

Abstract
Maize (Zea mays) root system architecture (RSA) mediates the key functions of plant anchorage and acquisition of nutrients and water. In this study, a set of 204 recombinant inbred lines (RILs) was derived from the widely adapted Chinese hybrid ZD958(Zheng58 × Chang7‐2), genotyped by sequencing (GBS) and evaluated as seedlings for 24 RSA related traits divided into primary, seminal and total root classes. Significant differences between the means of the parental phenotypes were detected for 18 traits, and extensive transgressive segregation in the RIL population was observed for all traits. Moderate to strong relationships among the traits were discovered. A total of 62 quantitative trait loci (QTL) were identified that individually explained from 1.6% to 11.6% (total root dry weight/total seedling shoot dry weight) of the phenotypic variation. Eighteen, 24 and 20 QTL were identified for primary, seminal and total root classes of traits, respectively. We found hotspots of 5, 3, 4 and 12 QTL in maize chromosome bins 2.06, 3.02‐03, 9.02‐04, and 9.05‐06, respectively, implicating the presence of root gene clusters or pleiotropic effects. These results characterized the phenotypic variation and genetic architecture of seedling RSA in a population derived from a successful maize hybrid.
Keywords: Maize, root system architecture, QTL, bin map, genotyping by sequencing (GBS)
Edited by: Tobias Baskin, University of Massachusetts Amherst, USA
INTRODUCTION
Root system architecture (RSA) is characterized by its role in anchorage and absorption of nutrients and water (Lynch 1995; Lynch 2013) and ability to respond dynamically to the soil environment (Williamson et al. 2001). Previous studies have demonstrated the ability of maize to alter growth patterns in order to explore for and utilize nutrient and water gradients (Lopez‐Bucio et al. [Link]). Root system development is therefore particularly sensitive to environmental factors. Although RSA plays a critical role in the growth and reproductive success of the mature plant, particularly under resource‐limiting conditions, relatively few root studies have been performed compared with those examining shoot and reproductive traits and a deeper understanding of the genetic control of RSA is required.
Maize RSA is comprised by embryonic and post‐embryonic roots (Feldman 1994). The embryonic root system is composed of a single primary root and seminal roots that emerge within several days of germination. Seminal root number has been observed to range between zero and 13 depending on the genotype (Hochholdinger et al. 2004). A variable number of lateral roots are also initiated from the primary and seminal roots, and together with root hairs, function to aid absorption by increasing surface area (Hochholdinger et al. 2004). The post‐embryonic root system is formed by crown roots, which initiate about 2 weeks after germination, brace roots and their lateral branches (Hochholdinger and Tuberosa 2009). The embryonic root system has an important role in seedling vigor, which is positively associated with final plant performance (Nass and Zuber 1971; Landi et al. 1998). Seedling RSA is also expected to be associated with survival of early season water stress (McCully and Canny 1988; Hochholdinger and Tuberosa 2009). A better understanding of the genetic basis of root traits will accelerate the progress of maize germplasm improvement for lodging and drought resistance, along with improved nutrient absorption (Giuliani et al. 2005; Wu et al. 2011; Li et al. 2015; Zhan and Lynch 2015).
Efficient and accurate phenotyping protocols are needed to improve identification of quantitative trait loci (QTL) controlling the RSA‐related traits. Direct root architecture phenotyping in the field is difficult and the variability of the soil environment within fields is a concern (Lynch 1995; Kumar et al. 2014). Paszkowski and Boller (2002) reported that the lateral rootless1 mutant could be complemented by symbiotic microbes and high fertility (Paszkowski and Boller 2002), indicating the magnitude of a root loci's effect is likely to be mediated by the soil environment. For greater control of the root environment and improvement of data quality, investigation of root architecture traits have been conducted using hydroponic, gel‐based and rolled paper systems (Tuberosa et al. 2003; Woll et al. 2005; Kumar et al. 2012; Abdel‐Ghani et al. 2013; Burton et al. 2014; Kumar et al. 2014; Pace et al. 2015). Among these, hydroponics has been favored for precise control of nutrient concentration and the ease of phenotyping. Using this method, many QTL controlling lateral root number, primary root length and other traits have been identified at various water and nutrient regimes (Mano et al. 2005; Zhu et al. 2006; Liu et al. 2008; Abdel‐Ghani et al. 2013; Burton et al. 2014; Kumar et al. 2014).
Although seedling RSA probably is a weak predictor of mature RSA in the field (Nass and Zuber 1971; Zhu et al. 2011), overlapping QTL for seedling and mature RSA and nitrogen utilization efficiency (NUE) were identified under different nitrogen levels and RSA was associated with NUE (Li et al. 2015). Previous QTL mapping studies using seedlings have shown that RSA‐related traits are controlled by a large number of minor‐effect loci that exhibit strong genotype‐environment (GE) interactions (de Dorlodot et al. 2007; Burton et al. 2014; Li et al. 2015). Burton et al. (2014) reported 15 QTL that individually explained 0.44% to 13.5% of the phenotypic variation among 21 root traits in a greenhouse study using three populations of recombinant inbred lines. Li et al. (2015) identified 147 QTL, including five QTL clusters, for RSA‐related traits using a recombinant inbred line population under different nitrogen levels. Backcrossing major RSA QTL into another genetic background increased grain yield (Li et al. 2015). A genome‐wide association study identified many candidate genes for 22 seedling RSA traits using 384 inbred lines (Pace et al. 2015). A meta‐analysis of RSA highlighted six clusters, and in particular, bin 1.07 where 11 QTL from three populations were mapped (Hund et al. 2011). This region was found to be influential in both seedlings and mature plants (Tuberosa et al. 2003; Hund et al. 2004; Zhu et al. 2005; Hund et al. 2011). Kumar et al. (2014) used candidate gene association mapping strategy to identify several molecular markers associated with root traits within the genes Rth1 (root hairless 1) and Rth3 (root hairless 3), and the mutant paralogs Rtcl (rootless concerning crown and seminal roots1‐like) and Rul1 (rootless with undetectable meristems1‐like). Genetic variation within classic mutant loci has been associated with root response to nitrogen and Rth3 was associated with grain yield under high nitrogen conditions, suggesting the potential of functional genetics to accelerate maize improvement (Kumar et al. 2014).
Single nucleotide polymorphism (SNP) markers have been favored for linkage and association studies, and high‐density genotyping of large populations has been done with Chip (Yan et al. 2009; Yan et al. 2010; Ganal et al. 2011; Li et al. 2014b) and genotype‐by‐sequencing (GBS) approaches (Gore et al. 2009; Lai et al. 2010; Chia et al. 2012; Jiao et al. 2012; Glaubitz et al. 2014). The objectives of this study were to: (i) construct a physically anchored genetic linkage map with ultra‐high density bin markers for a set of recombinant inbred lines (RILs) derived from the parents of a commercially successful Chinese maize hybrid; (ii) examine the phenotypic variation for seedling RSA traits; and (iii) map QTL associated with the seedling RSA traits.
RESULTS
Phenotypic variation among the parents and the population
The RIL population was grown hydroponically in a greenhouse and high throughput phenotyping of root trait on individual seedlings was performed as shown in Figure 1A. Phenotypic differences in the seedling root systems of the parents can be seen in Figure 1B. Scanned root images for Zheng58 (Figure 1C) and Chang7‐2 (Figure 1D) illustrate Zheng58's greater degree of lateral branching on the primary root (Table 2). Chang7‐2's equivalent lateral roots appear to be longer relative to Zheng58 (Table 2). A summary of the phenotyped RSA traits and their abbreviations are provided in Table 1. RSA data was collected on primary and seminal roots, along with the total root system, so the traits were divided into three classes. Significant differences between the means of the parental phenotypes were detected for 18 traits, including primary and seminal lateral root density (PLD and SLD), primary and seminal lateral root number (TNL_P and SRL_S), primary root length (PRL), root dry weight (RDW) and shoot dry weight (SDW). Parental means were not significantly different for the length of the total root system (TRL), total length of seminal lateral roots (STL), average length of seminal lateral roots (SLL), length of seminal axile and lateral roots (SAL), proportion of seminal lateral root length (SLP) and root to shoot ratio (RSR) (Table 2). The highly significant difference in root mass, but similar estimates of TRL and RSR for the parents suggests that Chang7‐2 may utilize less biomass per unit of root length. Extensive transgressive segregation in the RIL population was observed for all traits (Table 2; Figures S1–3).
Figure 1.
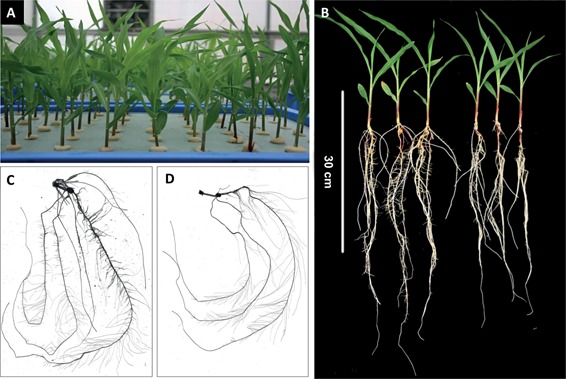
Seedling production using the hydroponic system and phenotyping (A) The parental inbreds and recombinant inbred lines (RILs) were cultivated in Styrofoam in boxes and natural light. (B) Root systems and shoots of the RIL parents Zheng58 (left) and Chang7‐2 (right). Scanned images of a representative (C) Zheng58 and (D) Chang7‐2 sample.
Table 2.
Statistical measures of parents and recombinant inbred line (RIL) phenotypes for 24 root system architecture (RSA) related traits
| RILs | |||||||
|---|---|---|---|---|---|---|---|
| Trait | Unit | Mean Chang7‐2 ± SE | Mean Zheng58 ± SE | t‐test | Mean ± SE | Min | Max |
| PRL | cm | 27.43 ± 0.83 | 29.71 ± 0.72 | * | 29.68 ± 0.36 | 13.48 | 43.38 |
| TNL_P | count | 137.10 ± 5.00 | 171.30 ± 5.10 | ** | 168.30 ± 2.80 | 45.2 | 316.8 |
| PLD | count/cm | 5.12 ± 0.14 | 5.93 ± 0.144 | ** | 5.74 ± 0.079 | 3.27 | 9.56 |
| ALL | cm | 1.53 ± 0.1 | 0.99 ± 0.05 | ** | 1.43 ± 0.03 | 0.43 | 2.97 |
| PLL | cm | 208.22 ± 8.5 | 167.04 ± 7.04 | ** | 218.66 ± 4.61 | 56.38 | 401.52 |
| PAL | cm | 241.99 ± 8.63 | 202.72 ± 7.67 | ** | 247.89 ± 4.66 | 84.35 | 417.7 |
| PLP | ratio | 0.88 ± 0.005 | 0.84±0.005 | ** | 0.86 ± 0.003 | 0.66 | 0.93 |
| SRN | count | 2.20 ± 0.10 | 2.80 ± 0.20 | ** | 3.30 ± 0.10 | 1.5 | 6.2 |
| TSL | cm | 37.12 ± 2.27 | 55.63 ± 2.93 | ** | 64.16 ± 1.30 | 23.25 | 115.11 |
| SRL_S | cm | 18.21 ± 0.98 | 21.59 ± 1.30 | * | 20.65 ± 0.37 | 8.95 | 36.92 |
| TNL_S | count | 166.7 ± 11.3 | 204.7 ± 9.3 | * | 288.1 ± 6.2 | 109.3 | 613.9 |
| SLD | count/cm | 4.546 ± 0.209 | 3.916 ± 0.18 | * | 4.64 ± 0.079 | 2.03 | 8.18 |
| SLL | cm | 0.54 ± 0.03 | 0.5 ± 0.01 | NS | 0.58 ± 0.01 | 0.24 | 1.06 |
| STL | cm | 96.79 ± 8.79 | 105.45 ± 6.04 | NS | 168.75 ± 4.97 | 36.07 | 388.85 |
| SAL | cm | 135.33 ± 11.02 | 161.92 ± 8.98 | NS | 234.44 ± 5.92 | 61.62 | 492.5 |
| SLP | ratio | 0.68 ± 0.01 | 0.64 ± 0.008 | NS | 0.70 ± 0.005 | 0.3 | 0.84 |
| TRL | cm | 375.09 ± 11.21 | 358.34 ± 10.34 | NS | 479.96 ± 8.24 | 244.95 | 850.85 |
| SDW | mg | 109.60 ± 2.00 | 135.60 ± 3.8 | ** | 143.90 ± 2.10 | 77.9 | 232.7 |
| RDW | mg | 25.40 ± 0.60 | 34.00 ± 0.7 | ** | 37.70 ± 0.50 | 22.3 | 57.8 |
| RSR | ratio | 0.26 ± 0.007 | 0.26 ± 0.006 | NS | 0.27 ± 0.003 | 0.15 | 0.45 |
| ARL | cm | 64.88 ± 2.43 | 86.6 ± 3.13 | ** | 93.84 ± 1.46 | 34.97 | 159.7 |
| LRL | cm | 308.50 ± 10.72 | 275.98 ± 8.89 | * | 386.93 ± 7.59 | 168.38 | 676.88 |
| TLP | ratio | 0.83 ± 0.005 | 0.76 ± 0.007 | ** | 0.80 ± 0.003 | 0.62 | 0.9 |
| SRL_T | g/cm | 7.03 ± 0.196 | 9.70 ± 0.20 | ** | 8.24 ± 0.12 | 4.75 | 15.15 |
SE, standard error (seedling observations: Chang7‐2, n = 66; Zheng58, n = 73); NS, not significant; *, p ≤ 0.05; ** p ≤ 0.01; significance of the analysis of variance test.
Table 1.
Summary of the root system architecture related traits and their measurements
| Classification | Trait | Abbreviation | Measurement and description |
|---|---|---|---|
| Primary | Length of the primary root | PRL | Measured using a ruler |
| Lateral root number | TNL_P | Count of lateral roots from the first emerged lateral root on the primary root | |
| Lateral root density | PLD | TNL_P/PRL; primary lateral roots/primary root length | |
| Total lateral root length | PLL | TRL – TSL – PRL; total root length ‐ total seminal length – primary root length | |
| Average lateral root length | ALL | PLL/TNL_P; primary lateral root length/number of lateral roots | |
| Axile and lateral length | PAL | PRL + PLL; length of primary axile root and its lateral roots | |
| Lateral root length proportion | PLP | PLL/PAL; primary lateral root length/primary axile and lateral length | |
| Seminal | Seminal root number | SRN | Count of the axile seminal roots |
| Total seminal root length | TSL | Total length of axile seminal roots | |
| Average seminal root length | SRL_S | TSL/SRN; total seminal root length/seminal root number | |
| Lateral root number | TNL_S | Count of the lateral roots on the seminal roots | |
| Lateral root density | SLD | TNL_S/TSL; seminal lateral roots/total seminal root length | |
| Axile and lateral root length | SAL | Length of seminal axile root(s) and their lateral roots | |
| Total lateral root length | STL | SAL – TSL; seminal axile and lateral length ‐ total seminal root length | |
| Average lateral root length | SLL | STL/TNL_S; total lateral root length/number of lateral roots | |
| Lateral proportion | SLP | STL/SAL; seminal lateral root length/seminal axile and lateral length | |
| Total | Total root length | TRL | Length of the whole root system |
| Total seedling shoot dry weight | SDW | Weight of the dried seedling shoot to 1/10 000 g | |
| Total seedling root dry weight | RDW | Weight of the dried seedling root to 1/10 000 g | |
| Total root dry weight to total shoot dry weight ratio | RSR | RDW/SDW; total root dry weight/total shoot dry weight | |
| Total axile root length | ARL | PRL + TSL; length of primary and all seminal roots, not including lateral roots | |
| Total lateral root length | LRL | TRL – ARL; total root length ‐ total axile root length | |
| Lateral proportion | TLP | LRL/ARL; total lateral root length/total axile and lateral length | |
| Average specific root length | SRL_T | RDW/TRL; Root mass divided by total root length |
Phenotypic correlation coefficients for the RILs are shown in Table 3. Correlations among the primary root traits indicate that RILs with a longer primary root (PRL) tended to have more lateral roots (TNL_P) (rp = 0.54), shorter lateral roots (ALL) (rp = −0.31) and slightly more total lateral root length (PLL) (rp = 0.23). Greater primary lateral root number was associated with greater lateral root density (PLD) (rp = 0.65), lateral root length (PLL) (rp = 0.34), and total length of the primary axile and lateral roots (PAL) (rp = 0.40). Average lateral root length (ALL) was moderately correlated with total lateral root length (PLL) (rp = 0.58). The large correlation between lateral root length (PLL) and axile and lateral length (PAL) (rp = 0.980), along with the mean of the RILs for the proportion of lateral roots (PLP) (0.86) shows that the preponderance of root length comes from the lateral roots.
Table 3.
Pearson correlation coefficients for 24 root system architecture (RSA) related traits in 204 ZD958 recombinant inbred lines (RILs)
| Traits | PRL | TNL_P | PLD | ALL | PLL | PAL | PLP | SRN | TSL | SRL_S | TNL_S | SLD | SLL | STL | SAL | SLP | TRL | SDW | RDW | RSR | ARL | LRL | TLP | SRL_T |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PRL | <2.20E‐16 | 0.003 | 9.37E‐06 | 1.21E‐03 | 4.62E‐05 | 8.47E‐05 | 0.096 | 5.36E‐05 | 2.77E‐09 | 0.140 | 0.007 | 0.006 | 0.683 | 0.576 | 9.59E‐05 | 0.005 | 0.240 | 0.240 | 0.791 | 1.21E‐13 | 0.131 | 1.03E‐05 | 0.029 | |
| TNL_P | 0.543 | <2.20E‐16 | 7.65E‐12 | 6.44E‐07 | 3.85E‐09 | 0.120 | 0.037 | 0.005 | 1.91E‐06 | 0.032 | 0.420 | 0.350 | 0.315 | 0.099 | 0.322 | 7.84E‐06 | 0.256 | 0.172 | 0.619 | 1.16E‐05 | 1.40E‐04 | 0.434 | 2.16E‐06 | |
| PLD | −0.204 | 0.646 | 5.44E‐05 | 0.003 | 8.14E‐04 | 5.67E‐07 | 0.099 | 0.625 | 0.549 | 0.293 | 0.107 | 0.180 | 0.092 | 0.160 | 0.022 | 0.005 | 0.846 | 0.882 | 0.967 | 0.259 | 0.001 | 4.47E‐07 | 4.64E‐05 | |
| ALL | −0.305 | −0.455 | −0.279 | <2.20E‐16 | 1.20E‐14 | <2.20E‐16 | 0.977 | 0.206 | 0.333 | 0.708 | 0.599 | 0.000 | 0.076 | 0.194 | 0.009 | 1.22E‐07 | 6.10E‐04 | 0.083 | 0.008 | 0.027 | 1.65E‐11 | <2.20E‐16 | 7.00E‐05 | |
| PLL | 0.225 | 0.340 | 0.209 | 0.576 | <2.20E‐16 | <2.20E‐16 | 0.005 | 0.585 | 9.62E‐04 | 0.365 | 0.679 | 0.010 | 0.042 | 0.042 | 0.211 | <2.20E‐16 | 1.89E‐06 | 0.002 | 0.022 | 0.188 | <2.20E‐16 | <2.20E‐16 | 3.46E‐18 | |
| PAL | 0.281 | 0.398 | 0.233 | 0.506 | 0.980 | <2.20E‐16 | 0.005 | 0.352 | 2.69E‐04 | 0.238 | 0.581 | 0.015 | 0.027 | 0.022 | 0.287 | <2.20E‐16 | 2.54E‐07 | 0.002 | 0.011 | 0.057 | <2.20E‐16 | <2.20E‐16 | 1.04E‐19 | |
| PLP | −0.272 | 0.109 | 0.342 | 0.639 | 0.771 | 0.721 | 0.026 | 0.121 | 0.540 | 0.951 | 0.247 | 1.31E‐04 | 0.026 | 0.087 | 0.002 | 6.88E‐13 | 2.95E‐04 | 0.045 | 0.010 | 0.023 | <2.20E‐16 | <2.20E‐16 | 4.04E‐12 | |
| SRN | −0.117 | −0.146 | −0.116 | 0.002 | −0.195 | −0.194 | −0.156 | 1.27E‐14 | 6.66E‐06 | 7.48E‐10 | 0.112 | 0.002 | 1.50E‐09 | 6.18E‐12 | 0.096 | 0.002 | 3.72E‐06 | 4.90E‐13 | 0.111 | 2.17E‐10 | 0.032 | 0.004 | 0.002 | |
| TSL | 0.279 | 0.196 | −0.034 | −0.089 | 0.038 | 0.066 | −0.109 | 0.505 | <2.20E‐16 | <2.20E‐16 | 4.02E‐09 | 1.34E‐05 | <2.20E‐16 | <2.20E‐16 | 0.533 | <2.20E‐16 | 2.29E‐13 | <2.20E‐16 | 0.021 | <2.20E‐16 | 2.34E‐10 | 2.16E‐06 | 0.331 | |
| SRL_S | 0.401 | 0.326 | 0.042 | −0.068 | 0.229 | 0.252 | 0.043 | −0.309 | 0.614 | 2.51E‐05 | 2.90E‐08 | 0.048 | 1.20E‐04 | 6.07E‐08 | 0.018 | 3.35E‐10 | 6.89E‐04 | 4.35E‐06 | 0.336 | <2.20E‐16 | 3.03E‐06 | 0.018 | 2.78E‐04 | |
| TNL_S | 0.104 | 0.150 | 0.074 | −0.026 | 0.064 | 0.083 | 0.004 | 0.414 | 0.639 | 0.290 | 1.19E‐09 | 5.24E‐04 | <2.20E‐16 | <2.20E‐16 | 2.14E‐13 | <2.20E‐16 | <2.20E‐16 | 1.47E‐14 | 0.212 | <2.20E‐16 | <2.20E‐16 | 0.122 | 3.16E‐05 | |
| SLD | −0.188 | −0.057 | 0.113 | 0.037 | −0.029 | −0.039 | 0.081 | −0.112 | −0.397 | −0.376 | 0.409 | 0.348 | 5.91E‐04 | 0.104 | <2.20E‐16 | 0.382 | 0.520 | 0.002 | 0.000 | 1.63E‐09 | 0.030 | 1.98E‐12 | 4.03E‐04 | |
| SLL | −0.191 | −0.066 | 0.094 | 0.281 | 0.180 | 0.170 | 0.265 | 0.213 | 0.300 | 0.139 | 0.241 | −0.066 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | 5.06E‐08 | 1.00E‐08 | 0.464 | 1.08E‐03 | <2.20E‐16 | 1.65E‐10 | 1.32E‐06 | |
| STL | −0.029 | 0.071 | 0.118 | 0.125 | 0.143 | 0.154 | 0.156 | 0.407 | 0.605 | 0.266 | 0.809 | 0.239 | 0.725 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | 0.219 | 2.22E‐16 | <2.20E‐16 | 2.13E‐06 | 8.84E‐09 | |
| SAL | 0.039 | 0.116 | 0.099 | 0.091 | 0.142 | 0.161 | 0.120 | 0.457 | 0.741 | 0.368 | 0.833 | 0.114 | 0.680 | 0.976 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | <2.20E‐16 | 0.575 | <2.20E‐16 | <2.20E‐16 | 0.002 | 1.78E‐07 | |
| SLP | −0.270 | −0.070 | 0.161 | 0.182 | 0.088 | 0.075 | 0.215 | 0.117 | −0.044 | −0.166 | 0.484 | 0.631 | 0.660 | 0.692 | 0.576 | 7.85E‐12 | 1.14E‐05 | 0.081 | 5.61E‐04 | 0.131 | 8.88E‐16 | <2.20E‐16 | 2.59E‐09 | |
| TRL | 0.196 | 0.307 | 0.195 | 0.360 | 0.646 | 0.670 | 0.475 | 0.220 | 0.577 | 0.422 | 0.658 | 0.062 | 0.592 | 0.804 | 0.819 | 0.455 | <2.20E‐16 | <2.20E‐16 | 0.046 | <2.20E‐16 | <2.20E‐16 | 3.86E‐14 | 1.89E‐21 | |
| SDW | 0.083 | 0.080 | 0.014 | 0.238 | 0.326 | 0.351 | 0.251 | 0.317 | 0.484 | 0.236 | 0.555 | 0.045 | 0.370 | 0.589 | 0.613 | 0.302 | 0.660 | <2.20E‐16 | 1.29E‐13 | 2.81E‐11 | <2.20E‐16 | 3.57E‐04 | 0.009 | |
| RDW | 0.083 | 0.096 | 0.010 | 0.122 | 0.211 | 0.214 | 0.141 | 0.478 | 0.677 | 0.315 | 0.504 | −0.218 | 0.388 | 0.567 | 0.633 | 0.122 | 0.591 | 0.630 | 1.97E‐06 | <2.20E‐16 | 6.00E‐15 | 0.979 | 8.83E‐04 | |
| RSR | 0.019 | 0.035 | −0.003 | −0.184 | −0.161 | −0.177 | −0.181 | 0.112 | 0.161 | 0.068 | −0.088 | −0.267 | −0.052 | −0.086 | −0.040 | −0.239 | −0.140 | −0.488 | 0.326 | 0.022 | 0.020 | 3.60E‐06 | 2.54E‐13 | |
| ARL | 0.489 | 0.302 | −0.079 | −0.155 | 0.092 | 0.133 | −0.159 | 0.426 | 0.960 | 0.648 | 0.588 | −0.406 | 0.227 | 0.535 | 0.672 | −0.106 | 0.568 | 0.444 | 0.627 | 0.160 | 4.57E‐10 | 5.68E‐08 | 0.173 | |
| LRL | 0.106 | 0.264 | 0.229 | 0.449 | 0.713 | 0.715 | 0.580 | 0.150 | 0.425 | 0.320 | 0.585 | 0.152 | 0.607 | 0.764 | 0.751 | 0.523 | 0.970 | 0.605 | 0.511 | −0.162 | 0.419 | <2.20E‐16 | 4.33E‐24 | |
| TLP | −0.303 | 0.055 | 0.345 | 0.581 | 0.632 | 0.599 | 0.782 | −0.203 | −0.325 | −0.165 | 0.109 | 0.467 | 0.428 | 0.325 | 0.213 | 0.618 | 0.497 | 0.248 | −0.002 | −0.318 | −0.369 | 0.642 | 5.02E‐21 | |
| SRL_T | −0.153 | −0.325 | −0.281 | −0.275 | −0.559 | −0.580 | −0.461 | 0.214 | −0.068 | −0.252 | −0.287 | −0.245 | −0.331 | −0.389 | −0.356 | −0.402 | −0.601 | −0.184 | 0.231 | 0.483 | −0.096 | −0.631 | −0.596 | |
| Colored scales: | ||||||||||||||||||||||||
| −0.8 < rp < −0.5 | −0.5 < rp < −0.3 | −0.3 < rp < 0.3 | 0.3 < rp < 0.5 | 0.5 < rp < 0.8 | rp > 0.8 | |||||||||||||||||||
Lower left: Pearson correlation coefficients; Upper right: P‐value for Pearson correlation test
The phenotypic correlations among the seminal root traits provide some similar insights. Total seminal root length (TSL) exhibited a moderate correlation with seminal root number (SRN) (rp = 0.51) and average seminal root length (SRL_S) (rp = 0.61), indicating the RIL population contains members with a mixture of SRN and SRL_S phenotypes. The average seminal root length (SRL_S) of RILs with more seminal roots (SRN) tended to be shorter (rp = −0.31). Longer axile seminal root length (TSL) was associated with greater lateral root number (TNL_S) (rp = 0.64) and total seminal lateral root length (STL) (rp = 0.61). A similar, but weaker, trend was observed between seminal root number (SRN) and lateral root number (TNL_S) (rp = 0.41) and lateral root length (STL) (rp = 0.41). The average lateral root length (SLL) tended to be longer when there were more lateral roots (TNL_S) (rp = 0.24). Most of the root length of the seminal roots (SAL) could be attributed to lateral root length (STL) (rp = 0.98), and STL was strongly associated with both number of lateral roots (TNL_S) (rp = 0.81) and their average length (SLL) (rp = 0.73).
Total root system length (TRL) had moderate positive correlations with shoot dry weight (SDW) (rp = 0.66), root dry weight (rp = 0.59) and total axile root length (rp = 0.57). Longer root systems were associated with less mass per unit root length (SRL_T) (rp = −0.60). Seedlings with more shoot mass (SDW) also tended to have more root mass (rp = 0.63), although the RIL population exhibited considerable variation for root to shoot ratio (RSR) (Table 2; Figure S3). Primary root length (PRL) was positively correlated with average seminal root length (SRL_S) (rp = 0.40), but the number of lateral roots on the primary root (TNL_P) was only weakly associated with the number of lateral roots on seminal roots (TNL_S) (rp = 0.15). The average lateral root length on the primary root (ALL) was longer than the average seminal lateral root length (SLL) (Table 2) and had a low, but significant correlation (rp = 0.28). The combined length of the primary root and its lateral roots (PAL) was moderately correlated with the total root system length (TRL) (rp = 0.67) and total lateral root length (LRL) (rp = 0.71), but less so with shoot and root dry weight (rp = 0.35 and 0.21). The total length of the seminal axile and lateral roots (SAL) and the number of seminal lateral roots (TNL_S) were moderately correlated with shoot and root dry weight (rp ≈ 0.5 to 0.63). Total axile root length (ARL) exhibited a greater correlation with root dry weight (RDW) (rp = 0.63) than total lateral root length (LRL) (rp = 0.51). Overall, the phenotypic correlations suggest the existence of independently segregating QTL for most traits, the possibility of loci with pleiotropic effects, and common QTL among highly correlated RSA traits.
SNP calling and bin map construction
A modified sliding window method was used for construction of the bin map, resulting in a map with 7,319 marker bins (Figure 2A; Table 4). The average physical interval of the adjacent bins was 278.7 kb, with a maximum of 7,700 kb and minimum of 100 kb. There were 63 bins larger than 2 Mb, 25 of which were located within 20 Mb of the centromeres. A linkage map was constructed by R/qtl, with a total length of 2,812.28 cM (Table 4). The largest and average intervals on the linkage map were 15.39 cM and 0.38 cM. Pairwise recombination fractions for all markers were calculated and there were no apparent issues with the map (Figure 2B).
Figure 2.
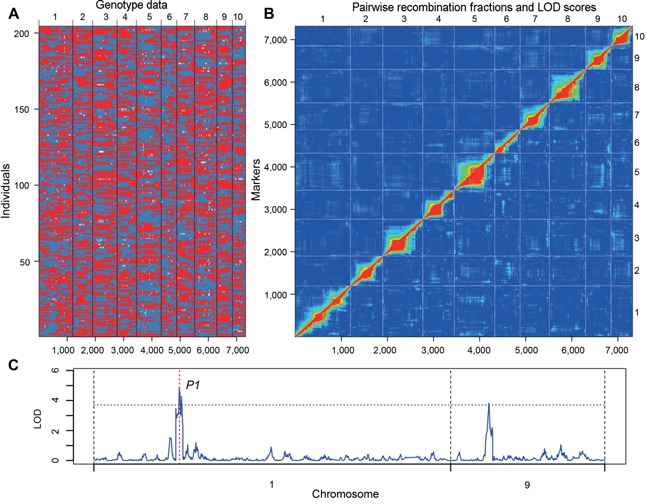
Recombination bin‐map of recombinant inbred line (RIL) population and quantitative trait loci (QTL) mapping of silk color P1 (pericarp1) (A) The bin‐map consists of 7 319 bin markers inferred from 248 168 high‐quality SNPs in the RIL population and has a total genetic distance of 2812 cM. The physical position of the markers is based on the B73 RefGen_V2 sequence. Red: Chang7‐2 genotype; Blue: Zheng58 genotype; White: heterozygote. (B) Pairwise recombination fractions (upper left) and logarithm (base 10) of odds (LOD) scores for tests of linkage (bottom right) for all markers were calculated and graphed as a heatmap. Marker and chromosome numbers are provided in the axis labels. Red corresponds to low recombination or a high LOD score, while blue represents unlinked markers. (C) The composite interval mapping (CIM) plot for silk color shows the QTL identified on chromosomes 1 and 9. The location of the pigment gene pericarp color1 (p1) is indicated by red dashed lines.
Table 4.
Summary of bin marker characteristics in the recombinant inbred line (RIL) population
| Chr | Genetic distance (cM) | Number of the bin markers |
|---|---|---|
| Chr1 | 512.70 | 1,197 |
| Chr2 | 257.50 | 702 |
| Chr3 | 298.56 | 869 |
| Chr4 | 204.49 | 683 |
| Chr5 | 398.72 | 878 |
| Chr6 | 205.98 | 548 |
| Chr7 | 212.11 | 635 |
| Chr8 | 314.98 | 787 |
| Chr9 | 221.59 | 551 |
| Chr10 | 185.65 | 469 |
| SUM | 2812.28 | 7,319 |
QTL analysis
In order to examine the power of the bin map and population, QTL mapping for silk color, which has high heritability, was performed. Two QTL were identified, the largest of which was located on chromosome 1 with a peak at 47.9 Mb (logarithm (base 10) of odds (LOD) = 4.90) (Figure 2C). The QTL peak is located in the tandem repeat region of the previously characterized and cloned P1 (pericarp color1) gene (Lechelt et al. 1989; Grotewold et al. 1994). The other QTL on chromosome 9 had a minor effect and has not been previously reported.
A total of 62 QTL were identified for the 24 RSA related traits (Figure 3; Table 5). The QTL were distributed across all of the chromosomes and had confidence intervals associated with their genomic position ranging from 0.6 Mb to 55.8 Mb (Table 5). Within the primary root class, the largest QTL detected were for the proportion of lateral roots PLP (Chr. 9, LOD = 5.00), lateral root number (TNL_P) (Chr. 1, LOD = 4.76), and length of axile and lateral roots (PAL) (Chr. 9, LOD = 4.13). A single QTL for primary root length (4.57% phenotypic variance explained) was mapped to a 3.6 Mb region of chromosome 7 and the positive allele was contributed by Zheng58. Two QTL for lateral root number (TNL_P) were identified on chromosomes 1 and 3 and Zheng58 carried the alleles for increased lateral root number. Four QTL were detected for lateral root density (PLD) located on chromosomes 1, 3 and 6. The effect size of these loci was similar, but Chang7‐2 and Zheng58 contributed alleles for increased density at two loci each. A QTL for TNL_P and PLD overlapped on chromosome 3 with Bin3_69 as the peak marker at 9.3Mb. Zheng58 contributed alleles for increased average lateral root length (ALL) at three of the four detected QTL on chromosomes 4, 5 and 6. The two QTL for the total length of lateral roots on the primary root (PLL) mapped to chromosome 9. The PLL QTL with a peak 135.6 Mb overlapped with QTL for total length of the axile and lateral roots (PAL) at 134.5 Mb and the proportion of lateral root length (PLP) at 135.6 Mb. Zheng58's alleles contributed the decreasing effect at these loci. A second PAL QTL was located on chromosome 10 at 145.2 Mb, the same location as another PLP QTL.
Figure 3.
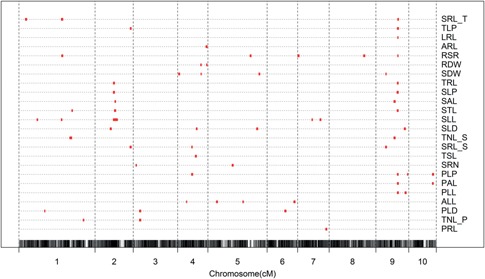
Composite interval mapping (CIM) plots for the 24 root system architecture related traits The x‐axis indicates the physical positions of bin markers. The y‐axis lists the root system architecture (RSA) traits. The red dots on the broken lines represent quantitative trait loci (QTL).
Table 5.
Summary of quantitative trait loci (QTL) identified in the recombinant inbred line (RIL) population
| Classification | QTLa | Chr | Peak_Binb | Bin interval | Peak_Pos(Mb)c | Physical_interval (Mb)c | Peak_Pos | Linkage interval (cM) | LOD | Var(%)d | Add | Bin locatione |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary | PRL | 7 | Bin7_597 | Bin7_587‐Bin7_609 | 170.70 | 168.7–172.3 | 192.47 | 188.26–197.92 | 2.44 | 4.57 | 2.20 | Bin7.05 |
| Primary | TNL_P | 1 | Bin1_1084 | Bin1_1080‐Bin1_1095 | 280.10 | 279.7–282.1 | 433.78 | 431.79–437.99 | 4.76 | 4.16 | 17.96 | Bin1.1 |
| Primary | TNL_P | 3 | Bin3_69 | Bin3_60‐Bin3_79 | 9.30 | 8.1–10.7 | 46.56 | 40.81–51.57 | 2.91 | 6.62 | 20.59 | Bin3.03 |
| Primary | PLD | 1 | Bin1_405 | Bin1_393‐Bin1_415 | 86.10 | 82.1–88.5 | 173.66 | 171.44–176.13 | 3.00 | 6.57 | −0.53 | Bin1.04 |
| Primary | PLD | 3 | Bin3_69 | Bin3_60‐Bin3_79 | 9.30 | 8.1–10.7 | 46.56 | 40.81–51.57 | 3.35 | 5.72 | 0.53 | Bin3.03 |
| Primary | PLD | 6 | Bin6_393 | Bin6_382‐Bin6_403 | 139.60 | 137.4–141.2 | 122.87 | 117.66–128.6 | 3.84 | 5.40 | −0.52 | Bin6.05 |
| Primary | ALL | 4 | Bin4_149 | Bin4_140‐Bin4_160 | 31.80 | 26.8–35.1 | 61.33 | 58.86–62.56 | 3.70 | 3.34 | 0.18 | Bin4.04 |
| Primary | ALL | 5 | Bin5_117 | Bin5_105‐Bin5_129 | 17.60 | 14.7–19.6 | 60.49 | 56.03–65.7 | 3.87 | 4.61 | −0.27 | Bin5.03 |
| Primary | ALL | 5 | Bin5_769 | Bin5_759‐Bin5_779 | 168.60 | 167.2–170.1 | 237.60 | 233.89–240.32 | 3.34 | 6.80 | 0.25 | Bin5.04 |
| Primary | ALL | 6 | Bin6_524 | Bin6_518‐Bin6_532 | 163.00 | 162–164.2 | 184.75 | 179.21–189.97 | 2.92 | 4.97 | 0.22 | Bin6.07 |
| Primary | PLL | 9 | Bin9_473 | Bin9_455‐Bin9_484 | 135.60 | 130.5–137.4 | 149.27 | 143.59–153.48 | 3.15 | 3.02 | −32.50 | Bin9.05/06 |
| Primary | PLL | 9 | Bin9_566 | Bin9_553‐Bin9_576 | 149.00 | 147.4–150.3 | 200.56 | 195.37–205.56 | 2.83 | 5.16 | 31.45 | Bin9.06 |
| Primary | PAL | 9 | Bin9_470 | Bin9_455‐Bin9_484 | 134.50 | 130.5–137.4 | 148.29 | 143.59–153.48 | 4.13 | 3.88 | −28.27 | Bin9.05/06 |
| Primary | PAL | 10 | Bin10_459 | Bin10_448‐Bin10_461 | 145.20 | 143.5–145.5 | 163.61 | 158.15–166.15 | 3.03 | 5.03 | −29.93 | Bin10.07 |
| Primary | PLP | 4 | Bin4_388 | Bin4_381‐Bin4_413 | 158.00 | 156.9–162.7 | 95.97 | 92.75–102.89 | 3.56 | 4.78 | 0.02 | Bin4.06 |
| Primary | PLP | 9 | Bin9_473 | Bin9_455‐Bin9_478 | 135.60 | 130.5–136.3 | 149.27 | 143.59–151.5 | 3.80 | 4.03 | −0.02 | Bin9.05 |
| Primary | PLP | 9 | Bin9_589 | Bin9_587‐Bin9_591 | 153.30 | 152.7–153.8 | 215.61 | 212.04–216.6 | 5.00 | 8.82 | 0.03 | Bin9.07 |
| Primary | PLP | 10 | Bin10_459 | Bin10_448‐Bin10_464 | 145.20 | 143.5–145.9 | 163.61 | 158.15–168.66 | 2.55 | 5.18 | −0.02 | Bin10.07 |
| Seminal | SRN | 3 | Bin3_30 | Bin3_25‐Bin3_33 | 4.00 | 3.5–4.6 | 20.55 | 17.3–22.82 | 4.16 | 4.66 | 0.39 | Bin3.02 |
| Seminal | SRN | 5 | Bin5_492 | Bin5_485‐Bin5_496 | 83.30 | 82.2–83.7 | 166.74 | 161.66–171.85 | 2.75 | 4.53 | −0.36 | Bin5.04 |
| Seminal | TSL | 4 | Bin4_468 | Bin4_457‐Bin4_483 | 178.20 | 175.1–180.8 | 123.45 | 118.24–128.64 | 3.32 | 4.93 | −8.22 | Bin4.07 |
| Seminal | SRL_S | 2 | Bin2_683 | Bin2_667‐Bin2_700 | 228.40 | 225.1–231.9 | 239.13 | 234.44–245.08 | 2.98 | 6.49 | −2.32 | Bin2.09 |
| Seminal | SRL_S | 4 | Bin4_401 | Bin4_384‐Bin4_405 | 160.40 | 157.3–161.6 | 98.93 | 93.74–100.17 | 4.03 | 6.85 | −2.63 | Bin4.06 |
| Seminal | SRL_S | 9 | Bin9_156 | Bin9_145‐Bin9_163 | 24.00 | 22.1–24.7 | 70.11 | 63.37–73.08 | 3.92 | 5.52 | −2.52 | Bin9.02/03 |
| Seminal | TNL_S | 1 | Bin1_894 | Bin1_886‐Bin1_897 | 247.30 | 246.4–247.7 | 351.65 | 341.4–356.53 | 3.00 | 5.26 | −42.82 | Bin1.08 |
| Seminal | TNL_S | 9 | Bin9_391 | Bin9_371‐Bin9_407 | 110.30 | 107.9–115.8 | 126.78 | 120.13–130.73 | 3.36 | 6.38 | −45.26 | Bin9.04 |
| Seminal | SLD | 2 | Bin2_284 | Bin2_249‐Bin2_317 | 100.90 | 62.3–118.1 | 106.07 | 100.88–111 | 2.69 | 4.04 | 0.49 | Bin2.05 |
| Seminal | SLD | 4 | Bin4_480 | Bin4_471‐Bin4_490 | 180.00 | 178.7–182.1 | 127.90 | 124.94–132.38 | 4.03 | 7.33 | 0.58 | Bin4.08 |
| Seminal | SLD | 5 | Bin5_980 | Bin5_975‐Bin5_988 | 201.30 | 200.8–202.4 | 332.69 | 326.85–336.92 | 2.82 | 3.85 | 0.47 | Bin5.06 |
| Seminal | SLD | 9 | Bin9_552 | Bin9_544‐Bin9_565 | 147.30 | 146.4–148.9 | 195.12 | 189.84–200.32 | 2.69 | 5.31 | −0.52 | Bin9.06 |
| Seminal | SLL | 1 | Bin1_269 | Bin1_263‐Bin1_282 | 48.20 | 47.2–51.2 | 123.22 | 120.99–126.92 | 3.38 | 2.04 | 0.07 | Bin1.03 |
| Seminal | SLL | 1 | Bin1_704 | Bin1_698‐Bin1_710 | 192.50 | 191.6–193.2 | 287.14 | 284.16–290.87 | 4.05 | 5.09 | −0.08 | Bin1.06 |
| Seminal | SLL | 2 | Bin2_475 | Bin2_390‐Bin2_507 | 187.50 | 153.6–195.4 | 141.36 | 121.86–151.99 | 2.50 | 4.64 | 0.05 | Bin2.06 |
| Seminal | SLL | 7 | Bin7_345 | Bin7_336‐Bin7_360 | 112.20 | 109.5–117.9 | 96.67 | 94.94–100.87 | 5.78 | 6.62 | −0.10 | Bin7.02 |
| Seminal | SLL | 7 | Bin7_510 | Bin7_498‐Bin7_519 | 156.20 | 153.8–157.5 | 154.03 | 149.08–157.74 | 2.86 | 4.51 | 0.07 | Bin7.03/04 |
| Seminal | STL | 1 | Bin1_899 | Bin1_896‐Bin1_904 | 248.00 | 247.6–248.7 | 358.54 | 354.75–361.54 | 4.21 | 4.79 | −27.21 | Bin1.08 |
| Seminal | STL | 2 | Bin2_454 | Bin2_437‐Bin2_471 | 181.10 | 177.4–185.6 | 134.94 | 129.76–140.13 | 3.09 | 6.32 | 34.48 | Bin2.06 |
| Seminal | STL | 9 | Bin9_468 | Bin9_450‐Bin9_478 | 134.30 | 128.5–136.3 | 147.54 | 142.36–151.5 | 2.68 | 4.52 | −30.38 | Bin9.05 |
| Seminal | SAL | 2 | Bin2_454 | Bin2_448‐Bin2_469 | 181.10 | 179.8–185.3 | 134.94 | 133.22–139.39 | 4.28 | 7.03 | 44.25 | Bin2.06 |
| Seminal | SAL | 9 | Bin9_390 | Bin9_371‐Bin9_408 | 110.10 | 107.9–116.1 | 126.53 | 120.13–131.23 | 2.82 | 5.02 | −38.25 | Bin9.04 |
| Seminal | SLP | 2 | Bin2_436 | Bin2_390‐Bin2_443 | 176.20 | 153.6–178.2 | 129.26 | 121.86–132.23 | 2.84 | 7.06 | 0.04 | Bin2.06 |
| Seminal | SLP | 9 | Bin9_467 | Bin9_449‐Bin9_480 | 134.10 | 128.2–136.6 | 147.30 | 141.61–152 | 3.02 | 7.50 | −0.04 | Bin9.05 |
| Total | TRL | 2 | Bin2_420 | Bin2_394‐Bin2_442 | 169.80 | 155.9–178 | 126.30 | 122.6–131.99 | 3.60 | 2.63 | 35.96 | Bin2.06 |
| Total | TRL | 9 | Bin9_470 | Bin9_455‐Bin9_478 | 134.50 | 130.5–136.3 | 148.29 | 143.59–151.5 | 3.45 | 5.99 | −58.22 | Bin9.05 |
| Total | SDW | 4 | Bin4_23 | Bin4_15‐Bin4_28 | 3.10 | 2.1–3.7 | 11.46 | 5.96–15.46 | 2.56 | 1.61 | 0.01 | Bin4.01 |
| Total | SDW | 4 | Bin4_576 | Bin4_568‐Bin4_584 | 213.70 | 209.3–217.6 | 159.12 | 157.39–160.86 | 6.04 | 9.24 | −0.02 | Bin4.09 |
| Total | SDW | 5 | Bin5_1005 | Bin5_999‐Bin5_1009 | 204.90 | 203.9–205.4 | 347.69 | 342.67–350.68 | 4.21 | 5.68 | 0.02 | Bin5.06/07 |
| Total | SDW | 9 | Bin9_152 | Bin9_148‐Bin9_157 | 23.40 | 22.9–24.1 | 67.87 | 66.14–70.6 | 3.56 | 6.41 | −0.02 | Bin9.03 |
| Total | RDW | 4 | Bin4_571 | Bin4_557‐Bin4_584 | 211.50 | 204.1–217.6 | 158.13 | 154.92–160.86 | 4.53 | 6.45 | 0.00 | Bin4.09 |
| Total | RDW | 4 | Bin4_673 | Bin4_670‐Bin4_677 | 239.20 | 238.9–239.6 | 195.72 | 194.23–199.73 | 2.82 | 5.02 | 0.00 | Bin4.1 |
| Total | RSR | 1 | Bin1_711 | Bin1_702‐Bin1_716 | 193.30 | 192.2–194.5 | 291.36 | 286.65–296.41 | 3.11 | 2.80 | 0.02 | Bin1.06 |
| Total | RSR | 5 | Bin5_900 | Bin5_894‐Bin5_907 | 190.00 | 189.3–190.8 | 286.74 | 283.25–292.5 | 3.97 | 4.43 | −0.03 | Bin5.05 |
| Total | RSR | 7 | Bin7_12 | Bin7_1‐Bin7_21 | 2.30 | 0‐3.7 | 5.48 | 0–10.69 | 2.85 | 4.07 | −0.02 | Bin7.00 |
| Total | RSR | 8 | Bin8_660 | Bin8_654‐Bin8_664 | 159.10 | 157.9–159.5 | 235.69 | 231.16–241.83 | 2.64 | 4.91 | −0.02 | Bin8.06 |
| Total | RSR | 9 | Bin9_470 | Bin9_465‐Bin9_477 | 134.50 | 133.7–136.1 | 148.29 | 146.31–150.76 | 5.90 | 11.58 | 0.03 | Bin9.05 |
| Total | ARL | 4 | Bin4_673 | Bin4_664‐Bin4_678 | 239.20 | 238.1–239.8 | 195.72 | 190.25–200.48 | 2.79 | 3.08 | 7.33 | Bin4.1 |
| Total | LRL | 9 | Bin9_472 | Bin9_465‐Bin9_477 | 135.20 | 133.7–136.1 | 148.78 | 146.31–150.76 | 4.22 | 6.76 | −56.93 | Bin9.05 |
| Total | TLP | 2 | Bin2_689 | Bin2_672‐Bin2_699 | 230.20 | 225.8–231.8 | 240.36 | 235.92–244.83 | 3.09 | 6.60 | 0.02 | Bin2.09 |
| Total | TLP | 9 | Bin9_473 | Bin9_455‐Bin9_486 | 135.60 | 130.5–137.9 | 149.27 | 143.59–153.97 | 3.03 | 6.05 | −0.02 | Bin9.05 |
| Total | SRL_T | 1 | Bin1_82 | Bin1_70‐Bin1_86 | 11.30 | 10.1–11.7 | 50.56 | 42.33–52.81 | 2.82 | 3.10 | 0.62 | Bin1.01 |
| Total | SRL_T | 1 | Bin1_708 | Bin1_699‐Bin1_716 | 193.00 | 191.7–194.5 | 289.88 | 284.66–296.41 | 4.07 | 5.97 | 0.85 | Bin1.06 |
| Total | SRL_T | 9 | Bin9_473 | Bin9_465‐Bin9_485 | 135.60 | 133.7–137.6 | 149.27 | 146.31–153.73 | 6.26 | 10.73 | 1.17 | Bin9.05 |
The abbreviations of trait names is listed in Table 1; bthe peak position with the highest LOD of each QTL; cthe positions of the identified QTL according to B73 reference sequence Version 5.60; dpercentage of phenotypic variation explained by additive effect of the identified QTL; ethe bins were defined according to the B73 physical map using the data from the website: http://www.maizegdb.org. LOD, logarithm (base 10) of odds
The most significant QTL for the seminal root class traits were average lateral root length (SLL) (Chr. 7, LOD = 5.78), total length of axile and lateral roots SAL (Chr. 2, LOD = 4.28), total length of lateral roots (STL) (Chr. 1, LOD = 4.21), and number of seminal roots (SRN) (Chr. 3, LOD = 4.16). Two QTL were identified for seminal root number with the positive effect contributed by Zheng58 at one loci and Chang7‐2 at the other. Only one QTL was detected for the total length of axile and lateral seminal roots (TSL), which explained 4.93% of the phenotypic variation, and Chang7‐2 alleles increased the length. This QTL's physical interval largely overlapped that of a larger QTL (LOD = 4.03) for lateral root density (SLD). Three QTL were detected for the average seminal root length on chromosomes 2, 4 and 9. Chang7‐2 alleles increased length at these loci. A total of two QTL were detected for lateral root number (TNL_S), located on chromosomes 1 and 9, with increasing alleles contributed by Chang7‐2. Four QTL for lateral root density (SLD) explained 20.54% of the total phenotypic variation and were located on chromosomes 2, 4, 5 and 9. None of these overlapped with QTL for lateral root density on the primary root (PLD). Five QTL identified for average lateral root length (SLL) explained 22.91% of the total phenotypic variation, but none co‐localized with QTL for average lateral root length on the primary root (ALL). Three QTL were detected for total lateral root length (STL) on chromosomes 1, 2 and 9. The QTL on chromosome 2 maps to the same bin as a QTL with a higher LOD score for total length of axile and lateral roots (SAL). The QTL on chromosome 9 maps to a bin adjacent to a QTL for the proportion of lateral roots (SLP). The second QTL for SAL on chromosome 9 and the first QTL for SLP on chromosome 2 appear to be distinct from the QTL for STL.
The largest QTL detected for the total class of traits were average specific root length (SRL_T) (Chr. 9, LOD = 6.26), shoot dry weight (SDW) (Chr. 4, LOD = 6.04), root to shoot ratio (RSR) (Chr. 9, LOD = 5.90) and root dry weight (RDR) (Chr. 4, LOD = 4.53). Two QTL were identified for total root length (TRL), one of which mapped to the same interval on chromosome 9 (Bin9_470) as primary axile and lateral root length (PAL). Chang7‐2 alleles increased root length at the loci for both traits. Four QTL for the seedling shoot dry weight (SDW) were identified across chromosomes 4, 5 and 9. The QTL on chromosome 9 overlapped with a QTL for the average length of seminal lateral roots (SRL_S). Two QTL were identified for root dry weight (RDW), the most significant of which on chromosome 4, broadly overlapped with a QTL for SDW and the only QTL for total axile root length (ARL). However, no QTL for the ratio of root to shoot mass were detected in this region. RSR QTL mapped to chromosomes 1, 5, 7, 8 and 9, and explained 27.79% of the total phenotypic variation. The RSR QTL on chromosome 9 mapped to a 2.4 Mb interval, explained 11.58% of the phenotypic variation, and alleles from Zheng58 increased the ratio. The only QTL for total lateral root length (LRL) also resided in a 2.4 Mb interval in the same region and alleles from Zheng58 decreased root length. QTL for total length of seminal lateral roots (STL), proportion of seminal lateral roots (SLP) total root length (TRL) and total proportion of lateral roots (TLP), and average specific root length (SRL_T) also map to this same genomic region from 130 Mb to 138 Mb on chromosome 9. Of these, the QTL for SRL_T had the largest LOD score and alleles for Zheng58 resulted in greater mass per unit root length. Two additional QTL were detected for SRL_T on chromosome 1 and the positive effect was also contributed from Zheng58 alleles at these loci. When the QTL from this study are examined in terms of the classical maize chromosome bins, QTL clusters are observed in bins 2.06, 3.02‐03, 9.02‐04 and 9.05‐06, with a total of 5, 3, 4 and 12 QTL, respectively (Figure 3).
DISCUSSION
Contribution of the high‐quality bin map to identification of QTL
Many QTL studies have been performed using sparse genetic maps constructed using restriction fragment length polymorphism and simple sequence repeat markers, resulting in large inter‐marker intervals (Beavis and Grant 1991; Gonzalo et al. 2010; Cai et al. 2012; Zheng and Liu 2013). High marker density is preferable for accurate identification of recombination breakpoints in mapping populations and improvement in the estimates of QTL positions. Advances in sequencing technology have enabled higher marker densities for identification of genetic loci associated with agronomic traits (Jones et al. 2007; Ganal et al. 2011; Tian et al. 2011; Li et al. 2014b). Use of GBS and construction of bin maps has proven to be a successful strategy for improved QTL detection in rice (Huang et al. 2009; Xie et al. 2010; Yu et al. 2011) and sorghum (Zou et al. 2012). This approach was recently applied to a large maize F2 population to produce better estimates of QTL positions for reproductive architecture in our previous study (Chen et al. 2014). In the present study, 7,319 bin markers filtered from 54,543 SNPs for the RILs were sufficient to accurately map p1, a major locus associated with silk color, indicating the capability of the bin map and population to fine‐map QTL.
Genetic architecture of maize seedling root phenotypes
Relatively few mutants controlling root architecture have been characterized, precisely mapped and functionally analyzed so far. An example of a major root architecture QTL is root‐ABA1, although it showed decreases on the yield (Giuliani et al. 2005), but successful cloning of a root architecture QTL is yet to be reported. Burton et al. (2014) recently mapped 15 QTL for 21 seedling root architecture traits using three recombinant inbred populations derived from public US inbreds. Effect sizes were small and individual QTL explained 0.44% to 13.5% of the total phenotypic variation. These results are similar to our findings using a population derived from an elite Chinese hybrid. The proportion of phenotypic variation explained by the QTL ranged from 1.61% to 11.58% and the total proportion explained per trait was low. Few consensus root length QTL were identified in a meta‐analysis of nine maize mapping populations testing different genetic backgrounds and environments (Hund et al. 2011). Of the QTL in our study, only a primary lateral root density (PLD) QTL in maize chromosome bin 6.05 co‐located with one of the root‐QTL clusters identified in the meta‐analysis (Hund et al. 2011). The QTL for seminal root number in chromosome 5 in our study was located near a QTL identified from the NyH RIL population (Burton et al. 2014). Overall, there was little overlap in QTL found between our study and previous works, and the root QTL clusters in chromosome bins 2.06, 3.02‐03, 9.02‐04 and 9.05‐06 may therefore be considered novel.
Traits with QTL in the bin 2.06 cluster include average length of lateral roots (SLL), total lateral root length (STL), total length of axile and lateral roots (SAL), proportion of lateral roots (SLP) and total root length (TRL). Zheng58 contributed the increasing alleles at all of these QTL. The peak bin for STL and SAL is the same and may be assumed to represent a single locus, considering the very high correlation between the two traits. The LOD score for SAL was the highest out of all QTL in the cluster (LOD = 4.28), and somewhat greater than that of STL (LOD = 3.09). QTL for seminal root number (SRN), total axile seminal root length (TSL) and average seminal root length (SRL_S) were not detected in the area, but the axial roots contribute little to the total seminal root length compared to lateral roots. Chang7‐2 is the source of increasing alleles: the identified TSL and SRL_S QTL. A total of four QTL were identified for average lateral root length (SLL), although the parents were not significantly different for this trait, but the SLL QTL in bin 2.06 had the lowest LOD of the QTL in the cluster. A QTL for lateral root number (TNL_S) was also absent from the cluster. Considering the moderate to large correlations among these traits, this cluster may represent the effect of a single locus or the composite effect of some subset. This cluster clearly influences total seminal lateral root length (and hence total root length (TRL), but not through the number of lateral roots or solely through the average length of the lateral roots. This may explain the presence of the QTL in the cluster for the proportion of lateral roots to total seminal root length (SLP). A large SLP value is obtained by a RIL with more lateral branching per unit axile length. For similar values of seminal lateral roots (TNL_S) and average lateral root length (SLL), lateral root density (SLD) is increased with decreasing total seminal root length (TSL). This combination of factors may be most clearly expressed by the SAL trait, which resides between the QTL for average lateral root length (SLL) and lateral root density (SLD).
QTL for the number of lateral roots on the primary root (TNL_P), lateral root density on the primary root (PLD), and seminal root number were found in bins 3.02‐03. The QTL for TNL_P and PLD map to the same location (9.30 Mb) and the increasing alleles are contributed by Zheng58. Considering TNL_P has the superior LOD score and PLD is derived from TNL_P, these two QTL likely represent a common locus. The QTL for seminal root number is somewhat distant and probably distinct. Among the QTL detected in maize bins 9.02‐04, Chang7‐2 alleles increased average seminal root length (SRL_S) and shoot dry weight (SDW) in bins 9.02‐03 and the number of seminal lateral roots (TNL_S) and total seminal axile and lateral length (SAL) in bin 9.04. The SAL locus maps to an adjacent bin marker as TNL_S, which has the higher LOD score, and so may be interpreted as a common locus given the large correlation between these traits. RSA phenotypes with QTL located in maize bins 9.05‐06 include total primary lateral root length (PLL), total primary axile and lateral root length (PAL), proportion of primary lateral roots (PLP), seminal lateral root density (SLD), total seminal lateral root length (STL), seminal lateral root proportion (SLP), total root length (TRL), root to shoot ratio (RSR), total lateral root length (LRL), total lateral root proportion (TLP) and average specific root length (SRL_T). Chang7‐2 alleles increased traits at all loci except the second of two PLL QTL, RSR and SRL_T. The detected PLL QTL are within 50 cM of each other, in repulsion phase, and have a similar effect size. Zheng58 alleles in the QTL cluster may be interpreted as conferring greater mass partitioning to the root system, specifically more mass per unit root length, and less primary and seminal lateral root length overall. At the stage phenotyped, Zheng58 has significantly more primary and secondary lateral roots (TNL_P and TNL_S) and Chang7‐2 has significantly longer average lateral root length on the primary root (ALL) and total lateral root length (LRL) (Table 2). A recent study showed that Chang7‐2 likely contributes a favorable crown root system to the ZD958 hybrid (Han et al. 2015), although Chang7‐2 is considered a small root branching line (Weimin 2004).
Maize root system architecture and seedling vigor
Although root system architecture is important for plant performance, the seedling and mature ideotype structures favorable for specific environments and wide adaptation remain to be elucidated. Seedling vigor in terms of vegetative area or mass has been routinely selected for in the breeding of new commercial maize hybrids, but most genetic mapping for seedling vigor has been performed in rice (Redona and Mackill 1996; Cui et al. 2002; Huang et al. 2004; Abe et al. 2012). Wu et al. (2011) investigated nitrogen response in seedlings of 11 Chinese commercial hybrids released from 1973 to 2009, including ZD958, using a solution culture system and reported improved relative shoot and root growth rates, total root length, lateral root length and axile root length at high nitrogen in newer hybrids (Wu et al. 2011). The practice of reciprocal recurrent selection for the production of improved hybrids may easily result in the source of favorable alleles differing between parental inbreds. Zheng58 and Chang7‐2 have rather large differences in seedling vigor in terms of shoot and root biomass accumulated, but similar partitioning (Table 2). Chang7‐2 appears to achieve a similar total root length (TRL) with shorter axile roots (ARL), fewer seminal roots (SRN), less lateral branching (TNL_P, TNL_S) and longer lateral roots (ALL) (Table 2).
Burton et al. (2014) observed a stronger allometric correlation between root length and mass than root number. We found similar results in our population. The correlation of TLN_P, SRN and TLN_S with RDW in our RIL population was rp ≈ 0.10, 0.48 and 0.50. The correlation of PLL, STL, LRL and TSL with RDW was rp ≈ 0.21, 0.57, 0.51 and 0.68. Two studies recently reported that RSA with fewer and longer lateral roots conveyed greater drought tolerance, ability to capture nitrogen from low‐N soils, and higher yield under environmental stresses than RSA with a greater number of shorter lateral roots (Zhan and Lynch 2015; Zhan et al. 2015). Li et al. (2015) reported co‐localization of RSA traits with QTL for nitrogen use efficiency (NUE). Positive alleles for SDW and RSR QTL emanated from both parents, while those for RDW QTL were inherited from Zheng58. Bin 4.09 contains QTL for both SDW and RDW, but the positive effects are inherited from different parents. The lack of an RSR QTL near this site at other SDW or RDW QTL may be related to the difference in the effect sizes of root and shoot mass QTL. The largest RSR QTL mapped to the same location as the only total lateral root length (LRL). Interestingly, Zheng58 alleles reduced total lateral root length and increased the ratio of root to shoot weight. The identification and cloning of favorable loci for seedling vigor and root growth lends itself to marker assisted selection (MAS), which has been previously used to improve root architecture in maize (Li et al. 2015) and rice (Steele et al. 2006; Steele et al. 2013).
In conclusion, we have analyzed the phenotypic variation and the corresponding QTLs of 24 RSA‐related traits using an RIL population. Significant correlations were observed among many of the traits as supported by the studied co‐localization of correlated traits. Our results will be useful in the future for maize breeders to reduce the amount of phenotyping effort and to improve the root architecture.
MATERIALS AND METHODS
Plant materials and phenotyping
A population of F7 Zea mays RILs was produced by single seed decent, derived from crossing Zheng58 and Chang7‐2, which forms the popular commercial hybrid Zhengdan958. Zheng58 is the female parent and is known to contribute a highly branched mature root system (Falin 2001). In contrast, the male parent Chang7‐2 is a source of root lodging susceptibility (Weimin 2004). Seeds of uniform size from the 204 lines were selected and sterilized in 10% H2O2 for 45 min, followed by washing, using distilled water three times, then were transferred to filter paper and germinated. Three days later, the geminated seeds were transferred to wet filter paper, covered with another piece of wet filter paper, rolled up, and placed in beakers with distilled water. After 6 d, seedlings with their endosperm detached and transferred to the revised Hoagland solution (Hoagland and Arnon 1938) in a greenhouse hydroponic system. The solution included 0.6 mmol/L MgSO4.7H2O, 0.1 mmol/L NaH2PO4, 4 mmol/L KNO3, 0.75 mmol/L K2SO4, 0.25 mmol/L KH2PO4, 1 µmol/L H3BO3, 0.5 mmol/L MnSO4.H2O, 0.5 mmol/L ZnSO4.7H2O, 0.18 mmol/L CuSO4.5H2O, 0.06 mmol/L Na2MoO4.2H2O, 0.1 mmol/L FeSO4.7H2O, 0.1 mmol/L Na2.EDTA and 0.4 mmol/L CaCl2.2H2O. The solution was replaced every 2 d, and air was added into the solution using pumps. The maize seedlings grew in a greenhouse with natural sunlight at 16/8 h d/night cycle at 26–28 °C.
Parental phenotypes were calculated using 60 seedling samples of each inbred. For the RILs, the trials were conducted using a completely randomized design with two replications. Three plants for each genotype were planted in each replication with plant spacing of 3 cm, row spacing of 5 cm. All of materials were simultaneously planted in the same greenhouse. The concentration of nitrate was kept at 4 mmol/L using potassium nitrate (KNO3). Roots and shoots of the seedlings were harvested separately after 2 weeks of growth. The shoots were transferred to an oven and dried for 72 h at 80 °C. The roots were placed into plastic bags with a little water and kept refrigerated at 4 °C until processed.
Root segments were imaged in a transparent plastic tray in water in a document scanner (Hp V700). The entire seedling root was scanned at one time using the tray sized 19 cm in width and 24 cm in length. Then, we cropped images of the entire seedling root into two parts (seminal roots and primary roots), which were edited using Photoshop software. The divided images were analyzed with WinRHIZO 2004b software (Regent Instruments, Canada). Trait abbreviations and calculations for seminal roots, primary roots and total roots are listed in Table 1. Root systems were then dried and weighed in the manner of the shoots.
DNA extraction, genotyping, SNP calling and bin map construction
The parents of the RILs, Zheng58 and Chang7‐2, have been previously deeply sequenced to 28.41× and 24.95× coverage, respectively (Jiao et al. 2012). Each RIL was planted in the greenhouse, the fresh seedling shoot tissues were harvested from 8–10 plants and ground using the Geno2010 machine. The DNA extraction and sequencing was by the GBS method as described by Chen et al. (2014). GBS of the entire RIL population yielded more than 354 million 100‐bp reads, which in average covered 0.09×genome sequences for each RIL. The sequence data was first aligned to the B73 RefGen_v2 by BWA software (Li and Durbin 2009), followed by SNP detection using Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Mapping quality and base quality were set to 20. SNPs were filtered strictly using three criteria: (i) homozygous; (ii) supported by more than five reads; and (iii) diversity between Zheng58 and Chang7‐2. This resulted in the retention of 2,231,331 high‐quality SNPs. On average, 54,543 SNPs were obtained per RIL, with a density of one SNP per 46 kb.
The bin map was constructed using a sliding window approach as described by Huang et al. (2009) with a little modification. Raw SNPs were scanned in 20‐SNP‐windows with a sliding step of two SNPs. In each window, the genotype was defined by the ratio of two kinds of SNPs: the widows was called homozygous Chang7‐2 genotype when SNPZheng58:SNPChang7‐2 ≤ 5:15, homozygous Zheng58 genotype when SNPZheng58:SNPChang7‐2 ≥ 15:5 and heterozygous genotype when SNPZheng58:SNPChang7‐2 was larger than 5:15 and less than 15:5. Regions with no more than five continuous heterozygous windows were set as breakpoint, since there will be several heterozygous windows for transition at the breakpoints, but these should not span more than five uninterrupted windows. Adjacent 100 kb segments with the same genotype across the 204 RILs were merged together as a bin marker. The heterozygous bins were set as missing and imputed by R/qtl software (Broman et al. 2003) using the “argmax” method. Bins with extreme distortion (Chi‐test, p‐value < 1.0 E‐7) were discarded. The linkage map was constructed with the est.map function of R/qtl.
QTL mapping
Composite interval mapping (CIM) was performed for the seven phenotypes using the R/qtl package (Broman et al. 2003) with a scanning window size of 10‐cM. QTL were considered significant at a LOD threshold of 2.5, which is commonly used for QTL mapping in maize (Melchinger et al. 1998; Ruta et al. 2010; Li et al. 2014a) and the confidence intervals were estimated using the 1.5 LOD‐drop method. R (R Core Team, 2014) was used to generate the reported statistical parameters and figures, along with QTL additive effects and phenotypic variation explained, as described by Yu et al. (2011). The agricolae package for R (http://CRAN.R-project.org/package=agricolae) was used to calculate Pearson phenotypic correlations and their significance.
AUTHOR CONTRIBUTIONS
W.S. and B.W. carried out phenotyping and genotyping of parental lines and the individuals of the population, data analysis and drafted the manuscript. A.H. participated in the data analysis. X.D. performed the SNP calling and bin‐map construction. J.P.L. performed root phenotyping of the RIL population. J.S.L. designed the experiment, supervised the study and revised the manuscript.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Phenotypic variation for the seven RSA related traits of primary root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.
Figure S2. Phenotypic variation for the seven RSA related traits of seminal root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.
Figure S3. Phenotypic variation for the seven RSA related traits of total root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.
ACKNOWLEDGEMENTS
This work was supported by 863 Project (2012AA10A305), Chinese Universities Scientific Fund (2014XJ036), NSF (31301321) and 948 Project (2011‐G15).
Song W, Wang B, Hauck AL, Dong X, Li J, Lai J ( 2015) Genetic dissection of maize seedling root system architecture traits using an ultra‐high density bin‐map and a recombinant inbred line population. J Integr Plant Biol 58: 266–279
Available online on Nov. 23. 2015 at www.wileyonlinelibrary.com/journal/jipp
REFERENCES
- Abdel‐Ghani A, Kumar B, Reyes‐Matamoros J, Gonzalez‐Portilla P, Jansen C, Martin J, Lee M, Lübberstedt T ( 2013) Genotypic variation and relationships between seedling and adult plant traits in maize (Zea mays L.) inbred lines grown under contrasting nitrogen levels. Euphytica 189: 123–133 [Google Scholar]
- Abe A, Takagi H, Fujibe T, Aya K, Kojima M, Sakakibara H, Uemura A, Matsuoka M, Terauchi R ( 2012) OsGA20ox1, a candidate gene for a major QTL controlling seedling vigor in rice. Theor Appl Genet 125: 647–657 [DOI] [PubMed] [Google Scholar]
- Beavis WD, Grant D ( 1991) A linkage map based on information from four F2 populations of maize (Zea mays L.). Theor Appl Genet 82: 636–644 [DOI] [PubMed] [Google Scholar]
- Broman KW, Wu H, Sen S, Churchill GA ( 2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890 [DOI] [PubMed] [Google Scholar]
- Burton AL, Johnson JM, Foerster JM, Hirsch CN, Buell CR, Hanlon MT, Kaeppler SM, Brown KM, Lynch JP ( 2014) QTL mapping and phenotypic variation for root architectural traits in maize (Zea mays L.). Theor Appl Genet 127: 2293–2311 [DOI] [PubMed] [Google Scholar]
- Cai H, Chen F, Mi G, Zhang F, Maurer HP, Liu W, Reif JC, Yuan L ( 2012) Mapping QTLs for root system architecture of maize (Zea mays L.) in the field at different developmental stages. Theor Appl Genet 125: 1313–1324 [DOI] [PubMed] [Google Scholar]
- Chen Z, Wang B, Dong X, Liu H, Ren L, Chen J, Hauck A, Song W, Lai J ( 2014) An ultra‐high density bin‐map for rapid QTL mapping for tassel and ear architecture in a large F(2) maize population. BMC Genomics 15: 433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia JM, Song C, Bradbury PJ, Costich D, de Leon N, Doebley J, Elshire RJ, Gaut B, Geller L, Glaubitz JC, Gore M, Guill KE, Holland J, Hufford MB, Lai J, Li M, Liu X, Lu Y, McCombie R, Nelson R, Poland J, Prasanna BM, Pyhajarvi T, Rong T, Sekhon RS, Sun Q, Tenaillon MI, Tian F, Wang J, Xu X, Zhang Z, Kaeppler SM, Ross‐Ibarra J, McMullen MD, Buckler ES, Zhang G, Xu Y, Ware D ( 2012) Maize HapMap2 identifies extant variation from a genome in flux. Nat Genet 44: 803–807 [DOI] [PubMed] [Google Scholar]
- Cui H, Peng B, Xing Z, Xu G, Yu B, Zhang Q ( 2002) Molecular dissection of seedling‐vigor and associated physiological traits in rice. Theor Appl Genet 105: 745–753 [DOI] [PubMed] [Google Scholar]
- de Dorlodot S, Forster B, Pages L, Price A, Tuberosa R, Draye X ( 2007) Root system architecture: Opportunities and constraints for genetic improvement of crops. Trends Plant Sci 12: 474–481 [DOI] [PubMed] [Google Scholar]
- Falin Z ( 2001) The development and application of the elite maize inbred line of Zheng58. Crops 4: 21 [Google Scholar]
- Feldman L ( 1994) The maize root The Maize Handbook. Springer, New York: pp. 29–37 [Google Scholar]
- Ganal MW, Durstewitz G, Polley A, Berard A, Buckler ES, Charcosset A, Clarke JD, Graner EM, Hansen M, Joets J, Le Paslier MC, McMullen MD, Montalent P, Rose M, Schon CC, Sun Q, Walter H, Martin OC, Falque M ( 2011) A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 6: e28334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani S, Sanguineti MC, Tuberosa R, Bellotti M, Salvi S, Landi P ( 2005) Root‐ABA1, a major constitutive QTL, affects maize root architecture and leaf ABA concentration at different water regimes. J Exp Bot 56: 3061–3070 [DOI] [PubMed] [Google Scholar]
- Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES ( 2014) TASSEL‐GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 9: e90346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalo M, Holland JB, Vyn TJ, McIntyre LM ( 2010) Direct mapping of density response in a population of B73 x Mo17 recombinant inbred lines of maize (Zea mays L.). Heredity (Edinb) 104: 583–599 [DOI] [PubMed] [Google Scholar]
- Gore MA, Chia JM, Elshire RJ, Sun Q, Ersoz ES, Hurwitz BL, Peiffer JA, McMullen MD, Grills GS, Ross‐Ibarra J, Ware DH, Buckler ES ( 2009) A first‐generation haplotype map of maize. Science 326: 1115–1117 [DOI] [PubMed] [Google Scholar]
- Grotewold E, Drummond BJ, Bowen B, Peterson T ( 1994) The myb‐homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell 76: 543–553 [DOI] [PubMed] [Google Scholar]
- Han J, Wang L, Zheng H, Pan X, Li H, Chen F, Li X ( 2015) ZD958 is a low‐nitrogen‐efficient maize hybrid at the seedling stage among five maize and two teosinte lines. Planta 24: 935–949 [DOI] [PubMed] [Google Scholar]
- Hoagland DR, Arnon DI ( 1938) The water‐culture method for growing plants without soil. California Agric Exp Sta Circ 357: 1–39 [Google Scholar]
- Hochholdinger F, Tuberosa R ( 2009) Genetic and genomic dissection of maize root development and architecture. Curr Opin Plant Biol 12: 172–177 [DOI] [PubMed] [Google Scholar]
- Hochholdinger F, Woll K, Sauer M, Dembinsky D ( 2004) Genetic dissection of root formation in maize (Zea mays) reveals root‐type specific developmental programmes. Ann Bot 93: 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T, Dong G, Sang T, Han B ( 2009) High‐throughput genotyping by whole‐genome resequencing. Genome Res 19: 1068–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Yu T, Su L, Yu SB, Zhang ZH, Zhu YG ( 2004) Identification of chromosome regions associated with seedling vigor in rice. Yi Chuan Xue Bao 31: 596–603 [PubMed] [Google Scholar]
- Hund A, Fracheboud Y, Soldati A, Frascaroli E, Salvi S, Stamp P ( 2004) QTL controlling root and shoot traits of maize seedlings under cold stress. Theor Appl Genet 109: 618–629 [DOI] [PubMed] [Google Scholar]
- Hund A, Reimer R, Messmer R ( 2011) A consensus map of QTLs controlling the root length of maize. Plant Soil 344: 143–158 [Google Scholar]
- Jiao Y, Zhao H, Ren L, Song W, Zeng B, Guo J, Wang B, Liu Z, Chen J, Li W, Zhang M, Xie S, Lai J ( 2012) Genome‐wide genetic changes during modern breeding of maize. Nat Genet 44: 812–815 [DOI] [PubMed] [Google Scholar]
- Jones ES, Sullivan H, Bhattramakki D, Smith JS ( 2007) A comparison of simple sequence repeat and single nucleotide polymorphism marker technologies for the genotypic analysis of maize (Zea mays L.). Theor Appl Genet 115: 361–371 [DOI] [PubMed] [Google Scholar]
- Kumar B, Abdel‐Ghani AH, Pace J, Reyes‐Matamoros J, Hochholdinger F, Lubberstedt T ( 2014) Association analysis of single nucleotide polymorphisms in candidate genes with root traits in maize (Zea mays L.) seedlings. Plant Sci 224: 9–19 [DOI] [PubMed] [Google Scholar]
- Kumar B, Abdel‐Ghani AH, Reyes‐Matamoros J, Hochholdinger F, Lubberstedt T ( 2012) Genotypic variation for root architecture traits in seedlings of maize (Zea mays L.) inbred lines. Plant Breed 131: 465–478 [Google Scholar]
- Lai J, Li R, Xu X, Jin W, Xu M, Zhao H, Xiang Z, Song W, Ying K, Zhang M, Jiao Y, Ni P, Zhang J, Li D, Guo X, Ye K, Jian M, Wang B, Zheng H, Liang H, Zhang X, Wang S, Chen S, Li J, Fu Y, Springer NM, Yang H, Wang J, Dai J, Schnable PS, Wang J ( 2010) Genome‐wide patterns of genetic variation among elite maize inbred lines. Nat Genet 42: 1027–1030 [DOI] [PubMed] [Google Scholar]
- Landi P, Albrecht B, Giuliani MM, Sanguineti MC ( 1998) Seedling characteristics in hydroponic culture and field performance of maize genotypes with different resistance to root lodging. Maydica 43: 111–116 [Google Scholar]
- Lechelt C, Peterson T, Laird A, Chen J, Dellaporta SL, Dennis E, Peacock WJ, Starlinger P ( 1989) Isolation and molecular analysis of the maize P locus. Mol Gen Genet 219: 225–234 [DOI] [PubMed] [Google Scholar]
- Li F, Jia HT, Liu L, Zhang CX, Liu ZJ, Zhang ZX ( 2014a) Quantitative trait loci mapping for kernel row number using chromosome segment substitution lines in maize. Genet Mol Res 13: 1707–1716 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R ( 2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K, Yan J, Li J, Yang X ( 2014b) Genetic architecture of rind penetrometer resistance in two maize recombinant inbred line populations. BMC Plant Biol 14: 152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Chen F, Cai H, Liu J, Pan Q, Liu Z, Gu R, Mi G, Zhang F, Yuan L ( 2015) A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J Exp Bot 66: 3175–3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Li J, Chen F, Zhang F, Ren T, Zhuang Z, Mi G ( 2008) Mapping QTLs for root traits under different nitrate levels at the seedling stage in maize (Zea mays L.). Plant Soil 305: 253–265 [Google Scholar]
- Lopez‐Bucio J, Cruz‐Ramirez A, Herrera‐Estrella L (2003) The role of nutrient availability in regulating root architecture. Curr Opin Plant Biol 6: 280–287 [DOI] [PubMed] [Google Scholar]
- Lynch J ( 1995) Root architecture and plant productivity. Plant Physiol 109: 7–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JP ( 2013) Steep, cheap and deep: An ideotype to optimize water and N acquisition by maize root systems. Ann Bot 112: 347–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano Y, Omori F, Muraki M, Takamizo T ( 2005) QTL mapping of adventitious root formation under flooding conditions in tropical maize (Zea mays L.) seedlings. Breed Sci 55: 343–347 [Google Scholar]
- McCully ME, Canny MJ ( 1988) Pathways and processes of water and nutrient movement in roots. Plant Soil 111: 159–170 [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA ( 2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchinger AE, Utz HF, Schon CC ( 1998) Quantitative trait locus (QTL) mapping using different testers and independent population samples in maize reveals low power of QTL detection and large bias in estimates of QTL effects. Genetics 149: 383–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass HG, Zuber MS ( 1971) Correlation of corn (Zea mays L.) roots early in development to mature root development. Crop Sci 11: 655–658 [Google Scholar]
- Pace J, Gardner C, Romay C, Ganapathysubramanian B, Lubberstedt T ( 2015) Genome‐wide association analysis of seedling root development in maize (Zea mays L.). BMC Genomics 16: 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszkowski U, Boller T ( 2002) The growth defect of lrt1, a maize mutant lacking lateral roots, can be complemented by symbiotic fungi or high phosphate nutrition. Planta 214: 584–590 [DOI] [PubMed] [Google Scholar]
- Redona ED, Mackill DJ ( 1996) Mapping quantitative trait loci for seedling vigor in rice using RFLPs. Theor Appl Genet 92: 395–402 [DOI] [PubMed] [Google Scholar]
- Ruta N, Liedgens M, Fracheboud Y, Stamp P, Hund A ( 2010) QTLs for the elongation of axile and lateral roots of maize in response to low water potential. Theor Appl Genet 120: 621–631 [DOI] [PubMed] [Google Scholar]
- Steele KA, Price AH, Shashidhar HE, Witcombe JR ( 2006) Marker‐assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor Appl Genet 112: 208–221 [DOI] [PubMed] [Google Scholar]
- Steele KA, Price AH, Witcombe JR, Shrestha R, Singh BN, Gibbons JM, Virk DS ( 2013) QTLs associated with root traits increase yield in upland rice when transferred through marker‐assisted selection. Theor Appl Genet 126: 101–108 [DOI] [PubMed] [Google Scholar]
- Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint‐Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES ( 2011) Genome‐wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43: 159–162 [DOI] [PubMed] [Google Scholar]
- Tuberosa R, Salvi S, Sanguineti MC, Maccaferri M, Giuliani S, Landi P ( 2003) Searching for quantitative trait loci controlling root traits in maize: A critical appraisal. Plant Soil 255: 35–54 [Google Scholar]
- Weimin S, Weimin, Wang, Ailian Zhao, Taiping Zhang. ( 2004) Excellent high general combining ability of maize inbred line Chang 7‐2 and its utilization. Seed 23: 73–76 [Google Scholar]
- Williamson LC, Ribrioux SP, Fitter AH, Leyser HM ( 2001) Phosphate availability regulates root system architecture in Arabidopsis . Plant Physiol 126: 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll K, Borsuk LA, Stransky H, Nettleton D, Schnable PS, Hochholdinger F ( 2005) Isolation, characterization, and pericycle‐specific transcriptome analyses of the novel maize lateral and seminal root initiation mutant rum1. Plant Physiol 139: 1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Chen F, Chen Y, Yuan L, Zhang F, Mi G ( 2011) Root growth in response to nitrogen supply in Chinese maize hybrids released between 1973 and 2009. Sci China Life Sci 54: 642–650 [DOI] [PubMed] [Google Scholar]
- Xie W, Feng Q, Yu H, Huang X, Zhao Q, Xing Y, Yu S, Han B, Zhang Q ( 2010) Parent‐independent genotyping for constructing an ultrahigh‐density linkage map based on population sequencing. Proc Natl Acad Sci USA 107: 10578–10583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J ( 2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 4: e8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Yang X, Shah T, Sanchez‐Villeda H, Li J, Warburton M, Zhou Y, Crouch JH, Xu Y ( 2010) High‐throughput SNP genotyping with the GoldenGate assay in maize. Mol Breed 25: 441–451 [Google Scholar]
- Yu H, Xie W, Wang J, Xing Y, Xu C, Li X, Xiao J, Zhang Q ( 2011) Gains in QTL detection using an ultra‐high density SNP map based on population sequencing relative to traditional RFLP/SSR markers. PLoS ONE 6: e17595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan A, Lynch JP ( 2015) Reduced frequency of lateral root branching improves N capture from low‐N soils in maize. J Exp Bot 66: 2055–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan A, Schneider H, Lynch J ( 2015) Reduced lateral root branching density improves drought tolerance in maize. Plant Physiol 168: 1603–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZP, Liu XH ( 2013) Genetic analysis of agronomic traits associated with plant architecture by QTL mapping in maize. Genet Mol Res 12: 1243–1253 [DOI] [PubMed] [Google Scholar]
- Zhu J, Ingram PA, Benfey PN, Elich T ( 2011) From lab to field, new approaches to phenotyping root system architecture. Curr Opin Plant Biol 14: 310–317 [DOI] [PubMed] [Google Scholar]
- Zhu J, Kaeppler SM, Lynch JP ( 2005) Mapping of QTLs for lateral root branching and length in maize (Zea mays L.) under differential phosphorus supply. Theor Appl Genet 111: 688–695 [DOI] [PubMed] [Google Scholar]
- Zhu J, Mickelson SM, Kaeppler SM, Lynch JP ( 2006) Detection of quantitative trait loci for seminal root traits in maize (Zea mays L.) seedlings grown under differential phosphorus levels. Theor Appl Genet 113: 1–10 [DOI] [PubMed] [Google Scholar]
- Zou G, Zhai G, Feng Q, Yan S, Wang A, Zhao Q, Shao J, Zhang Z, Zou J, Han B, Tao Y ( 2012) Identification of QTLs for eight agronomically important traits using an ultra‐high‐density map based on SNPs generated from high‐throughput sequencing in sorghum under contrasting photoperiods. J Exp Bot 63: 5451–5462 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Figure S1. Phenotypic variation for the seven RSA related traits of primary root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.
Figure S2. Phenotypic variation for the seven RSA related traits of seminal root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.
Figure S3. Phenotypic variation for the seven RSA related traits of total root class in the RIL population
The y‐axis shows number of the individuals corresponding to the histogram bins on the x‐axis. P1 and P2 represent Chang7‐2 and Zheng58, respectively. The units and measurements are listed in Table 1.