

Abstract
Protein translation can be affected by changes in the secondary structure of mRNA. The dinQ gene in Escherichia coli encodes a primary transcript (+1) that is inert to translation. Ribonucleolytic removal of the 44 first nucleotides converts the +1 transcript into a translationally active form, but the mechanism behind this structural change is unknown. Here we present experimental evidence for a mechanism where alternative RNA secondary structures in the two dinQ mRNA variants affect translation initiation by mediating opening or closing of the ribosome binding sequence. This structural switch is determined by alternative interactions of four sequence elements within the dinQ mRNA and also by the agrB antisense RNA. Additionally, the structural conformation of +1 dinQ suggests a locking mechanism comprised of an RNA stem that both stabilizes and prevents translation initiation from the full-length dinQ transcript. BLAST search and multiple sequence alignments define a new family of dinQ-like genes widespread in Enterobacteriaceae with close RNA sequence similarities in their 5′ untranslated regions. Thus, it appears that a whole new family of genes is regulated by the same mechanism of alternative secondary RNA structures.
Keywords: DinQ, E. coli, RNA processing, RNA structure, translation initiation
INTRODUCTION
Initiation of translation of mRNA can be controlled by a number of mechanisms and allows the cell to express the protein from mRNA only when needed. This post-transcriptional regulation of gene expression can be intricate, and elucidating the basic regulatory mechanisms is important to gain a full understanding of how gene expression is regulated. In bacteria, protein translation is initiated by positioning and binding of the small (30S) ribosomal subunit to the ribosome binding sequence (RBS) that contains a Shine-Dalgarno sequence (SD) in close proximity to the translational start codon (Grunberg-Manago et al. 2014). The SD sequence, located in the 5′ untranslated region (5′ UTR) of most bacterial mRNAs, base pairs to the 3′ end of the 16S RNA in the translational initiation complex formed by 30S and SD. Mechanistically, translation initiation is often regulated via control of SD availability and involves regulating access of the 30S ribosome subunit to the RBS by a strong secondary structure around the SD sequence that prevents binding of the 30S subunit. The RNA sequence directly masking the SD sequence in the closed SD structure is frequently involved in regulating the switch from accessible to inaccessible SD and typically involves regulatory RNAs, temperature, proteins, and the small ligands in riboswitches (Henkin 2008; Geissmann et al. 2009; Smith et al. 2010; Storz et al. 2011; Kortmann and Narberhaus 2012; Serganov and Nudler 2013; Brantl and Jahn 2015).
The dinQ gene in the arsR-gor intergenic region of Escherichia coli is translated into a small hydrophobic protein of 27 aa (Fig. 1A,B). The DinQ protein locates to the inner membrane and when overexpressed, modulates nucleoid compaction and inhibits conjugal recombination (Weel-Sneve et al. 2013). Expression of DinQ is regulated at several levels. First, the dinQ gene is transcriptionally repressed by the LexA repressor that upon exposure to DNA damaging agents is cleaved, and as a result, dinQ repression is alleviated and dinQ mRNA levels increase (Fernandez De Henestrosa et al. 2000; Weel-Sneve et al. 2013). Second, the dinQ mRNA is regulated by the antisense agrB RNA (Fig. 1A,C) that binds to a region upstream of the RBS. The ability and necessity of the antisense agrB RNA to block the DNA damage-induced effects from dinQ overexpression is demonstrated by the high DNA damage sensitivity of an agrB mutant (Weel-Sneve et al. 2013). In addition to dinQ and agrB, the locus expresses the agrB homologous agrA RNA which in contrast to agrB, did not inhibit DNA damage-induced toxicity from dinQ. A final level of regulation occurs post-transcriptionally. The primary dinQ mRNA transcript (+1 dinQ mRNA) is inert to translation and is only translated after cleavage by an unknown cellular process into a translationally active +44 dinQ transcript. However, no mechanistic explanation for the cleavage-mediated activation of +1 dinQ has been demonstrated (Weel-Sneve et al. 2013).
FIGURE 1.

(A) A schematic representation of the dinQ, agrA, and agrB genes and transcripts. Drawing is to scale. The positions of the LexA binding site and the flanking gor and arsR genes are shown. (B) dinQ mRNA. Transcriptional initiation site +1 and processing site +44 are indicated (red letters). RNA sequences 1, 2, 3, and 4 are indicated by red, purple, blue, and green letters, respectively. Underlined are the agrB antisense region, the Shine-Dalgarno (SD) sequence, and the dinQ terminator. DinQ start and stop codons (asterisks) are shown in red. DinQ translation sequence is shown above the nucleotide sequence. All numbering of the dinQ sequence and RNA structures are relative to the +1 transcription initiation site. (C) agrB and agrA RNA transcripts. Antisense sequence is underlined (agrB). Rho-independent transcriptional terminator is indicated in bold. Red letters indicate nucleotides in agrA that are different from agrB. (D) Potential base-pairing of four RNA sequences and agrB antisense RNA: (red) sequence 1 (U16–C28); (purple) sequence 2 (G94–G106); (blue) sequence 3 (U167–U178); (green) sequence 4 (A182–G187). The 36 first nucleotides of agrB antisense RNA are shown base-paired with G88–A119 encompassing sequence 2 of dinQ. The +44 processing site, the Shine-Dalgarno (SD) sequence (G184–G187), and the GUG start codon starting at G195 are indicated. (E) +1 dinQ mRNA. Base-pairing of sequences 1 and 2 allow sequences 3 and 4 to form the closed SD hairpin. (F) +44 dinQ mRNA. Elimination of sequence 1 allows formation of the competing duplex 2:3, thus excluding sequences 3 and 4 to form the closed SD hairpin. (G) Binding of agrB to G88–A119 in +44 dinQ sequestrates sequence 2, allowing the closed SD hairpin to form.
The dinQ-agrB locus shares many similarities to the three type I toxin–antitoxin loci shoB-ohsC (Kawano et al. 2005; Fozo et al. 2008), zor-orz (Fozo et al. 2010; Wen et al. 2014), and the LexA regulated tisB-istR1 (Vogel et al. 2004; Darfeuille et al. 2007) in E. coli. The three toxin–antitoxin loci encode small hydrophobic peptides of 26 (ShoB) and 29 (Zor and TisB) amino acids. Typically, toxin mRNA stability or translation is repressed by their antisense RNA, but when overproduced, the peptides are toxic and lead to cell stasis and death (Wen and Fozo 2014). The dinQ, tisB, shoB, and zor genes all have long 5′ UTRs where their respective antisense RNAs affect translation by base-pairing to a target sequence upstream of the RBS (Darfeuille et al. 2007; Fozo 2012; Weel-Sneve et al. 2013; Wen et al. 2014).
To understand the mechanism behind the switch from a translationally inactive +1 dinQ to a translationally active +44 dinQ, we used selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE), RNA folding algorithms, covariance information, and mutations of key structural elements to demonstrate the formation of alternative secondary RNA structures in the 5′ UTR of +1 and +44 RNA. The results support a mechanism where alternative secondary RNA structures are determined by alternative interactions of four sequence elements (within the 5′ UTR), resulting in a closed SD stem–loop or an open SD in +1 and +44 dinQ, respectively. We found that the closed SD stem–loop in +1 dinQ RNA correlates with the lack of DinQ translation and vice versa; the open SD in +44 dinQ RNA correlates with considerable DinQ translation. We also present evidence for an interaction between +44 dinQ and the agrB antisense RNA resulting in structural sequestration of the SD sequence and cleavage of the +44 dinQ-agrB duplex by ribonuclease III (RNase III). In addition, a BLAST search in databases reveals a whole family of dinQ-like genes encoding small single transmembrane peptides that appear to be similarly regulated.
RESULTS
The primary sequence of dinQ allows for mutually exclusive base-pairings in the 5′ UTR of +1 and +44 dinQ
Assisted by folding algorithms (Zuker 2003), dot plot analysis, and manual inspection, four sequences were identified in the 5′ UTR of dinQ with the potential to form mutually exclusive base-pairings resulting in alternative RNA secondary structures of +1 and +44 dinQ. We suggest the following model for dinQ and agrB where the secondary RNA structures of +1 and +44 dinQ are determined by alternative interactions between the four RNA sequences: 1 (U16-C28), 2 (G94-G106), 3 (U167-U178), 4 (A182-G187) and antisense agrB RNA in the case of +44 (Fig. 1B,D). RNA duplex 1:2 in +1 dinQ mRNA is formed by the complementary sequences 1 and 2 while hairpin structure 3:4 containing the sequestrated SD sequence is formed by RNA sequences 3 and 4 (Fig. 1E). Translation initiation of +1 dinQ mRNA is predicted to be inhibited because the sequestrated SD sequence GGAG (G184–G187) in hairpin 3:4 is unavailable for ribosome binding. In +44 dinQ RNA, sequence 1 (U16–C28) is eliminated by an unknown cellular process and can no longer form the duplex with RNA sequence 2 (G94–G106), allowing the complementary sequences 2 and 3 to form duplex 2:3 (Fig. 1F). Duplex 2:3 in +44 dinQ RNA prevents sequences 3 and 4 from forming RNA hairpin structure 3:4, thus leaving the SD sequence open and accessible for ribosome binding and translation initiation (Fig. 1F). According to our model, this is the only situation where translation of DinQ is initiated.
The folding model suggests an inhibitory role for the agrB antisense RNA in translation initiation of +44 dinQ RNA. The situation is outlined in Figure 1G where closing of hairpin 3:4 is mediated by antisense binding of agrB RNA to nucleotides G88–A119 in the dinQ RNA region containing sequence 2 (G94–G106). Another possibility is the analogous situation where agrB mRNA binds to +1 dinQ RNA. However, binding of agrB RNA to the antisense region of +1 dinQ RNA should not affect the formation of hairpin 3:4 and the sequestration of the SD sequence (Fig. 1D).
Destabilization of the SD hairpin is mediated by secondary structure changes in the dinQ 5′ UTR
To test the alternative folding model, we used SHAPE analysis to probe the secondary structures of +1 dinQ and +44 dinQ RNA (Mortimer and Weeks 2009). In vitro transcripts of 331 nt (starting at +1) and 288 nt (starting at +44) were synthesized, and the RNAs were folded and reacted with the SHAPE reagent benzoyl cyanide (BzCN). After primer extension, discrete SHAPE adducts were represented as bands resolved on a sequencing gel. In SHAPE analysis, nucleotides constrained by base-pairing and tertiary interactions show low SHAPE reactivity, whereas single-stranded and unconstrained nucleotides show higher reactivities (Weeks and Mauger 2011). SHAPE reactivity was assessed with different primers throughout the +1 and +44 dinQ transcripts (Supplemental Fig. 1A–E). SHAPE reactivities could not be assessed for nucleotides G1–C14 in +1 and U44–G56 in +44 dinQ due to a strong primer extension signal from the 5′ end, and nucleotides A287–U331 due to lack of primer extension information close to the 3′ end. The observed patterns of SHAPE reactivity are in good agreement with the secondary RNA structure models of +1 dinQ (Fig. 2A) and +44 dinQ 5′ UTR (Fig. 2B) as well as the dinQ open reading frame (ORF) and 3′ UTR (Fig. 2C). High-reactivity nucleotides correspond largely to regions predicted to contain single-stranded sequences, hairpin loops, internal loops, and bulges. Correspondingly, we see little reactivity in regions predicted to form stems or duplexes. One exception is the SHAPE reactivity of nucleotide G100 in the middle of RNA sequence 2 of +1 dinQ (Supplemental Fig. 1D,E). The presence of a SHAPE reactive nucleotide predicted to be buried in the middle of an RNA duplex appears to be contradictory and could mean that G100 for a structural reason is flexible or locked in a SHAPE reactive conformation. The predicted 5′ UTR structure of +1 and +44 dinQ differs in all structural elements except for stem–loop 2 (SL2) (G57–U87) and stem–loop 4 (SL4) (A129–U167) (Fig. 2A,B). Similar SL2 and SL4 structures in +1 and +44 dinQ are supported by identical SHAPE reactivity patterns in these regions (Supplemental Fig. 1C–E). In addition to SL2 and SL4, the 5′ UTR structure of +44 dinQ includes stem S2 composed of G97–A105 and U168–C176, stem–loop 3 (SL3) (U110–A127), and a single-stranded region (A177–A191) containing the SD sequence (Fig. 2B). Stem S2 corresponds to the RNA duplex formed by sequences 2 and 3 in Figure 1F. The 5′ UTR mRNA structure of +1 dinQ includes, in addition to SL2 and SL4, the long stem S1 made up of G1–C28 and G94–C120, stem–loop 1 (SL1) (G32–C43), and the SD stem–loop 5 (SL5) (C173–G187) (Fig. 2A). The upper part of the long stem S1 corresponds to the RNA duplex formed by sequences 1 and 2 in Figure 1E.
FIGURE 2.

Secondary structure models of dinQ RNA. (A) +1 dinQ 5′ UTR. (B) Secondary structure of +44 dinQ 5′ UTR. (C) Secondary structure of the dinQ-ORF and 3′ UTR region in +1 dinQ and +44 dinQ RNA. Different colored bars correspond to SHAPE reactive regions and match Supplemental Figure 1. Nucleotides in red, purple, blue, and green correspond to nucleotide sequences 1, 2, 3, and 4, respectively. Sequence interaction a/a′ supported by sequence conservation is indicated. Nucleotides encircled indicate the agrB antisense region in dinQ. Stem–loops (SL) and stems (S) are numbered, the Shine-Dalgarno (SD) region is shaded green, and the GUG translational start, UGA stop, and U44 cleavage site are shown in red.
The SHAPE reactivity pattern downstream from the SD sequence indicates that +1 and +44 dinQ RNA have identical secondary structures in the 3′ domain (Fig. 2C). The 3′ domain includes four stem–loops, SL6 (G194–C215), SL7 (G216–C246), SL8 (U250–A285), and the terminator stem–loop SL9 (A295–U325). No SHAPE information was obtained for the transcriptional terminator (SL9) and hence the terminator structure is shown as calculated.
To further test the validity of the predicted interactions of RNA sequences 1 (U16–C28), 2 (G94–G106), 3 (U167–U178), and 4 (A182–G187), we made in vitro transcripts of +1 dinQ and +44 dinQ RNA with the destabilizing mutations m2+44 and m3 in RNA sequences 2 and 3 of +44 dinQ, and m1 and m2+1 in RNA sequences 1 and 2 of +1 dinQ (Supplemental Fig. 2A–C). In addition, we combined m2+44 and m3 in +44 dinQ, and m1 and m2+1 in +1 dinQ in order to introduce compensatory mutations that changed the primary sequence but preserved the interaction of the RNA sequences. In agreement with the secondary structure model of +44 dinQ RNA (Fig. 2B), we observed enhanced SHAPE reactivity in RNA sequences m2+44 and m3 and their respective interacting sequences 3 and 4 (Supplemental Figs. 3A,B, lanes 6 and 7 [in each]). In agreement with our SHAPE data and consistent with our model, mutation m2+44 in +44 dinQ disrupts the interaction of sequences 2 and 3, permitting sequence 3 to form a hairpin with sequence 4 that sequestrates the SD sequence (Supplemental Fig. 3A, lane 7). When we combined the compensatory mutations m2+44 and m3 in +44 dinQ, SHAPE reactivity in the 5′ UTR was restored to a pattern indistinguishable from wild-type +44 dinQ RNA, indicating that the suggested interaction of sequences 2 and 3 is valid (Supplemental Fig. 3A,B, lanes 5 and 8 [in each]). A closer examination of our SHAPE data suggests that the G to C mutations in positions 97, 98, 100, 102, and 104 of m2+44 have created an unintentional RNA sequence interaction with G68–C77 in SL2. Such an interaction is in agreement with the lack of SHAPE reactive nucleotides in C102–G94 of m2+44 and G68–C77 of SL2 (Supplemental Fig. 3B, lane 7).
Consistent with the role of RNA sequences 1 and 2 in the +1 dinQ structure, SHAPE reactivity in the mutant regions m1 and m2+1 was enhanced (Supplemental Fig. 3C, lanes 2 and 3). Somewhat surprisingly, the increased SHAPE reactivity is more pronounced in the region containing the four consecutive uracil residues U16–U19, suggesting that this part of the 1:2 duplex is easily affected by the destabilizing mutations m1 and m2+1. Our SHAPE data confirmed that mutation m2+1 in +1 dinQ weakened the RNA duplex 1:2, resulting in RNA sequence 2 and 3 interaction and partial opening of the SD hairpin (Supplemental Fig. 3A, lane 3). When we combined the compensatory mutations m1 and m2+1 in +1 dinQ, the SHAPE reactivity was restored to a pattern very similar to wild-type +1 dinQ RNA, indicating that the suggested sequence interactions are valid (Supplemental Fig. 3A,B,C, lanes 1 and 4 [in each]).
In total, the SHAPE reactivity patterns of wild-type and mutated dinQ RNA support our model of alternative RNA sequence interactions in the secondary RNA structures of +1 dinQ and +44 dinQ. However, the structures presented do not comprise structural elements often found in RNAs, like longer distance interactions, pseudoknots, metastable structures, or other unusual RNA structures. Such structural elements may be present in dinQ, but their prediction would require a more detailed mutational and structural analysis.
Identification of dinQ-like genes
The accuracy of RNA secondary structure predictions can be aided by observing covariance in the internal base-pairing of closely related RNA sequences. With a sufficiently large alignment of a given RNA family, it is possible to directly infer secondary structure from alignment data (Gorodkin et al. 2010). To search for dinQ nucleotide sequence homologs, we performed a BLAST search in nucleotide sequence databases. Many of the hits from E. coli and Shigella species are almost 100% similar to E. coli MG1655 dinQ and must be considered true dinQ genes. A different set of hits are less similar to dinQ but clearly are homologs of dinQ based on a striking similarity to both the dinQ mRNA 5′ UTR sequence and signature amino acids in the DinQ protein sequence (Fig. 3A,B). This second class of dinQ homologs was found in chromosomes and naturally occurring plasmids of the Enterobacteriaceae family, and we collectively name the genes dql (dinQ like). The dql genes were further grouped into dqlA and dqlB based on differences in the amino acid sequence (Fig. 3B). The majority of Dql peptides found in sequence databases are unannotated or annotated with a longer N-terminal region. The dql genes appear to be relatively widespread in Enterobacteriaceae, and some strains have more than one dql gene and also dql genes together with dinQ. The transcriptional and translational functionality of all the dql genes is strongly indicated by the presence of σ70 recognized promoters, Rho-independent transcription terminators, and a plausible ribosome binding site containing an SD sequence correctly positioned relative to the start codon of the Dql peptide (Supplemental Fig. 4).
FIGURE 3.

Conserved sequence and structure elements in the 5′ UTR of dinQ and dql genes. (A) Nucleotide positions are relative to the dinQ mRNA sequence. Conserved −10 promoter elements, Shine-Dalgarno (SD) sequences, and start codons are indicated. Nucleotide regions colored red, purple, blue, and green correspond to nucleotide sequences 1, 2, 3, and 4, respectively. Conservation of stem–loops 2 and 4 (SL2 and SL4) is indicated with a dot-bracket notation. Conserved upper- and lower stems are indicated with a dot-bracket representation and colored nucleotides according to standard IUPAC nucleotide annotation. Sequence interaction a/a′ supported by sequence conservation is indicated. (B) Sequence alignment of DinQ and 14 Dql peptides. Hydrophobic amino acids (AILMV) are blue; polar amino acids (DEKR) are red; aromatic amino acids (FY) are magenta; small amino acids (G) are green; all others (CNPQST) are black.
A multiple sequence alignment of dinQ and 14 dql RNA sequences demonstrates a close structural similarity in their 5′ UTRs. Covarying bases in regions predicted to interact are found in the stems of SL2 and SL4 and the a/a′ interaction in the base of S1 (Fig. 3A). Like dinQ, the majority of dql sequences contain four sequence elements within their 5′ UTR with the potential to form mutually exclusive base-pairing of sequence elements 1:2, 2:3, and 3:4 (Fig. 3A; Supplemental Fig. 5).
The largest sequence variation was found in the first part of the 5′ region of the dql genes corresponding to nucleotides G5–A40 in dinQ. Other dql genes were omitted from the alignment due to an even greater length variation in this region.
In total, the secondary RNA structure inferred from the dinQ and dql nucleotide alignment together with our SHAPE data supports the predicted +1 dinQ and +44 dinQ RNA structures.
In vitro translation of dinQ correlates with SD stem–loop stability
The differences in secondary RNA structure around the SD sequence in +1 and +44 dinQ indicated that the two transcripts have differences in translational activity. However, regulation of DinQ translation has proven difficult to test in vivo. Toxicity from DinQ overexpression in combination with the low abundance of +44 dinQ RNA compared to +1 dinQ RNA made it difficult to compare +1 and +44 dinQ translation. To circumvent the problem, we chose to test the translational capacity of in vitro transcribed +1 dinQ and +44 dinQ RNA in S30 cell-free translation extracts from E. coli. In agreement with our earlier studies of DinQ translation (Weel-Sneve et al. 2013), only +44 dinQ RNA gave rise to distinct protein bands corresponding in size to the DinQ peptide (Fig. 4A, upper panel, right). Initiation of +44 dinQ translation appears to be fast and increased steadily throughout the 4 min incubation time. In contrast, +1 dinQ RNA was not translated under the same conditions and only a faint band was observed after 4 min of incubation (Fig. 4A, upper panel, left).
FIGURE 4.

Translational capacity of dinQ transcripts in the presence of agrB RNA. (A) In vitro translation in S30 extracts of +1 dinQ and +44 dinQ RNA in the absence or presence of agrA or agrB antisense RNA. Amount of RNA and incubation time is indicated. (B) Complex formation between labeled +44 dinQ and unlabeled agrB RNA (upper autoradiograph), and labeled +1 dinQ and unlabeled agrB RNA (lower autoradiograph). The electrophoretic mobility shift assay was performed as described in Materials and Methods using 1.3 pmol of labeled +1 dinQ or +44 dinQ RNA mixed with different concentrations of agrB RNA in a final volume of 15 µL. The ratios of agrB and dinQ RNA are indicated. (C) SHAPE reactivity of the Shine-Dalgarno (SD) region of +1 dinQ and +44 dinQ RNA monitored with or without a fivefold excess of agrB or agrA RNA. The SD sequence G184–G187 is indicated in green. Two red bars correspond to SHAPE reactive regions of the SD region in +1 dinQ and +44 dinQ. (D) In vitro translation in S30 extracts of +44 dinQ RNA mixed with increasing amounts of agrB antisense RNA. Incubation time is 4 min.
The SHAPE data from mutated +1 dinQ and +44 dinQ RNA demonstrated that sequestration of the SD sequence is sensitive to mutations in RNA sequences 1, 2, and 3. We found that the translation efficiency of in vitro transcripts of +1 dinQ carrying mutations m1 and m2+1, and +44 dinQ carrying mutations m2+44 and m3 closely mirrored the SHAPE-predicted stability of the SD hairpin (Supplemental Fig. 6A,B). The somewhat more open SD hairpin in +1 dinQ carrying mutation m1 correlates with increased translation efficiency of m1 transcripts in S30 extracts (Supplemental Fig. 6A). Likewise, the closed SD hairpin in the +44 dinQ mutant m2+44 correlates with the lack of translation in S30 extracts (Supplemental Fig. 6B). Taken together, the in vitro translation data supports our model where an open versus closed structure around the SD sequence determines the translational state of +1 and +44 dinQ RNA.
Binding of agrB RNA stabilizes the SD stem–loop in +44 dinQ and inhibits translation initiation
Binding of agrB RNA to the antisense region of +44 dinQ was predicted to close the otherwise open SD structure (Fig. 1G). To test the prediction, we monitored the pattern of SHAPE reactivity around the SD region of +1 dinQ and +44 dinQ RNA, either alone or mixed with a fivefold excess of agrA or agrB RNA. Prior to the SHAPE experiments, an in vitro mobility shift experiment confirmed that agrB RNA binds effectively to +44 dinQ RNA (Fig. 4B). The SHAPE results clearly indicated that agrB RNA binding closes the open SD region in +44 dinQ, and the SHAPE reactivity pattern in the SD region was similar to the pattern in +1 dinQ RNA (Fig. 4C). agrA has a few mismatches in the dinQ antisense region compared to agrB. In accordance with this sequence difference, agrA RNA did not affect the SHAPE reactivity pattern in the SD region of +44 dinQ. The SHAPE reactivity pattern around the SD region in +1 dinQ RNA was unaffected by a fivefold excess of agrA or agrB RNA (Fig. 4C).
To test if agrA and agrB RNA affects dinQ translation, in vitro transcripts of +1 and +44 dinQ in combination with agrA or agrB transcripts were translated in S30 extracts. In agreement with the SHAPE reactivity pattern, +44 dinQ translation was inhibited by a fivefold excess of agrB RNA but was not inhibited by agrA RNA (Fig. 4A). Furthermore, we tested the translational capacity of +44 dinQ with increasing agrB: +44 dinQ RNA ratios. A threefold excess of agrB RNA was required to achieve full translational repression of +44 dinQ (Fig. 4D). The mobility shift experiment indicated that agrB RNA binds completely to the +44 dinQ RNA at 1:1 ratio (Fig. 4B). The reason why a 1:1 ratio was insufficient to repress translation is unclear, but may reflect instability or dissociation of agrB RNA in the S30 extracts and that a threefold excess is required to fully prevent the ribosome from binding. These results suggest that the structural transition from an open to a closed SD structure, followed by inhibition of +44 dinQ translation, is a consequence of agrB RNA binding to the 5′ UTR of +44 dinQ.
agrB RNA preferentially mediates RNase III processing of +44 dinQ RNA
Many antisense RNAs mediate cleavage of target mRNAs by generating a double-stranded RNA (dsRNA) site for RNase III (Thomason and Storz 2010). Perfectly complementary dsRNA substrates are cleaved by RNase III to yield a 2-nucleotide (nt) 3′ overhang (Court et al. 2013). In addition, RNase III has the ability to degrade long dsRNA duplexes to ∼11–15 bp products (Nicholson 2014). The fact that agrB RNA binds to and affects folding and translation of +44 dinQ (Fig. 4) demonstrates that agrB and +44 dinQ forms an RNA duplex which in turn could generate a dsRNA substrate for RNase III. To test this hypothesis, we subjected 3′ labeled +1 dinQ or +44 dinQ RNA, alone or mixed with agrB RNA, to in vitro RNase III cleavage (Fig. 5). The +44 dinQ/agrB RNA duplex was efficiently cleaved to completion within 4 min, whereas a small but stable fraction of +1 dinQ RNA was processed under the same conditions (Fig. 5A,B). The agrB RNA-dependent RNase III processing of +1 dinQ is in agreement with our finding that a fraction of +1 dinQ RNA forms a complex with agrB RNA (Fig. 4B). Vice versa, agrB RNA was rapidly processed in the presence of +44 dinQ RNA but more slowly processed in the presence of +1 dinQ RNA (Fig. 5C,D), supporting that the +44 dinQ/agrB duplex is preferentially cleaved by RNase III. RNase III processing of free +1 dinQ, +44 dinQ, or agrB RNA could not be detected, even in reaction mixtures containing up to 0.1 U RNase III.
FIGURE 5.
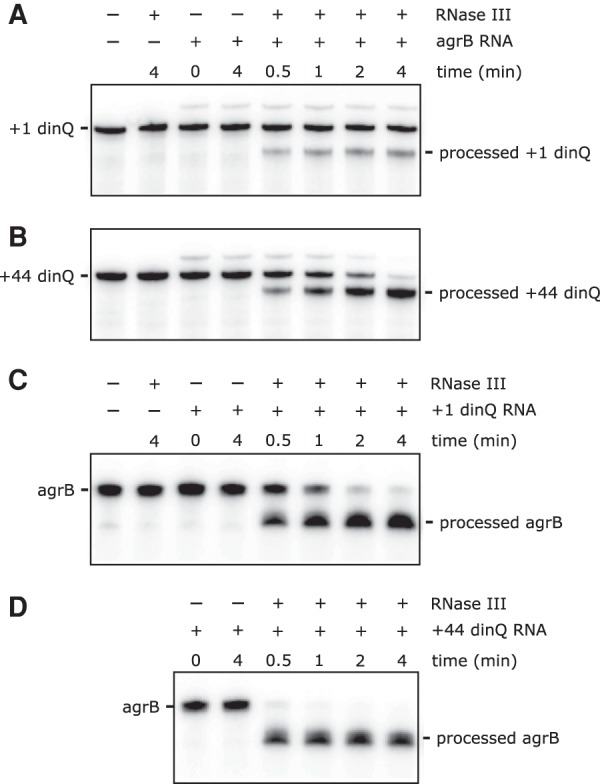
RNase III-mediated cleavage of dinQ and agrB RNA. (A,B) 4.4 pmol of 3′ end labeled +1 dinQ or +44 dinQ RNA, alone or mixed with a twofold excess of antisense agrB RNA in a final volume of 17.5 µL, was treated with 0.001 U of RNase III for different times as indicated. (C,D) 6.6 pmol of 3′ end labeled agrB RNA, alone or mixed with an equimolar amount of +1 dinQ or +44 dinQ RNA in a final volume of 26.3 µL, was treated with 0.005 U of RNase III for different times as indicated.
Primer extension analysis of RNase III processed fragments of +1 dinQ, +44 dinQ, and agrB indicated several 5′ ends in the duplex region (Supplemental Fig. 7). Two initial cleavage events in dinQ were mapped 5′ of G104 and G106 with secondary processing 5′ of C116 and A118 (Supplemental Fig. 7A,C). Primer extension of processed agrB RNA generated multiple extension stops, and their exact positioning was difficult to interpret due to their close proximity to the extension primer (Supplemental Fig. 7B,C). However, the pattern of primer extension products observed during the time course of agrB RNA processing suggests primary cleavage events 5′ of U15 and C16 with secondary processing 5′ of A24 and G25 (Supplemental Fig. 7C). The origin of multiple RNase III cleavage products in +1 and +44 dinQ is not clear, but suggests that RNase III can process the 34 bp dinQ/agrB duplex region in more than one position in vitro, and may reflect the ability of RNase III to process longer RNA duplexes into 11–15 bp fragments (Nicholson 2014).
DISCUSSION
In this work, we present evidence for a structural RNA switch triggered by 5′ processing of the full-length dinQ mRNA. The switch is mediated by structural rearrangements involving mutually exclusive base-pairing of four RNA sequences in the 5′ UTR of dinQ that effectively converts a translationally inactive +1 dinQ to a translationally active +44 dinQ RNA. In agreement with SHAPE data, mutation analysis and in vitro translation experiments, a secondary structure model is presented illustrating how processing of +1 dinQ at nucleotide 44 followed by reshuffling of sequence elements 2, 3, and 4 switches the translationally inert +1 dinQ to a translationally active +44 dinQ (Fig. 6A,B). Our SHAPE and in vitro translation data also demonstrate that the translationally active +44 dinQ RNA upon binding of antisense agrB undergoes a conformational change that sequestrates the SD sequence in a hairpin resulting in translational repression of the DinQ peptide (Fig. 6C). Additionally, dinQ-agrB duplexes that preferentially form with +44 dinQ RNA are rapidly cleaved in vitro by RNase III.
FIGURE 6.

A model of dinQ translation regulation based on the data presented. (A) Secondary structure of the 5′ UTR of +1 dinQ. The identical 3′ domain structure of +1 dinQ and +44 dinQ RNA is indicated with a labeled box. The ribosome is drawn as an orange sphere. The SL2, SL4, SD, and SD-SL are labeled. SL = Stem–loop, SD = Shine-Dalgarno. (B) Secondary structure of the 5′ UTR of +44 dinQ RNA. (C) The translationally inactive state of +44 dinQ RNA when hybridized to agrB RNA. The site of RNase III cleavage of dinQ is indicated with a red triangle.
Binding of bacterial regulatory RNAs to their complementary sequences in the 5′ UTR of their target mRNA often represses translation initiation by directly masking the SD sequence and inhibiting ribosome binding (Frohlich and Vogel 2009; Brantl and Jahn 2015). In contrast, translational repression in vitro of +44 dinQ is mediated by antisense agrB binding to a target site relatively far from the SD sequence. In effect, agrB RNA binding restores the RNA architecture found in +1 dinQ by effectively replacing the function RNA sequence 1 has in sequestrating RNA sequence 2 (Fig. 6C). A second consequence of agrB binding is RNase III-mediated processing of +44 dinQ in the duplex region. Previously, we could not detect processing of dinQ in the antisense region, most likely due to formation of a short-lived cleavage intermediate (Weel-Sneve et al. 2013). We could, however, detect accumulation of +44 dinQ RNA in an agrB deletion mutant which is expected to occur if RNase III-mediated degradation of cellular +44 dinQ ceases due to lack of antisense agrB.
There are similarities in the way other antisense RNAs repress translation of their target mRNAs. The type I toxin–antitoxin locus tisB/istR-1 in E. coli is clearly similar to dinQ/agrB. Both tisB and dinQ are repressed by the LexA repressor, and their translationally inactive full-length mRNAs are activated by a cleavage event at nucleotides 42 and 44, respectively (Darfeuille et al. 2007; Weel-Sneve et al. 2013). In addition, their respective antisense RNAs mediate both translational repression and RNase III processing of their target mRNA. Interestingly, translation of TisB is regulated by a ribosome standby mechanism that appears to be different from the mechanism we have suggested for dinQ (Darfeuille et al. 2007). TisB translation is normally inhibited by an inaccessible secondary structure around the SD site that blocks ribosome binding. Access to the tisB SD sequence is then promoted by a ribosome that slides from an upstream standby site into a transiently open SD secondary structure. Binding of a ribosome to the standby site also involves processing at +42 and structural rearrangements of the 5′ UTR of tisB mRNA and in addition is inhibited by the antisense RNA istR-1 that competes with ribosomes for the standby site (Darfeuille et al. 2007). Contrary to the situation in dinQ, in vitro experiments suggested that cleavage at +42 in tisB 5′ UTR is not enough to mediate opening of the secondary SD structure necessary for translation initiation to occur (Darfeuille et al. 2007). Due to differences in the composition of the folding buffer used, we reanalyzed the 5′ UTR structure of tisB with SHAPE under the same conditions used in this work and confirmed that the secondary SD structure is closed in both +1 and +42 tisB RNA (data not shown). These results suggest, therefore, that there are differences in how SD sequence accessibility is regulated in tisB and dinQ. Nevertheless, we cannot exclude the possibility that a standby mechanism also operates in dinQ.
Another example is the type I toxin–antitoxin locus bsrG/SR4 in Bacillus subtilis. Like +44 dinQ/agrB, the RNA duplex formed by bsrG/SR4 is cleaved by RNase III, but translation of the BsrG toxin is also repressed by the SR4 antitoxin RNA in the absence of RNase III (Jahn and Brantl 2013; Brantl and Jahn 2015). However, a difference between dinQ/agrB and bsrG/SR4 comes from the way the duplexes are formed. While dinQ/agrB interacts at their 5′ end, the bsrG/SR4 duplex is formed at their 3′ end.
A regulatory mechanism similar to dinQ could operate in shoB. The shoB gene in Escherichia coli encodes the short hydrophobic peptide ShoB that is highly lethal upon overexpression and contains a long and structured 5′ UTR as seen in dinQ (Fozo et al. 2008). Multiple 5′ ends were mapped for the shoB transcript. The longest fragment fused to a lacZ reporter gene gave no reporter activity, possibly due to inhibitory structures formed by the long 5′ UTR. A second 5′ end was mapped 41 nt downstream from the longest fragment, and this fragment fused to the lacZ reporter gave measurable reporter activity (Fozo et al. 2008). We note that the shoB gene contains four sequence elements in the 5′ UTR allowing for a folding very similar to dinQ (Supplemental Fig. 5). The second 5′ end of shoB maps between sequence 1 and sequence 2 of shoB and could result in translation initiation by reshuffling sequences 2, 3, and 4 analogous to dinQ.
The large and stable S1 structure formed by G1–C28 and G94–C120 in +1 dinQ appears to be important for both translation repression and agrB interaction (Figs. 2A, 6). The S1 structure encompasses the 13 bp RNA duplex 1:2 and also the 34 nt agrB antisense RNA recognition sequence. The 1:2 duplex is calculated to be the energetically most stable duplex in dinQ with a ΔG of −23.9 kcal/mol at 37°C (Zuker 2003) and the role of duplex 1:2 in translation repression was demonstrated by increased translation from +1 dinQ RNA containing a mutation destabalizing RNA sequence 1 (Supplemental Fig. 6).
Inefficient duplex formation between agrB and +1 dinQ RNA was demonstrated by mobility shift and in vitro RNase III processing experiments (Figs. 4B, 5). Given the length difference between the agrB/dinQ and 1:2 duplexes (34 bp versus 13 bp), it is likely that structural factors in addition to duplex 1:2 contribute to inefficient agrB binding. In this respect, the phylogenetically conserved a/a′ duplex and additional base-pairing in the region between a/a′ and duplex 1:2 likely add stability to S1 (Fig. 2A). Formation of stable RNA–RNA complexes in natural antisense-target RNA pairs involves an initial interaction of accessible single-stranded seed regions, which subsequently is rapidly converted to a stable antisense-target RNA hybrid (for review, see Updegrove et al. 2015). The agrB antisense regions in +1 and +44 dinQ have different secondary structures. Thus, it is likely that accessible regions in +44 dinQ involved in initial agrB RNA interaction are inaccessible in +1 dinQ. Future work should focus on narrowing down functional segments in dinQ required for agrB RNA interaction. A comparable inhibitory strategy exists in the well-characterized type I TA system hok/sok. Antisense RNA binding and translation initiation of full-length hok mRNA is inhibited by a fold-back-inhibition element (fbi) (Gerdes et al. 1997). Contrary to the S1 structure in dinQ mRNA, the fbi element in hok is formed by long-range 5′–3′ pairing. Exonucleolytic processing of 3′ hok disrupts the fbi element and activates translation and antisense RNA binding (Gerdes et al. 1997). Consequently, we propose that the stable S1 structure in +1 dinQ is a double locking structure that protects the full-length dinQ transcript from agrB RNA binding and RNase III cleavage and also prevents opening of the SD stem–loop that would lead to translation initiation from full-length dinQ RNA. In this way, the S1 double-lock has a key function in ensuring both stability and translation repression of the +1 dinQ mRNA.
Small polypeptides (<50 amino acids) have been overlooked due to the challenges in annotation, biochemical detection, and functional characterization (Storz et al. 2014). However, comparative sequence analysis has predicted hundreds of small peptides and a subset of these peptides has been characterized (Hobbs et al. 2011). Many of the small peptides, including DinQ, are localized in the inner membrane and mechanistically they impact diverse processes (Alix and Blanc-Potard 2009; Weel-Sneve et al. 2013). Typically, small membrane-associated peptides are tightly regulated to avoid overexpression leading to lethal toxicity characterized by phenotypes such as depolarization and reduced ATP concentration. In sum, our discovery reveals new insight into the transcriptional and post-transcriptional regulation of a new class of dql genes encoding small hydrophobic single transmembrane peptides.
MATERIALS AND METHODS
In vitro transcription
Of note, +1 dinQ, +44 dinQ, agrA, and agrB RNA for SHAPE and in vitro translation experiments were transcribed with MEGAscript T7 Transcription Kit (Ambion/Life Technologies) from PCR fragments containing a T7 promoter followed by the sequence of the RNA (oligonucleotides listed in Supplemental Table 1). PCR fragments containing site-directed mutations in +1 and +44 dinQ were made by splicing PCR products with overlap extension (SOEing PCR) (Higuchi et al. 1988). All primers used are listed in Supplemental Table 1. Reaction assembly, purification of RNA, and quality control were performed according to the MEGAscript T7 Transcription Kit protocol. The 5′ start of +1 dinQ and +44 dinQ RNA sequence was changed to GG and the 5′ start of agrA and agrB RNA was changed to G in order to improve T7 polymerase initiation, otherwise the RNA 5′ start and 3′ stop were as reported earlier (Weel-Sneve et al. 2013).
Chemical probing of RNA
Benzoyl cyanide (BzCN) RNA 2′-O-adduct formation was performed as previously described (Mortimer and Weeks 2009). Twelve picomoles of RNA was heated to 95°C for 2 min and placed on ice. The RNA was folded at 37°C for 10 min in folding buffer (final concentration: 40 mM MOPS, pH 8.0, 80 mM potassium acetate, pH 8.0, 20 mM MgCl2). Freshly prepared BzCN (BzCN dissolved in DMSO) was added to 6 pmol of folded RNA to a final BzCN concentration of 18 mM followed by incubation for 5 sec before the RNA was recovered by ethanol precipitation. The RNA was redissolved in 0.5× TE buffer (pH 7.5). Control RNA without BzCN was prepared with DMSO.
3′ end labeling and RNase III cleavage
In vitro transcribed +1 dinQ, +44 dinQ, and agrB RNA was 3′ end labeled with T4 RNA Ligase (Ambion) and equimolar amounts of [32P]pCp (PerkinElmer, NEG019A) according to the protocol. Unincorporated [32P]pCp was removed by applying the mixture to an RNase-free Sephadex G25 column. Folding of RNA prior to RNase III cleavage was performed as described for chemical probing of RNA; the RNA was heated to 95°C for 1 min and placed on ice before folding at 37°C for 10 min in folding buffer followed by addition of RNase III (Ambion). The reaction was stopped by adding an equal volume of Gel Loading Buffer II (Ambion) before being analyzed on a denaturing 5% polyacrylamide/8 M urea gel, and visualized on Typhoon 9410 (Amersham). An aliquot of the RNase III reaction was left for primer extension analysis.
Primer extension
Primer extension was performed as previously described (Mortimer and Weeks 2009). 0.4 pmol of a 5′-radiolabeled primer (Supplemental Table 1) was annealed to 1.5 pmol of RNA by heating at 65°C for 6 min and 35°C for 15 min. cDNA was synthesized using Superscript III (Invitrogen) and incubated at 52°C for 5 min. RNA templates were removed by adding NaOH to a final concentration of 200 mM at 95°C for 5 min and neutralized with an equal amount of HCl.
Sequence markers were generated with Sequenase DNA Sequencing Kit v.2.0 (USB Corporation). For each sequencing reaction, 0.8 pmol 5′-radiolabeled primer and 80 pmol single-stranded DNA templates were used. Single-stranded sequencing templates were made by asymmetric PCR of the T7 promoter containing in vitro transcription templates.
Primer extension products alongside with sequence markers were analyzed on an 8% polyacrylamide/7 M urea sequencing gel and visualized on Typhoon 9410 (Amersham).
Band intensities of the primer extension products represent the level of ribose 2′-hydroxyl acylation. Reactive ribose 2′-hydroxyl positions are associated with flexible nucleotides more likely to be unpaired, whereas unreactive and low reactive 2′-hydroxyl positions are more likely to be paired or otherwise constrained (Weeks and Mauger 2011).
Primer extension stops 1 nt prior to the position of a particular ribose 2′-hydroxyl adduct and is accounted for during interpretation of SHAPE reactivity.
In vitro translation with S30 extracts
In vitro translation of folded +1 dinQ and +44 dinQ RNAs was performed with E. coli T7 S30 Extract System for Linear Templates according to protocol (Promega, catalog # L1030) with [14C]-leucine as radiolabeled amino acid. Briefly, 10 pmol of +1 or +44 dinQ RNA either alone or in combination with 10–50 pmol antisense agrA or agrB RNA was incubated with S30 extracts components and [14C]-Leucine in a final volume of 50 µL. The translation products were analyzed by SDS–PAGE and visualized on Typhoon 9410.
Electrophoretic mobility shift assay
In vitro transcribed +1 dinQ, +44 dinQ, and agrB RNA were labeled and purified as described for 3′ end labeling and RNase III cleavage. Folding of RNA prior to electrophoretic mobility shift was performed as described for chemical probing of RNA; the RNA was heated to 95°C for 1 min and placed on ice before folding at 37°C for 10 min in 1× folding buffer. Different concentrations of unlabeled agrB RNA were mixed with 1.3 pmol of labeled +1 dinQ or +44 dinQ RNA in a final volume of 15 µL of 1× folding buffer and incubated for 10 min at 37°C. Samples were mixed with one volume loading buffer (1× folding buffer, 50% glycerol) and analyzed at 4°C on a 4% native polyacrylamide gel with 1× TBE and visualized on Typhoon 9410.
RNA secondary structure modeling and multiple sequence alignment
Secondary structure figures were prepared with VARNA (Darty et al. 2009) and were further refined and annotated with the vector drawing program Inkscape (www.inkscape.org). Multiple sequence alignment was made with MEGA version 6 (Tamura et al. 2013).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the Research Council of Norway (BIOTEK 2021).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.058461.116.
REFERENCES
- Alix E, Blanc-Potard AB. 2009. Hydrophobic peptides: novel regulators within bacterial membrane. Mol Microbiol 72: 5–11. [DOI] [PubMed] [Google Scholar]
- Brantl S, Jahn N. 2015. sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol Rev 39: 413–427. [DOI] [PubMed] [Google Scholar]
- Court DL, Gan J, Liang YH, Shaw GX, Tropea JE, Costantino N, Waugh DS, Ji X. 2013. RNase III: genetics and function; structure and mechanism. Annu Rev Genet 47: 405–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darfeuille F, Unoson C, Vogel J, Wagner EG. 2007. An antisense RNA inhibits translation by competing with standby ribosomes. Mol Cell 26: 381–392. [DOI] [PubMed] [Google Scholar]
- Darty K, Denise A, Ponty Y. 2009. VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics 25: 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez De Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. 2000. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol 35: 1560–1572. [DOI] [PubMed] [Google Scholar]
- Fozo EM. 2012. New type I toxin-antitoxin families from “wild” and laboratory strains of E. coli: Ibs-Sib, ShoB-OhsC and Zor-Orz. RNA Biol 9: 1504–1512. [DOI] [PubMed] [Google Scholar]
- Fozo EM, Kawano M, Fontaine F, Kaya Y, Mendieta KS, Jones KL, Ocampo A, Rudd KE, Storz G. 2008. Repression of small toxic protein synthesis by the Sib and OhsC small RNAs. Mol Microbiol 70: 1076–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozo EM, Makarova KS, Shabalina SA, Yutin N, Koonin EV, Storz G. 2010. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res 38: 3743–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich KS, Vogel J. 2009. Activation of gene expression by small RNA. Curr Opin Microbiol 12: 674–682. [DOI] [PubMed] [Google Scholar]
- Geissmann T, Marzi S, Romby P. 2009. The role of mRNA structure in translational control in bacteria. RNA Biol 6: 153–160. [DOI] [PubMed] [Google Scholar]
- Gerdes K, Gultyaev AP, Franch T, Pedersen K, Mikkelsen ND. 1997. Antisense RNA-regulated programmed cell death. Annu Rev Genet 31: 1–31. [DOI] [PubMed] [Google Scholar]
- Gorodkin J, Hofacker IL, Torarinsson E, Yao Z, Havgaard JH, Ruzzo WL. 2010. De novo prediction of structured RNAs from genomic sequences. Trends Biotechnol 28: 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunberg-Manago M, Studer SM, Joseph S. 2014. Protein synthesis initiation in bacteria. In eLS. 10.1002/9780470015902.a0000542.pub3. [DOI] [Google Scholar]
- Henkin TM. 2008. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev 22: 3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. 1988. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res 16: 7351–7367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs EC, Fontaine F, Yin X, Storz G. 2011. An expanding universe of small proteins. Curr Opin Microbiol 14: 167–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn N, Brantl S. 2013. One antitoxin-two functions: SR4 controls toxin mRNA decay and translation. Nucleic Acids Res 41: 9870–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M, Reynolds AA, Miranda-Rios J, Storz G. 2005. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res 33: 1040–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortmann J, Narberhaus F. 2012. Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 10: 255–265. [DOI] [PubMed] [Google Scholar]
- Mortimer SA, Weeks KM. 2009. Time-resolved RNA SHAPE chemistry: quantitative RNA structure analysis in one-second snapshots and at single-nucleotide resolution. Nat Protoc 4: 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AW. 2014. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscip Rev RNA 5: 31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A, Nudler E. 2013. A decade of riboswitches. Cell 152: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Fuchs RT, Grundy FJ, Henkin TM. 2010. Riboswitch RNAs: regulation of gene expression by direct monitoring of a physiological signal. RNA Biol 7: 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz G, Wolf YI, Ramamurthi KS. 2014. Small proteins can no longer be ignored. Annu Rev Biochem 83: 753–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomason MK, Storz G. 2010. Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet 44: 167–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Updegrove TB, Shabalina SA, Storz G. 2015. How do base-pairing small RNAs evolve? FEMS Microbiol Rev 39: 379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel J, Argaman L, Wagner EG, Altuvia S. 2004. The small RNA IstR inhibits synthesis of an SOS-induced toxic peptide. Curr Biol 14: 2271–2276. [DOI] [PubMed] [Google Scholar]
- Weeks KM, Mauger DM. 2011. Exploring RNA structural codes with SHAPE chemistry. Acc Chem Res 44: 1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weel-Sneve R, Kristiansen KI, Odsbu I, Dalhus B, Booth J, Rognes T, Skarstad K, Bjørås M. 2013. Single transmembrane peptide DinQ modulates membrane-dependent activities. PLoS Genet 9: e1003260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Fozo EM. 2014. sRNA antitoxins: more than one way to repress a toxin. Toxins (Basel) 6: 2310–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Won D, Fozo EM. 2014. The ZorO-OrzO type I toxin–antitoxin locus: repression by the OrzO antitoxin. Nucleic Acids Res 42: 1930–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31: 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.