (GTEx Consortium 2015), which measured mRNA expression profiles of 8555 samples across 53 human tissues using mRNA sequencing. Gene-level expression data were downloaded from the GTEx data portal and the expression values were quantile normalized. On average, the 83 TFs in our network are expressed in 90% of the samples in the GTEx data at an expression cutoff of 0.1 RPKM (cutoff used by the GTEx consortium). Seventy-six of the 83 TFs are expressed in >50% of the samples.
The gene-level protein expression profiles of 30 human tissues generated by high-resolution mass spectrometry were downloaded from the Human Proteome Map (HPM) (Kim et al. 2014). The HPM data set includes proteins encoded by 17,294 genes, which covers ∼84% of all protein-coding genes in humans. On average, the protein expression of each TF is detected in 40% of the samples. All TFs are expressed in at least one sample in the protein expression data.
Pearson correlation coefficient (PCC) was calculated for all possible pairs of genes at the mRNA and the protein levels using a massively parallel Java program (Das et al. 2012). Two genes are considered to be coexpressed at the mRNA/protein level if their mRNA/protein expression profiles have a PCC of 0.6 or greater (approximately the top 1% of all possible interactions ranked by PCC).
Protein–protein interaction and disease-gene association data
High-quality, binary protein–protein interactions were obtained from HINT (Das and Yu 2012), a protein interaction database with high-quality interactions collated from literature-curation and high-throughput experiments.
A comprehensive list of disease-associated genes was compiled from the Human Gene Mutation Database (HGMD) (Stenson et al. 2003, 2009) and the Online Mendelian Inheritance in Man (OMIM) (Amberger et al. 2009, 2011) database. miRNA-disease associations were collated from two databases: the human microRNA disease database (HMDD) (Lu et al. 2008) and miR2Disease database (Jiang et al. 2009). In total, we collected 2712 manually curated associations between 263 miRNAs and 184 diseases. To standardize the disease nomenclature across databases, unique disease identifiers were assigned to each phenotypically distinct disease through computational and manual curation.
Calculating the similarity score of two regulators
The similarity score of two regulators was calculated based on (i) the number of gene targets they share, (ii) the number of targets of each regulator that directly interact with target proteins of the other regulator in the protein–protein interaction network, and (iii) the number of targets of each regulator that are functionally similar to the targets of the other regulator based on biological process terms from Gene Ontology. Functional similarity of two genes based on the Gene Ontology biological process was calculated using the total ancestry measure as previously described (Yu et al. 2007; Das et al. 2012). Functional similarities between all genes were calculated using a massively parallel Java program (Das et al. 2012).
The similarity score is calculated as
where
A = {Shared targets of the two regulators}
B = {Interacting targets of two regulators}
C = {Functionally similar targets of two regulators}
Ω = {All targets of the two regulators}
The most stringent criterion for functional similarity of two regulators is the number of targets they share, followed by the number of interacting targets, and then by the number of functionally similar targets in GO. On average, for any two regulators, targets identified in sets A, B, and C contribute to about 5%, 10%, and 85%, respectively, to the similarity score between the two regulators. Although the major component of the similarity score is the set of functionally similar targets of two regulators, our results remain the same when we evaluate the similarity of two regulators based solely on the number of targets they share (Fig. 4), or the number of targets of one regulator that directly interacts with targets of the other regulator (Supplemental Fig. 5).
Statistical analyses
We performed randomization tests to evaluate the enrichment of coexpression, protein–protein interaction, and disease association among co-regulated genes. We generated 100 randomized networks by permuting gene identifiers of both regulators and target genes. The randomized networks generated have the same degree distribution, network topology, and motif structures as the original network.
For each functional relationship, we compared the fraction of co-regulated gene pairs sharing a specific functional relationship in the real network (p1) to the average fraction of co-regulated gene pairs sharing the same functional relationship in random networks (p2). The enrichment of each functional relationship among co-regulated genes with respect to random expectations was measured by log odds ratio (LOD).
The standard error of the LOD is calculated as
The statistical significance of the enrichment was evaluated by the Z-test. To avoid large errors in the estimation of enrichments, we only performed the enrichment calculation when there were at least 10 pairs of genes with an observed functional relationship. Also, we limited the enrichment calculations to up to ≥4 common regulators because <1% of all gene pairs share five or more common regulators.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anders Skanderup for insightful scientific discussions. This work was supported by National Institute of General Medical Sciences grants R01 GM097358, R01 GM104424, and R01 GM108716, National Cancer Institute grant R01 CA167824, National Institute of Child Health and Human Development grant R01 HD082568, and Simons Foundation Autism Research Initiative grant 367561 to H.Y.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.048025.114.
REFERENCES
- Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger J, Bocchini CA, Scott AF, Hamosh A. 2009. McKusick's Online Mendelian Inheritance in Man (OMIM). Nucleic Acids Res 37: D793–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger J, Bocchini C, Hamosh A. 2011. A new face and new challenges for Online Mendelian Inheritance in Man (OMIM(R)). Hum Mutat 32: 564–567. [DOI] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297. [DOI] [PubMed] [Google Scholar]
- Betel D, Koppal A, Agius P, Sander C, Leslie C. 2010. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol 11: R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. 2003. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113: 25–36. [DOI] [PubMed] [Google Scholar]
- Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, Jen J, Isaacs WB, Bova GS, Sidransky D. 1997. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res 57: 4997–5000. [PubMed] [Google Scholar]
- Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. 2008. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 40: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Chen ST, Fuh CS, Juan HF, Huang HC. 2011. Coregulation of transcription factors and microRNAs in human transcriptional regulatory network. BMC Bioinformatics 12Suppl 1: S41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Min R, Gerstein M. 2011. TIP: a probabilistic method for identifying transcription factor target genes from ChIP-seq binding profiles. Bioinformatics 27: 3221–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce CM. 2009. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10: 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV. 2012. MYC on the path to cancer. Cell 149: 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Yu H. 2012. HINT: high-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol 6: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das J, Mohammed J, Yu H. 2012. Genome-scale analysis of interaction dynamics reveals organization of biological networks. Bioinformatics 28: 1873–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. 2011. Non-coding RNAs in human disease. Nat Rev Genet 12: 861–874. [DOI] [PubMed] [Google Scholar]
- Feldman I, Rzhetsky A, Vitkup D. 2008. Network properties of genes harboring inherited disease mutations. Proc Natl Acad Sci 105: 4323–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynt AS, Lai EC. 2008. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat Rev Genet 9: 831–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. 2003. Global analysis of protein expression in yeast. Nature 425: 737–741. [DOI] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. 2007. The human disease network. Proc Natl Acad Sci 104: 8685–8690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. 2007. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27: 91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. 2015. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Nagaraj SH, Hudson NJ, Dalrymple BP, Reverter A. 2011. Genome-wide patterns of promoter sharing and co-expression in bovine skeletal muscle. BMC Genomics 12: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurel B, Iwata T, Koh CM, Jenkins RB, Lan F, Van Dang C, Hicks JL, Morgan J, Cornish TC, Sutcliffe S, et al. 2008. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod Pathol 21: 1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, et al. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. 2008. Gene regulation by transcription factors and microRNAs. Science 319: 1785–1786. [DOI] [PubMed] [Google Scholar]
- Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, et al. 2014. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res 42: D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Greenbaum D, Gerstein M. 2002. Relating whole-genome expression data with protein-protein interactions. Genome Res 12: 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. 2009. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res 37: D98–D104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. 2004. Human microRNA targets. PLoS Biol 2: e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. 2007. The role of site accessibility in microRNA target recognition. Nat Genet 39: 1278–1284. [DOI] [PubMed] [Google Scholar]
- Kim RS, Ji H, Wong WH. 2006. An improved distance measure between the expression profiles linking co-expression and co-regulation in mouse. BMC Bioinformatics 7: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. 2014. A draft map of the human proteome. Nature 509: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. 2011. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget 2: 669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. 2005. Combinatorial microRNA target predictions. Nat Genet 37: 495–500. [DOI] [PubMed] [Google Scholar]
- Krol J, Loedige I, Filipowicz W. 2010. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet 11: 597–610. [DOI] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298: 799–804. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120: 15–20. [DOI] [PubMed] [Google Scholar]
- Liang H, Li WH. 2007. MicroRNA regulation of human protein–protein interaction network. RNA 13: 1402–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Chen YJ, Chen CY, Oyang YJ, Juan HF, Huang HC. 2012. Crosstalk between transcription factors and microRNAs in human protein interaction network. BMC Syst Biol 6: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. 2008. An analysis of human microRNA and disease associations. PLoS One 3: e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco A, Konikoff C, Karr TL, Kumar S. 2009. Relationship between gene co-expression and sharing of transcription factor binding sites in Drosophila melanogaster. Bioinformatics 25: 2473–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NJ, Walhout AJ. 2009. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 31: 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maston GA, Evans SK, Green MR. 2006. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet 7: 29–59. [DOI] [PubMed] [Google Scholar]
- Nepusz T, Yu H, Paccanaro A. 2012. Detecting overlapping protein complexes in protein-protein interaction networks. Nat Methods 9: 471–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. 2005. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 435: 839–843. [DOI] [PubMed] [Google Scholar]
- Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455: 58–63. [DOI] [PubMed] [Google Scholar]
- Shalgi R, Lieber D, Oren M, Pilpel Y. 2007. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput Biol 3: e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen-Orr SS, Milo R, Mangan S, Alon U. 2002. Network motifs in the transcriptional regulation network of Escherichia coli. Nat Genet 31: 64–68. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN. 2003. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21: 577–581. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, Cooper DN. 2009. The Human Gene Mutation Database: 2008 update. Genome Med 1: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su N, Wang Y, Qian M, Deng M. 2010. Combinatorial regulation of transcription factors and microRNAs. BMC Syst Biol 4: 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang J, Zhu J, van Oudenaarden A. 2007. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol Cell 26: 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergoulis T, Vlachos IS, Alexiou P, Georgakilas G, Maragkakis M, Reczko M, Gerangelos S, Koziris N, Dalamagas T, Hatzigeorgiou AG. 2012. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res 40: D222–D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Li CX, Li YS, Lv JY, Ma Y, Shao TT, Xu LD, Wang YY, Du L, Zhang YP, et al. 2011. MiRNA-miRNA synergistic network: construction via co-regulating functional modules and disease miRNA topological features. Nucleic Acids Res 39: 825–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Gerstein M. 2006. Genomic analysis of the hierarchical structure of regulatory networks. Proc Natl Acad Sci 103: 14724–14731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Luscombe NM, Qian J, Gerstein M. 2003. Genomic analysis of gene expression relationships in transcriptional regulatory networks. Trends Genet 19: 422–427. [DOI] [PubMed] [Google Scholar]
- Yu H, Jansen R, Stolovitzky G, Gerstein M. 2007. Total ancestry measure: quantifying the similarity in tree-like classification, with genomic applications. Bioinformatics 23: 2163–2173. [DOI] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. 2008a. High-quality binary protein interaction map of the yeast interactome network. Science 322: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Lin J, Zack DJ, Mendell JT, Qian J. 2008b. Analysis of regulatory network topology reveals functionally distinct classes of microRNAs. Nucleic Acids Res 36: 6494–6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.