

Abstract
PTC299 is a novel small molecule that specifically blocks the production of protein from selected mRNAs that under certain conditions use noncanonical ribosomal translational pathways. Hypoxia, oncogenic transformation, and viral infections limit normal translation and turn on these noncanonical translation pathways that are sensitive to PTC299. Vascular endothelial cell growth factor (VEGF) is an example of a transcript that is posttranscriptionally regulated. Single doses of PTC299 (0.03 to 3 mg/kg) were administered orally to healthy volunteers in a phase 1 single ascending‐dose study. In a subsequent multiple ascending‐dose study in healthy volunteers, multiple‐dose regimens (0.3 to 1.2 mg/kg twice a day or 1.6 mg/kg 3 times a day for 7 days) were evaluated. PTC299 was well tolerated in these studies. As expected in healthy volunteers, mean plasma VEGF levels did not change. Increases in Cmax and AUC of PTC299 were dose‐proportional. The target trough plasma concentration associated with preclinical efficacy was achieved within 7 days at doses of 0.6 mg/kg twice daily and above. These data demonstrate that PTC299 is orally bioavailable and well tolerated and support clinical evaluation of PTC299 in cancer, certain viral infections, or other diseases in which deregulation of translational control is a causal factor.
Keywords: PTC299, safety and tolerability, noncanonical mRNA translation, phase 1 clinical trial, pharmacokinetics
The production of tumor suppressors and proto‐oncogene proteins in normal cells during development is highly regulated. One key mechanism of protein regulation occurs through the interaction of regulatory elements found in messenger ribonucleic acid (mRNA) and the translation machinery that generates protein. Pathological conditions such as oncogenic transformation and viral infection have been shown to deregulate translational control.1, 2 For example, during conditions of cellular stress including hypoxia, oncogenesis, viral infection, and nutrient depletion, cap‐dependent translation of mRNA is greatly reduced leading to cell‐cycle arrest, senescence, and apoptosis. Cancer cells and viruses use noncanonical pathways to overcome the reduction in normal cap‐dependent translation of mRNA. Specifically, cancer cells use alternative ribosomal initiation mechanisms such as internal ribosomal entry sites of selected mRNA of proteins to overcome this reduction in mRNA translation and promote aberrant cell proliferation. The expression of c‐myc and vascular endothelial cell growth factor (VEGF) is regulated by multiple levels of transcriptional and posttranscriptional controls.3, 4, 5 Translation of VEGF mRNA in tumor cells under hypoxic conditions occurs through alternative mechanisms that allow the robust production of VEGF protein, which results in angiogenesis that supports further tumor growth.6, 7 Similarly, viruses use similar noncanonical pathways to avoid cellular inhibition of cap‐dependent translation, to more efficiently use the viral genome through the translation of multiple open‐reading frames, and because viral mRNA may not have the structures usually involved in normal cap‐dependent translation.8, 9
We optimized and developed PTC299 (Figure 1A; 4‐chlorophenyl 6‐chloro‐1‐[4‐methoxyphenyl]‐1,3,4,9‐tetrahydro‐2H‐pyrido[3,4‐b]indole‐2‐carboxylate), which reduces the stress‐induced protein synthesis of proteins that include VEGF, c‐myc, and a subset of other factors that, under certain conditions, rely on noncanonical translation pathways for their production. Preclinical studies have shown that PTC299 does not inhibit constitutive cap‐dependent translation but does inhibit the translation of VEGF mRNA through a novel mechanism involving the 5′‐untranslated region of the mRNA.10 Preclinical studies have also demonstrated that PTC299 acts to reduce hypoxia‐induced tumor VEGF production without reducing normal cap‐dependent VEGF production. PTC299 has shown similar or greater preclinical antitumor activity than that of drugs that target global VEGF.10 Specific inhibition of production tumor‐derived VEGF by PTC299 spares homeostatic expression of this angiogenic protein, potentially avoiding side effects such as bleeding, hypertension, and proteinuria associated with global VEGF‐targeted therapies.
Figure 1.
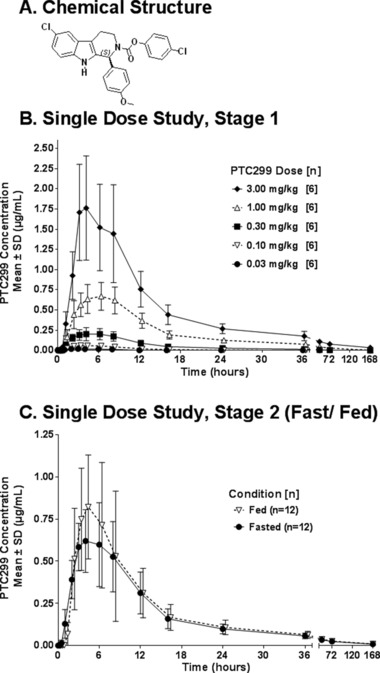
Chemical structure of PTC299 and plasma concentrations of PTC299 by dose level after a single dose of PTC299: stage 1 and stage 2. (A) Chemical structure. (B) Plasma concentrations of PTC299 by dose level in fasting subjects receiving single doses of PTC299. (C) Plasma concentrations of PTC299 by fed‐fasting status in subjects receiving single doses of 1 mg/kg PTC299. Fed subjects received PTC299 within 30 minutes after a high‐fat, high‐calorie meal. Fasted subjects had not eaten for ≥10 hours before PTC299 administration. A value of zero was used for calculating mean values if the measured concentration was below the lower limit of quantification.
Although PTC299 is poorly water soluble, adequate exposure of compound was obtained in preclinical studies when administered orally in a lipid‐based formulation. In support of the clinical development of PTC299, comprehensive, IND‐enabling toxicology studies in rats and dogs were performed, and safety pharmacology studies showed no adverse off‐target effects and no adverse effects on neurological or cardiopulmonary organ systems. No evidence for increased bleeding, hypertension, or proteinuria was noted in rats or dogs. PTC299 is tolerated at doses and exposures that are greater than 10‐fold excess of those predicted to be efficacious. Additional studies showed no evidence of genotoxic effects. Based on the safety profile of PTC299 in preclinical studies, Phase 1 studies in healthy volunteers were conducted, the results of which are reported here. Currently, the clinical development of PTC299 is on hold pending the evaluation of 2 cases of hepatotoxicity that were seen in phase 1b and phase 2 studies.11 The nonclinical toxicology studies did not indicate that PTC299 had any potential to cause hepatotoxicity. PTC has suspended the program, but is reevaluating the potential use of PTC299 in specific cancer indications including leukemia and lymphoma and nononcology indications.
Methods
The first study was conducted at SGS Life Sciences Services, Research Unit Stuivenberg (Antwerp, Belgium), and the second study was conducted at SGS Life Sciences Services Research Unit Aster (Paris, France), according to ICH and Good Clinical Practice guidelines. Local institutional review boards reviewed and approved the study protocols and the informed consent documents. Before any study‐related procedures were performed, all subjects provided written informed consent for participation in the study.
Study Design
Study 1 (PTC299‐ONC‐001‐HV) was an escalating single‐dose, randomized, double‐blind, placebo‐controlled safety, pharmacokinetic, and food‐effect study conducted in 2 stages in healthy volunteers. In stage 1, 40 subjects were enrolled in 5 cohorts of 8 subjects each, with each cohort comprising 4 men (3 PTC299, 1 placebo) and 4 women (3 PTC299, 1 placebo). Within each panel, subjects were randomly assigned to PTC299 or placebo. Successive cohorts received progressively higher doses of PTC299. Subjects and clinical staff were blinded to treatment but not to dose level because a placebo for each dose level was added. Five dose levels were evaluated: 0.03, 0.1, 0.3, 1.0, and 3.0 mg/kg. In stage 2, 12 additional subjects were randomized to receive 1 mg/kg of PTC299 after an overnight fast (3 men, 3 women) or within 30 minutes after consuming a standardized, high‐fat, high‐calorie meal (3 men, 3 women). One to 2 weeks later, subjects were crossed over to the opposite food‐intake regimen. In both stages, subjects were confined to the clinic on the evening before the administration of study drug. They remained under observation for 72 hours and returned for a follow‐up visit 7 days after each administration of study drug.
Study 2 (PTC299‐ONC‐002‐HV) was an escalating multiple‐dose, open‐label safety and pharmacokinetic study in healthy volunteers. In stage 1, 24 subjects were enrolled in 3 cohorts of 8 subjects each (4 men, 4 women). The 3 cohorts underwent sequential dose escalations across 3 dose levels (0.3, 0.6, and 1.2 mg/kg/dose twice daily). Subjects were administered the study medication with food for 7 days, with only the morning dose given on day 7. In stage 2, 8 additional subjects (4 men, 4 women) were administered PTC299 at a dose of 1.6 mg/kg 3 times a day with food for 7 consecutive days. In both stages, subjects were confined to the clinic on the evening before the initial administration of study drug. The subjects remained under observation throughout the assigned dosing period and for a 3‐day follow‐up period.
Study Population
Entry criteria were comparable in both studies. Subjects were healthy male or healthy nonpregnant, nonlactating female volunteers ranging in age from 18 to 55 years (study 1) or 18 to 65 years (study 2) weighing from 50 to 99 kg. Subjects with a history of severe adverse drug reactions were excluded from participation in the study.
Formulation and Administration
PTC299 drug substance and drug product were manufactured in accordance with current Good Manufacturing Practices. The drug product was a solubilized lipid formulation composed of 2.667% w/w PTC299, 49.869% w/w Gelucire 44/14 (lauroyl polyoxylglycerides), 47.459% w/w Solutol HS 15 (macrogol 15 hydroxystearate EP), and 0.005% w/w butylated hydroxytoluene (2,6‐di‐tert‐butyl‐4‐methylphenol) and filled in size 00 hard‐gelatin capsules. Capsules were provided in 3 dosage strengths containing 2, 10, or 20 mg of active drug substance with increasing drug loading for each strength. Matching placebo for each dose was provided for study 1. PTC299 capsules and the matching placebos were administered orally.
Safety Assessments
In both studies, data regarding adverse events, vital signs, hematology, biochemistry, coagulation, urinalyses, physical examinations, and ECGs were collected. Assessments were performed throughout the 72‐hour confinement period and at the follow‐up visit in study 1 and throughout the 10‐ or 17‐day confinement periods in study 2.
Quantification of Plasma and Serum VEGF Levels
The concentrations of VEGF in plasma and serum were determined using a commercially available enzyme‐linked immunosorbent assay (ELISA).
Blood Collection and Determination of PTC299 Plasma Concentrations
In study 1, blood samples for the determination of PTC299 plasma concentrations were collected immediately before administration of study drug and ∼0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, and 72 hours after administration and at the follow‐up visit on day 7 (168 hours).
In stage 1 of study 2, blood samples for the determination of PTC299 plasma concentrations were collected on day 1 and day 7 immediately prior to and ∼1, 2, 3, 4, and 10 hours after administration of each dose (morning and evening) and on days 2, 3, 4, 5, and 6 immediately prior to and 4 hours after administration of each dose (morning and evening). In stage 2 of study 2, blood samples for the determination of PTC299 plasma concentrations were collected on day 1 and day 7 immediately prior to and ∼1, 2, 3, and 4 hours after administration of each dose (morning, afternoon, and evening) and on days 2, 3, 4, 5, and 6 immediately prior to and 4 hours after each dose (morning, afternoon, and evening). PTC299 concentrations in plasma were quantified using a validated high‐performance liquid chromatography with tandem mass spectrometry (HPLC‐MS/MS). The lower limit of quantification was 5 ng/mL.
All blood samples were collected in tubes containing potassium ethylene diamine triacetic acid (K3EDTA) as the anticoagulant and were centrifuged. Plasma concentrations of PTC299 were quantified using HPLC‐MS/MS. PTC299 and its internal standard (deuterated PTC299) were recovered by liquid–liquid extraction using methyl‐tert‐butyl ether from K3EDTA‐treated human plasma. The mixture was vortexed and centrifuged and the organic layer transferred to a clean glass culture tube. After evaporating the organic layer under nitrogen in a 50°C water bath, the residue was reconstituted, and 10 μL of the extract was injected into an HPLC system with a Varian Polaris C18 column (Waters) and quantified using a tandem mass spectrometer. Chromatography was performed using 3 mobile phases: (A) 100% Millipore H2O, (B) 100% acetonitrile, and (C) 1% acetic acid. The transition ions m/z 465.4 → 127.1 and m/z 470.5 → 128.9 were monitored for PTC299 and the internal standard, respectively. Calibration standards were prepared at concentrations of 5, 10, 50, 200, 1000, and 2000 ng/mL. Quality control (QC) samples were prepared at 3 concentrations of PTC299 (15, 1000, and 1500 ng/mL). The peak areas for PTC299 were determined using an Agilent 1100 Series liquid chromatograph with an Applied Biosystems/MDS Sciex Series 4000 mass spectrometer for quantification. Calibration curves were obtained by performing linear regression (weighted 1/x*x) on the calibration standards, using an Analyst v. 1.3.1 data reduction software package. For the present study, the overall method precision (% coefficient of variation) for QC samples in the assay of PTC299 in plasma ranged from 2.73% to 4.85%, and the overall accuracy (% relative error) ranged from –2.00% to 2.50%. The precision of calibration standards was 5.86%, 5.81%, and 5.28% for the low (15 mg/mL), medium (1000 ng/mL), and high (1500 ng/mL) concentrations, respectively. The mean correlation coefficient for the analytical batches was >0.995.
Data Analysis
Pharmacokinetic parameters were estimated from plasma concentration data using standard noncompartmental methods. For samples in which PTC299 concentrations were below the lower limit of quantification, a value of zero was used for subsequent calculations. To provide a more uniform method of assessment across all subjects, the mean noncompartmental half‐life (t1/2) values were calculated from least‐squares linear regression of log‐transformed individual subject concentration–time data using all available times from 6 through 72 hours in the single‐dose study and from 4 through 12 hours in the multiple‐dose study. Dose‐proportionality of pharmacokinetic parameters and potential sex effects were examined by analysis of variance (ANOVA).12 In the food‐effect comparison, the ratios of the geometric means and their 90% confidence intervals for AUC and Cmax of the fed and fasted populations were calculated from log‐transformed data and compared. In addition, data for each subject were fitted to a 2‐compartment model by weighted nonlinear regression. All pharmacokinetic and statistical analyses were performed using WinNonlin (version 4.0) and SAS (version 8.2).
Results
Subject Disposition and Demography
A total of 52 healthy volunteers participated in study 1 (40 in stage 1 and 12 in stage 2), and 32 healthy volunteers participated in study 2 (24 in stage 1 and 8 in stage 2). No subject discontinued participation in either study prematurely.
In stage 1 of study 1, there were an equal number of male (20) and female (20) subjects. Of the 40 subjects, 39 were white, and 1 was Asian. The median age across the dose groups in stage 1 ranged from 43.0 to 49.5 years, with individual subject ages ranging from 20 to 55 years. Body mass index values were typical of adult healthy volunteers. Demographic characteristics were generally similar between the PTC299 and placebo groups.
In stage 2 of study 1, there were equal numbers of male (6) and female (6) subjects. Of the 12 subjects, 11 were white, and 1 was Hispanic. The median age was 45.5 years, with individual ages ranging from 18 to 52 years. Body mass index values were typical of adult healthy volunteers.
In stage 1 of study 2, there were equal numbers of male (12) and female (12) subjects. Of the 24 subjects, 17 were white, 4 were African/West Indian, and 1 was reported as “other.” The median age across the dose groups in stage 1 ranged from 44.5 to 59.0 years, with individual subject ages ranging in age from 21 to 64 years. Body mass index values were typical of adult healthy volunteers. Demographic characteristics were generally similar between the PTC299 and placebo treatment groups. In stage 2 of study 2, there were equal numbers of male (4) and female (4) subjects. Of the 8 subjects, 7 were white, and 1 was reported as “other.” The median age was 50.0 years, with individual ages ranging from 31 to 65 years. Body mass index values were typical of adult healthy volunteers. Demographic characteristics were generally similar between the PTC299 and placebo groups.
Clinical Safety
In study 1, PTC299 was well tolerated (Table 1). All adverse events were mild (grade 1) in severity, except for 1 case of moderate (grade 2) diarrhea in a subject in stage 2 receiving 1 mg/kg of PTC299 in the fasted state. Among the 40 subjects dosed in stage 1, the most frequent treatment‐emergent adverse events were headache (9 episodes in 8 subjects, all receiving PTC299) and nausea (5 episodes in 5 subjects, 4 receiving PTC299 and 1 receiving placebo). Other types of adverse events occurred in fewer than 5 subjects (10%). During stage 2, the most frequent adverse events were headache (3 episodes in 3 subjects) and back pain (2 episodes in 2 subjects); other adverse events were each noted in only 1 subject. The incidence, relationship to study drug, and severity of adverse events were not clearly dose dependent, although the number of headaches increased slightly with dose. All adverse events resolved by the end of the study. No deaths or serious adverse events occurred during the study. No subject discontinued participation in the study for safety reasons.
Table 1.
Treatment‐Related Adverse Events: Study 1—Stage 1 and Stage 2
| Study 1, Single Dose | ||||||||
| Stage 1 | Stage 2 | |||||||
| Fasted | Fed | Fasted | ||||||
| PTC299 Dose, mg/kg | PTC299 Dose, mg/kg | |||||||
| Placebo | 0.03 | 0.1 | 0.3 | 1.0 | 3.0 | 1.0 | 1.0 | |
| n = 10 | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | n = 12 | n = 12 | |
| Number (%) of Subjects With Adverse Event | ||||||||
| Gastrointestinal disorders | ||||||||
| Diarrhea | — | — | — | — | — | 1 (17) | — | 1— (8)a |
| Epigastric discomfort | — | 1 (17) | — | — | — | — | — | — |
| Nausea | — | — | 1 (17) | — | — | — | — | 1 (8) |
| Stomach discomfort | — | 1 (17) | — | — | — | 2 (33) | — | — |
| Vomiting | — | — | — | — | 1 (17) | — | — | — |
| General disorders | ||||||||
| Fatigue | — | — | — | — | — | 1 (17) | — | 1 (8) |
| Feeling hot | — | 2 (33) | — | — | — | — | — | — |
| Nervous system disorders | ||||||||
| Dizziness | — | 1 (17) | — | — | — | — | — | — |
| Headache | — | 1 (17) | — | — | 1 (17) | 1 (17) | — | — |
| Study 2, Multiple Dose | ||||||||
| Stage 1 | Stage 2 | |||||||
| PTC299 mg/kg/dose Twice Daily | Placebo n = 2 | PTC299 mg/kg/ dose 3 Times Daily | ||||||
| 0.3 n = 6 | 0.6 n = 6 | 1.2 n = 6 | 1.6 n = 6 | |||||
| Gastrointestinal disorders | ||||||||
| Diarrhea | 1a (17) | — | — | — | 1 (50) | 2 (33) | ||
aGrade 2 (moderate) event; all other events were grade 1 (mild).
In study 2, during the dose‐escalation stage of the multiple‐dose study, PTC299 was generally well tolerated (Table 1). Sporadic episodes of diarrhea, constipation, nausea, eye pruritus, back pain, productive cough, and insomnia were noted in subjects receiving PTC299. Sporadic episodes of diarrhea, constipation, and headache were also noted in subjects receiving placebo. The incidence and severity of the treatment‐emergent adverse events were not dose dependent. All adverse events resolved by the end of the study.
Vital signs (systolic blood pressure [SBP], diastolic blood pressure [DBP], heart rate [HR], respiratory rate, and temperature) were recorded at several times during both studies. Mean values and mean changes from baseline at each time in the PTC299 groups did not differ from those in the placebo group. Only 1 subject, a 56‐year‐old woman receiving 1.6 mg/kg 3 times daily of PTC299 during stage 2 of study 2, had evidence of hypertension (grade 1). SBP and DBP values for this subject were 151/93 mm Hg at screening, were elevated at several times after PTC299 administration (highest value, of 155/103 mm Hg, 4 hours after the day 1 morning dose), and remained elevated, with a value of 149/97 mm Hg, at the day 21 end‐of‐study visit.
No medically significant changes in hematology, biochemistry, or coagulation parameters occurred during either study. No significant urinary abnormalities were noted in any subject in either study. There were no safety concerns based on physical examinations, vital sign measurements, or ECGs of the subjects.
Pharmacokinetics
In study 1, following administration of single escalating doses of PTC299 (0.03 to 3 mg/kg), quantifiable concentrations of PTC299 were present in plasma within 30 minutes to 1 hour after dosing. The peak PTC299 plasma concentration (Tmax) generally occurred with a median time of ∼3 to ∼6 hours after administration (Table 2, Figure 1B). At dose levels ≥0.30 mg/kg, PTC299 concentrations persisted in plasma through 72 hours (mean values of 2, 27, and 76 ng/mL with doses of 0.3, 1, and 3 mg/kg, respectively, 72 hours postdose); at the dose of 3.0 mg/kg, low concentrations of PTC299 (mean, 32 ng/mL) were still measurable 168 hours after administration of drug. The plasma concentration–time course of PTC299 was well described by a 2‐compartment model. Fitting of individual subject data resulted in a mean half‐life during the distribution phase (t1/2α) of ∼3 hours and a terminal half‐life (t1/2β) in the range of 28 to 56 hours.
Table 2.
Mean (SD) PTC299 Pharmacokinetic Parameters: Study 1—Stage 1 and Stage 2
| PTC299 Dose, mg/kg | |||||||
|---|---|---|---|---|---|---|---|
| Stage 1 | Stage 2 | ||||||
| 0.03 Fasted | 0.10 Fasted | 0.30 Fasted | 1.00 Fasted | 3.00 Fasted | 1.0 Fasted | 1.0 Fed | |
| Parameter, Units | n = 6 | n = 6 | n = 6 | n = 6 | n = 6 | n = 12 | n = 12 |
| Tmax,a h | 4.34 | 3.84 | 5.17 | 5.33 | 3.50 | 4.34 | 3.58 |
| (2.17) | (2.13) | (1.33) | (1.03) | (0.55) | (1.57) | (1.38) | |
| Cmax,a μg/mL | 0.0195 | 0.0632 | 0.214 | 0.682 | 1.87 | 0.663 | 0.933 |
| (0.004) | (0.016) | (0.078) | (0.166) | (0.64) | (0.223) | (0.348) | |
| C24 h,a μg/mL | 0.0 | 0.002 | 0.03 | 0.12 | 0.27 | 0.10 | 0.11 |
| (0.0) | (0.004) | (0.01) | (0.03) | (0.06) | (0.03) | (0.04) | |
| AUC0–24,a μg·h/mL | 0.132 | 0.574 | 2.33 | 8.39 | 19.7 | 7.39 | 8.25 |
| (0.044) | (0.131) | (0.814) | (2.11) | (6.16) | (2.39) | (3.56) | |
| AUC0–t,a μg·h/mL | 0.116 | 0.569 | 2.75 | 12.9 | 31.4 | 11.4 | 12.3 |
| (0.045) | (0.135) | (1.11) | (3.29) | (7.17) | (3.66) | (4.86) | |
| Dose‐normalized Cmax,a μg/mL/mg/kg | 0.65 | 0.63 | 0.71 | 0.68 | 0.62 | 0.66 | 0.93 |
| (0.13) | (0.16) | (0.26) | (0.17) | (0.21) | (0.22) | (0.35) | |
| Dose‐normalized AUC0–24,a μg·h/mL/mg/kg | 4.4 | 5.7 | 7.8 | 8.4 | 6.6 | 7.4 | 8.2 |
| (1.5) | (1.3) | (2.7) | (2.1) | (2.1) | (2.4) | (3.6) | |
| t1/2, α,b h | NE | NE | 2.96 | 3.33 | 3.50 | 3.37 | 3.18 |
| (NE) | (NE) | (0.72) | (0.67) | (1.27) | (1.13) | (1.13) | |
| t1/2, β,b h | NE | NE | 28.3 | 42.2 | 55.8 | 53.4 | 43.4 |
| (NE) | (NE) | (15.1) | (17.6) | (16.9) | (27.0) | (25.8) | |
AUC, area under the concentration–time curve; Cmax, maximum compound concentration; t1/2, half‐life; Tmax, time of maximum compound concentration; NE, not estimatable.
Values represent male and female subjects combined.
Noncompartmental.
Based on 2‐compartment model fitted with time lag and weighted with 1/concentration.
Increases in mean Cmax and AUC0–24 values were generally dose‐proportional (Table 2); although the increases in mean AUC0‐24 values tended to be greater than dose‐proportional through a dose of 1.00 mg/kg and then less than dose‐proportional between doses of 1.00 and 3.00 mg/kg, these differences across groups in dose‐proportionality did not reach statistical significance (ANOVA following log transformation of PK parameters). Ingestion of a high‐fat, high‐calorie meal just prior to administration of 1 mg/kg of PTC299 in stage 2 increased the mean Cmax by ∼40%, from 633 to 933 ng/mL, but did not significantly change other PK parameters. The difference in the Cmax values in fasted versus fed patients was outside the 90% confidence intervals, but the difference in the AUC0–24 values was not. AUC0–24 values were not significantly different between male and female subjects in stage 1 or stage 2.
In study 2, in subjects receiving PTC299 for 7 days twice or three times per day, frequent sampling was performed after the morning dose on days 1 and 7 (Table 3, Figure 2B) of both stage 1 (twice‐daily dosing) and stage 2 (3‐times‐a‐day dosing). On day 1, mean maximum concentration (Cmax) values after the second dose were almost twice those after the first dose. By day 7, the mean Cmax values after the first and second doses were similar.
Table 3.
Mean (SD) PTC299 Pharmacokinetic Parameters: Study 2—Stage 1 and Stage 2
| Stage 1 | Stage 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| PTC299 Dose mg/kg/dose Twice Daily | PTC299 Dose mg/kg/dose 3 Times Daily | |||||||
| 0.3 n = 6 | 0.6 n = 6 | 1.2 n = 6 | 1.6 n = 6 | |||||
| Parameter, Units | Day 1 | Day 7 | Day 1 | Day 7 | Day 1 | Day 7 | Day 1 | Day 7 |
| Tmax (after pm dose), h | 3.2 | 3.3 | 3.2 | 3.3 | 3.0 | 3.3 | 2.5 | 2.3 |
| (0.41) | (0.52) | (0.41) | (0.52) | (0.00) | (0.52) | (1.1) | (1.4) | |
| Cmax (after pm dose), μg/mL | 0.48 | 0.59 | 0.97 | 1.2 | 2.0 | 2.5 | 2.4 | 4.7 |
| (0.15) | (0.18) | (0.24) | (0.27) | (0.29) | (0.57) | (0.46) | (1.9) | |
| C24h, μg/mL | 0.094 | 0.21 | 0.26 | 0.54 | 0.41 | 0.85 | 1.3 | 2.4 |
| (0.04) | (0.09) | (0.10) | (0.21) | (0.17) | (0.32) | (0.40) | (0.62) | |
| AUC0–24, μg·h/mL | 4.3 | 8.4 | 10 | 19 | 18 | 33 | 37 | 79 |
| (1.2) | (2.8) | (2.6) | (4.9) | (4.0) | (9.4) | (5.9) | (19) | |
| Dose‐normalized Cmax, μg/mL/mg/kg | 0.79 | 0.99 | 0.81 | 0.97 | 0.82 | 1.03 | 0.51 | 0.98 |
| (0.24) | (0.29) | (0.20) | (0.22) | (0.12) | (0.24) | (0.10) | (0.38) | |
| Dose‐normalized AUC0–24, μg·h/mL/mg/kg | 7.2 | 14 | 8.4 | 16 | 7.5 | 14 | 7.7 | 16 |
| (2.0) | (4.7) | (2.2) | (4.1) | (1.6) | (3.9) | (1.2) | (4.0) | |
| t1/2, h | NC | 164 (9.7)a | NC | 210 (35)b | NC | 228 (58)b | NC | 225 (55)b |
| CL/F (mL/min) | NC | 88 (23) | NC | 83 (28) | NC | 81 (14) | NC | 69 (20) |
AUC0–24 h, area under the concentration–time curve from time zero to 24 hours postdose; C24, concentration at 24 hours after first daily dose; Cmax, maximum compound concentration; NC, not calculated; Tmax, time of maximum compound concentration; CL/F, nominal clearance (normalized for bioavailability).
Values represent male and female subjects combined.
n = 4. bn = 3.
Figure 2.
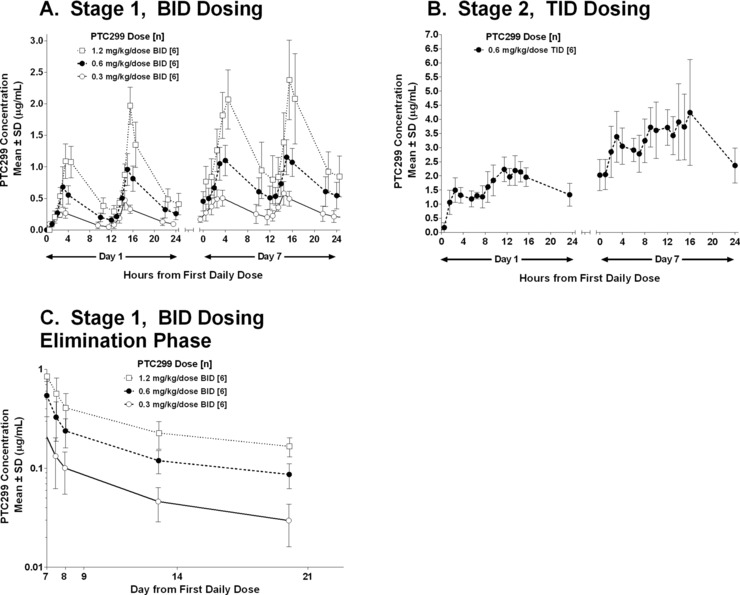
Plasma concentrations of PTC299 by dose. (A) Twice‐daily dosing of PTC299 for 7 days. (B) Three‐times‐daily dosing of PTC299 for 7 days. (C) Plasma concentrations of PTC299 by dose after cessation of twice‐daily dosing. A value of zero was used for calculating mean values if the measured concentration was below the lower limit of quantification.
After stage 1, a log‐linear elimination phase was not observed after PTC299 administration at any dose, and therefore the estimate of the half‐life may not be accurate (Figure 2C,D). Mean drug concentrations on day 21 (14 days after the last dose) were 39, 90, and 167 ng/mL for 0.3, 0.6, and 1.2 mg/kg twice daily, respectively. The calculated mean half‐life based on elimination after 7 days of dosing was 6.8, 8.8, and 9.5 days for 0.3, 0.6, and 1.2 mg/kg twice daily, respectively.
The Cmax and AUC0–24 were generally dose‐proportional in stage 1. When comparing day 1 with day 7, there were increases in mean Cmax and AUC0–24 at all doses, indicating accumulation (∼2‐fold) on continuous dosing of PTC299. A 2‐compartment model could be readily fitted to all the individual subject data throughout the 7‐day course of treatment.
Potential sex‐related differences were analyzed by ANOVA. No significant differences in Cmax or AUC0–24 values were observed between men and women.
Evaluation of Plasma and Serum VEGF Levels
Plasma and serum levels of VEGF were evaluated in stage 1 of study 1 in subjects receiving placebo or PTC299 at a dose of 3 mg/kg. When adjusted to baseline concentrations, changes in mean VEGF levels were similar in subjects receiving PTC299 at a dose of 3 mg/kg or placebo (data not shown).
In study 2, plasma and serum VEGF concentrations were quantified in all subjects. Mean absolute values and changes from baseline in plasma and serum VEGF concentrations are plotted in Figure 3A for stage 1 and in Figure 3B for stage 2. Overall, no consistent effect of PTC299 on concentrations of circulating physiological VEGF was noted at any dose.
Figure 3.
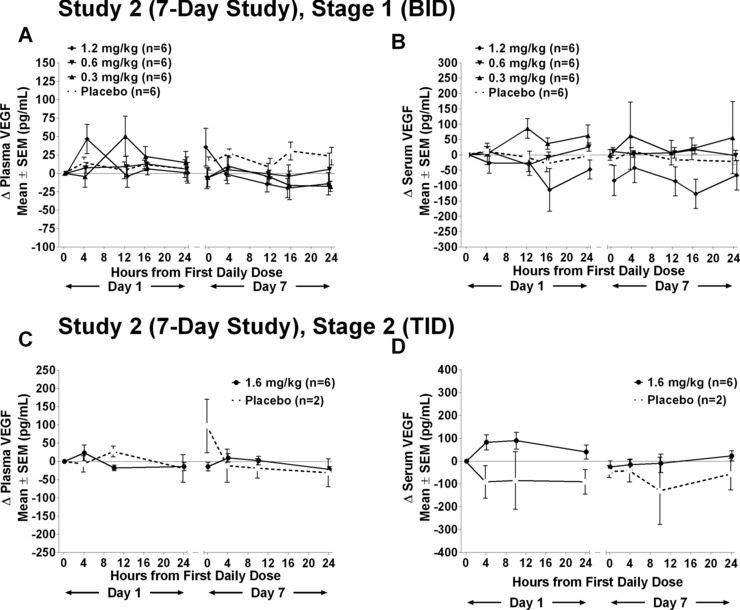
Change in VEGF (A, C) plasma and (B, D) serum concentrations: 7‐day study—stage 1 and stage 2. Levels of plasma or serum levels of VEGF measured by ELISA on day 1 and day 7 at specified times in subjects dosed daily for 7 days with placebo, 0.3, 0.6, or 1.2 mg/kg twice daily.
Discussion
PTC299 is a novel small molecule that inhibits the production of protein from selected mRNAs that use noncanonical ribosomal translational initiation sites. Under conditions of stress such as hypoxia, viral infection, or oncological transformation, the normal mRNA cap‐dependent translational processes are inhibited. However, the translation of mRNAs coding for cellular proteins including VEGF, c‐myc, survivin, and a number of other proto‐oncogenes and viral proteins can proceed through noncanonical pathways for translation under such conditions enriching for their expression. PTC299 selectively inhibits mRNA translated through these noncanonical pathways. For example, PTC299 does not interfere with normal cap‐dependent translation of VEGF mRNA but does inhibit VEGF produced from tumor cells cultured under hypoxic conditions. Therefore, PTC299 will normalize stress‐induced VEGF in cancer patients, but should not reduce the levels of VEGF in healthy subjects.
PTC299 as tested in the dose range (0.03 to 3 mg/kg) in the single‐dose study was within allometrically scaled no‐observed‐adverse‐effect levels determined in rats and dogs; the range was intentionally broad to assess symptomatic or laboratory events as a prelude to phase 2 testing in cancer patients. Food‐effect information and pharmacokinetic modeling of the single‐dose data informed the selection of twice‐daily dosing at mealtimes in the subsequent multiple‐dose study. The multiple‐dose trial was intended to explore a dose range that would rapidly achieve the target plasma concentrations observed in preclinical studies and to develop an appropriate safety evaluation for later phase 2 trials. For both studies, PTC299 was generally well tolerated, and no safety parameters of note were identified, consistent with the lack of target organ toxicities identified in the Good Laboratory Practices toxicology evaluation of PTC299.
Treatment‐emergent adverse events and laboratory abnormalities were generally grade 1. The incidence and severity of these findings were not greater in the PTC299 group than in the placebo group, and no dose dependence was apparent.
The solubilized formulation of PTC299 provides increases in exposure with increasing doses from 0.03 to 3 mg/kg (Table 2). The pharmacokinetics of PTC299 in both studies indicated dose proportionality for Cmax and AUC, and revealed no relevant sex effects on any parameter. In the multiple‐dose study, pharmacokinetic parameters were consistent with those observed in the single‐dose study. Accumulation of PTC299 was observed with multiple dosing, and there was no evidence of substantial autoinduction of drug metabolism.
In solubilized formulations, there is no barrier for drug dissolution. The dissolved drug in the formulation remains in the supersaturated state until the solubilized fraction gets absorbed. Typically, such formulations demonstrate a reduced food effect, consistent with the minimal effect seen here. Alterations in the pharmacokinetic profile of PTC299 when taken after a meal were minor, with a 40% increase in Cmax but no significant change in the AUC.
Levels of PTC299 were quantifiable 14 days after the 7 days of twice‐daily dosing. The calculated terminal half‐life after a single dose was ∼2.5 days. After 7 days of twice‐daily dosing, the calculated terminal half‐life was between 7 and 10 days; the apparent half‐life increased with increasing dose. Collectively, the results indicate that trough plasma concentrations exceeding target values associated with efficacy in preclinical model systems (∼300 ng/mL) can be achieved and maintained safely. Application of the 2‐compartment model suggests that twice‐daily dosing will result in target plasma concentrations quickly while minimizing the Cmax.
Studies in cancer patients at doses used in these studies demonstrated that PTC299 reduces levels of VEGF in cancer patients.13, 14, 15, 16 Because the healthy subjects in these studies were not under physiological stress and based on the mode of activity of PTC299, as expected, we did not observe a PTC299‐induced pharmacodynamic effect on circulating VEGF. The phase 1 studies in healthy volunteers reported here provide an important understanding of the safety and pharmacokinetics of PTC299 and will allow us to model dosing and predict exposures more accurately if PTC299 goes forward in nononcological indications or for specific oncological indications.
Funding
The development of PTC299 was supported in part by grants from the Department of Defense and the NIH Small Business Innovation Research (SBIR) program.
Declaration of Conflicting Interests
PTC299 is under development by PTC Therapeutics, Inc. As employees or consultants of PTC Therapeutics, Inc., all authors receive compensation in the form of salary, consulting fees, and/or stock options. SH is a former employee of PTC Therapeutics, Inc.
Acknowledgments
We thank the volunteer subjects who committed their time and effort. We thank Langdon Miller, Kerri Donnelly, Elizabeth Colacino, Nicolas P. Lamontagne, and the clinical staff who were involved in the conduct of this study. We thank Tamilarasu Nadarajan for his medicinal chemistry efforts. We thank Christopher Trotta for reading and editing the article.
References
- 1. Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003;3:179–192. [DOI] [PubMed] [Google Scholar]
- 2. Ruggero D. Translational control in cancer etiology. Cold Spring Harb Perspect Biol. 2013;5:a012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bornes S, Boulard M, Hieblot C, et al. Control of the vascular endothelial growth factor internal ribosome entry site (IRES) activity and translation initiation by alternatively spliced coding sequences. J Biol Chem. 2004;279:18717–18726. [DOI] [PubMed] [Google Scholar]
- 4. Bornes S, Prado‐Lourenco L, Bastide A, et al. Translational induction of VEGF internal ribosome entry site elements during the early response to ischemic stress. Circ Res. 2007;100:305–308. [DOI] [PubMed] [Google Scholar]
- 5. Yoo PS, Mulkeen AL, Cha CH. Post‐transcriptional regulation of vascular endothelial growth factor: implications for tumor angiogenesis. World J Gastroenterol. 2006;12:4937–4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levy AP, Levy NS, Goldberg MA. Post‐transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem. 1996;271:2746–2753. [DOI] [PubMed] [Google Scholar]
- 7. Akiri G, Nahari D, Finkelstein Y, Le SY, Elroy‐Stein O, Levi BZ. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene. 1998;17:227–236. [DOI] [PubMed] [Google Scholar]
- 8. Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nat Rev Microbiol. 2011;9:860–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Firth AE, Brierley I. Non‐canonical translation in RNA viruses. J Gen Virol. 2012;93:1385–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davis T, Cao L, Tamilarasu, N , et al. Preclinical development of PTC299: an orally bioavailable small molecule drug that selectively inhibits the production of VEGF protein, tumor growth, and microvessel density. In 18th EORTC‐NCI‐AACR Symposium on Molecular Targets and Cancer Therapeutics. EJC Supplements 2006;4:20. [Google Scholar]
- 11. Packer RJ, Rood BR, Turner DC, et al. Phase I and pharmacokinetic trial of PTC299 in pediatric patients with refractory or recurrent central nervous system tumors: a PBTC study. J Neurooncol. 2015;121:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith BP, Vandenhende FR, DeSante KA, et al. Confidence interval criteria for assessment of dose proportionality. Pharm Res. 2000;17:1278–1283. [DOI] [PubMed] [Google Scholar]
- 13. Dickler MN, Schneider BP, Volm M, et al. Reduction of serum VEGF and IL‐6 levels in patients with metastatic breast cancer: Results of a study of PTC299, an oral inhibitor of tumor VEGF synthesis, and aromatase inhibitors. J Clin Onc. 2011;29:abstract 1100. [Google Scholar]
- 14. Luke JJ, Callahan LA, Darby CH, et al. Results of a study of the oral inhibitor PTC299 on tumor VEGF synthesis. J Clin Onc. 2011;29:abstract 3069. [Google Scholar]
- 15. George CP, Luke JJ, Winkelman J, et al. Circulating VEGF levels as a biomarker to clinical benefit to PTC299, the first inhibitor of VEGF synthesis. J Clin Onc. 2012;30:abstract 13588. [Google Scholar]
- 16. Schwartz GK, Luke J, Dickler M, et al. In Abstracts of the EORTC ‐ NCI ‐ ASCO Annual Meeting on Molecular Markers in Cancer. Eur J Cancer. 2011;47:S15. [Google Scholar]