

Abstract
Eukaryotic transfer RNAs contain on average 14 modifications. Investigations of their biological functions require the determination of the modification sites and the dynamic variations of the modification fraction. Base methylation represents a major class of tRNA modification. Although many approaches have been used to identify tRNA base methylations, including sequencing, they are generally qualitative and do not report the information on the modification fraction. Dynamic mRNA modifications have been shown to play important biological roles; yet, the extent of tRNA modification fractions has not been reported systemically. Here we take advantage of a recently developed high-throughput sequencing method (DM-tRNA-seq) to identify and quantify tRNA base methylations located at the Watson-Crick face in HEK293T cells at single base resolution. We apply information derived from both base mutations and positional stops from sequencing using a combination of demethylase treatment and cDNA synthesis by a thermophilic reverse transcriptase to compile a quantitative “Modification Index” (MI) for six base methylations in human tRNA and rRNA. MI combines the metrics for mutational and stop components from alignment of sequencing data without demethylase treatment, and the modifications are validated in the sequencing data upon demethylase treatment. We identify many new methylation sites in both human nuclear and mitochondrial-encoded tRNAs not present in the RNA modification databases. The potentially quantitative nature of the MI values obtained from sequencing is validated by primer extension of several tRNAs. Our approach should be widely applicable to identify tRNA methylation sites, analyze comparative fractional modifications, and evaluate the modification dynamics between different samples.
Keywords: high-throughput sequencing, methylation, modification, quantification, tRNA
INTRODUCTION
Over 100 types of post-transcriptional RNA modifications have been identified in biology (Grosjean 2005). RNA modifications are a source of cellular and biological tuning of RNA function (Phizicky and Hopper 2010; Wei et al. 2011; Fu et al. 2014; Li and Mason 2014). The most extensively modified cellular RNAs include rRNA and tRNA, each with multifaceted functions. rRNA modifications affect ribosome maturation and numerous aspects of protein synthesis (Decatur and Fournier 2002; Liang et al. 2009; Higa-Nakamine et al. 2012). Mammalian tRNAs are the most highly modified RNA molecule in the cell, containing on average 14 modified nucleotides per molecule, or one modification present for approximately every five residues (Phizicky and Hopper 2010; Cantara et al. 2011; Machnicka et al. 2013; Zheng et al. 2015). tRNA modifications are known to affect all aspects of tRNA biology including decoding and charging efficiency and fidelity, in vivo stability, and intracellular localization (Bjork et al. 1989; Gerber and Keller 1999; Ohira and Suzuki 2011; Whipple et al. 2011; Zaborske et al. 2014). The human tRNAome consists of ∼500 tRNA genes distributed among 49 isoacceptors, i.e., tRNAs with different anticodon sequences (Chan and Lowe 2009). Furthermore, each isoacceptor family contains many distinct sequences in the tRNA body, so that ∼300 tRNA sequences are encoded in a human genome (Goodenbour and Pan 2006; Chan and Lowe 2009). These tRNAs may have different modification patterns depending on sequence, anticodon stem–loop context, and other factors. Although it is commonly assumed that many modification sites in tRNA are fully modified, previous literature provides scattered evidence that certain modification sites in specific tRNAs can be fractionally modified (Kuchino et al. 1981; Chan et al. 1982; Saikia et al. 2010; Vinayak and Pathak 2010; Hauenschild et al. 2015), suggesting that dynamic differences in tRNA modification may be a potentially useful parameter for biological regulation.
Extensive studies have been performed to identify tRNA modifications in a site-specific manner (e.g., Chan et al. 2010; Suzuki and Suzuki 2014). The most common method was to first separate a tRNA of interest from total cellular RNA, followed by digestion to oligonucleotides and the determination of modification site by either thin-layer chromatography (TLC) or liquid chromatography and mass spectrometry (LC/MS). These methods have the ability to access all modification types and have been the standards in establishing the full modification patterns in individual tRNAs. Major drawbacks of these methods are the difficulty and the very large amount of material needed to isolate an individual tRNA from cellular RNA. Another method of identifying tRNA modification sites relies on reverse transcriptase (RT) stops and/or mutations that occur at several specific modification types at the Watson-Crick face of the nucleobase (Motorin et al. 2007; Ryvkin et al. 2013). This “modification-detection-by-synthesis” method can be applied transcriptome-wide for tRNA. However, previous tRNA sequencing methods were inefficient and not quantitative due to the low quality of sequencing reads derived from poorly characterized RT stop and mutation efficiencies at individual modification types and sites. It was also shown that RT stop and mutation are strongly context dependent for a modification type such as N1-methyladenosine (m1A) (Hauenschild et al. 2015). In TLC, LC/MS, and previous sequencing studies, it has not been feasible to establish quantitative information on tRNA modification fractions at individual sites.
Recent studies on N6-methyladenosine (m6A) and N1-methyladenosine (m1A) in mRNAs indicate that dynamic RNA modifications play important biological roles (Dominissini et al. 2012, 2016; Meyer et al. 2012; Li et al. 2016). Dynamic mRNA modification has a cell-type and cell-state-dependent pattern that includes the location of the modification sites and the modification fractions at each site. tRNA modifications are generally present in the same locations derived from the specificity of the modification enzymes and tRNA sequence/structure. Therefore, dynamic tRNA modifications would most likely be derived from the variations in the modification fractions at each site. Indeed, a previous study using low-resolution microarrays on m1A58 modification shows that m1A58 sites in some tRNAs are hypomodified (Saikia et al. 2010). Despite previous efforts, the transcriptome-wide method has been lacking to systematically quantify tRNA modifications at single-base resolution.
Recently, our group and Lowe/Phizicky laboratories published Illumina sequencing methods for tRNA: DM-tRNA-seq (demethylase tRNA sequencing) and ARM-seq (AlkB-facilitated RNA methylation sequencing) (Cozen et al. 2015; Zheng et al. 2015). The principle of both methods is to use the E. coli AlkB demethylase and its mutant as central components to remove m1A, N3-methyl-cytosine (m3C), and N1-methyl-guanosine (m1G) modifications at the Watson-Crick face in tRNA prior to cDNA synthesis. While the initial results mainly emphasized the increased frequency in the full-length tRNA or tRNA fragment reads due to demethylation, neither publication provided a detailed analysis on the tRNA modification landscape, especially the ability of using tRNA-seq to quantify modification fractions. In our published DM-tRNA-seq results (Zheng et al. 2015), we showed that the application of a thermophilic RT (TGIRT) enabled a large number of reads derived from readthroughs of these modifications. Furthermore, TGIRT leaves a very strong mutation and stop signature for different modification types and sites. However, a thorough analysis on the applicability of the DM-tRNA-seq method on tRNA modifications was not carried out.
Here, we provide a comprehensive, detailed study on applying DM-tRNA-seq to identify specific base methylations as well as to generate quantitative information of modification fractions at these sites in the human tRNA transcriptome. We were able to thoroughly analyze six base methylations in tRNA and rRNA [m1A, m3C, m1G, N2,N2-dimethyl-guanosine (m22G), 1-methylinosine (m1I), and N3-methyl-uridine (m3U), Fig. 1]. Using known positions of modification in tRNA, we can infer identification of five other modifications [inosine (I), dihydrouridine (D), methyl-2-thio-N6-threonylcarbamoyl-adenosine (ms2t6A), methyl-2-thio-N6-isopentenyl-adenosine (ms2i6A), 1-methyl-3-(3-amino-3-carboxypropyl)pseudouridine (m1acp3Ψ)]. We provide a complete map of the human tRNA transcriptome on four methylations (m1A, m3C, m1G, m22G) in both nuclear and mitochondrial-encoded tRNAs in HEK293T cells, many of which were unknown or unvalidated previously. We apply a quantitative metric, the “Modification Index” (MI), to describe each modification site that includes frequencies of mutations and stops to further characterize the modification landscape in human tRNAs. MI values enable access of quantitative or semiquantitative information regarding the modification levels at individual tRNA positions. We validate the quantitative nature of the MI analysis for several tRNAs by primer extension. Our results establish DM-tRNA-seq as a high-throughput method to detect and quantify or semiquantify numerous tRNA modifications and its applicability for the studies of dynamic tRNA modifications.
FIGURE 1.
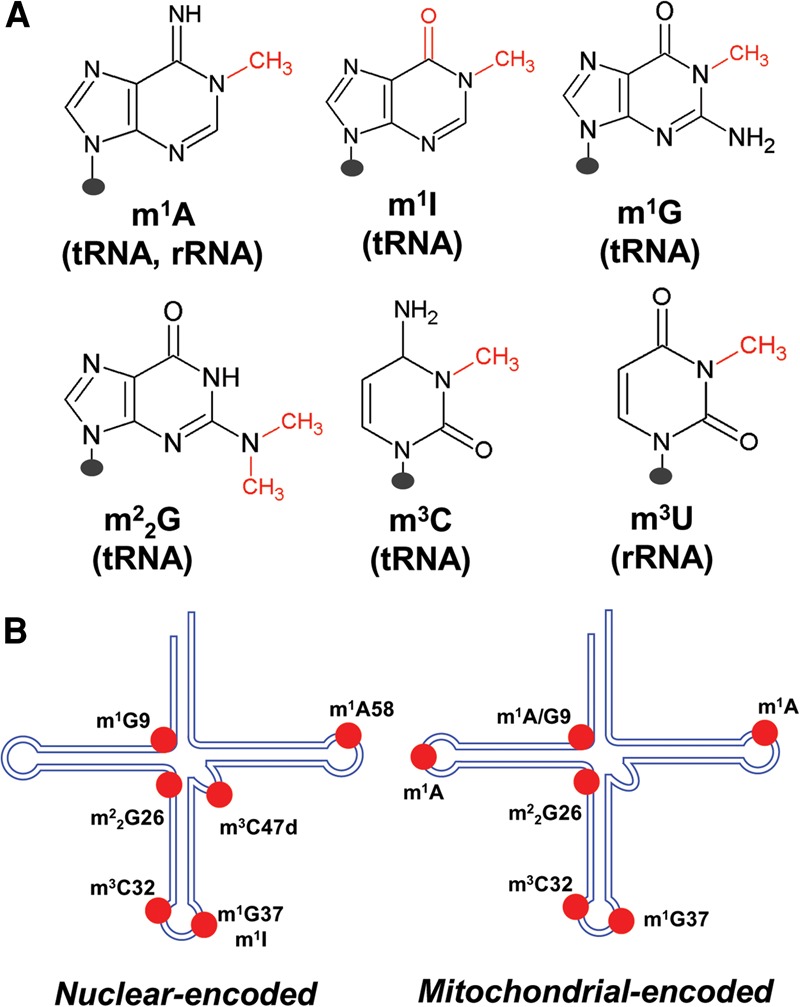
Methylations in human tRNA and rRNA investigated in this study. (A) Chemical structure of the six modifications where the methyl group is shown in red. The presence of each modification in tRNA and/or rRNA is listed beneath each base. (B) Schematic tRNA cloverleaf structure where the methylations for nuclear-encoded (left) and mitochondrial-encoded (right) tRNAs investigated in this work are located.
RESULTS
Identification of tRNA methylation sites
Nextgen tRNA sequencing had been difficult to be performed efficiently and quantitatively due to the presence of a large number of base modifications at the Watson-Crick face and the stable tRNA structure. Our laboratory and the Lowe/Phizicky laboratories independently developed tRNA-seq methods that first removed many base methylations using AlkB-derived enzymes before cDNA synthesis (Cozen et al. 2015; Zheng et al. 2015). We also used a thermophilic reverse transcriptase (TGIRT) that could more efficiently read through base methylations than the commonly used superscript RTs. Hence, more mutation signatures are present in our sequencing data using cellular RNA from HEK293T cells. Our sequencing strategy split each sample into two, one directly sequenced and the other first treated with the demethylase enzymes before cDNA synthesis. In this work, the untreated sequencing data are used for modification analysis, whereas the demethylase-treated sequencing data are used for validating the presence of modifications.
To identify potential sites of modifications that result in RT readthrough mutations and stops, all sequencing reads were aligned to mature tRNA sequences to obtain maximal coverage of modified molecules. A decision tree is shown for the flagged modification indices ≥0.15 at each position (Supplemental Fig. S1). We first look for the effect of demethylase treatment at each flagged site. m1A, m3C, m1G, m22G, and m3U sites show decreased MI values upon demethylase treatment (left branch). Sites that are unaffected by demethylase treatment are further assessed based on whether the MI is composed of only mutations, only stops, or a mixture of mutations and stops (right branch). Among the right branch, mutations only correspond to A-to-inosine modification or isodecoder misalignment, stops only correspond to the presence of bulky modifications in the Watson-Crick face and dihydrouridine (D), and a mixture of mutations and stops corresponds to other, uncommon modifications in the Watson-Crick face. This type of analysis enables us to identify the specifics of the modification using the MI values.
Biological tRNAs are made of type I's which have a 4–5 nucleotide (nt) variable loop, and type II's which have a longer variable loop that folds into a short hairpin. We plotted the cumulative MI for all type I tRNAs comprised of 18 of the 20 amino acids, and type II tRNA comprised of the amino acids leucine and serine (Fig. 2A). For type I tRNAs, high MI peaks are apparent around nucleotides 58, 37, 34, 26, 20, 9, and 4–7. Upon demethylase treatment, MI values are reduced to near background levels for the peak around 58, >50% for the peak around 37, and moderately reduced for the peak around 9. As we have already shown previously (Zheng et al. 2015), this result validates the peaks around position 58 as m1A58, 37 as m1G37, 9 as m1G9; m1A and m1G are known tRNA modification targets of the AlkB and AlkB-D135S enzymes used in the sequencing experiment. The same conclusion can be applied to the type II tRNAs for the peak around 67 as m1A at the same location as m1A58 in the type I tRNA, 50 as m3C located in the loop of the variable loop hairpin (m3C47d in the standard tRNA nomenclature), 37 as m1G37, 32 as m3C32, and 9 as m1G9. As expected from known tRNA modifications, some positions are not affected by demethylase treatment including the peak around 34 as inosine, around 20 that can be found commonly in tRNAs containing two consecutive dihydrouridines in the D loop.
FIGURE 2.

Cumulative modification index and mutation and stop plots for type I and type II tRNAs. The x-axis corresponds to the position along the tRNA. (Black line) untreated, (red line) demethylase-treated samples. (A) MI for type I tRNA on the left and type II tRNA on the right. The modifications assigned to each peak are also indicated in the graph. Demethylase-treated samples show a significant decrease for the m1A, m1G, and m3C modifications. (B) Mutation fractions for type I tRNA on the left and type II tRNA on the right. (C) Stop fractions for type I tRNA on the left and type II tRNA on the right.
The demethylase treatment significantly reduced the MI values of the m1G37 peak, but only moderately reduced the MI value of the m1G9 peak, even though both are made of the same chemical structure (Fig. 2A). This result can be interpreted as the extent of tRNA structure interfering with the demethylase reaction: m1G37 is located in the anticodon loop and easily accessible to the demethylase enzyme, whereas m1G9 forms a base triple with the 23–12 base pair of the D stem and is much more buried. Our demethylase enzyme reactions were performed under conditions where the modified tRNAs are still folded. At this time, the demethylase treatment cannot yet be performed under conditions where modified tRNAs are denatured which would require the removal or chelation of all divalent metal ions. The demethylase enzymes require divalent metal ions for their enzymatic activity.
Interestingly, the demethylase reaction also slightly reduced the MI values of the type II tRNAs at the peak around 26, corresponding to m22G26 modifications. This result suggests that our demethylase mixture is also reactive toward the m22G26 modification. m22G26 pairs with nucleotide 44, and this pair is sandwiched between the D and the anticodon stems, making it even less accessible than m1G9 which may explain the poor reactivity of the demethylase toward the m22G26 modification.
We further plotted the two parameters that compose the MI values, mutation fraction and stop fraction, separately for the cumulative type I and II tRNAs (Fig. 2B,C). Dominant mutations are all attributed to the four known types and locations of base methylations in tRNA, m1A, m1G, m22G, and m3C, as well as the well-known A34 to inosine modifications. Dominant stops are attributed to the known m1G, m22G, m3C, ms2t6A, and consecutive dihydrouridine (DD) modifications. Strong stops are present around nucleotides 4–7, which are moderately reduced upon demethylase treatment. This region is not known to be modified; yet the RT appears to fall off frequently during cDNA synthesis. At this time, we do not understand why these stops are present. In some cases, it is likely due to the presence of m1G9 in the RNA template of the RT reaction, since removal of m1G9 by demethylase prior to cDNA synthesis simultaneously reduces the extent of stops in this region (see the graph for several mitochondrial tRNAs in Fig. 3D,E). In other cases, it may be due to a reduction of RT processivity after encountering many modified nucleotides in the RNA template, or some kind of RNA–cDNA structure effects. Reverse transcriptase stops at m1A58 are likely underestimated in this sequencing data set due to the short cDNA products generated that can be missed in the sequencing reaction.
FIGURE 3.

Plots for individual tRNAs with known and unknown methylations. (Black line) Untreated; (red line) demethylase-treated samples. Dashed line shows the MI value of 15% which we use here as the threshold for detectable modifications with high confidence. (A) LeuCAG c1t34 (chromosome 1, tRNA34) isodecoder contains all four demethylase reactive modifications of m1A, m3C, m1G, and m22G. MI plot is on the left, mutation plot in the middle, and stop plot on the right. (B) LeuCAG c1t34 sequence with annotated modifications identified in panel A. The brackets mark the acceptor, D, anticodon, and T stems of this tRNA. (C) MI plot of AspGTC showing a newly identified m1A9 modification. (D) MI plot of mitochondrial tRNAAla showing a newly identified m1G37 modification. (E) MI plot of mitochondrial tRNAArg showing a newly identified m1A modification in the D loop whose sequence is also shown.
A representative example of how MI, mutation, and stop fraction plots look for a specific tRNA with multiple methylations at W-C face is shown in Figure 3A. In these graphs, the x-axis designates the nucleotide position and the y-axis the fraction of mutated and stopped (left panel), just mutated (middle), or just stopped (right) reads aligned to each position in this tRNA, LeuCAG isodecoder from chromosome 1, tRNA34 (CAG-c1t34) according to the hg19 genomic tRNA database (Chan and Lowe 2009). Because of the background, we choose to interpret only these peaks with an MI value ≥0.15 as a confidence threshold (dashed line in Fig. 3A). Three known methylations, m1A, m1G, and m22G (Fig. 3B) can be readily identified from these graphs that all have reduced MI values upon demethylase treatment. The large stop peak toward the 5′ end of the tRNA is likely derived from difficulties of RT retaining processivity as discussed for the cumulative type I and type II plots (Fig. 2C).
For LeuCAG, we found another peak located at C47d that was also removed upon demethylase treatment, consistent with a previously unidentified m3C at this position. We also found an unknown adenosine modification at position 9 in AspGTC (Fig. 3C). For position 9 modifications in human tRNA, m1G9 is common among cytoplasmic tRNAs, whereas m1A9 is common among mitochondrial tRNAs. We were surprised to find that the cytoplasmic tRNAAsp also contains a modification that responded to demethylase treatment, which is consistent with an m1A modification. We performed additional experiments to independently validate these two new methylation sites (see below).
In order to validate the usefulness of our alignment strategy with respect to available aligners, we compared mapping using either Bowtie1 or Bowtie2 aligner (Supplemental Fig. S2). A major difference between Bowtie1 and Bowtie2 is that Bowtie2 allows for insertions and deletions (indels). For the highly abundant tRNAs shown in Supplemental Figure S2, the modification sites are easily identified with both alignment programs. The alignments using Bowtie2 seem to be a little noisier than the alignments using Bowtie1, which may be in part derived from the presence of a small amount of indels shifting the mapped positions as arbitrary misincorporations.
We also adjusted for the number of mismatches in the seed sequence, or v = 0, 1, 2, and 3 mismatches, a common feature in the Bowtie1 aligner (Supplemental Fig. S3). Allowing for fewer mismatches reduced the overall number of reads due to the abundant mutations located close to the 3′ end of tRNAs such as m1A58. However, even for a heavily modified tRNA such as the LeuCAG, we were able to analyze MIs at v = 2, 3 mismatches. Comparing the MI for the Bowtie1 maps relative to the indel-containing Bowtie2 maps also showed a relatively small difference in MI across all of the modifications. Average MI across these highly abundant tRNAs also did not change significantly, nor were there any substantial differences in the plots for cumulative MI across the whole tRNA. These results gave us confidence in the method of alignment and further analysis.
Validation of m1A9 in tRNAAsp (AspGTC) and m3C47d in tRNALeu (LeuCAG)
We performed additional experiments to validate the two new modifications at A9 in AspGTC and C47d in LeuCAG. Since the sequencing was performed using reverse transcription, we made every effort to avoid any RT reaction in our validation.
For AspGTC, our approach was to use an AspGTC-specific oligo and RNase H cleavage to first isolate the 5′ fragments of the AspGTC RNA from gel purified total tRNA from HEK293T cells, followed by nuclease T1 digestion to detect a modified fragment containing A9 by MALDI or nuclease P1 and alkaline phosphatase treatment to detect a methylated A residue by LC-MS (Supplemental Fig. S4A). We tested a variety of conditions and applied the most stringent condition for fragment isolation (Supplemental Fig. S4B). We detected all three expected nuclease T1 fragments in the MALDI assay, including the UUAG/UU(A*)G peaks that differ by 15 Da, indicating a presence of a methyl group (Supplemental Fig. S4C). We also readily detected m1A using the established QQQ LC-MS method for m1A RNA methylation studies (Supplemental Fig. S4D; Dominissini et al. 2016). These results, together with the mutation and stop signatures at this position (Supplemental Fig. S4E), indicate that the modified A9 residue in AspGTC corresponds to N1-methyladenosine.
For LeuCAG, we also used the approach of isolating RNase H cleaved LeuCAG fragments from gel purified total tRNA from HEK293T cells, followed by nuclease P1 and alkaline phosphatase treatment to specifically identify m3C (Supplemental Fig. S5A). Again, stringent RNase H cleavage conditions were used to obtain LeuCAG fragments (Supplemental Fig. S5B), and QQQ LC-MS showed the appropriate amount of m3C as well as m1A (from m1A58 of LeuCAG) from the isolated fragments (Supplemental Fig. S5C). We also utilized an m3C-specific base chemistry to validate the LeuCAG site (Supplemental Fig. S5D). Hydrazine reacts with 3-methylcytosine and renders the site abasic, which can be detected as a strand scission after aniline treatment (Peattie 1979; Behm-Ansmant et al. 2011). Using 3′ 32P-labeled total tRNA from HEK293T cells, hydrazine/aniline treatment yielded three major fragments that are consistent with the cleavage products of tRNASer at m3C32, tRNAThr at m3C32, and tRNASer at m3C47d, and the LeuCAG site at m3C47d (Supplemental Fig. S5E; Parisien et al. 2013). These specific tRNA products were confirmed by tRNA microarrays (Supplemental Fig. S5F). These results, together with the mutation and stop signatures at this position (Supplemental Fig. S5G), indicate that the modified C47d residue in LeuCAG corresponds to N3-methylcytosine.
Assessment of quantification of methylation fractions
We calculated the MI value for all tRNA isodecoders among the top 25% expressed tRNAs according to the number of aligned reads in the untreated sample. Our DM-tRNA-seq data are of sufficient quality that the mapped reads for each of these top quartile tRNAs range from 20,000 to 700,000. Although the MI values can be precisely measured, there are several inherent caveats to interpret these as the precise modification fraction. In particular, the reverse transcription-based method is prone to underestimate the modification fraction to varying degrees in a context-dependent manner (Ryvkin et al. 2013; Hauenschild et al. 2015). In our case, this underestimation can be derived from either the inadequacy of RT stops or the ability of RT to also incorporate the correct nucleotide opposite to the modified base. The m1A58 stop fraction is likely underestimated in our sequencing data; these RT stop reads are only 18 residues and can be easily missed in our experimental design which was aimed for sequencing longer RT products. This effect may be mitigated in future experiments by expanding the size range of RT products to be sequenced. The quantitative variation of incorporating the correct nucleotide, however, is a large obstacle in obtaining strictly quantitative information as this effect can be highly dependent on the chemical structure and the sequence context of the modified base (Hauenschild et al. 2015). For these reasons, the MI values cannot yet be used to fully represent the precise modification fraction without further study. In this work, we choose to group the MI values in three regimes (0.15–0.50, 0.50–0.80, and 0.80–1.0) as a semiquantitative metric for ease of description and discussion. Table 1 shows the MI groups of four methylation types in the tRNA transcriptome.
TABLE 1.
Methylations identified in the human tRNAome by DM-tRNA-seq

m1A58: Forty of 46 (87%) sites are in the high group, suggesting that most tRNAs are fully modified at this position. Aside from AspGTC tRNA with MI < 0.15 (Fig. 3C), the only other tRNA in the low group is GluCTC. Both Asp and Glu tRNA results are consistent with literature (Kuchino et al. 1981; Chan et al. 1982) that these tRNAs are hypomodified at m1A58. We also validate the low m1A58 modification in tRNAAsp by primer extension (Fig. 4 below). As described above, we were unable to obtain stop information for the m1A58 modifications in the previous sequencing experiment.
FIGURE 4.
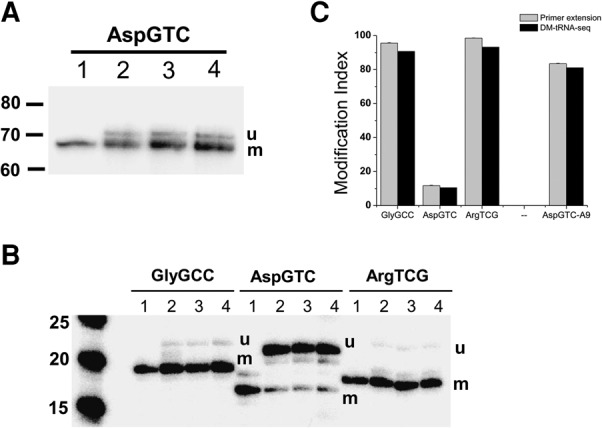
Validation of modification index by primer extension stops with AMV RT. All experiments were designed in such a way that the lower band corresponds to the amount of modified tRNA (m) and the upper band the amount of unmodified tRNA (u); see Supplemental Figure S4. (Lane 1) Extension using only the respective α-32P-dNTP to indicate the product location. (Lanes 2–4) Extension from biological triplicates. (A) AspGTC for position 9, a putative m1A identified in DM-tRNA-seq. (Lane 1) α-32P-dCTP only; (lanes 2–4) dATP, α-32P-dCTP, ddGTP, dTTP. (B) GlyGCC, AspGTC, ArgTCG for position m1A58. (Lanes 2–4 for GlyGCC and for AspGTC) α-32P-dATP, dCTP, ddGTP, dTTP. (Lanes 2–4 for ArgTCG) ddATP, α-32P-dCTP, dGTP, dTTP. (C) Quantitative comparison of modification fraction determined by primer extension (gray) and by DM-tRNA-seq (black).
The only other tRNA of note in this group is the initiator tRNA (Met-i) with MI value of 0.77 without the consideration of stops. In yeast, m1A58 is known to be required for the stability of the initiator-tRNA, but not for other tRNAs (Kadaba et al. 2004). It is possible that m1A58 is also required for Met-i stability in mammalian cells. If this is the case, the MI of 0.77 is close to the high regime (≥0.80), so Met-i could still be fully modified in HEK293T cells.
m1G37: Nine of 15 (60%) sites are high. This modification is known to prevent ribosome frameshifting in specific tRNA and codon contexts (Bjork et al. 1989; Gamper et al. 2015), so high levels of modifications are expected. Surprisingly, six of 15 tRNAs are in the intermediate and low groups. This result is not fully consistent with the known function m1G37 modification. It may be derived from the strong context dependence of incorporating higher fractions of correct nucleotides opposite the m1G in these particular tRNAs.
m22G26: Without exception, all 25 sites belong to the high group. This modification is known to provide rigidity of the coaxially stacked D and anticodon stems (Steinberg and Cedergren 1995) and to prevent misfolding of at least one tRNA (Edqvist et al. 1995; Pallan et al. 2008). Full modification at these sites would ensure that these tRNAs all have proper conformations and rigidity for translation.
m1G9: Fifteen of 19 (79%) sites are high. This modification is known to play a role in maintaining the structure of mitochondrial tRNAs and possibly cytosolic tRNAs as well (Helm et al. 1999). Hence, the modification fraction is expected to be high for most of these tRNAs.
m1A9: To our surprise, AspGTC (Fig. 3B) has a high MI at position 9 which is reduced upon demethylase treatment, suggesting the presence of m1A9 modification which we validated by mass spectrometry (Supplemental Fig. S4) and by primer extension below. m1A9 is not known previously to be present in cytosolic tRNAs, but is common among mitochondrial-encoded tRNAs (Suzuki and Suzuki 2014).
m3C: These include nine sites at C32, of which only one is in the high group (11%); among the five sites at C47d located in the loop region of the hairpin in the variable arm of type II tRNAs, only one is in the high group (20%); the single site at C20 in the elongator tRNAMet, also predicted by HAMR analysis (Ryvkin et al. 2013) is in the high group. The functions of these m3C modifications are unclear; they may perform structural roles for tRNA. However, the prevalence of many m3C32 and m3C47d modifications present in the low and intermediate groups suggests that m3C modification fractions may be useful for regulatory purposes.
We also analyzed the modification indexes for the 22 human mitochondrial tRNAs (Table 2). The complete mitochondrial tRNA modifications have been mapped in the bovine liver (Suzuki and Suzuki 2014), and several human mitochondrial tRNA modifications are also known (Machnicka et al. 2013). We were able to detect all 19 expected m1A/G9 modifications, and 16 of 19 (84%) belong to the high MI group. M1A/G9 in mitochondrial tRNAs are very efficiently removed by the demethylases (Fig. 3C,D), presumably due to the lower structural stability of mitochondrial versus cytosolic tRNAs. On the basis of the bovine mitochondrial tRNA modifications, we detected m1A58 or equivalent in mtLeu(TAA), mtLys, and mtSer(TGA), but not in mtGlu, mtIle, and mtCys. We detected all three m1G37 modifications in mtGln, mtLeu(TAG), mtPro, m22G26 in mtIle, m3C32 in mtSer(TGA), and mtThr, as predicted from bovine mt-tRNAs.
TABLE 2.
Methylations identified in the human mitochondrial tRNA by DM-tRNA-seq
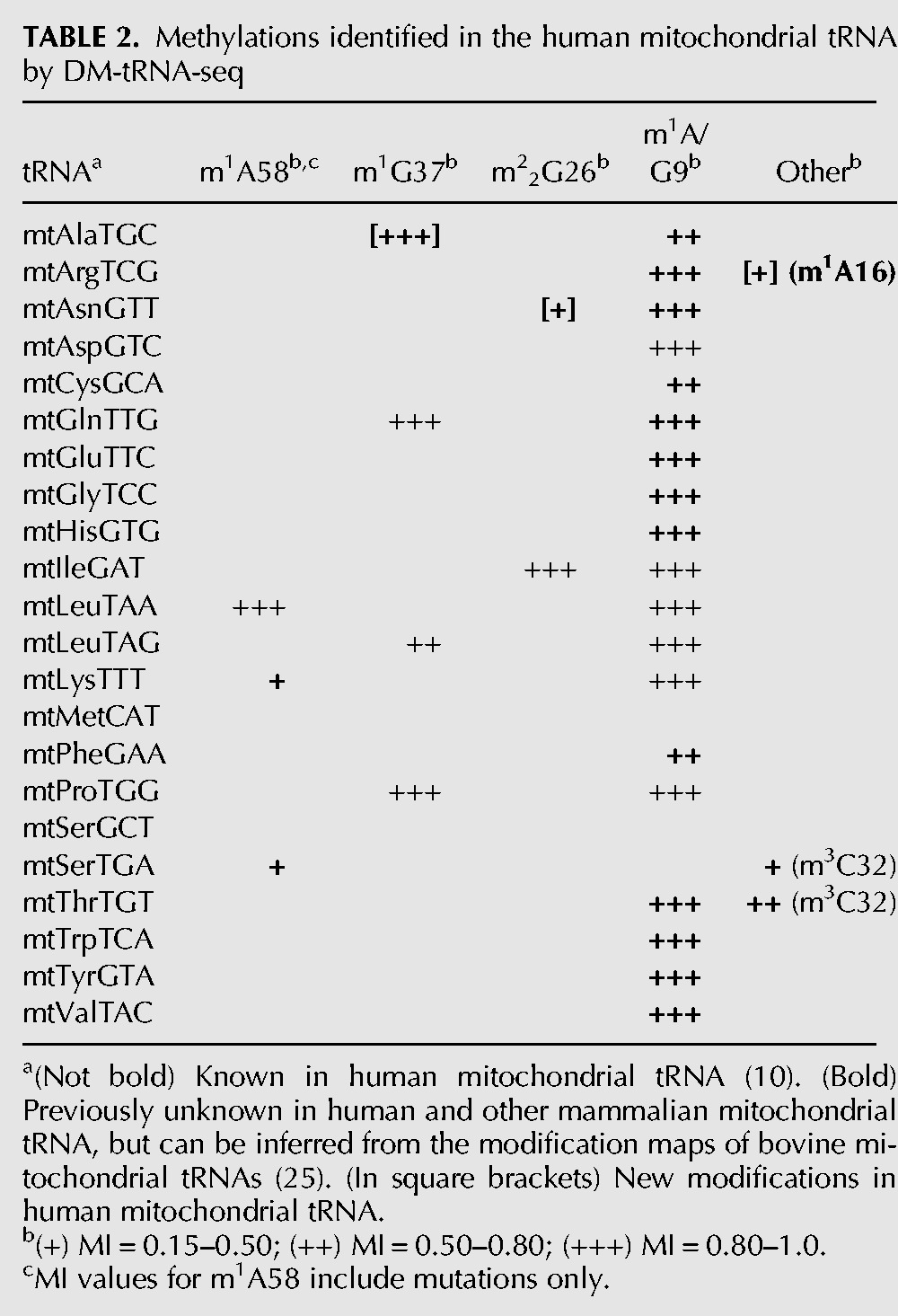
We also found three new mitochondrial tRNA modifications that could not be inferred from the bovine mitochondrial tRNAs. Human mtAla has a G after the anticodon UGC, and this G is highly modified to m1G (Fig. 3D). We also observed above threshold MI values for G26 in mtAsn, which could be modified to m22G and A16 in mtArg which could be modified to m1A (Fig. 3E).
MI value correlations by primer extension
We applied the standard primer extension method to correlate the quantitative nature of the MI values in assessing tRNA methylations (Fig. 4). Primer extension relies on using retroviral reverse transcriptases such as AMV RT that stop at methylated nucleotides such as m1A. Our experimental design involves a primer that ends 1 nt away from the modification site such as m1A58 (Supplemental Fig. S6). Primer extension reaction is visualized by using the appropriate α-32P-dNTP incorporated at position 59. When the reaction mixture contains three dNTPs and one specific ddNTP, the short reaction product corresponds to the stops at the modified nucleotide, and the long reaction product corresponds to the amount of unmodified tRNA. In this way, we validated the presence and the correlation of MI values from sequencing of m1A9 in AspGTC tRNA (Fig. 4A), as well as the low fraction of m1A58 in AspGTC (Fig. 4B). We also correlated the MI values for m1A58 of GlyGCC tRNA, a tRNA known to have m1A58 in the Modomics database and of ArgTCG tRNA, a tRNA not present in the Modomics database.
In all four cases, the modification indices obtained by primer extension and sequencing are similar (Fig. 4C). The MI values from DM-tRNA-seq seem to slightly underestimate the MI values from AMV RT primer extension by up to ∼10%. For m1A58, this result may be derived from the under-counting of reads derived from RT stops at this position in our sequencing data, as discussed above. Our results clearly confirm the very low m1A58 modification levels of AspGTC at ∼12%, as well as the presence of m1A9 in AspGTC. These results indicate that the MI values from DM-tRNA-seq are excellent parameters for estimating modification fractions.
Other modifications
We also looked for peaks with >15% MI values that do not change upon demethylase treatment (Fig. 5). AlaAGC is known to contain two inosine modifications at 34 (first anticodon nucleotide) and 37, and three W-C face methylations at m1A58, m1I37, and m22G26 (Grosjean et al. 1996; Machnicka et al. 2013). I34 is readily apparent in the mutation graph as essentially 100% of this residue has been converted from A to I, which reads as G, and it does not respond to demethylase treatment (Fig. 5B). m1A58 and m22G26 can be easily picked out due to the reduction in peak height upon demethylase treatment. m1I37 leads to mostly stops in the untreated sample; demethylase fully removes the methyl group so that the RT stop is fully eliminated; at the same time, the mutation fraction goes up to nearly 100% because of the A37 to I conversion. AsnGTT is known to contain two consecutive dihydrouridines in the D-loop (Machnicka et al. 2013). Each D modification is known to weaken W-C base pairing by ∼1 kcal/mol (Dalluge et al. 1996), so that the presence of two consecutive D's leads to strong RT stops that do not respond to demethylase treatment (Fig. 5C). LysTTT is known to contain a bulky modification, 2-methylthio-6-threonylcarbamoyl-A (ms2t6A) at A37 (Machnicka et al. 2013). This modification leads to an ∼98% stop in the RT reaction which does not respond to the demethylase treatment (Fig. 5D). Because we are starting out with ∼300,000 reads for this tRNA (Fig. 5D inset), we are still able to obtain ∼6000 reads that process past the modification, and we can assess potential modifications upstream of ms2t6A37 after this strong stop. Mitochondrial tRNATyr is known to contain another bulky modification, 2-methylthio-6-isopentenyl-A (ms2i6A) at A37 (Machnicka et al. 2013). This modification leads to an ∼95% stop in the RT reaction which does not respond to the demethylase treatment (Fig. 5E). Similarly, the high number of read counts for this tRNA (Fig. 5E inset) still enables the analysis of m1G9 modification in mtTyr.
FIGURE 5.

Plots for individual tRNAs with other methylations from DM-tRNA-seq. The MI value of these modifications does not change upon demethylase treatment. (A) Chemical structure of the five modifications where the chemical group is shown in red. The presence of each modification in tRNA and/or rRNA is listed beneath each base. (B) Mutation and stop plots showing AlaAGC with the known m1I37 and I34 modifications. (C) Stop plot showing the consecutive dihydrouridine modifications (DD) in AsnGTT. (D) Stop plot for LysTTT showing the ∼98% stop at ms2t6A37. Inset (y-axis = log10 counts along the LysTTT tRNA) shows that even with such strong stop, sufficient number of counts is still present for the analysis of modifications in this tRNA upstream of this stop. (E) MI plot for mitochondrial tRNATyr showing an ∼95% stop at ms2i6A37. Inset (y-axis = log10 counts along the mtTyr tRNA) shows that even with such strong stop, sufficient number of counts is still present for the analysis of the m1G9 modifications in this tRNA upstream of this stop.
Modification index heat maps for all abundant tRNAs
We also show mutation and stop fractions across all tRNA isodecoders that are present in the top 25% abundant tRNAs as heat maps (tRNALeu isodecoders in Fig. 6; tRNA isodecoders for other amino acids in Supplemental Fig. S7). To enhance presentation, all nucleotide positions in the heat map (x-axis) are converted to standard tRNA nomenclature so that the anticodon nucleotides are always numbered 34–36, D loop nucleotides 14–20, and variable loop nucleotides 44–48. Human tRNALeu’s contain five distinct isoacceptors with the anticodons of AAG, CAG, TAG, CAA, and TAA. In the mutation graph, the untreated samples show high mutation values at m1A58, m1G37, m22G26 for all isodecoders, m3C47d for the two CAG-isodecoders, and I for the two AAG-isodecoders. As expected, the m1A58, m1G37, and m3C47d modifications are significantly reduced upon demethylase treatment. M1G37 and m22G26 also show significant stops in the untreated sample, but the stops are substantially reduced for m1G37, and moderately reduced for m22G26.
FIGURE 6.

Mutation and stop heat maps for all abundant isodecoders in the tRNALeu family. The x-axis indicates the tRNA position according to the standard tRNA nomenclature so that the anticodons are always numbered 34–36, and the last nucleotide is always 76. Nucleotides not present according to this nomenclature are shown in gray. Stops can only be analyzed up to nucleotide 61 so nucleotides 62–76 are shown in gray. The scale of the heat map is shown on the lower left. Modifications are annotated in red or in blue according to their identification through mutation or stop component, respectively. Only the isodecoders that are among the top 25% most abundant tRNA species are shown.
Applying the above criteria, all identified m1A, m1G, m22G, and m3C sites in the tRNA transcriptome for 47 isoacceptors from HEK293T cells are shown in Table 1 (the remaining two annotated isoacceptors in the genomic tRNA database are excluded here because they are present at very low levels). They include 12/46 new m1A58 (26%), 4/15 new m1G37 (27%), 10/25 new m22G (40%), 6/21 new m1G9 (29%), and 7/15 new m3C (47%) sites not present in the human and other mammalian tRNAs in the Modomics database (Machnicka et al. 2013).
rRNA modifications
To extend the application of DM-RNA-seq, we applied it to sequence human rRNAs from HeLa cells. Human rRNA has four known modifications present at the Watson-Crick face of nucleobases: 1-methyl-3-(3-amino-3-carboxypropyl) pseudouridine (m1acp3Ψ, Fig. 5A) and N6,N6-dimethyladenosine (m62A) in the 18S rRNA, m1A and 3-methyl uridine (m3U, Fig. 1A) in the 28S rRNA. The DM-RNA-seq method was able to detect three of these modifications (Fig. 7). The m62A site was not accessible in our present study because it is located 19 nt away from the 3′ end of 18S rRNA, such that very short sequencing reads will be needed to obtain stop fraction. The m1A site shows a very high MI value of 0.976 contributed about equally from mutations and stop; this high MI value is reduced to background levels upon demethylase treatment as expected (Fig. 7A). The rRNA m1A result indicates that both mutations and stop can contribute significantly to the m1A modification signature. The m3U site shows an MI value of 0.713 mostly derived from mutations; it is also reduced to background levels upon demethylase treatment (Fig. 7B), indicating that our demethylase mixture also acts on m3U. The m1acp3Ψ site shows an MI value of 0.885 contributed about equally from mutations and stop; it does not respond to demethylase treatment (Fig. 7C).
FIGURE 7.

MI plots for rRNA modifications. (Black line) Untreated, (red line) demethylase-treated samples. The mutation and stop component of each modification is shown below the graph. (A) The 100 nt region in the 28S rRNA around m1A1322. (B) The 100 nt region in the 28S rRNA around m3U4530. For both A and B, demethylase treatment reduces the MI to background levels. (C) The 100 nt region in the 18S rRNA around the m1acp3-Ψ modification at 1248. The black and red lines overlap, indicating that the demethylases do not act on this modification.
Both the m1A site in the 28S rRNA and the m1acp3Ψ site in the 18S rRNA are previously known to be fully modified (Liang et al. 2009; Meyer et al. 2011). The very high MI values of 0.885–0.976 at these two sites nicely reflect these high modification fractions.
DISCUSSION
This work demonstrates that RNA methylations at the Watson-Crick face can be precisely identified and importantly, their modification fractions assessed by the newly developed DM-tRNA-seq method. We show that the high extent of readthrough of the modifications by the thermophilic, group II intron RT (TGIRT) generates an adequate amount of reads to enable transcriptome-wide analysis of five tRNA and two rRNA methylations. The tRNA methylations we detect and assess quantitatively cover approximately one-third of all human tRNA modification sites. We were able to identify many previously known sites in the widely used RNA modification databases as well as new sites for which no prior experimental data exist. Examples of completely novel sites include the m3C47d site in LeuCAG and the m1A9 in AspGTC, which we validated by sequencing-independent methods (Supplemental Figs. S4, S5).
Comparing the parallel sequenced, demethylase-treated and untreated data lends high confidence regarding the m1A, m3C, m1G, and m22G methylations that are widespread in the tRNA transcriptome. Interestingly, the particular mutation and stop patterns can also allow for initial assessment of the modification types for the same base such as m1G and m22G: m1G is manifested by more significant stop than mutation fractions, whereas m22G is manifested by more mutation than stop signatures (Fig. 2B,C).
Of course, vast prior knowledge of these methylations in defined positions in many tRNAs significantly aided the validation and strengthening of our method. On the other hand, human tRNA modifications are still inadequately represented in the widely used modification databases such as Modomics and the RNA Modification Database (Cantara et al. 2011; Machnicka et al. 2013). Of the 47 human tRNA isoacceptors shown in Table 1, 11 do not have any data among the human and mammalian tRNAs, and 20 have data only among other mammalian tRNAs. Of the 22 human mitochondrial tRNAs shown in Table 2, 17 do not have data among the human mitochondrial tRNAs. Our results clearly help fill some of these large gaps.
A unique and new feature of our method is the use of the modification index (MI) to assess the quantitative nature of each detectable modification site. The MI values from DM-tRNA-seq also correlate well with the classical primer extension stops using AMV RT for several m1A sites (Fig. 4). The slightly lower modification fraction obtained in sequencing compared to primer extension stops may be explained by TGIRT inserting the “correct” base some of the time at the methylation sites. Furthermore, the MI values for m1A58 in tRNAs likely underevaluated the m1A58 modification fraction in this study due to the lack of stop information in our experimental design. As we have seen for the m1A9 sites for mitochondrial tRNAs and the m1A1322 site in rRNA, TGIRT does not read through m1A all the time. In the future, the m1A58 stop fraction should be obtained by using a longer TGIRT sequencing template for short RNA fragments derived from m1A58 stops.
Surprisingly, we found that numerous methylation sites seem to fall into low and medium modified categories in the HEK293T tRNA transcriptome (Tables 1, 2). This result seems to suggest that dynamic ranges exist for tRNA methylations to have a cell-type and cell-state-dependent pattern. However, a confounding factor not investigated in detail here is the effect of sequence context of the modification site on the MI value. A recent work by Hauenschild et al. (2015) shows clear differences in mutation and stop fraction for m1A with a different +1 nucleotide sequence, indicating that sequence context is an important parameter in determining the ration of mutations and stops at each site. Without additional studies of context dependence for each modification type and site, MI values cannot yet be fully used to precisely identify modification fractions. Despite these potential caveats, MI values can be interpreted as providing a lower bound on the modification fraction for each methylation site.
Biologically, quantitative differences in specific tRNA modifications have been well documented for the 5-methoxycarbonylmethyl (mcm5) and 5-methoxy-carbonyl-methyl-2-thio (mcm5s2) U34 modifications in tRNAArg(UCU) and tRNAGlu(UUC) and 5-methyl (m5C)-C34 in tRNALeu(CAA) to enhance stress response (Begley et al. 2007; Chan et al. 2012; Patil et al. 2012a,b). In yeast, global levels of many modification types such as m5C, m22G, and 2′ O-methyl-C (Cm) can change significantly when cells are exposed to distinct types of chemical stressors (Chan et al. 2010; Gu et al. 2014).
In summary, using high-throughput DM-tRNA-seq data, we identified and semiquantified six base methylations and evaluated five other modifications in the human tRNA transcriptome and rRNA. Using the modification index metric, we assessed quantitative information on methylations at the Watson-Crick face among all isoacceptors in nuclear-encoded tRNAs and all mitochondrial-encoded tRNAs. While the functional investigation on the consequence of potential differences in modification fractions between cells is beyond the scope of this work, our result demonstrates the feasibility of using DM-tRNA-seq to investigate dynamic tRNA modification patterns.
MATERIALS AND METHODS
DM-tRNA-seq of HEK293T and HeLa cells
Experimental details of the DM-tRNA-seq method were described in Zheng et al. (2015). The same method was applied for HeLa rRNA sequencing except the input RNA was isolated rRNA that was subjected to chemical fragmentation before library preparation. Our DM-tRNA-seq data are composed of biological replicates of total tRNA from HEK293T cells without and with demethylase treatment. Both sets of replicates map with r2 ≥ 0.99. For the analysis here, we used the untreated sample 2 which has 9,002,637 mapped reads to nuclear-encoded tRNA and 2,269,204 reads to mitochondrial-encoded tRNA, and treated sample 1 with 15,740,700 mapped reads to nuclear-encoded tRNA and 2,851,034 reads to mitochondrial-encoded tRNA. The rRNA-seq data contains 7,531,718 mapped reads.
Sequencing alignments
All reads were sequenced on an Illumina HiSeq 2000 with paired-end mapping with read lengths of 100 base pairs. Standard quality control via FastQC was performed after sequencing and after subsequent trimming mentioned next.
Sequencing reads were aligned using Bowtie to a modified tRNA genome file containing nuclear-encoded tRNAs, mitochondrially encoded tRNAs, and human rRNAs (found at the GEO accession for this paper). Splicing of tRNAs was accounted for in the modified genome file and only mature tRNAs were aligned against. Briefly, the tRNA library was adapted from the tRNAScan-SE library (http://gtrnadb.ucsc.edu/Hsapi19/; Chan and Lowe 2009) by appending 3′CCA to tRNAs from the genomic tRNA database. Isodecoders with identical SEscan scores from the genomic tRNA database were consolidated for ease of identity assignment, so the total number of annotated tRNA genes and pseudogenes were reduced from 625 to 462. Prior to mapping, reads were processed using Trimmomatic v0.32 as well as further trimming using custom Python scripts to remove any further artifacts from demultiplexing and removal of primers, adapters, or any other low-quality sequences. Sequences >15 bp were then aligned to the library using Bowtie 1.0 with sensitive options using the highest allowed mismatch settings for Bowtie 1.0. Mapping to all references occurred simultaneously. Only one alignment (best with Bowtie1 or k = 1 with Bowtie2, k refers to the number of allowed distinct alignments per raw read) declared as valid by the respective mapping software was reported for each read. Analysis was also performed using k = 3 to allow for up to three alignments per read and also v = 0, 1, 2, 3 (how many mismatches allowed in the seed sequence per raw read) in Bowtie1 to allow for fewer mismatches.
Modification index (MI)
The mapping pipeline proceeded by conversion from the SAM output from Bowtie using custom C and Python scripts to separate isodecoders based on previous alignment. From here, further cleanup based on redundancy (any read that could inherently map due to misalignment) was also discarded. For each position in the reference file, the following was calculated: at each position = n, how many counts existed (total counts) = c, how many misincorporations or mutations = m, and how many aligned reads stopped at the position = s. This leads to a full modification index (MI) calculated at each position by (mutations at position + stops at position directly 3′ to position)/(total counts at position), or (m + s)/c, and individual mutation and stop components to the metric can be further reduced by calculating m/c and s/c, respectively.
For high stringency detection purposes for this work, sites corresponding to an MI value of 15% or below were discarded as noise due to sequencing error or basal level of misincorporation from the RT reaction. For simplicity and ease of discussion, sites with MI ≥ 15% were categorized in the range of 15%–50% as low, 50%–80% as medium, and 80%–100% as highly or completely modified. Positional shifts of modifications due to tRNA length (either due to variations in length of type I tRNAs or type I versus type II tRNAs) were recognized and adjusted for in calculations for heat maps. Modification index in regions of variable loop are accounted for accordingly. Modification index 3′ to m1A57/58/59/68 (m1A58 in standard tRNA nomenclature) is only derived from mutations due to the lack of stop information at these positions because of the short reads needed for stops at this m1A position.
Reverse transcription primer extension
Total RNA was isolated from HEK293T cells in biological triplicates using TRIzol (Life Technologies). tRNA was gel purified by 8% denaturing PAGE (7M urea, 1× TBE). Seven hundred nanograms of total RNA or 200 ng of total tRNA was annealed to 5 pmol of specific primer in 30 mM Tris-Cl (pH 7.5), 2 mM KCl at 90°C for 90 sec. The mixture was cooled at room temperature for 3 min. The reverse transcription reaction was carried out in 1× AMV buffer, 0.1 mM each cold (d)dNTP, 1.25 µM radiolabeled dNTP, and 0.2 U/µL AMV RT (New England Biolabs). The reaction was performed at 37°C for 30 min. To degrade the RNA after the RT reaction, 10 U of RNaseH (Epicentre) was added, and the reaction was incubated at 37°C for 5 min. RNA loading dye (9 M urea, 100 mM Na2EDTA, bromophenol blue, xylene cyanol) was added and the reaction was resolved by denaturing gels (7 M urea, 1× TBE).
Validation of m1A9 in tRNAAsp (AspGTC) and m3C47d in tRNALeu (LeuCAG)
Total RNA was isolated from HEK293T cells using TRIzol (Life Technologies). Total tRNA was gel purified by 8% denaturing PAGE (7 M urea, 1× TBE).
RNase H cleavage and fragment isolation
For AspGTC, 5′ 32P-labeled tRNA was spiked into 20 µg of total tRNA and annealed to 20 pmol DNA oligo (5′-TGGCTCCCCGTCGGGGAATCGAACCCCGGTCTCCCGCGTGACAGGCGGGG) complementary to nucleotides 26–76. For LeuCAG, 3′ 32P-labeled tRNA was spiked into 20 µg of total tRNA and annealed to 20 pmol DNA oligo (5′-CTGCGACCTGAACGCAGCGCCTTAGACCGCTCGGCCATCCTGAC) complementary to nucleotides 1–42. The tRNA/oligo was annealed in 100 mM Tris-HCl (pH 7.5), 0.5 mM EDTA at 90°C for 90 sec. The reaction was cooled at room temperature for 3 min. Digestion was performed by adding NaCl to 100 mM, MgCl2 to 10 mM, and RNase H (Epicenter) to a final concentration of 1 U/µL. The reaction was incubated at 37°C for 30 min. 0.1 U/µL DNase I (NEB) was added and the reaction was incubated at 37°C for 30 min. The reaction was then ethanol precipitated, resuspended in 1× RNA loading dye (4.5 M urea, 50 mM EDTA), and resolved on 10% denaturing PAGE (7 M urea, 1× TBE). The digestion products were eluted in 200 mM NH4Cl, 50 mM NH4OAc overnight. Glycogen was added to aid precipitation.
MALDI-TOF
Approximately 2 pmol of eluted fragements was digested with 0.5 U/µL T1 (Ambion) in 10 mM NH4OAc for 30 min at 37°C. One pico mole of the digested fragments was mixed with an equal amount of MALDI matrix, composed by 9:1 (v:v) ratio of 20-, 40-, 60-trihydroxyacetophenone (THAP, 10 mg/mL in 50% CH3CN/H2O): diammonium citrate (50 mg/mL in H2O). The mixture was spotted on a MALDI sample plate, dried under vacuum, and analyzed by a Bruker Ultra-flextreme MALDI-TOF Mass Spectrometer in reflector, positive mode.
LC/MS-QQQ
Approximately 1 pmol of RNase T1-digested fragments was digested with nuclease P1 in 100 mM ammonium acetate at 37°C for 2 h. Three microliters of freshly prepared 1 M ammonium carbonate was added with 1 U of alkaline phosphatase (Roche) and further incubated at 37°C for 2 h. Mixture was centrifuged through a 0.22 m PVDF syringe filter (Millex-GV) to remove contaminants. Remaining sample was then separated by reverse phase ultra-performance liquid chromatography (RP-UPLC) on a C18 column (Agilent's ZORBAX Eclipse XDB-C18, Rapid Resolution HT, 2.1 × 50 mm, 1.8 µm, max 600 bar; mobile phase A: water + 0.1% (v/v) formic acid, mobile phase B: methanol + 0.1% (v/v) formic acid, with a gradient of 2%–11% B in 4.5 min) with on-line mass spectrometry detection using an Agilent 6460 triple-quadrupole (QQQ) mass spectrometer in positive electrospray ionization mode. The nucleosides were identified via dynamic multiple reaction monitoring (DMRM) by using the nucleoside-to-base ion mass transitions of 282–150 (m1A) and 258–126 (m3C).
Hydrazine/analine cleavage and tRNA microarray
3′ 32P-labeled tRNA was spiked into 0.2 µg/µL total tRNA and incubated with 10% hydrazine in 3 M NaCl at 4°C for 10 min, quenched with 0.3 M NaOAc/HOAc, pH 5.5, and ethanol precipitated. The pellet was then resuspended in 1 M aniline, 1 M HOAc. The reaction was incubated at 60°C for 10 min, quenched with 0.3 M NaOAc/HOAc, pH 5.5, and ethanol precipitated. The pellet was resuspended in 1× RNA loading buffer (4.5 M urea, 50 mM EDTA) and resolved on 15% denaturing PAGE (7 M urea, 1× TBE). Fragments were eluted in 200 mM KCl, 50 mM KOAc overnight. Poly(A) RNA and salmon sperm DNA were added to aid in precipitation. The fragments were then hybridized to custom-made tRNA microarrays at 60°C for 16 h and exposed to phosphorimaging as previously described (Parisien et al. 2013).
DATA DEPOSITION
DM-tRNA-seq data are from GSE66550. The rRNA-seq data with and without demethylase treatment are from GSE76434.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. Qing Dai for his guidance in MALDI experiments. This work was supported by the National Institutes of Health (DP1GM105386 to T.P.). W.C.C. and M.E.E. were recipients of a National Institutes of Health Chemistry and Biology Training grant (T32 GM008720). M.E.E. is a recipient of the National Science Foundation predoctoral fellowship (DGE-1144082).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.056531.116.
REFERENCES
- Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ. 2007. Trm9-catalyzed tRNA modifications link translation to the DNA damage response. Mol Cell 28: 860–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behm-Ansmant I, Helm M, Motorin Y. 2011. Use of specific chemical reagents for detection of modified nucleotides in RNA. J Nucleic Acids 2011: 408053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Wikstrom PM, Bystrom AS. 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986–989. [DOI] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. 2011. The RNA modification database, RNAMDB: 2011 update. Nucleic Acids Res 39: D195–D201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM. 2009. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37: D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JC, Yang JA, Dunn MJ, Agris PF, Wong TW. 1982. The nucleotide sequence of a glutamate tRNA from rat liver. Nucleic Acids Res 10: 4605–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ. 2010. A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6: e1001247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. 2012. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun 3: 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozen AE, Quartley E, Holmes AD, Hrabeta-Robinson E, Phizicky EM, Lowe TM. 2015. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Methods 12: 879–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalluge JJ, Hashizume T, Sopchik AE, McCloskey JA, Davis DR. 1996. Conformational flexibility in RNA: the role of dihydrouridine. Nucleic Acids Res 24: 1073–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decatur WA, Fournier MJ. 2002. rRNA modifications and ribosome function. Trends Biochem Sci 27: 344–351. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. 2016. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530: 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edqvist J, Straby KB, Grosjean H. 1995. Enzymatic formation of N2,N2-dimethylguanosine in eukaryotic tRNA: importance of the tRNA architecture. Biochimie 77: 54–61. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Gamper HB, Masuda I, Frenkel-Morgenstern M, Hou YM. 2015. Maintenance of protein synthesis reading frame by EF-P and m1G37-tRNA. Nat Commun 6: 7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. 1999. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149. [DOI] [PubMed] [Google Scholar]
- Goodenbour JM, Pan T. 2006. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res 34: 6137–6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H. 2005. Modification and editing of RNA: historical overview and important facts to remember. In Fine-tuning of RNA functions by modification and editing (ed. Grosjean H), Vol. 12, pp. 1–22. Springer, Berlin/Heidelberg, Germany. [Google Scholar]
- Grosjean H, Auxilien S, Constantinesco F, Simon C, Corda Y, Becker HF, Foiret D, Morin A, Jin YX, Fournier M, et al. 1996. Enzymatic conversion of adenosine to inosine and to N1-methylinosine in transfer RNAs: a review. Biochimie 78: 488–501. [DOI] [PubMed] [Google Scholar]
- Gu C, Begley TJ, Dedon PC. 2014. tRNA modifications regulate translation during cellular stress. FEBS Lett 588: 4287–4296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauenschild R, Tserovski L, Schmid K, Thuring K, Winz ML, Sharma S, Entian KD, Wacheul L, Lafontaine DL, Anderson J, et al. 2015. The reverse transcription signature of N-1-methyladenosine in RNA-seq is sequence dependent. Nucleic Acids Res 43: 9950–9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Giege R, Florentz C. 1999. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 38: 13338–13346. [DOI] [PubMed] [Google Scholar]
- Higa-Nakamine S, Suzuki T, Uechi T, Chakraborty A, Nakajima Y, Nakamura M, Hirano N, Kenmochi N. 2012. Loss of ribosomal RNA modification causes developmental defects in zebrafish. Nucleic Acids Res 40: 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. 2004. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18: 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchino Y, Shindo-Okada N, Ando N, Watanabe S, Nishimura S. 1981. Nucleotide sequences of two aspartic acid tRNAs from rat liver and rat ascites hepatoma. J Biol Chem 256: 9059–9062. [PubMed] [Google Scholar]
- Li S, Mason CE. 2014. The pivotal regulatory landscape of RNA modifications. Annu Rev Genomics Hum Genet 15: 127–150. [DOI] [PubMed] [Google Scholar]
- Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C. 2016. Transcriptome-wide mapping reveals reversible and dynamic N-methyladenosine methylome. Nat Chem Biol 12: 311–316. [DOI] [PubMed] [Google Scholar]
- Liang XH, Liu Q, Fournier MJ. 2009. Loss of rRNA modifications in the decoding center of the ribosome impairs translation and strongly delays pre-rRNA processing. RNA 15: 1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. 2013. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res 41: D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B, Wurm JP, Kotter P, Leisegang MS, Schilling V, Buchhaupt M, Held M, Bahr U, Karas M, Heckel A, et al. 2011. The Bowen-Conradi syndrome protein Nep1 (Emg1) has a dual role in eukaryotic ribosome biogenesis, as an essential assembly factor and in the methylation of Ψ1191 in yeast 18S rRNA. Nucleic Acids Res 39: 1526–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Muller S, Behm-Ansmant I, Branlant C. 2007. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol 425: 21–53. [DOI] [PubMed] [Google Scholar]
- Ohira T, Suzuki T. 2011. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc Natl Acad Sci 108: 10502–10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallan PS, Kreutz C, Bosio S, Micura R, Egli M. 2008. Effects of N2,N2-dimethylguanosine on RNA structure and stability: crystal structure of an RNA duplex with tandem m22G:A pairs. RNA 14: 2125–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M, Wang X, Perdrizet G II, Lamphear C, Fierke CA, Maheshwari KC, Wilde MJ, Sosnick TR, Pan T. 2013. Discovering RNA-protein interactome by using chemical context profiling of the RNA-protein interface. Cell Rep 3: 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A, Chan CT, Dyavaiah M, Rooney JP, Dedon PC, Begley TJ. 2012a. Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol 9: 990–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil A, Dyavaiah M, Joseph F, Rooney JP, Chan CT, Dedon PC, Begley TJ. 2012b. Increased tRNA modification and gene-specific codon usage regulate cell cycle progression during the DNA damage response. Cell Cycle 11: 3656–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peattie DA. 1979. Direct chemical method for sequencing RNA. Proc Natl Acad Sci 76: 1760–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK. 2010. tRNA biology charges to the front. Genes Dev 24: 1832–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryvkin P, Leung YY, Silverman IM, Childress M, Valladares O, Dragomir I, Gregory BD, Wang LS. 2013. HAMR: high-throughput annotation of modified ribonucleotides. RNA 19: 1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saikia M, Fu Y, Pavon-Eternod M, He C, Pan T. 2010. Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs. RNA 16: 1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Cedergren R. 1995. A correlation between N2-dimethylguanosine presence and alternate tRNA conformers. RNA 1: 886–891. [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Suzuki T. 2014. A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs. Nucleic Acids Res 42: 7346–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayak M, Pathak C. 2010. Queuosine modification of tRNA: its divergent role in cellular machinery. Biosci Rep 30: 135–148. [DOI] [PubMed] [Google Scholar]
- Wei FY, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, et al. 2011. Deficit of tRNALys modification by Cdkal1 causes the development of type 2 diabetes in mice. J Clin Invest 121: 3598–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipple JM, Lane EA, Chernyakov I, D'Silva S, Phizicky EM. 2011. The yeast rapid tRNA decay pathway primarily monitors the structural integrity of the acceptor and T-stems of mature tRNA. Genes Dev 25: 1173–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborske JM, DuMont VL, Wallace EW, Pan T, Aquadro CF, Drummond DA. 2014. A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol 12: e1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T. 2015. Efficient and quantitative high-throughput tRNA sequencing. Nat Methods 12: 835–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.