

Summary
Viral replication and spreading are fundamental events in the viral life cycle, accounting for the assembly and egression of nascent virions, events that are directly associated with viral pathogenesis in target hosts. These processes occur in cellular compartments that are modified by specialized viral proteins, causing a rearrangement of different cell membranes in infected cells and affecting the ER, mitochondria, Golgi apparatus, vesicles and endosomes, as well as processes such as autophagic membrane flux. In fact, the activation or inhibition of membrane trafficking and other related activities are fundamental to ensure the adequate replication and spreading of certain viruses. In this review, data will be presented that support the key role of membrane dynamics in the viral cycle, especially in terms of the assembly, egression and infection processes. By defining how viruses orchestrate these events it will be possible to understand how they successfully complete their route of infection, establishing viral pathogenesis and provoking disease. © 2015 The Authors Reviews in Medical Virology Published by John Wiley & Sons, Ltd.
Abbreviations used
- ADP
adenosine diphosphate
- ALIX
ALG‐2 (apoptosis‐linked gene 2)‐interacting protein X
- APOBEC3
apolipoprotein B mRNA‐editing enzyme‐catalytic, polypeptide‐like 3
- Arf
ADP‐ribosylation factor
- Arf‐GEF
Arf‐GTP exchange protein
- ASFV
African swine fever virus
- Atg
autophagy‐related protein
- BFA
Brefeldin A
- CCR
C‐C chemokine receptor
- CD
cluster of differentiation
- COPI I and II
clathrin, coatomer protein complex I and II
- CPV
cytopathic vacuole
- CVB3
Coxsackievirus B3
- CXCR
C‐X‐C chemokine receptor
- C3‐PI3K
lipid class III phosphatidylinositol 3‐kinase complex
- DC
dendritic cell
- DENV
dengue virus
- DMVs
double‐membrane vesicles
- dsDNA
double‐stranded DNA
- dsRNA
double‐stranded RNA
- Env
envelope
- ERGIC
ER–Golgi intermediate compartment
- ESCRT
endosomal‐sorting complex required for transport
- GALT
gut‐associated lymphoid tissue
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- GTPase
guanosine triphosphatase
- HDAC6
histone deacetylase 6
- ISG‐15
interferon‐stimulated gene 15 protein
- LC3‐I
microtubule‐associated protein 1A/1B‐light chain 3
- LC3‐II
the phosphatidylethanolamine‐conjugated from LC3‐I
- LHBs
large HBV surface protein
- MA
matrix viral protein
- MLV
murine leukaemia virus
- MVB
multivesicular body
- MT
microtubule
- MTOC
microtubule organizing centre
- NCLDVs
nucleocytoplasmic large DNA viruses
- Nef
negative factor
- NS5A
non‐structural 5A protein
- NS5B
non‐structural 5B protein
- PI4P5‐K Iα
phosphatidylinositol‐4‐phosphate 5‐kinase Iα
- PIP2
phosphatidylinositol‐4,5‐bisphosphate
- prM
precursor membrane
- RC
replication complex
- RUBV
rubella virus
- SAMHD1
sterile alpha motif (SAM) and histidine‐aspartate (HD) domain‐containing protein 1
- SFV
Semliki forest virus
- SQSTM1
sequestosome‐1 (or p62)
- ssRNA
single‐stranded RNA
- SVP
spherical or filamentous envelope particles
- TGN
trans‐Golgi network
- Tsg101
tumour susceptibility gene 101
- UPR
unfolded protein response
- VAMP
vesicle‐associated membrane protein
- VAP
VAMP‐associated protein
- Vif
viral infectivity factor
- Vps
vacuolar protein sorting‐associated protein
- VF
viral factory
- VS
Virological synapse
- 5ptaseIV
polyphosphoinositide 5‐phosphatase IV
Introduction
Viruses are small structures that lack the metabolic pathways and structures necessary to ensure their own survival, relying on their host's machinery to replicate their genome and spread their progeny. Accordingly, viruses have developed strategies to enter cells and exploit their structures to replicate. These strategies also serve to evade immune responses, such as those involving toll‐like receptors and autophagic‐mediated antigen presentation 1, 2, 3, 4. Similarly, viruses use the target cell's main trafficking pathways to ensure their propagation, exploiting the endosome or vesicular compartments by recruiting the clathrin, coatomer protein complex (COPI) I and II (Figure 1), the endosomal‐sorting complex required for transport (ESCRT) and their accessory proteins (reviewed by 1, 2, 5: Figure 2), as well as small guanosine triphosphatases (GTPases) 2. This is evident during neutrophil‐mediated phagocytosis, where microorganisms can be cleared by granule and vesicle secretion 6. Therefore, determining how viruses use and rearrange intracellular organelles during their biological cycle is an important goal that will aid the development of new antiviral strategies, and our understanding of these pathologies. Indeed, there is growing evidence that cell's modify their membranes to defend themselves against pathogens and infection, altering their spatial reorganization and vesicle trafficking. In this review, we focus on the importance of the membrane flux triggered by viruses to achieve replication and egression, and to ensure their propagation.
Figure 1.
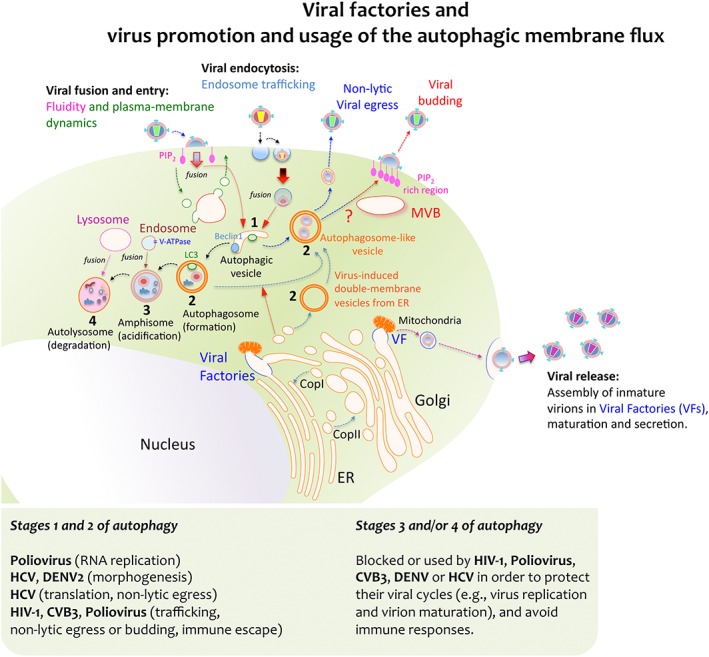
Viral factories and virus‐triggered autophagic membrane flux for replication and egression. Some viruses achieve replication by exploiting the cell's membrane transport pathways, thereby generating membrane organelles named Viral Factories (VFs). These VFs are organised by different viral proteins, and they represent specialized compartments for viral‐gene replication, morphogenesis, export, maturation and release. Moreover, these compartments also serve to override or evade the immune responses directed against viral genomes. Viral proteins can enter secretory pathways by co‐translational translocation into the ER in order for them to be further transported to the Golgi complex, either in vesicles or in a coatomer protein complex (COP) II‐dependent manner. Viral complexes formed inside the VFs communicate with vesicles, mitochondria, Golgi cisternae and ER membranes. This interaction allows viral complexes to be transported through the Golgi network to the plasma membrane and it promotes their final release as viral particles. Alternatively, some viruses take advantage of the host's autophagic machinery for their own replication and pathogenesis. Viruses first initiate the formation of vesicles that bear key autophagic proteins, such as Beclin‐1 and LC3, capturing portions of membranes from the ER and other cytoplasmic elements. This assembly evolves toward an immature double‐membrane vesicle (DMV) that serves as an aggresome compartment to recruit viruses or newly formed viral replication complexes. Several RNA viruses induce the formation of these autophagosome‐like vesicles (also referred to as DMVs) to enhance viral replication and non‐lytic egression, such as poliovirus and CVB3, HIV‐1 and HCV. How these viruses trigger the accumulation of autophagosome‐like vesicles and DMVs remains unclear. Some theories involve blocking the fusion of nascent autophagosomes with late endosomes and lysosomes, as in the case of HIV‐1 Nef, which appears to cause autophagosome accumulation by inhibiting their progression towards more mature stages. Indeed, autophagosome‐like vesicles may represent a trafficking pathway for these viruses, connecting to multivesicular bodies (MVBs), and assuring virus assembly and budding at the cell surface while protecting them from intrinsic antiviral factors and immune responses. The morphogenesis and release of mature and infectious HBV particles also require Tsg101 and depend on the ESCRT‐MVB system. Under standard conditions the lumen of autophagosomes acidifies after fusion with endosomes that carry vacuolar (H+)‐ATPase (V‐ATPase) to form amphisomes. The autophagic membrane flux progresses by fusing with lysosomes in order to form the autolysosome that contains the former's proteinases. Poliovirus inhibition of autophagosome formation attenuates viral replication while inhibiting autolysosome formation, and thus, catalytic activity does not affect the virus. However, degradation of cellular triglycerides by autophagy benefits DENV replication and autolysosome degradation dampens IFN activation following HCV infection
Figure 2.
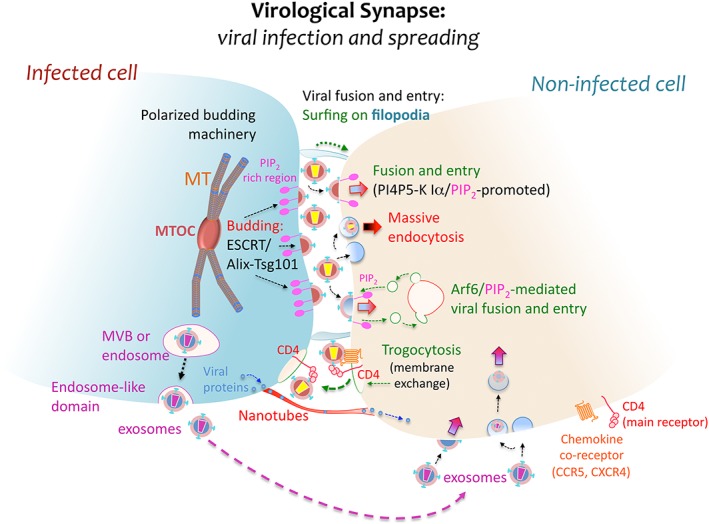
Virological synapse and spreading. At the virological synapse (VS), some viruses attach structural polyproteins to PIP2‐rich membrane regions of the infected cell for further budding and release into the intercellular space. PIP2 confers fluidity to the cell membrane and favours virus–cell fusion. These virions then bind to specific receptors in order to infect the neighbouring target cell at the VS, fusing with its plasma membrane directly or after surfing on actin‐structured filopodia, or being internalized by endocytosis as is believed to occur with HIV‐1. The VS represents an efficient environment for viral budding. It typically arises in PIP2‐enriched plasma membrane domains, where the membrane of the infected cell is polarized towards the synaptic junction through the movement of vesicles governed by the ESCRT/Alix‐Tsg101 machinery or by MVBs coordinating the translocation of the MTOC. This scaffolding facilitates subsequent viral infection and spread from the infected to the nearby uninfected cell. In addition, long membrane nanotubes may also form between neighbouring cells, promoting viral protein trafficking. Other dynamic membrane events involved in viral infection and spreading are trogocytosis, Arf6/PIP2‐mediated membrane dynamics and exosomal transport. Trogocytosis involves the exchange of cell surface membrane patches that may contain receptor clusters associated to viral particles, while exosomes are vesicles formed from MVBs that could participate in viral infection and spreading between cells
Membrane dynamics during viral replication
Several of the cell's organelles and membrane structures are involved in viral replication and in fact, many viruses use specific cellular compartments to replicate, referred to as viral factories (VFs: Figure 1). These VFs provide a physical scaffold that brings together elements required for genome replication and morphogenesis 1, 7. VFs are usually formed by rearranging the host's cell membranes, reorganizing the cytoskeleton and recruiting specific organelles, like mitochondria (reviewed in 8). These viral driven events involve the association of replication complexes (RCs) with ER derived membranes to form a VF. Hence, intracellular membrane dynamics appear to be crucial for viral replication and survival.
A well‐known example of a VF is that used by vaccinia virus, an enveloped pathogen of the Poxvirus family that replicates in the cytoplasm by assembling small rough ER‐derived cisternae into a microenvironment that resembles a cytoplasmic mini‐nucleus for viral replication 9. Similarly, the RCs of Togaviruses associate with endocytic membranes, while Nodavirus RCs associate with mitochondrial membranes (reviewed in 1). Thus, specific membrane compartments can be used as VFs by RNA viruses to concentrate viral replicases and key cofactors, and ensure efficient viral genome replication 10. In this context, both rubella virus (RUBV), a relevant human teratogenic Togaviridae virus 11, and Semliki forest virus (SFV), a member of the Alphavirus group of this family 12, couple their RNA synthesis to endosome and lysosome membranes modified by the association of virus specific components. The subsequent fusion of these late endosomes and lysosomes generates cytopathic vacuoles (CPVs) 13, 14 that are lined with small vesicular invaginations or spherules (viral RNA replication sites) 13, 14. CPVs establish complex and reversible contact with endocytotic vesicles through internal membranes interconnected with transport endosomes 8. For example RUBV forms VFs around CPVs via the recruitment of membrane structures from the ER cisternae, Golgi stacks and mitochondria 8 (Figure 1). The Golgi apparatus is a highly dynamic organelle with a sustained, functional flux of membrane proteins 15, and it can serve as a morphogenic mould for Rubiviruses, Coronaviruses, Arteriviruses and Bunyaviruses 1, 8, 16, 17. These RUBV factories connect viral replication with the assembly and maturation of nascent virions at Golgi membranes, contributing to the virus escaping from the host cell's defences 16.
Some viruses induce the formation of double‐membrane vesicles (DMVs) and/or autophagosomes for replication 1, 2, 3, 4, 18, 19, such as the positive RNA viruses of the Flaviviridae family and Nidovirales order 20, 21 (Figure 1). The RNA polymerase of the human poliovirus, a Picornaviridae family member responsible for poliomyelitis, can also assemble DMVs 22. Infection by this virus triggers the modification of different intracellular membrane structures and organelles (but not mitochondria), converting them into virus replication vesicles. In fact, poliovirus‐associated DMVs resemble autophagosomes, as also described for another Picornaviridae family member, Coxsackievirus B3 (CVB3 23, 24: Figure 1). Autophagosomes are DMVs generated by membrane trafficking and they are related to the catabolic process of autophagy, which involves the degradation of cytoplasmic components within lysosomes 25, 26. Autophagy maintains the organism's homeostasis by sequestering undesired intracellular elements for lysosomal degradation and recycling 25, 26. Viruses often use autophagy to complete their lifecycle and evade immune responses, even though it is based on catalytic pathways 3, 23, 24. Poliovirus, like other positive RNA viruses, has evolved the capacity to convert autophagy into a key cellular motor for replication 3, 10, 23. During autophagy, a cytosolic form of the microtubule‐associated protein 1A/1B light chain 3 (LC3‐I) conjugates with phosphatidylethanolamine to form LC3‐II and associate with autophagosomal membranes, ultimately producing the degradation of LC3‐II during the late steps of autophagy 27. Conversely, the p62 protein (or sequestosome‐1, SQSTM1) interacts with ubiquitinated proteins, LC3 and other proteins to ensure the correct degradation of undesired material. LC3‐II augments during active autophagy when p62 is degraded 28, 29. In this context, Poliovirus or CVB3 infection triggers the generation of autophagosomes with a higher LC3‐II/LC3‐I ratio and with LC3 foci, structures that support the RNA RC without promoting lysosome degradation (evident through p62 stabilization 23, 24: Figure 1). However, it is unclear whether these viruses block autophagosome maturation into amphisomes, avoiding autophagosome fusion with endosomes 30. Such events override the appearance of degradative autolysosomes 31 or they may provoke the formation of autophagosome‐like structures disconnected from catalytic pathways. It is also thought that these autophagosomes may ultimately serve as a membrane scaffold to permit the egression of nascent virions from infected cells, preventing cell lysis 30 (Figure 1).
All HCV viral genotypes (1a, 1b and 2a), positive RNA flaviviruses that are a major cause of chronic liver disease 32, induce autophagosome accumulation 33, 34. This involves regulation of the unfolded protein response (UPR), which relieves ER stress and prevents the formation of catalytic autolysosomes by suppressing their fusion with lysosomes 33, 34 (Figure 1). Apparently, the success of viral replication relies on the recruitment of membrane‐trafficking proteins to ER‐derived membrane scaffolds 35, 36, 37, 38, 39, 40, 41. Hence, domain 1 of the non‐structural 5A (NS5A) protein and the helicase domain of NS3 are sufficient to achieve efficient DMV formation, which also depends on tightly regulated cis cleavage of the HCV‐polyprotein precursor 35 and requires cyclophilin A isomerase activity 36. NS5A associates with NS5B, a RNA‐dependent RNA polymerase, a complex that interacts with VAMP (vesicle‐associated membrane protein)‐associated proteins (VAPs) 37, 38 and recruits Ras‐like small GTPases (e.g. Rab1, Rab5 and Rab7), enlarging the viral replication compartment by docking membrane vesicles 39, 40, 41. This process also regulates autophagy 42, given that HCV‐induced autophagosomes support viral replication and the delivery of incoming viral RNA to the translation apparatus, and/or the recruitment of cellular factors for translation. However, some controversy still surrounds this issue, autophagosomes can mature into acidic amphisomes in HCV‐infected cells 43, 44, and subsequently fuse with late endosomes or lysosomes 44 (Figure 1). Autophagic membrane flux appears to be necessary to translate the HCV genome, yet it appears to be dispensable once viral infection has begun 45. Moreover, no changes in either p62 or the degradation of long‐lived proteins are observed 33, despite the enhanced autolysosome formation in cells expressing HCV replicons 46. While specific silencing of autophagy genes does not affect viral translation and RNA replication, it does apparently alter HCV morphogenesis 47. However, the silencing of factors critical for autophagosomes formation, like LC3 or Atg7, appears to suppress HCV RNA replication 48, while HCV replication is apparently potentiated when the UPR promotes autophagy 49. Conversely, HCV infection seems to promote autophagy without concomitant stimulation of the UPR and autophagy does not appear to be required as a platform for HCV RNA replication 50. Thus, doubts remain about the role of autophagy and the UPR in HCV replication, although the distinct interactions between autophagy and HCV replication suggest that such membrane flux promotes viral replication.
The dengue virus (DENV) is a mosquito‐borne single positive‐stranded RNA virus of the Flaviviridae family that causes dengue fever 51. There are five antigenically related but distinct DENV virus genotypes (DENV‐1 to DENV‐5) 51, 52. Like HCV, there is evidence that autophagy may be implicated in DENV replication. Following cell entry and nucleocapsid uncoating, DENV RNA is translated into a single polyprotein that passes into the ER lumen where the different viral proteins are processed. In fact, DENV‐2 proteins involved in translation and replication are found in or in close proximity to autophagosomes during viral infection 53, 54. Accordingly, inhibition of autophagosome formation dampens the production of infectious DENV‐2 particles 53, while stabilizing autophagosomes and/or amphisomes by impeding their fusion with lysosomes enhances viral egression 55. Indeed, DENV‐3 seems to promote autophagy during early infection 54, while inhibition of autophagosome formation also dampens the production of infectious DENV‐3 54. Hence, DENV‐2 and ‐3 appear to interact with the autophagy machinery in a different manner, and while it is conceivable that amphisomes or autophagosomes represent the site of DENV‐2 translation/replication 54, 55, autophagolysosomes could be the crucial site for DENV‐3 viral replication 54. The distribution of NS1 or DENV‐2 and DENV‐3 double‐stranded RNA (dsRNA) in the different autophagy‐associated membrane structures confirms these observations (Figure 1). Moreover, nascent viral particles are formed and mature in these structures, then travelling through the trans‐Golgi network (TGN) to egress 56, 57. Remarkably, the precursor membrane (prM) protein of the DENV‐1–4 genotypes behaves similarly and it is cleaved by the TGN‐protease furin in the secretory pathway 58, assuring viral assembly and the infectivity of nascent viral particles 59.
HIV is a single‐stranded RNA virus (Lentivirus genus of the Retroviridae family) that causes AIDS. HIV type 1 (HIV‐1) alters the autophagic membrane flux of the host cell's organelles, thereby modulating the intracellular milieu in favour of viral replication and propagation 3 (Figure 1). When CD4+ T cells, monocytes and dendritic cells (DCs) are infected with HIV‐1, autophagic vacuole formation is blocked and the expression of autophagy proteins down‐regulated (e.g. LC3 and Beclin1 19, 60: Figure 1). Remarkably, the HIV‐1 protein Nef (negative factor) blocks the autophagic flux of membranes, especially during the autolysosome stage of autophagy, resulting in an accumulation of autophagosomes and LC3 in macrophages (Figure 1). Thus, Nef prevents autophagic degradation of HIV‐1 biosynthetic intermediates of virions by targeting the lipid class III phosphatidylinositol 3‐kinase (C3‐PI3K) complex and by associating with Beclin1 (Atg6—autophagy‐related protein 6—in yeast). Significantly, Beclin1 is actually part of the C3‐PI3K complex, together with the vacuolar protein sorting‐associated proteins 34 (Vps34) and 15 (p150). Nef therefore alters the sub‐cellular distribution of Vps34, potentially ensuring the survival of the viral progeny 3, 61. Indeed, Nef is thought to promote the appropriate HIV‐1 Gag membrane localization and processing, thereby facilitating viral cell‐to‐cell transfer 62. Although the catalytic activity of autophagy appears to be impeded by HIV‐1, autophagosome formation or accumulation is still promoted. Hence, the HIV‐1 Gag protein promotes early stages of autophagosome formation by directly interacting with LC3 in macrophages, enhancing HIV‐1 yields and Gag processing, a critical step in virion assembly and release 61 (Figure 1). Notably, newly identified components of the ubiquitin‐like conjugation system all seem to be involved in HIV‐1 replication (e.g. Atg7, Atg8—LC3 is its best characterized mammalian homologue—Atg12 and Atg16L2—responsible for vesicle elongation) 63. However, it remains unclear how these factors actually affect HIV‐1 replication, which occurs in the nucleus of infected cells. Moreover, while autophagic vacuoles would appear to be fundamental for HIV‐1 morphogenesis and egression, how HIV‐1 overrides or uses autophagy to persist remains poorly understood. Hence, the infectious capacity of nascent HIV‐1 virions depends on the uptake of the viral infectivity factor (Vif) during viral budding, a process influenced by histone deacetylase 6 (HDAC6), which promotes autophagic clearance of Vif 64. Other positive RNA viruses exploit the formation of ER‐derived membrane scaffolds and membrane autophagic flux to replicate (e.g. the Norwalk virus), because the membrane‐bound nsp48 protein also binds to VAP‐A 65.
RNA replication may occur in endosomes, lysosomes (Togaviruses), peroxisomes and chloroplasts (Tombusvirus), or mitochondria (Nodaviruses), shielded from immune responses. All positive RNA viruses transform cytoplasmic membranes into specialized viral replication sites 10. The antiviral effect of Brefeldin A (BFA), an inhibitor of anterograde ER–Golgi network membrane dynamics, suggests that membrane trafficking must be active for enterovirus replication, as reported for Picornaviruses, poliovirus and Coxsackievirus 66, 67. BFA prevents the membrane flux required to form replication compartments, blocking virus secretion from infected cells 68 by inhibiting ADP (adenosine diphosphate)‐ribosylation factor (Arf)‐GTP exchange proteins (Arf‐GEFs). This blockade negatively affects COPI coat generation at the Golgi by diminishing and sequestering Arf1‐GTP 69. For several Picornaviruses, COPII‐coated vesicles may provide membranes suitable for replication 70, although autophagosomes may also contribute at this point 23 (Figure 1). Reovirus and SFV also promote coated‐pit formation 71. Moreover, the small GTPase Rab7 is soon recruited for SFV internalization when associated to intermediate endosomes 72, which in turn induces the formation of CPVs that is an important event for viral RNA synthesis in target cells 13.
An important biological process common to the recently proposed Megavirales order is viral replication within cytoplasmic VFs 73. Giant viruses (also called nucleocytoplasmic large DNA viruses—CLDVs) belonging to this order are double‐stranded DNA (dsDNA) viruses with a genome and particle size comparable to those of small bacteria 74. African swine fever virus (ASFV; from the Asfarviridae family), poxviruses and iridoviruses are the three families of NCLDVs that terminate or undergo their entire replication cycle in the cytoplasm 75, 76, 77. This feature is not observed in herpes viruses or baculoviruses, other large DNA viruses of eukaryotes that undergo nuclear DNA replication and transcription 78. Giant viruses provoke VF formation in the cytoplasm of infected cells to permit genome replication and morphogenesis 73, 79. ASFV factories are similar to the aggresomes formed at the MTOC (microtubule organizing centre) 80, and they provoke a re‐arrangement of the intermediate vimentin cytoskeleton at the MTOC into a star shaped structure that resembles the microtubule aster formed during mitosis, a structure required for late gene expression 81. Together with an ASFV chaperone, the hsp70 cell chaperone is recruited to ASFV factories, along with mitochondria, facilitating the folding of viral structural proteins like the major capsid protein p72 82, 83. Nascent ASFV virions are formed from VF membranes through the assembly and recruitment of viral proteins in VFs. Thus, the viral membranes in VFs may be connected to cellular organelles, particularly given that resident ER markers are detected with the viral p17, p54 and pB318L proteins in new viral particles 84, 85, 86, 87. ASFVs are thought to reorganize cell membranes through viral proteins that contain a KDE motif, inducing the redistribution of ER‐associated proteins 88 and the viral p54 protein. The latter is required for the correct VF localization of the membranes and the collapse of the ER‐derived cisternae 89. ASFV infection is achieved by redistributing membranes from the secretory pathway and TGN 90. Therefore, these common biological features of giant virus replication and virion architecture could reflect a common origin, and the sharing of a large set of ancestral genes 74, 91.
Membrane dynamics during viral assembly and budding
Budding is an important event in the life cycle of enveloped viruses, influencing their morphology and infectiousness. During budding, successful infection is achieved by adjustment and distortion of the target cell's plasma membrane 4. The structural Gag polyprotein is common to several retroviruses, like HIV‐1 and the murine leukaemia virus (MLV), representing the minimal plasma membrane component required for viral assembly 92. HIV‐1 Gag localizes to phosphatidylinositol‐4,5‐bisphosphate (PIP2) rich plasma membrane regions, where PIP2 plays a critical role in HIV‐1 virion assembly 93 (Figures 1 and 2). In fact, the matrix viral protein (MA) within the unprocessed HIV‐1 Gag polypeptide drives Gag towards these PIP2 membrane domains 92, 94 in a myristoylation‐dependent manner 95, raft domains where HIV‐1 buds 95, 96, 97. Phosphate hydrolysis by polyphosphoinositide 5‐phosphatase IV (5ptaseIV) diminishes the plasma membrane PIP2 98, causing the Gag polypeptide to translocate from HIV‐1 budding sites at the membrane to CD63 rich compartments, thereby inhibiting viral release 93. Similarly, Arf6/Q67L expression, a GTP‐bound mutant of Arf6, alters the trafficking of Arf6/PIP2‐associated vesicles, provoking their accumulation in the cytoplasm to where Gag is redirected. These complexes lie far from the budding sites at the membrane, thereby dampening virus release 93. Although the assembly of HIV‐1 at the cell surface is only partially understood, several key steps in the membrane trafficking of viral proteins have been defined, shedding light on both the viral assembly and budding processes 92, 99.
Enveloped viruses like HIV‐1, Vesicular stomatitis virus (VSV), Ebola virus (EBOV) and Hepatitis B virus (HBV), and other RNA and DNA viruses, mainly emerge from cells by co‐opting the host's ESCRT machinery 100, 101, which plays a vital role in cellular abscission and in multivesicular body (MVB) biogenesis (a process by which ubiquitinated misfolded or damaged proteins enter endosomes to be destroyed). In addition, MVBs are important intermediates in endolysosomal transport 102 (Figures 1 and 2). Gag activity drives ESCRT‐III complex formation at the budding site of HIV‐1, which binds to and recruits the ESCRT‐I complex and the ALG‐2 (apoptosis‐linked gene 2)‐interacting protein X (ALIX). This ESCRT‐III complex promotes the excision of nascent virions at the cell surface, an event potentially equivalent to the cleavage of intraluminal vesicles from MVBs 103, 104 (Figures 1 and 2). Moreover, the tumour susceptibility gene 101 (Tsg101) is a subunit of the ESCRT‐I complex that drives viral RNA transport and envelope fusion to late endosomes, processes required for infection and RNA release 105 (Figure 2). However, the interaction of viral Gag protein with the ESCRT machinery appears not to be absolutely required for HIV‐1 viral budding 106, 107, 108. Nevertheless, interferon‐stimulated gene 15 protein (ISG‐15) inhibits HIV‐1 egression by interfering with ESCRT‐III protein membrane flux during budding 109, 110.
Remarkably, morphogenesis and the release of HBV particles also require Tsg101 111, although this DNA virus lacks a viral protein bearing the late (L) domain necessary to interact with the ESCRT‐machinery 101. However, α‐taxilin interacts directly with Tsg101 and with the large HBV surface protein (LHBs), thereby recruiting the viral capsids to ESCRT complexes, thus permitting correct viral formation and egression 111. Therefore, HBV maturation and egression depends on the ESCRT‐MVB system. Notably, HBV infected cells also produce large amounts of non‐infectious spherical or filamentous envelope particles (SVPs). These SVPs are a mixture of lipids and viral surface proteins that accumulate in an ER–Golgi intermediate compartment (ERGIC), budding into the lumen and provoking release through the general secretory pathway 112.
Many other enveloped RNA viruses bud in an ESCRT‐dependent manner 5, 100, 113, as do most negative‐strand non‐segmented single‐stranded RNA (ssRNA) viruses, such as Rhabdoviruses, Filoviruses and most Paramyxoviruses, all of which recruit ESCRTs for viral egression 114, 115. Even the budding of negative‐strand segmented‐ssRNA Arena viruses involves an ESCRT‐dependent pathway 116, 117. However, no evidence for the participation of ESCRTs has yet been reported in Nipah, Measles, HRSV or Bornaviridae budding (5). Indeed, the enveloped influenza virus buds in an ESCRT‐independent manner as its matrix protein is devoid of an ESCRT‐binding domain 118, 119. Other viruses are also released from the host's plasma membrane through their MAs, such as the Newcastle disease virus or VSV. In these cases, bud formation and excision from the membrane are matrix‐dependent processes 120, 121, as for influenza virus. However, much work is still required to determine how membrane dynamics affect the trafficking and assembly of these viruses, particularly in terms of the cellular factors that control the trafficking of the structural proteins of these viruses to the plasma membrane 92, 122.
Given all of these findings, membrane dynamics has a crucial influence on the assembly and budding of numerous viruses, and it may represent an important and complex target to limit the viral life cycle.
Membrane dynamics and viral spreading
Viruses use various cell communication pathways to achieve effective cell‐to‐cell dissemination 123. First described for type 1 HTLV (HTLV‐1) 124, the virological synapse (VS) is a complex structure found at the interface of infected:uninfected cells. Viral receptors and the egression machinery accumulate at the VS 125, making the infection and spread of HTLV‐1 through T lymphocytes cell–cell dependent. Direct cell‐to‐cell transmission facilitated by the formation of stable cellular junctions has several advantages, including faster replication rates 126, successful transmigration of infected cells across mucosal barriers 123 and viral protection from host responses. However, such transmission is still to be confirmed for HIV 127, 128.
Cell‐to‐cell spreading of HIV‐1 (Figure 2) is considered to involve microtubule‐mediated polarization and substantial budding, followed by the entry of free viral particles into target cells 129. Thus, it involves pathways that regulate cell‐free virus entry by modifying membrane dynamics. In this regard, most HIV‐1‐infected GALT (gut‐associated lymphoid tissue) cells in intestinal crypts are infected by concentrated pools of free HIV‐1 viral particles in HIV‐1‐infected humanized mice. Fewer infected cells are found in the mucosal regions and the lamina propria, where VS presumably occur 130, explaining why infection of permissive cells by free viral particles is crucial for HIV‐1 replication and pathogenesis in vivo. This is consistent with the recent identification of the key cell signals required for efficient early HIV‐1 infection and the establishment of latency in CD4+ T cells 131, 132, 133, 134, 135, 136. Interference with retroviral cell‐to‐cell transmission is not only produced by blocking cytoskeletal motility 137 and depleting membrane‐cholesterol 138 but also, by interfering with Arf6‐governed plasma membrane dynamics. Moreover, restricting plasma membrane fluidity caused by altering early HIV‐1‐triggered phosphatidylinositol‐4‐phosphate 5‐kinase (PI4P5‐K) Iα activation and the ensuing detrimental effects PIP2 production on HIV‐1 transmission 4, 133, 135. In fact, Arf6‐coordinated membrane trafficking is required for efficient HIV‐1 fusion, entry and infection of CD4+ T lymphocytes 135 (Figures 1 and 2). The flux and turnover of PIP2‐enriched vesicles from the plasma membrane, driven and coordinated by the Arf6‐GTP/GDP cycle, ensures cell surface membrane regeneration and it allows membrane exchange between the viral and target cell surfaces. This type of membrane trafficking, coupled with enhanced fluidity, is in strong synergy with the key HIV‐1/receptor (CD4 and C–X–C or C–C chemokine receptor type 4 or 5—CXCR4 or CCR5), interactions that promote fusion pore formation in target cells. These interactions take place between the non‐regenerative HIV‐1 viral membrane and the dynamic cell surface membrane, favouring efficient virus–cell fusion, entry and infection, both for the free virus and in the context of the VS 4, 133, 135 (Figures 1 and 2).
Despite these similarities, some contact‐specific events that affect membrane flux should also be considered. During cell–cell HIV transmission, intense viral endocytosis drives entry into neighbouring cells even if they are in contact 139 (Figure 2). Remarkably, biofilm‐like structures at the surface of infected cells concentrate HTLV‐1 viruses for their efficient transmission to target cells 140 and cellular projection is used to transmit pseudo‐rabies virus. Retroviruses also travel along membrane protrusions that contact adjacent cells and MLV surfs on the filopodia of fibroblasts before entering cells 141 (Figure 2). HIV‐1 also takes advantages of filopodia for cell‐to‐cell transmission 141, similarly surfing on the narrower membranous nanotubes that connect cells separated no more than 100 µm 142 and facilitating the transfer of viral proteins to the inner side of the membrane. These actin structures extend from HIV‐infected cells to target cells irrespective of receptor‐envelope interactions 142 (Figure 2).
The take up of larger membrane invaginations at the VS of connected cells 129 is known as trogocytosis, an event that may also control the extent and stability of the synapse, regulating its duration 143. HIV particles, like CD4 molecules and other membrane components, are transferred by trogocytosis from uninfected to infected cells in a manner triggered by the HIV‐1 envelope (Env)/CD4 128 (Figure 2). This mechanism could be very significant and render cells permissive to HIV infection, as recently proposed 144.
The cell–cell contacts and signalling induced by the HIV‐1 Env complex that occur at the VS can activate autophagic membrane flux, leading to apoptotic cell death of uninfected CD4+ T cells 60, 145. This lethal autophagy may provoke or enhance immunodeficiency, as observed in vivo where the majority of CD4+ T cells undergoing apoptosis, as well as the peripheral blood and lymph nodes of HIV patients, remain uninfected 146. Simultaneously, autophagy can be suppressed in infected CD4+ T cells 60, thereby antagonizing Env‐mediated apoptosis and allowing viral replication to occur in infected CD4+ T cells. In this context, HIV‐1 evades immune responses in an HIV‐1 Env‐CD4‐dependent manner by efficiently impairing autophagy in DCs when early contacts are established 19. Taking into account the role autophagy plays in viral replication, HIV‐1 can enhance or suppress autophagy at different stages of its viral cell cycle, favouring persistence and the evasion of immune responses, and therefore, its pathogenesis 19 (Figure 1). Finally, like other viruses (e.g. CMV), HIV‐1 stably associates with professional APCs during infection (such as DCs) to further infect lymphocytes during T‐cell scanning or antigen presentation 147. In fact, HIV‐1 enters DCs by exploiting exosomal trafficking during antigen presentation 148 (Figure 2).
Concluding Remarks
This review examines the intracellular trafficking of viruses that occurs in association with cell‐membrane structures, some of which may be newly assembled by viruses to ensure their replication and budding. Membranes derived from the ER, mitochondria, lysosomes and endosomes are sculpted by viruses to generate VFs, acquiring their own functional morphology. These structures help ensure RNA replication is accomplished without alerting the host's defence mechanisms.
Although the importance of membrane dynamics during viral infection has been established, several questions remain unanswered. It remains unclear how proteins from distinct viruses and host cells use the same intracellular membrane compartments or events (e.g. autophagy) to achieve viral replication, without affecting important cellular processes. Conversely, it is not clear why viruses replicate in different subcellular membrane compartments, how they move across membranes and which host factors are involved in these events. Similarly, we still do not know how these changes in membrane dynamics enable viruses to avoid immune responses. Indeed, it remains unclear whether rearranging intracellular organelles enables viruses to escape the anti‐replicative activity of natural restriction factors, such as apolipoprotein B mRNA‐editing enzyme‐catalytic, polypeptide‐like 3 (APOBEC3) proteins, Tetherin (BST‐2/CD317/HM1.24) or SAMHD1 (sterile alpha motif (SAM) and histidine‐aspartate (HD) domain‐containing protein 1) for HIV‐1 149. Resolving these issues will help decipher how viruses rearrange membranes during their infection cycle, thereby aiding the design of new antiviral strategies that target these dynamic viral‐cell interactions and combat viral infection. These findings may also produce innovations in non‐viral gene delivery systems to tackle tumours and immune diseases.
New technical developments, such as more powerful microscopy systems 4, 8, 135, 150, will allow dynamic viral trafficking and egression to be studied in cells with better spatial and temporal resolution. Such information will further our understanding of the viral infection process and of how viruses succeed in deceiving the host's immune responses.
Conflict of Interest
The authors have no competing interests to declare.
Acknowledgements
This work and A.V.‐F. are supported by the European Regional Development Fund (ERDF), SAF2008‐01729 and SAF2011‐24671 (MICINN and MINECO, respectively, Spain), UNLL10‐3E‐783 (ERDF) and Fundación CajaCanarias, and by the Project RD12/0017/0034 integrated in the “Plan Nacional I + D + i” and co‐funded by ISCIII‐“Subdirección General de Evaluación” and ERDF (RIS‐RETIC) grants. M‐S.V and L.A‐R are supported by RD12/0017/0034‐(RIS‐RETIC) and SAF2011‐24671‐FPI‐associated grants and fellowships, respectively. We thank Dr Mark Sefton (Biomedred SL) for his linguistic revision of the manuscript. We apologize for all research studies and reviews that we have not discussed or cited in this Review. We have tried to avoid any and all such omissions but space limitations have surely made this an impossible endeavour.
de Armas‐Rillo, L. , Valera, M.‐S. , Marrero‐Hernández, S. , and Valenzuela‐Fernández, A. (2016) Membrane dynamics associated with viral infection. Rev. Med. Virol., 26: 146–160. doi: 10.1002/rmv.1872.
References
- 1. Miller S, Krijnse‐Locker J. Modification of intracellular membrane structures for virus replication. Nature Reviews Microbiology 2008; 6: 363–374 nrmicro1890 [pii]. DOI: 10.1038/nrmicro1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gruenberg J. Viruses and endosome membrane dynamics. Current Opinion in Cell Biology 2009; 21: 582–588 S0955‐0674(09)00091‐X [pii]. DOI: 10.1016/j.ceb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 3. Kim HJ, Lee S, Jung JU. When autophagy meets viruses: a double‐edged sword with functions in defense and offense. Seminars in Immunopathology 2010; 32: 323–341. DOI: 10.1007/s00281-010-0226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barroso‐Gonzalez J, Garcia‐Exposito L, Puigdomenech I, et al. Viral infection: moving through complex and dynamic cell‐membrane structures. Communicative & Integrative Biology 2011; 4: 398–408. DOI: 10.4161/cib.4.4.16716 1942‐0889‐4‐4‐8 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weissenhorn W, Poudevigne E, Effantin G, Bassereau P. How to get out: ssRNA enveloped viruses and membrane fission. Current Opinion in Virology 2013; 3: 159–167 S1879‐6257(13)00035‐7 [pii]. DOI: 10.1016/j.coviro.2013.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes and Infection 2003; 5: 1317–1327. [DOI] [PubMed] [Google Scholar]
- 7. Netherton C, Moffat K, Brooks E, Wileman T. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Advances in Virus Research 2007; 70: 101–182 S0065‐3527(07)70004‐0 [pii]. DOI: 10.1016/S0065-3527(07)70004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harak C, Lohmann V. Ultrastructure of the replication sites of positive‐strand RNA viruses. Virology 2015; 479‐480: 418–433 S0042‐6822(15)00075‐6 [pii]. DOI: 10.1016/j.virol.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tolonen N, Doglio L, Schleich S, Krijnse LJ. Vaccinia virus DNA replication occurs in endoplasmic reticulum‐enclosed cytoplasmic mini‐nuclei. Molecular Biology of the Cell 2001; 12: 2031–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salonen A, Ahola T, Kaariainen L. Viral RNA replication in association with cellular membranes. Current Topics in Microbiology and Immunology 2005; 285: 139–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Frey TK. Molecular biology of rubella virus. Advances in Virus Research 1994; 44: 69–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kujala P, Ikaheimonen A, Ehsani N, Vihinen H, Auvinen P, Kaariainen L. Biogenesis of the Semliki Forest virus RNA replication complex. Journal of Virology 2001; 75: 3873–3884. DOI: 10.1128/JVI.75.8.3873-3884.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Froshauer S, Kartenbeck J, Helenius A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. The Journal of Cell Biology 1988; 107: 2075–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee JY. Rubella virus replication complexes are virus‐modified lysosomes. Virology 1998; 240: 57–63 S0042‐6822(97)98906‐6 [pii]. DOI: 10.1006/viro.1997.8906. [DOI] [PubMed] [Google Scholar]
- 15. James Morre D, Mollenhauer HH. Microscopic morphology and the origins of the membrane maturation model of Golgi apparatus function. International Review of Cytology 2007; 262: 191–218 S0074‐7696(07)62004‐X [pii]. DOI: 10.1016/S0074-7696(07)62004-X. [DOI] [PubMed] [Google Scholar]
- 16. Risco C, Carrascosa JL, Frey TK. Structural maturation of rubella virus in the Golgi complex. Virology 2003; 312: 261–269 S0042682203003842 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Salanueva IJ, Novoa RR, Cabezas P, et al. Polymorphism and structural maturation of bunyamwera virus in Golgi and post‐Golgi compartments. Journal of Virology 2003; 77: 1368–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson DC, Baines JD. Herpesviruses remodel host membranes for virus egress. Nature Reviews Microbiology 2011; 9: 382–394 nrmicro2559 [pii]. DOI: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 19. Blanchet FP, Moris A, Nikolic DS, et al. Human immunodeficiency virus‐1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 2011; 32: 654–669 S1074‐7613(10)00160‐3 [pii]. DOI: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Meer Y, van Tol H, Locker JK, Snijder EJ. ORF1a‐encoded replicase subunits are involved in the membrane association of the arterivirus replication complex. Journal of Virology 1998; 72: 6689–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hall RA, Scherret JH, Mackenzie JS. Kunjin virus: an Australian variant of West Nile? The Annals of the New York Academy of Sciences 2001; 951: 153–160. [PubMed] [Google Scholar]
- 22. Hobson SD, Rosenblum ES, Richards OC, Richmond K, Kirkegaard K, Schultz SC. Oligomeric structures of poliovirus polymerase are important for function. The EMBO Journal 2001; 20: 1153–1163. DOI: 10.1093/emboj/20.5.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jackson WT, Giddings TH Jr, Taylor MP, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biology 2005; 3: e156 03‐PLBI‐RA‐0370R3 [pii]. DOI: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong J, Zhang J, Si X, et al. Autophagosome supports coxsackievirus B3 replication in host cells. Journal of Virology 2008; 82: 9143–9153 JVI.00641‐08 [pii]. DOI: 10.1128/JVI.00641-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eskelinen EL. Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005; 1: 1–10 1270 [pii]. [DOI] [PubMed] [Google Scholar]
- 26. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nature Cell Biology 2007; 9: 1102–1109 ncb1007‐1102 [pii]. DOI: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 27. Mizushima N. Autophagy: process and function. Genes and Development 2007; 21: 2861–2873 21/22/2861 [pii]. DOI: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 28. Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin‐induced cell death. The Journal of Cell Biology 2005; 171: 603–614 jcb.200507002 [pii]. DOI: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. The Journal of Biological Chemistry 2007; 282: 24131–24145 M702824200 [pii]. DOI: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 30. Richards AL, Jackson WT. That which does not degrade you makes you stronger: infectivity of poliovirus depends on vesicle acidification. Autophagy 2013; 9: 806–807 23962 [pii]. DOI: 10.4161/auto.23962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dunn WA Jr. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. The Journal of Cell Biology 1990; 110: 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoofnagle JH. Course and outcome of hepatitis C. Hepatology 2002; 36: S21–29 S0270913902001684 [pii]. DOI: 10.1053/jhep.2002.36227. [DOI] [PubMed] [Google Scholar]
- 33. Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 2008; 48: 1054–1061. DOI: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guevin C, Manna D, Belanger C, Konan KV, Mak P, Labonte P. Autophagy protein ATG5 interacts transiently with the hepatitis C virus RNA polymerase (NS5B) early during infection. Virology 2010; 405: 1–7 S0042‐6822(10)00376‐4 [pii]. DOI: 10.1016/j.virol.2010.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Romero‐Brey I, Berger C, Kallis S, et al. NS5A domain 1 and polyprotein cleavage kinetics are critical for induction of double‐membrane vesicles associated with hepatitis C virus replication. MBio 2015; 6: e00759 mBio.00759‐15 [pii]. DOI: 10.1128/mBio.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chatterji U, Bobardt M, Tai A, Wood M, Gallay PA. Cyclophilin and NS5A inhibitors, but not other anti‐hepatitis C virus (HCV) agents, preclude HCV‐mediated formation of double‐membrane‐vesicle viral factories. Antimicrobial Agents and Chemotherapy 2015; 59: 2496–2507 AAC.04958‐14 [pii]. DOI: 10.1128/AAC.04958-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tu H, Gao L, Shi ST, et al. Hepatitis C virus RNA polymerase and NS5A complex with a SNARE‐like protein. Virology 1999; 263: 30–41. DOI: 10.1006/viro.1999.9893 S0042‐6822(99)99893‐8 [pii]. [DOI] [PubMed] [Google Scholar]
- 38. Hamamoto I, Nishimura Y, Okamoto T, et al. Human VAP‐B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. Journal of Virology 2005; 79: 13473–13482 79/21/13473 [pii]. DOI: 10.1128/JVI.79.21.13473-13482.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sklan EH, Serrano RL, Einav S, Pfeffer SR, Lambright DG, Glenn JS. TBC1D20 is a Rab1 GTPase‐activating protein that mediates hepatitis C virus replication. The Journal of Biological Chemistry 2007; 282: 36354–36361 M705221200 [pii]. DOI: 10.1074/jbc.M705221200. [DOI] [PubMed] [Google Scholar]
- 40. Stone M, Jia S, Heo WD, Meyer T, Konan KV. Participation of rab5, an early endosome protein, in hepatitis C virus RNA replication machinery. Journal of Virology 2007; 81: 4551–4563 JVI.01366‐06 [pii]. DOI: 10.1128/JVI.01366-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Manna D, Aligo J, Xu C, et al. Endocytic Rab proteins are required for hepatitis C virus replication complex formation. Virology 2009; 398: 21–37 S0042‐6822(09)00770‐3 [pii]. DOI: 10.1016/j.virol.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death and Differentiation 2014; 21: 348–358 cdd2013187 [pii]. DOI: 10.1038/cdd.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent‐tagged LC3. Autophagy 2007; 3: 452–460 4451 [pii]. [DOI] [PubMed] [Google Scholar]
- 44. Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. The Journal of Clinical Investigation 2011; 121: 37–56 41474 [pii]. DOI: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dreux M, Chisari FV. Autophagy proteins promote hepatitis C virus replication. Autophagy 2009; 5: 1224–1225 10219 [pii]. [DOI] [PubMed] [Google Scholar]
- 46. Taguwa S, Kambara H, Fujita N, et al. Dysfunction of autophagy participates in vacuole formation and cell death in cells replicating hepatitis C virus. Journal of Virology 2011; 85: 13185–13194 JVI.06099‐11 [pii]. DOI: 10.1128/JVI.06099-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K. Knockdown of autophagy‐related gene decreases the production of infectious hepatitis C virus particles. Autophagy 2009; 5: 937–945 9243 [pii]. [DOI] [PubMed] [Google Scholar]
- 48. Sir D, Kuo CF, Tian Y, et al. Replication of hepatitis C virus RNA on autophagosomal membranes. The Journal of Biological Chemistry 2012; 287: 18036–18043 M111.320085 [pii]. DOI: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shinohara Y, Imajo K, Yoneda M, et al. Unfolded protein response pathways regulate Hepatitis C virus replication via modulation of autophagy. Biochemical and Biophysical Research Communications 2013; 432: 326–332 S0006‐291X(13)00193‐9 [pii]. DOI: 10.1016/j.bbrc.2013.01.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mohl BP, Tedbury PR, Griffin S, Harris M. Hepatitis C virus‐induced autophagy is independent of the unfolded protein response. Journal of Virology 2012; 86: 10724–10732 JVI.01667‐12 [pii]. DOI: 10.1128/JVI.01667-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Holmes EC, Twiddy SS. The origin, emergence and evolutionary genetics of dengue virus. Infection, Genetics and Evolution 2003; 3: 19–28 S1567134803000042 [pii]. [DOI] [PubMed] [Google Scholar]
- 52. Mustafa MS, Rasotgi V, Jain S, Gupta V. Discovery of fifth serotype of dengue virus (DENV‐5): a new public health dilemma in dengue control. Medical Journal Armed Forces India 2015; 71: 67–70. DOI: 10.1016/j.mjafi.2014.09.011 S0377‐1237(14)00172‐5 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lee YR, Lei HY, Liu MT, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008; 374: 240–248 S0042‐6822(08)00112‐8 [pii]. DOI: 10.1016/j.virol.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khakpoor A, Panyasrivanit M, Wikan N, Smith DR. A role for autophagolysosomes in dengue virus 3 production in HepG2 cells. The Journal of General Virology 2009; 90: 1093–1103 vir.0.007914‐0 [pii]. DOI: 10.1099/vir.0.007914-0. [DOI] [PubMed] [Google Scholar]
- 55. Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. Co‐localization of constituents of the dengue virus translation and replication machinery with amphisomes. The Journal of General Virology 2009; 90: 448–456 90/2/448 [pii]. DOI: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 56. Zybert IA, van der Ende‐Metselaar H, Wilschut J, Smit JM. Functional importance of dengue virus maturation: infectious properties of immature virions. The Journal of General Virology 2008; 89: 3047–3051 89/12/3047 [pii]. DOI: 10.1099/vir.0.2008/002535-0. [DOI] [PubMed] [Google Scholar]
- 57. Welsch S, Miller S, Romero‐Brey I, et al. Composition and three‐dimensional architecture of the dengue virus replication and assembly sites. Cell Host & Microbe 2009; 5: 365–375 S1931‐3128(09)00098‐5 [pii]. DOI: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murray JM, Aaskov JG, Wright PJ. Processing of the dengue virus type 2 proteins prM and C‐prM. The Journal of General Virology 1993; 74(Pt 2): 175–182. DOI: 10.1099/0022-1317-74-2-175. [DOI] [PubMed] [Google Scholar]
- 59. Hsieh SC, Zou G, Tsai WY, et al. The C‐terminal helical domain of dengue virus precursor membrane protein is involved in virus assembly and entry. Virology 2011; 410: 170–180 S0042‐6822(10)00711‐7 [pii]. DOI: 10.1016/j.virol.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Espert L, Varbanov M, Robert‐Hebmann V, et al. Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV‐1 infection. PLoS One 2009; 4: e5787 DOI: 10.1371/journal.pone.0005787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kyei GB, Dinkins C, Davis AS, et al. Autophagy pathway intersects with HIV‐1 biosynthesis and regulates viral yields in macrophages. The Journal of Cell Biology 2009; 186: 255–268 jcb.200903070 [pii]. DOI: 10.1083/jcb.200903070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Malbec M, Sourisseau M, Guivel‐Benhassine F, et al. HIV‐1 Nef promotes the localization of Gag to the cell membrane and facilitates viral cell‐to‐cell transfer. Retrovirology 2013; 10: 80 1742‐4690‐10‐80 [pii]. DOI: 10.1186/1742-4690-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brass AL, Dykxhoorn DM, Benita Y, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008; 319: 921–926 1152725 [pii]. DOI: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 64. Valera MS, de Armas‐Rillo L, Barroso‐Gonzalez J, et al. The HDAC6/APOBEC3G complex regulates HIV‐1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015; 12: 53 DOI: 10.1186/s12977-015-0181-5 10.1186/s12977-015-0181-5 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ettayebi K, Hardy ME. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP‐A and prevents cell surface expression of vesicular stomatitis virus G protein. Journal of Virology 2003; 77: 11790–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maynell LA, Kirkegaard K, Klymkowsky MW. Inhibition of poliovirus RNA synthesis by brefeldin A. Journal of Virology 1992; 66: 1985–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lanke KH, van der Schaar HM, Belov GA, et al. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. Journal of Virology 2009; 83: 11940–11949 JVI.01244‐09 [pii]. DOI: 10.1128/JVI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Doedens JR, Kirkegaard K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. The EMBO Journal 1995; 14: 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Belov GA, Ehrenfeld E. Involvement of cellular membrane traffic proteins in poliovirus replication. Cell Cycle 2007; 6: 36–38. [DOI] [PubMed] [Google Scholar]
- 70. Rust RC, Landmann L, Gosert R, et al. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. Journal of Virology 2001; 75: 9808–9818. DOI: 10.1128/JVI.75.20.9808-9818.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ehrlich M, Boll W, Van Oijen A, et al. Endocytosis by random initiation and stabilization of clathrin‐coated pits. Cell 2004; 118: 591–605. DOI: 10.1016/j.cell.2004.08.017 S0092867404007901 [pii]. [DOI] [PubMed] [Google Scholar]
- 72. Vonderheit A, Helenius A. Rab7 associates with early endosomes to mediate sorting and transport of Semliki forest virus to late endosomes. PLoS Biology 2005; 3: e233 04‐PLBI‐RA‐0777R2 [pii]. DOI: 10.1371/journal.pbio.0030233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mutsafi Y, Zauberman N, Sabanay I, Minsky A. Vaccinia‐like cytoplasmic replication of the giant Mimivirus. Proceedings of the National Academy of Sciences of the United States of America 2010; 107: 5978–5982 0912737107 [pii]. DOI: 10.1073/pnas.0912737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Katzourakis A, Aswad A. The origins of giant viruses, virophages and their relatives in host genomes. BMC Biology 2014; 12: 51 s12915‐014‐0051‐y [pii]. DOI: 10.1186/s12915-014-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McAuslan BR, Armentrout RW. The biochemistry of icosahedral cytoplasmic deoxyviruses. Current Topics in Microbiology and Immunology 1974; 77–105. [DOI] [PubMed] [Google Scholar]
- 76. Garcia‐Beato R, Salas ML, Vinuela E, Salas J. Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 1992; 188: 637–649 0042‐6822(92)90518‐T [pii]. [DOI] [PubMed] [Google Scholar]
- 77. Goorha R. Frog virus 3 DNA replication occurs in two stages. Journal of Virology 1982; 43: 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fuchs LY, Woods MS, Weaver RF. Viral transcription during Autographa californica nuclear polyhedrosis virus infection: a novel RNA polymerase induced in infected Spodoptera frugiperda cells. Journal of Virology 1983; 48: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nunes JF, Vigario JD, Terrinha AM. Ultrastructural study of African swine fever virus replication in cultures of swine bone marrow cells. Archives of Virology 1975; 49: 59–66. [DOI] [PubMed] [Google Scholar]
- 80. Heath CM, Windsor M, Wileman T. Aggresomes resemble sites specialized for virus assembly. The Journal of Cell Biology 2001; 153: 449–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stefanovic S, Windsor M, Nagata KI, Inagaki M, Wileman T. Vimentin rearrangement during African swine fever virus infection involves retrograde transport along microtubules and phosphorylation of vimentin by calcium calmodulin kinase II. Journal of Virology 2005; 79: 11766–11775 79/18/11766 [pii]. DOI: 10.1128/JVI.79.18.11766-11775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cobbold C, Windsor M, Wileman T. A virally encoded chaperone specialized for folding of the major capsid protein of African swine fever virus. Journal of Virology 2001; 75: 7221–7229. DOI: 10.1128/JVI.75.16.7221-7229.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rojo G, Chamorro M, Salas ML, Vinuela E, Cuezva JM, Salas J. Migration of mitochondria to viral assembly sites in African swine fever virus‐infected cells. Journal of Virology 1998; 72: 7583–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rouiller I, Brookes SM, Hyatt AD, Windsor M, Wileman T. African swine fever virus is wrapped by the endoplasmic reticulum. Journal of Virology 1998; 72: 2373–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Andres G, Garcia‐Escudero R, Simon‐Mateo C, Vinuela E. African swine fever virus is enveloped by a two‐membraned collapsed cisterna derived from the endoplasmic reticulum. Journal of Virology 1998; 72: 8988–9001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Chlanda P, Carbajal MA, Cyrklaff M, Griffiths G, Krijnse‐Locker J. Membrane rupture generates single open membrane sheets during vaccinia virus assembly. Cell Host & Microbe 2009; 6: 81–90 S1931‐3128(09)00219‐4 [pii]. DOI: 10.1016/j.chom.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 87. Chlanda P, Carbajal MA, Kolovou A, et al. Vaccinia virus lacking A17 induces complex membrane structures composed of open membrane sheets. Archives of Virology 2011; 156: 1647–1653. DOI: 10.1007/s00705-011-1012-1. [DOI] [PubMed] [Google Scholar]
- 88. Netherton C, Rouiller I, Wileman T. The subcellular distribution of multigene family 110 proteins of African swine fever virus is determined by differences in C‐terminal KDEL endoplasmic reticulum retention motifs. Journal of Virology 2004; 78: 3710–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Windsor M, Hawes P, Monaghan P, et al. Mechanism of collapse of endoplasmic reticulum cisternae during African swine fever virus infection. Traffic 2012; 13: 30–42. DOI: 10.1111/j.1600-0854.2011.01293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Netherton CL, Wileman TE. African swine fever virus organelle rearrangements. Virus Research 2013; 173: 76–86 S0168‐1702(12)00476‐5 [pii]. DOI: 10.1016/j.virusres.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 91. Colson P, De Lamballerie X, Yutin N, et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Archives of Virology 2013; 158: 2517–2521. DOI: 10.1007/s00705-013-1768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kerviel A, Thomas A, Chaloin L, Favard C, Muriaux D. Virus assembly and plasma membrane domains: which came first? Virus Research 2013; 171: 332–340 S0168‐1702(12)00310‐3 [pii]. DOI: 10.1016/j.virusres.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 93. Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV‐1 Gag targeting to the plasma membrane. Proceedings of the National Academy of Sciences of the United States of America 2004; 101: 14889–14894 0405596101 [pii]. DOI: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Inlora J, Chukkapalli V, Derse D, Ono A. Gag localization and virus‐like particle release mediated by the matrix domain of human T‐lymphotropic virus type 1 Gag are less dependent on phosphatidylinositol‐(4,5)‐bisphosphate than those mediated by the matrix domain of HIV‐1 Gag. Journal of Virology 2011; 85: 3802–3810 JVI.02383‐10 [pii]. DOI: 10.1128/JVI.02383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Saad JS, Loeliger E, Luncsford P, et al. Point mutations in the HIV‐1 matrix protein turn off the myristyl switch. Journal of Molecular Biology 2007; 366: 574–585 S0022‐2836(06)01627‐5 [pii]. DOI: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Nguyen DH, Hildreth JE. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid‐enriched membrane lipid rafts. Journal of Virology 2000; 74: 3264–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proceedings of the National Academy of Sciences of the United States of America 2006; 103: 2641–2646 0511136103 [pii]. DOI: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid‐specific inositol polyphosphate 5‐phosphatase. The Journal of Biological Chemistry 2000; 275: 20110–20116. DOI: 10.1074/jbc.M910119199 M910119199 [pii]. [DOI] [PubMed] [Google Scholar]
- 99. Sundquist WI, Krausslich HG. HIV‐1 assembly, budding, and maturation. Cold Spring Harbor Perspectives in Medicine 2012; 2: a006924 DOI: 10.1101/cshperspect.a006924 a006924 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Bieniasz PD. Late budding domains and host proteins in enveloped virus release. Virology 2006; 344: 55–63 S0042‐6822(05)00599‐4 [pii]. DOI: 10.1016/j.virol.2005.09.044. [DOI] [PubMed] [Google Scholar]
- 101. Chen BJ, Lamb RA. Mechanisms for enveloped virus budding: can some viruses do without an ESCRT? Virology 2008; 372: 221–232 S0042‐6822(07)00756‐8 [pii]. DOI: 10.1016/j.virol.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Piper RC, Katzmann DJ. Biogenesis and function of multivesicular bodies. Annual Review of Cell and Developmental Biology 2007; 23: 519–547. DOI: 10.1146/annurev.cellbio.23.090506.123319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Carlton JG, Martin‐Serrano J. The ESCRT machinery: new functions in viral and cellular biology. Biochemical Society Transactions 2009; 37: 195–199 BST0370195 [pii]. DOI: 10.1042/BST0370195. [DOI] [PubMed] [Google Scholar]
- 104. Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT machinery: it's all in the neck. Nature Reviews Molecular Cell Biology 2010; 11: 556–566 nrm2937 [pii]. DOI: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Luyet PP, Falguieres T, Pons V, Pattnaik AK, Gruenberg J. The ESCRT‐I subunit TSG101 controls endosome‐to‐cytosol release of viral RNA. Traffic 2008; 9: 2279–2290 TRA820 [pii]. DOI: 10.1111/j.1600-0854.2008.00820.x. [DOI] [PubMed] [Google Scholar]
- 106. Zhang Y, Qian H, Love Z, Barklis E. Analysis of the assembly function of the human immunodeficiency virus type 1 gag protein nucleocapsid domain. Journal of Virology 1998; 72: 1782–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fujii K, Munshi UM, Ablan SD, et al. Functional role of Alix in HIV‐1 replication. Virology 2009; 391: 284–292 S0042‐6822(09)00363‐8 [pii]. DOI: 10.1016/j.virol.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Popova E, Popov S, Gottlinger HG. Human immunodeficiency virus type 1 nucleocapsid p1 confers ESCRT pathway dependence. Journal of Virology 2010; 84: 6590–6597 JVI.00035‐10 [pii]. DOI: 10.1128/JVI.00035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Pincetic A, Kuang Z, Seo EJ, Leis J. The interferon‐induced gene ISG15 blocks retrovirus release from cells late in the budding process. Journal of Virology 2010; 84: 4725–4736 JVI.02478‐09 [pii]. DOI: 10.1128/JVI.02478-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Seo EJ, Leis J. Budding of enveloped viruses: interferon‐induced ISG15‐antivirus mechanisms targeting the release process. Advances in Virology 2012; 2012: 532723 DOI: 10.1155/2012/532723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Hoffmann J, Boehm C, Himmelsbach K, et al. Identification of alpha‐taxilin as an essential factor for the life cycle of hepatitis B virus. Journal of Hepatology 2013; 59: 934–941 S0168‐8278(13)00437‐6 [pii]. DOI: 10.1016/j.jhep.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 112. Patient R, Hourioux C, Roingeard P. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cellular Microbiology 2009; 11: 1561–1570 CMI1363 [pii]. DOI: 10.1111/j.1462-5822.2009.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Freed EO. Viral late domains. Journal of Virology 2002; 76: 4679–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hartlieb B, Weissenhorn W. Filovirus assembly and budding. Virology 2006; 344: 64–70 S0042‐6822(05)00587‐8 [pii]. DOI: 10.1016/j.virol.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 115. Okumura A, Harty RN. Rabies virus assembly and budding. Advances in Virus Research 2011; 79: 23–32 B978‐0‐12‐387040‐7.00002‐0 [pii]. DOI: 10.1016/B978-0-12-387040-7.00002-0. [DOI] [PubMed] [Google Scholar]
- 116. Groseth A, Wolff S, Strecker T, Hoenen T, Becker S. Efficient budding of the tacaribe virus matrix protein z requires the nucleoprotein. Journal of Virology 2010; 84: 3603–3611 JVI.02429‐09 [pii]. DOI: 10.1128/JVI.02429-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Emonet SE, Urata S, de la Torre JC. Arenavirus reverse genetics: new approaches for the investigation of arenavirus biology and development of antiviral strategies. Virology 2011; 411: 416–425 S0042‐6822(11)00018‐3 [pii]. DOI: 10.1016/j.virol.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chen BJ, Leser GP, Morita E, Lamb RA. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid‐derived virus‐like particles. Journal of Virology 2007; 81: 7111–7123 JVI.00361‐07 [pii]. DOI: 10.1128/JVI.00361-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bruce EA, Medcalf L, Crump CM, et al. Budding of filamentous and non‐filamentous influenza A virus occurs via a VPS4 and VPS28‐independent pathway. Virology 2009; 390: 268–278 S0042‐6822(09)00304‐3 [pii]. DOI: 10.1016/j.virol.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 120. Irie T, Licata JM, Jayakar HR, Whitt MA, Bell P, Harty RN. Functional analysis of late‐budding domain activity associated with the PSAP motif within the vesicular stomatitis virus M protein. Journal of Virology 2004; 78: 7823–7827. DOI: 10.1128/JVI.78.14.7823-7827.2004 78/14/7823 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Shnyrova AV, Ayllon J, Mikhalyov II, Villar E, Zimmerberg J, Frolov VA. Vesicle formation by self‐assembly of membrane‐bound matrix proteins into a fluidlike budding domain. The Journal of Cell Biology 2007; 179: 627–633 jcb.200705062 [pii]. DOI: 10.1083/jcb.200705062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. El Najjar F, Schmitt AP, Dutch RE. Paramyxovirus glycoprotein incorporation, assembly and budding: a three way dance for infectious particle production. Viruses 2014; 6: 3019–3054 v6083019 [pii]. DOI: 10.3390/v6083019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sattentau Q. Avoiding the void: cell‐to‐cell spread of human viruses. Nature Reviews Microbiology 2008; 6: 815–826. [DOI] [PubMed] [Google Scholar]
- 124. Igakura T, Stinchcombe JC, Goon PK, et al. Spread of HTLV‐I between lymphocytes by virus‐induced polarization of the cytoskeleton. Science 2003; 299: 1713–1716. DOI: 10.1126/science.1080115 1080115 [pii]. [DOI] [PubMed] [Google Scholar]
- 125. Jolly C, Sattentau QJ. Retroviral spread by induction of virological synapses. Traffic 2004; 5: 643–650. [DOI] [PubMed] [Google Scholar]
- 126. Dimitrov DS, Willey RL, Sato H, Chang LJ, Blumenthal R, Martin MA. Quantitation of human immunodeficiency virus type 1 infection kinetics. Journal of Virology 1993; 67: 2182–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Martin N, Sattentau Q. Cell‐to‐cell HIV‐1 spread and its implications for immune evasion. Current Opinion in HIV and AIDS 2009; 4: 143–149. DOI: 10.1097/COH.0b013e328322f94a 01222929‐200903000‐00012 [pii]. [DOI] [PubMed] [Google Scholar]
- 128. Massanella M, Puigdomenech I, Cabrera C, et al. Antigp41 antibodies fail to block early events of virological synapses but inhibit HIV spread between T cells. Aids 2009; 23: 183–188. [DOI] [PubMed] [Google Scholar]
- 129. Martin N, Welsch S, Jolly C, Briggs JA, Vaux D, Sattentau QJ. Virological synapse‐mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. Journal of Virology 2010; 84: 3516–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Archer KA, Durack J, Portnoy DA. STING‐dependent type I IFN production inhibits cell‐mediated immunity to Listeria monocytogenes . PLoS Pathogen 2014; 10: e1003861 DOI: 10.1371/journal.ppat.1003861 PPATHOGENS‐D‐13‐01967 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Jimenez‐Baranda S, Gomez‐Mouton C, Rojas A, et al. Filamin‐A regulates actin‐dependent clustering of HIV receptors. Nature Cell Biology 2007; 9: 838–846. [DOI] [PubMed] [Google Scholar]
- 132. Yoder A, Yu D, Dong L, et al. HIV envelope‐CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 2008; 134: 782–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Barrero‐Villar M, Barroso‐Gonzalez J, Cabrero JR, et al. PI4P5‐kinase Ialpha is required for efficient HIV‐1 entry and infection of T cells. The Journal of Immunology 2008; 181: 6882–6888. [DOI] [PubMed] [Google Scholar]
- 134. Barrero‐Villar M, Cabrero JR, Gordon‐Alonso M, et al. Moesin is required for HIV‐1‐induced CD4–CXCR4 interaction, F‐actin redistribution, membrane fusion and viral infection in lymphocytes. Journal of Cell Science 2009; 122: 103–113. [DOI] [PubMed] [Google Scholar]
- 135. Garcia‐Exposito L, Barroso‐Gonzalez J, Puigdomenech I, Machado JD, Blanco J, Valenzuela‐Fernandez A. HIV‐1 requires Arf6‐mediated membrane dynamics to efficiently enter and infect T lymphocytes. Molecular Biology of the Cell 2011; 22: 1148–1166 mbc.E10‐08‐0722 [pii]. DOI: 10.1091/mbc.E10-08-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Garcia‐Exposito L, Ziglio S, Barroso‐Gonzalez J, et al. Gelsolin activity controls efficient early HIV‐1 infection. Retrovirology 2013; 10: 39 1742‐4690‐10‐39 [pii]. DOI: 10.1186/1742-4690-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Jolly C, Mitar I, Sattentau QJ. Requirement for an intact T‐cell actin and tubulin cytoskeleton for efficient assembly and spread of human immunodeficiency virus type 1. Journal of Virology 2007; 81: 5547–5560 JVI.01469‐06 [pii]. DOI: 10.1128/JVI.01469-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Jolly C, Sattentau QJ. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. Journal of Virology 2005; 79: 12088–12094 79/18/12088 [pii]. DOI: 10.1128/JVI.79.18.12088-12094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Puigdomenech I, Massanella M, Cabrera C, Clotet B, Blanco J. On the steps of cell‐to‐cell HIV transmission between CD4 T cells. Retrovirology 2009; 6: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pais‐Correia AM, Sachse M, Guadagnini S, et al. Biofilm‐like extracellular viral assemblies mediate HTLV‐1 cell‐to‐cell transmission at virological synapses. Nature Medicine 2010; 16: 83–89 nm.2065 [pii]. DOI: 10.1038/nm.2065. [DOI] [PubMed] [Google Scholar]
- 141. Sherer NM, Lehmann MJ, Jimenez‐Soto LF, Horensavitz C, Pypaert M, Mothes W. Retroviruses can establish filopodial bridges for efficient cell‐to‐cell transmission. Nature Cell Biology 2007; 9: 310–315 ncb1544 [pii]. DOI: 10.1038/ncb1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sowinski S, Jolly C, Berninghausen O, et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV‐1 transmission. Nature Cell Biology 2008; 10: 211–219 ncb1682 [pii]. DOI: 10.1038/ncb1682. [DOI] [PubMed] [Google Scholar]
- 143. Joly E, Hudrisier D. What is trogocytosis and what is its purpose? Nature Immunology 2003; 4: 815 DOI: 10.1038/ni0903-815 ni0903‐815 [pii]. [DOI] [PubMed] [Google Scholar]
- 144. Aucher A, Puigdomenech I, Joly E, Clotet B, Hudrisier D, Blanco J. Could CD4 capture by CD8+ T cells play a role in HIV spreading? Journal of Biomedicine and Biotechnology 2010; 2010: 907371 DOI: 10.1155/2010/907371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV‐1 envelope proteins to CXCR4. The Journal of Clinical Investigation 2006; 116: 2161–2172. DOI: 10.1172/JCI26185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Debatin KM, Fahrig‐Faissner A, Enenkel‐Stoodt S, Kreuz W, Benner A, Krammer PH. High expression of APO‐1 (CD95) on T lymphocytes from human immunodeficiency virus‐1‐infected children. Blood 1994; 83: 3101–3103. [PubMed] [Google Scholar]
- 147. Izquierdo‐Useros N, Naranjo‐Gomez M, Erkizia I, et al. HIV and mature dendritic cells: Trojan exosomes riding the Trojan horse? PLoS Pathogen 2010; 6: e1000740 DOI: 10.1371/journal.ppat.1000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Izquierdo‐Useros N, Puertas MC, Borras FE, Blanco J, Martinez‐Picado J. Exosomes and retroviruses: the chicken or the egg? Cellular Microbiology 2011; 13: 10–17. DOI: 10.1111/j.1462-5822.2010.01542.x. [DOI] [PubMed] [Google Scholar]
- 149. Santa‐Marta M, de Brito PM, Godinho‐Santos A, Goncalves J. Host factors and HIV‐1 replication: clinical evidence and potential therapeutic approaches. Frontiers in Immunology 2013; 4: 343 DOI: 10.3389/fimmu.2013.00343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Brandenburg B, Zhuang X. Virus trafficking—learning from single‐virus tracking. Nature Reviews Microbiology 2007; 5: 197–208 nrmicro1615 [pii]. DOI: 10.1038/nrmicro1615. [DOI] [PMC free article] [PubMed] [Google Scholar]