

Abstract
Accounting for subject nonadherence and eliminating inappropriate subjects in clinical trials are critical elements of a successful study. Nonadherence can increase variance, lower study power, and reduce the magnitude of treatment effects. Inappropriate subjects (including those who do not have the illness under study, fail to report exclusionary conditions, falsely report medication adherence, or participate in concurrent trials) confound safety and efficacy signals. This paper, a product of the International Society for CNS Clinical Trial Methodology (ISCTM) Working Group on Nonadherence in Clinical Trials, explores and models nonadherence in clinical trials and puts forth specific recommendations to identify and mitigate its negative effects. These include statistical analyses of nonadherence data, novel protocol design, and the use of biomarkers, subject registries, and/or medication adherence technologies.
Keywords: nonadherence, professional subjects, duplicate subjects, adherence, clinical trials
Nonadherence, the extent to which patients fail to take medications and/or to follow treatment recommendations as prescribed by their health care providers, is a major public health concern. Nonadherence affects patient safety, increases health care costs, and contributes to global problems such as antibiotic resistance.1 Adherence in general clinical practice (real‐world adherence) is variable, with partial adherence (eg, forgetting to take a daily dose of aspirin or stopping antibiotics after a few days) occurring more often than complete nonadherence (eg, never filling a prescription).2 Nonadherence in real‐world settings can exceed 50% in some populations, and this situation can also pertain to nonmedication treatment recommendations such as monitoring blood glucose or exercising regularly.3, 4
Multiple factors contribute to real‐world nonadherence, including cost, complexity and duration of the regimen, disruption of lifestyle, the patient's perception of benefits and risks, and poor communication between doctor and patient.2, 5 Treatment factors, particularly side effects such as weight gain or sexual dysfunction, patient factors, such as the desire to be independent and eschew the healthcare system, and illness factors, including psychosis, depression, or cognitive impairment, are also important contributors to nonadherence.6
Nonadherence during the conduct of a clinical trial may include most types of real‐world nonadherence as well as several behaviors unique to clinical trials that we term artifactual nonadherence. When adherence is not monitored (or not monitored stringently), there is a general assumption that adherence is almost ideal in clinical trial settings.7 However, there is extensive evidence to the contrary: both real‐world and unique forms of nonadherence abound in clinical trials.8 Artifactual nonadherence is fundamentally different from real‐world nonadherence described above; it is also contrary to both the clinical trial protocol and the agreements in the informed consent process. Examples of these specific and intentional behaviors include denying previous or ongoing study participation while enrolling in multiple studies to collect stipends, pretending to have the medical disorder under study, and removing and discarding pills from a blister card or bottle while reporting perfect (or near perfect) adherence. Although the motivation for such nonadherence is usually a desire for financial gain, it may also occur for other reasons; for example, a subject may enroll in multiple studies to increase the chances of obtaining effective treatment.9 Regardless of motive, these types of intentional and covert nonadherence create data that are false or misleading (ie, misinformative), violate the processes of hypothesis testing, and subvert efforts to determine the true safety and efficacy of investigational compounds.9, 10, 11, 12
Although we cannot eliminate all forms of real‐world nonadherence from clinical trials, artifactual nonadherence and misinformative data must be recognized and, when feasible, eliminated from studies.8 When unrecognized, artifactual nonadherence can result in miscalculations of safety signals and effect sizes in the intended patient population.4, 8, 9, 13 Artifactual nonadherence may therefore needlessly expose patients to adverse events, prevent potentially important medications from reaching patients, and cost hundreds of millions of dollars annually in research and development spending.
Duplicate and Professional Subjects
A potentially significant source of artifactual nonadherence may be attributable to duplicate or professional subjects.11 Duplicate enrollers simultaneously participate in multiple studies, although not necessarily for remuneration. The primary motivation for professional subjects to enroll in a study is money.14 Professional subjects may also be more likely to respond to placebo.11 , 15, 16, 17 These subjects may fabricate a disease state, inflate ratings, or falsely claim to have the severity or acuity of the condition under study. Conversely, they may conceal comorbid medical or psychiatric conditions or previous study participation in order to avoid exclusionary protocol criteria.9
Although it is likely that the majority of duplicate and professional subjects (that we collectively term professional subjects for the remainder of the article) participate for financial gain, subjects may also attempt multiple enrollments for reasons other than collecting stipends.14 Professional subjects may participate more than once in the same study (or studies of the same compound) or may simultaneously participate in multiple studies. Professional subjects can therefore be viewed as either polygamous (ie, participating in multiple studies at the same time) or serially monogamous (ie, immediately starting a new study when a study ends, sometimes while still in the follow‐up phase of the previous study). It has been estimated that professional subjects represent upward of 5% to 10% of subjects in CNS studies, depending on the indication.18, 19 These subjects may travel to distant sites (even locations more than 100 miles apart) in order to participate in multiple studies and frequently change presenting indications or diagnoses between sites.17, 20
Professional subjects are a major source of artifactual nonadherence. In phase 2 to 4 studies, professionals may participate in multiple studies, consistently feigning perfect or near‐perfect medication adherence while never taking study medication (or being grossly underadherent).7 In some phase 1 studies or certain efficacy studies with observed dosing or involving depot injections, professionals may take multiple investigational products (IPs) simultaneously (or within a time frame that could be dangerous). Some volunteers have participated in up to 80 phase 1 studies and have a financial incentive to conceal recent study participation.21
Case Example: A 42‐year‐old screening for a schizophrenia study was found to be concurrently participating in another study. He had denied any previous study participation but laughed and said “you caught me” when confronted. He admitted to taking investigational product (IP) “only when it made my head feel clearer” but reported 100% adherence by pill count.
He was found by a subject registry to have screened or prescreened at a minimum of 7 unique sites (and was a dual enroller on at least 3 occasions) just in the past 12 months.22
Not all nonadherent clinical trial subjects are professionals, and not all professionals will be nonadherent. A monogamous professional may take IP as directed, although he or she may still be deceptive about the number, types, and timing of trials in which he or she has participated. Alternatively, nonprofessional subjects can have second thoughts about taking IP or develop an adverse event (AE) and be reluctant to disclose this to the investigator. We must be careful not to eliminate honestly nonadherent subjects (or subjects who are nonadherent due to an effect of the protocol or IP) from clinical studies. It is also important to distinguish between professionals in phase 2 to 4 studies and professionals in phase 1 studies, where it is expected that money is the primary motivation. Phase 1 professionals, as long as they are honest about medical conditions, observe washout periods, are monitored in clinic, know what to expect, and may be ideal subjects.8
Nonadherence and Its Impact on Clinical Trials
Deceptive or artifactual nonadherence can produce outcomes on study endpoints unrelated to treatment assignment.4, 9 Such data provide no meaningful information relevant to the hypothesis being tested (ie, it is noninformative). Such data may come from subjects who take no study medication, falsely report severe symptoms at baseline, or those that report clinical improvement regardless of actual response. In such cases, the impact on the apparent treatment effect size, study power, and the sample size needed to overcome these deleterious effects may be modeled.
We start with a 2‐sample t‐test, with N subjects per group and for which change scores for drug (Δdrug) and placebo (Δpbo) are known and the standard deviation of the change score (σ) is equivalent for both groups. The mean change score for the data that come from noninformative subjects, ΔNI, will be the same regardless of the treatment group. We assume that combining data from the noninformative and legitimate subjects within a group does not affect its standard deviation. There can be significant consequences if the standard deviation for noninformative subjects is significantly larger than that of the legitimate subjects or if ΔNI is significantly better than that that of drug or worse than that of placebo. However, in both cases the conservative assumption used in these examples underestimates the adverse effects of noninformative data on study power and sample size.
For a study with no noninformative subjects, the expected value for the t statistic would be approximated as shown in Equation 1:
| (1) |
If we now include subjects with noninformative data with the previous sample and assume that the proportion of subjects contributing noninformative data to the study (pNI) is equal for the treatment groups due to randomization, test statistics for the study subjects are shown in Equation 2.
Compared to Equation 1, Equation 2 shows that including noninformative subjects in the analysis decreases the magnitude of the resulting test statistic by the proportion of noninformative subjects included in the study and decreases the true effect size by the same proportion. The magnitude of the change for noninformative subjects is unimportant as long as randomization ensures that noninformative data are equally distributed.
Extending this simulation, Figure 1 shows the impact that noninformative subjects have on study power. For example, if 20% of subjects provide noninformative data, studies intended to be powered at 90% and 80% based on a true effect size would have actual power of 74% and 61%, respectively. One common response, as suggested by Equation 2, is to increase the sample size to recover statistical power. The increased sample size necessary, however, makes this inefficient, and the ability to maintain quality in detecting drug‐placebo differences and controlling variance may be compromised.16 This is especially true as new sites are added in the push to accelerate enrollment.23 By underestimating the magnitude of the true treatment effect, misinterpretations of the clinical importance of study results (and the feasibility of future trials) may occur.
Figure 1.
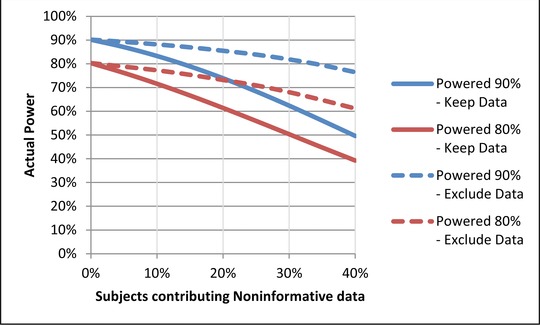
The impact of noninformative subjects on study power. If noninformative data are not excluded, then a study intended to be powered at 90% would have an actual power between 50% and 87% depending on the percentage of subjects (10%–40%) contributing noninformative data. A study intended to be powered at 80% would have an actual power between 39% and 72%.
Given the impact that noninformative data have on studies and the limitations of attempting to power through the problem, it is important to understand how eliminating noninformative data might benefit study analysis. Figure 1 shows the effect on statistical power when data from noninformative subjects are simply excluded, without replacing them with data from additional informative subjects. Although there is some loss of statistical power originally intended for the study, power remains substantially better than it would be if noninformative data were retained in the analysis. For the example of 20% noninformative data in studies with intended power of 90% and 80% as discussed above, actual power falls only to 86% and 73%, respectively, when noninformative data are excluded. If one wanted to recover the original study power by increasing the sample size, Figure 2 demonstrates that eliminating noninformative subject data and replacing them with informative subject data is more efficient than increasing sample size while retaining noninformative data.
Figure 2.
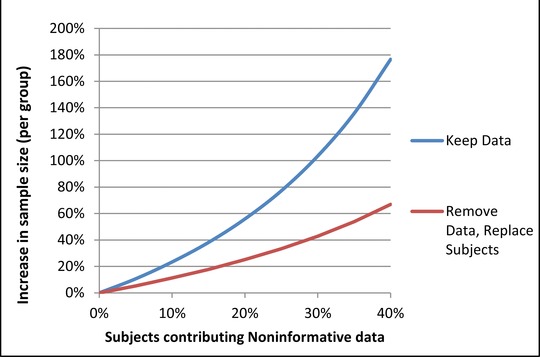
Increase in sample size necessary to recover power lost from noninformative data. As a greater percentage of subjects contribute noninformative data, the increased sample size required to maintain study power increases in a nonlinear fashion. When noninformative data are not excluded, the sample size required to maintain study power is greatly increased.
The impact of noninformative data has also been examined in the setting of outcomes for dichotomous endpoints. In a recent study modeling the impact of inappropriate subjects on clinical trial outcomes, 2 types of subjects were considered: those who are “destined to succeed” (such as a subject who feigns depression at study entry and then “recovers”) and those “destined to fail” (eg, a smoker in a smoking cessation trial who has no intention of taking study drugs or trying to quit).11 Although both types of inappropriate subjects were found to adversely impact statistical power, subjects who are “destined to succeed” appeared to have the more devastating impact (Figure 3). Under the conditions modeled (5% and 15% success rates in appropriate subjects for placebo and active treatment, respectively), it was found that a study population entirely composed of appropriate subjects required 141 subjects per group for 80% power, and the apparent odds ratio was 3.35. In contrast, when the study population included 10% “destined to succeed” professional subjects, 298 subjects per group were required for 80% power, and the apparent odds ratio was 1.81. Thus, although statistical power could be regained though a compensatory increase in sample size, there was an impact on apparent effect size that could not be overcome.11, 16
Figure 3.
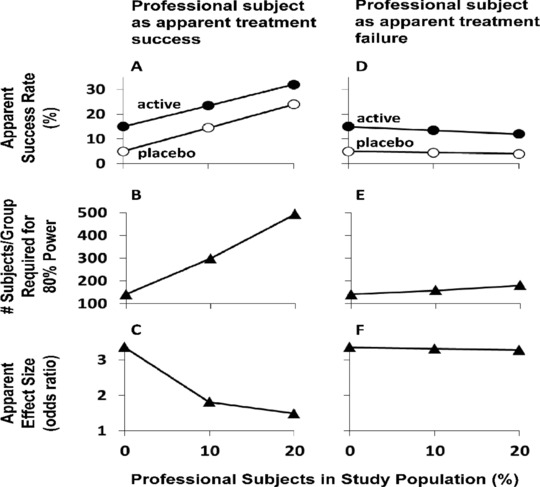
Impact of professional subjects who appear to achieve treatment success (A‐C) or treatment failure (D‐F) on an efficacy trial with a binary success/failure outcome. A and D: Apparent success rate, with success of appropriate subjects set at 15% for active medication (filled circles) and at 5% for placebo (open circles). B and E: Number of subjects/group required to achieve 80% power in detecting a significant treatment effect (α = 0.05; 2‐sided). C and F: Apparent effect size (odds ratio); in all cases there is an equal distribution of professional subjects among the placebo and active medication groups. This figure was reproduced from McCann et al11 with permission of the publisher.
Methods
The ISCTM Nonadherence Working Group was established in June 2014; ISCTM members and NIH scientists were invited by the co‐chairs to participate and investigate aspects of nonadherence based on their experience. The Working Group had e‐mail or Web discussions monthly and met biannually for face‐to‐face workshops. Literature searches were conducted using PubMed, SCOPUS, JAMA, and Google Scholar. Reports were brought back to the group, where findings were discussed and Working Group recommendations were formulated.
Discussions began with examinations of currently available and newly proposed methods to detect nonadherence.
Medication Adherence Biomarkers
Medication adherence in clinical trials is generally reported as >90% when based on subject self‐report and pill count.4, 8 However, multiple studies have demonstrated that there is very low agreement between the values obtained by self‐report and pill count when compared to medication adherence evaluated by periodic sampling of drug in plasma or urine.8, 11 , 24, 25, 26 Periodic monitoring of drug in biological fluids only represents a “snapshot” of medication adherence, whereas stratification of subjects based on either the presence or absence of drug (ie, above [ALQ] or below [BLQ] the limit of quantification) has revealed differential safety and efficacy signals in phase 1, 2, and 3 studies that are not seen when the entire sample is analyzed.8, 11
Riboflavin has been incorporated into both active and placebo arms as an adherence measure.27 However, riboflavin has several significant limitations, including a short half‐life and interference from natural sources, such as vitamin supplements. Recently, the National Institute on Drug Abuse (NIDA) has developed several strategies to monitor adherence, such as substituting the placebo arm with an ultralow dose of study drug. This “homeopathic dosing” strategy can be used if the study drug has a suitable half‐life and there is an assay sufficiently sensitive to detect study drug (or metabolite) in biological matrices. The informed consent is modified to advise the subject that he or she will receive 1 of x doses of study drug without mention of placebo. NIDA has incorporated this strategy into a proof of concept study (clinicaltrials.gov # NCT02401022), with the “adherence arm” receiving <4% of the dose administered to the active arm. An alternative strategy has been to identify adherence markers that can be readily incorporated into both the placebo and active arms. An ideal adherence marker would have a desirable pharmacokinetic profile (once‐ or twice‐a‐day dosing) with low intra‐ and intersubject variability, few drug‐drug or food interactions, and urinary or salivary excretion. It should be generally recognized as safe (GRAS) or FDA approved and not be commonly found in dietary sources, supplements, or pharmaceuticals. In order to be useful as an adherence marker, the molecule must not produce any pharmacological actions at doses that permit detection in biological matrices. Both acetazolamide and quinine have been identified as molecules that exhibit some or all of the properties of a useful adherence marker.28 For example, the carbonic anhydrase inhibitor acetazolamide is 100% orally bioavailable, and can readily be detected in blood and urine at doses of 15 mg, well below therapeutic doses, which can range up to 1 g/day. Preliminary data indicate that both urine and plasma acetazolamide levels will be sufficiently sensitive to provide an evaluation of the interval elapsed since the last dose of drug was administered. Although these strategies present only a snapshot of adherence and are therefore imperfect, incorporation of adherence markers may be useful in proof‐of‐concept trials to enable go/no‐go decisions based on solid hypothesis testing.28
Medication Adherence Monitoring Technologies
Testing new pharmacotherapies requires participants to take the study medication consistently and correctly in order to ensure that any changes in health outcomes can be directly attributed to treatment. Partial adherence or nonadherence can lead to study failure by interfering with proper interpretation of trial results.4, 11, 12, 26, 29 During early‐phase clinical trials, when dosing is often observed and administered on site, adherence is optimal. However, as soon as participants are allowed to take their medication on an outpatient basis, adherence rates decrease.30 It has been estimated that it takes only 30% of participants to be less than fully adherent to require doubling the number of participants necessary to produce an equally significant study.31 Suboptimal adherence prevents trials from determining exact dose‐response curves and optimal dosing for real‐world conditions.12 Failure to determine drug efficacy and safety in phase 2 is the leading reason for trial failures in phase 3.32
Traditionally, monitoring methods have been divided into 2 categories: direct methods (directly observed treatment [DOT], bioassays) and indirect methods (self‐reports, patient diaries, pill counts, prescription refills, clinical response, or third‐party [eg, caregivers] observation).33 The direct methods are considered to be more accurate and reliable but expensive and resource intensive.1, 26, 30 Some types of DOT may be carried out using cell phone technology, providing cost‐effective, real‐time information (Dawn I. Velligan, PhD, email communication, October, 2015). The indirect methods are viewed as subjective, cheaper, and practical for routine use.34 However, both direct and indirect methods have proven to overestimate adherence.11, 26, 33 A third category, comprised of electronic monitoring systems (such as MEMS, a conventional medicine bottle fitted with an electronic cap that date stamps each opening and closing) are more accurate than indirect measures but lack the ability to confirm ingestion.12, 26, 33, 35, 36 As a result, partial adherence and “drug holidays” frequently go undetected.11, 33
Three newer technologies are addressing the problem of directly confirming medication ingestion11: (1) Proteus Digital Health and e‐Tect are developing ingestible sensors that are embedded into each dose. The sensor is activated on ingestion and transmits a signal to a separate patch worn by the patient; (2) Xhale uses a custom breathalyzer to record the results of a breath test based on the ingestion of a chemical‐coated medication which transforms into a gas on ingestion; and (3) technology pioneered by AiCure uses artificial intelligence software on mobile devices to automatically confirm medication ingestion. A combination of facial recognition and motion‐sensing technology is able to determine if the patient is taking the medication correctly. Each method presents unique benefits and challenges to implementation based on cost, patient acceptance, ease of use, scalability, and changes to the manufacturing process of the investigational product.11
Subject Registries
The problem of research subjects overenrolling in phase 1 studies has been recognized for more than 25 years. Participation in multiple studies can lead to potentially dangerous drug interactions and skew safety data. France, the United Kingdom, and parts of Switzerland recognize the problem and mandate registration of phase 1 volunteers.14, 37, 38 In 2008, a network of sites in Florida began to use a fingerprinting method (now ClinicalRSVP) to attempt to identify duplicate volunteers.39
Discussions on the need for some kind of subject registry in efficacy studies (ie, phase 2‐4 patients) have paralleled the recognition of the growing problem of duplicate and professional subjects. Over the last decade there have been various attempts to track professional subjects within certain pharmaceutical companies, using patient identifiers gathered by vendors (eg, IVRS, ECG, central laboratory) to look for duplicate enrollers.40 Although these methods are capable of detecting duplicate subjects within a specific study or within a single sponsor, they do not track professionals across sponsors.
In 2011, Verified Clinical Trials and CTSdatabase each began to collect and compare subject identifiers within their HIPAA‐compliant databases in order to identify duplicate subjects.5, 20 Dupcheck followed in 2013.41 These systems are designed to be integrated into protocols as a simple procedure during the screening process (ie, access a secure website and get a report). Professional subjects may be eliminated prior to randomization by an exclusion criterion that has been added to the protocol.26 Alternatively, investigative sites can elect to use a subject registry, independent of sponsors or study, to enter all potential subjects during the prescreen process.20, 21, 40 This option, popular in such high‐duplicate metropolitan areas as Southern California and New York City, involves having every potential subject sign a generic, IRB‐approved database authorization in the waiting room.18
The registries differ mainly on what and how information is collected and what reporting and other services are provided. In addition, some registries continue to track subjects during the course of studies and notify sponsor and investigator if a subject tries to screen elsewhere.
To date, all registries function independently and do not “talk” to one another, although plans are under way to allow interoperability and integration of the major registries.41 A potential benefit of integrating the major registries is that phase 1 participants can be better compared with phase 2 to 4 participants to ensure healthy normals are not pretending to be patients (and vice versa).
Discussion
Where to Act: Approaches to Mitigate the Effects of Nonadherence
Appropriate trial design may be the first and best defense against artifactual nonadherence, but overly stringent and complex protocols (which attempt to enrich samples or answer additional safety/efficacy questions) may have unintended consequences.42 Thus, protocol complexity, increasing at rates up to 6% to 10% per year, may influence the enrollment of inappropriate phase 2 to 4 subjects. Professionals can adapt to complex visits and more stringent eligibility criteria, particularly when the criteria are subjective, by modifying their presentation and devoting substantial time to clinic visits.5, 9, 43 Increasing the complexity of scientific studies, even outside the clinical trial environment, has been associated with a greater likelihood of cheating.5, 44 Increased study complexity may be beneficial, however, when it is specifically targeted to address professional subjects or related problems. For example, the “RAMPUP” study design was recently proposed to address the problem of nonadherent subjects by shunting them away from efficacy evaluations based on data collected during a run‐in period with adherence monitoring.11
The following section examines the various stages of a protocol (see Figure 4) and discusses where and how steps may be taken to identify and mitigate the effects of nonadherence.
Figure 4.
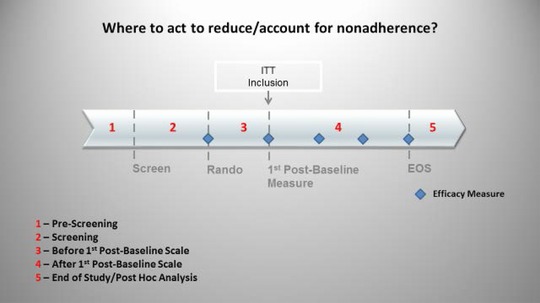
Where to act to account for and/or reduce nonadherence. Steps may be taken to address nonadherence during the prescreen process, before and after randomization, and during post hoc analysis. Discontinuing subjects prior to the first efficacy measurement may allow subjects to be excluded from the ITT.
Prescreen Methods to Address Nonadherence
Before a potential subject ever sees a consent form, where and how we obtain study participants should be considered. Do they come from advertising, the investigator's practice, or a research database? What motives incentivized these future subjects to respond? Altruism? Compensation? Desire for treatment of their condition?
The potential risks associated with posting overly detailed inclusion and exclusion criteria on the clinicaltrials.gov website must also be considered. Trials relying on subjective assessments as primary measures of efficacy are especially vulnerable to “gaming” by professional subjects, and it may imprudent to allow professional subjects access to very detailed information about the trial prior to screening. In such situations, investigators may choose to post inclusion criteria, including characteristics that cannot be manipulated (such as gender and age), while restricting sensitive information to exclusion criteria that are not posted on the website.
Stipends must be sufficient to compensate for time and travel to avoid biasing the sample by including only subjects with resources adequate to allow for participation in (increasingly complex) protocols.42 Stipends must also not be excessive, encouraging professional subjects or coercing subjects who would not otherwise participate.11, 42 Other appropriate incentives, such as providing standard of care treatments during or at the conclusion of the study, may also be considered. One important safeguard is the role of the Institutional Review Board (IRB) in the oversight of patient stipends, although significant variations exist in the stipends approved.
As a prerequisite for site selection, a sponsor may require investigators to recruit at least 50% of subjects from internal databases.45 This assures a known source of potential subjects that meet inclusion/exclusion (I/E) criteria and speeds enrollment.23 However, when this is done repeatedly, a repeat subject population is selected rather than a patient population. One consequence of this selection process may be the inadvertent conversion of known patients into professional subjects. One solution is to set limits on the number of previous studies that a subject has participated in (eg, no more than 2 in the last 2 years). However, without all past medical records or a subject registry, investigators must rely on subject self‐report to assess in how many past studies he or she has really participated.
It is also important to address the source of subjects recruited outside a practice or internal database.11, 46 Are they recruited from Craigslist or perhaps posted under “job listings” (and if so, should we be surprised if subjects looking for jobs respond primarily for the money?). If a recruitment vendor is used, are the same referrals recycled to multiple sites or for multiple indications? This leads investigators, sponsors, and vendors to repeatedly draw subjects from the same small pool (investigator databases preferred advertising venues) without access to the greater pool of patients.45 The significant quantity of repeat subjects thus selected is more likely to exhibit artifactual nonadherence than research‐naive subjects (who may be more likely to be adherent or real‐world nonadherent).46 Repeat subjects may be excellent subjects provided they are not “overused” and the investigator has access to PK data and treatment assignments from previous studies. These data can be used to inform decisions about future study participation. Pharmaceutical sponsors are therefore encouraged to provide treatment codes to sites, and investigators should check for previous placebo response or nonadherence in subjects under their watch prior to enrolling them in future studies.
Investigators also have financial and time pressures that can impact the quality of subjects recruited. For example, investigators are often not reimbursed for extensive preconsent activities such as phone screening, evaluating potential subjects in person, using a subject registry, or requesting records. These pressures may incentivize investigators to get subjects to sign consent forms before they are fully vetted in order to offset these unreimbursed costs. In addition, inexperienced or inappropriately motivated investigators must be carefully monitored by study sponsors.
Finally, there are subject characteristics, such as history of recurrent illness and documented adherence to previous treatment regimens that may help select more adherent subjects.11
Methods to Detect and Eliminate Nonadherence Prior to Randomization
Inclusion/exclusion criteria that permit real‐world patients to enter studies should lead to a greater percentage of authentic (vs artifactual) subjects.8 Subject reports can be confirmed with outside sources, such as a subject registry to detect prior study enrollment, a study partner to confirm history and symptoms, or a requirement for medical and/or pharmacy records prior to randomization. Certainly, reimbursing investigators at an unrealistically low screen‐fail ratio (eg, 1 screen‐failure paid for every 3 randomized) can lead to a situation in which sites may be forced to choose between not being paid and enrolling an inappropriate subject.
Case Example: A 60‐year‐old man with memory loss confirmed by a study partner and a Mini Mental State Exam score of 25 was enrolled into a study of mild Alzheimer's disease. He completed a diagnostic interview, physical exam, all labs and ratings, and an MRI. He was scheduled for a PET scan when records received from his cardiologist noted an HIV diagnosis and a 3‐drug treatment regimen, both of which he denied at study entry. When told he could not be in the study because of this exclusionary condition, he asked if he still got paid.
An independent review of diagnosis and ratings may be obtained during the screen.47 In addition, medication and protocol (eg, diary) adherence as well as consistency of ratings responses could be checked during a placebo lead‐in period, and prespecified nonadherent (eg, <80% or >120%) or inconsistent subjects (ie, ratings outliers) can be removed.46, 48
Adherence data during a placebo lead‐in phase may be used to exclude nonadherent subjects or to prespecify subjects to be excluded from the primary analysis.46, 48 By assessing subjects based only on their behavior prior to randomization, decisions to exclude subject data will not be biased across treatment groups. Reliance on pill counts and diary entries assumes that subjects are honestly reporting and correctly taking medications (or filling out diaries). Those subjects intending to deceive may alter their diaries (unless they are date and time stamped e‐diaries) or their blister cards just prior to the next visits to show perfect adherence. Ironically, the most dishonest subjects may appear to be the best and “easiest” subjects, with perfect adherence and lack of deviations from the protocol leading to increased enrollment and less effort for sites, monitors, and statisticians. This is where PK sampling, adherence markers, and adherence technologies can play a critical role in trial outcome.49, 50 Although they add some complexity and per‐subject cost to studies, it is likely more cost‐effective to identify and exclude sources of noninformative data than to attempt to overcome their effects with a larger sample size.11, 16 Although similar methods and criteria for nonadherence may be used to exclude subjects or to censor their data from analysis, the 2 courses may lead to different results. If subjects are aware of the risk of discontinuation due to nonadherence, they may be more likely to change behavior to avoid being discontinued from the study. This could have positive or negative consequences. Subjects may improve adherence as desired, or they may make dedicated efforts to more effectively conceal nonadherent behavior.9 Given the latter possibility, it may be preferable in some cases to censor nonadherent subject data without discontinuing the subject. Subjects may be identified for censoring based on criteria that are not applied or adjudicated until after the study has ended.
To illustrate this point, three theoretical examples are provided:
The protocol specifies that the intent‐to‐treat (ITT) analysis includes only subjects who have PK sampling that show quantifiable levels of background antidepressant treatment immediately prior to randomization. Despite a history and medical records showing 6 months of continuous fluoxetine treatment, a completed subject who was later found to have no fluoxetine in the PK sample drawn just prior to randomization was excluded from the primary analysis.
All subjects were given microdoses of acetazolamide as an adherence marker during a placebo run‐in period. Those who had no detectable levels of acetazolamide in at least 1 of the 2 urine samples taken during the run‐in were excluded from the primary analysis by prespecified criteria.
All subjects were required to use a medication adherence technology during a 14‐day placebo run‐in period. Those who were shown to be <75% compliant with their twice‐a‐day (BID) dosing regimen during this period were not randomized. Alternately, all subjects were randomized, but an adherence committee (blinded to treatment assignment and efficacy and safety data) identified subjects to be excluded from the primary analysis based on a review of all adherence data during the placebo run‐in.
It is important to note that professional subjects may be incentivized to learn about placebo run‐in designs and may feign perfect adherence during this period.9 Site staff, also incentivized to produce randomized subjects, may provide cues about the importance of adherence during this crucial period.32 A protocol design that is partially masked to both investigators and subjects may ameliorate this effect.46, 48 Even though more costly in the short run, it may be also be worthwhile to randomize subjects regardless of how they perform during the lead‐in period and eliminate them later from the primary analysis based on their lead‐in performance.
Methods to Mitigate the Effects of Nonadherence After Randomization
Due to the potential to create bias between treatment groups that could affect the validity of inferences drawn from study results, any actions to mitigate the effects of nonadherence taken after a subject has been randomized must be carefully considered. Any aspect of a strategy—including the source of information used to detect nonadherence, the intervention made once nonadherence is detected, and decisions on how to analyze the nonadherent subject's data—must be considered as a potential source of bias.26 Such considerations support the conservative use of an ITT population. However, there are also problems with overemphasizing the risk of potential bias. Focusing only on potential bias (and not the detection of nonadherence) may hurt the drug's apparent efficacy and increase its apparent safety profile.
Despite the potential bias that postrandomization interventions may introduce, a nonadherence mitigation strategy may be considered if the negative effects of nonadherence clearly outweigh the negative effects of introducing a potential bias. For example, it may be justifiable to censor a subject's data from analysis if it could be proven that not even a single dose of study drug was taken by the subject. Adherence technologies may assess such nonadherence in real time. Detecting subjects who should be encouraged to improve adherence could identify more drug‐treated subjects; however, when the drug is taken as prescribed, the resulting data are more informative.
It is generally accepted practice to discontinue subjects for safety and tolerability reasons. Study drug–related discontinuations may create bias but are accepted because safety concerns are paramount, outweighing evaluation of drug efficacy. The impact of dropping a subject depends on how missing data are handled in the analysis (last observation carried forward [LOCF], mixed‐effect model repeated measure [MMRM]). Discontinuation for issues of protocol nonadherence deemed unsafe is also acceptable (eg, not adhering to safety monitoring, enrollment in multiple studies, taking prohibited medications). For example, discontinuation prior to the first efficacy measurement would allow exclusion from the modified intent‐to‐treat (mITT) analysis.
Interventions may be initiated at the level of the study, site, or subject. A subject registry initially accessed at screen may continue to be utilized after randomization to identify subjects who attempt to screen at other sites during the course of a study. These subjects may be counseled, removed, and/or flagged for post hoc analyses. The exclusion of duplicate subjects from the ITT (ie, those that have participated in more than 1 study site in a program) has been reported in phase 3 studies.51 Centralized monitoring may also be employed to close sites where there is evidence of protocol nonadherence or suspicious data. On a subject level, prespecified nonadherence could trigger an intervention strategy. Investigators, once aware of true levels of nonadherence, can intervene with participants directly to explore why it occurred and make a determination of whether a subject should be counseled or dropped, in concert with the study medical monitor.
Real‐time medication adherence data allow sponsors to adopt interventions based on dosing profiles. Figure 5 demonstrates how study participants may have differing dosing patterns over the course of a trial.52 Custom interventions may be offered or triggered by predefined dosing patterns. Patient B, after a few months of consistent behavior, demonstrated highly variable dosing. Patient C discontinued use of the medication after a few months. Patient D had dosing holidays (stopped using the medication for a period of 2 weeks or more). In each case a custom intervention strategy could have minimized potential disruptions to the power of the study.
Figure 5.
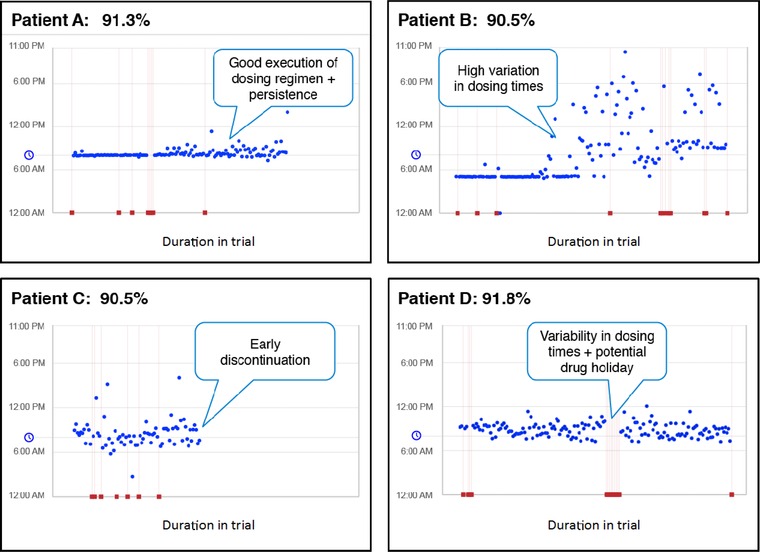
Different dosing patterns for study participants over the course of the trial. Patient A demonstrated appropriate adherence. Patient B demonstrated a highly variable dosing regimen. Patient C discontinued medication after a few months. Patient D demonstrated variability in dosing and periods of extended nonadherence during the study. AiCure data.52
Post Hoc Analysis: Using Adherence Data From Efficacy Trials to Inform Development Decisions
Post hoc analyses are routinely used in sensitivity analyses or in assessing the potential value of a drug for further clinical development. A signal in a subpopulation of informative subjects could result in a “go” decision rather than the “no‐go” decision that would likely result from the absence of an efficacy signal in the ITT population. Data on adherence to study medication and other protocol procedures may be used in similar ways. A typical per‐protocol analysis may include only subjects who completed the entire study while demonstrating adherence to study treatment and other procedures that meet a given standard (eg, detectable urine drug levels at ≥9 of 12 weekly visits). Alternatively, one might choose to handle different forms of nonadherence differently, depending on whether or not the nonadherence is artifactual and the data are misinformative. Positive results for a population combining adherent subjects and nonadherent but informative subjects suggest a greater likelihood of success if steps are taken to reduce misinformative data in subsequent trials.
Nonadherence can also affect decisions in the opposite direction: understanding nonadherence might also prevent missing an important safety signal that could lead to a no‐go decision.8 A detailed and careful analysis of adherence data, such as identifying subjects who show good medication adherence early in treatment and then become nonadherent after reporting an adverse event, might reveal important safety signals.
On the other hand, if other subjects show early and sustained medication nonadherence with a monitoring technology despite pill counts and self‐reports suggesting full adherence, those data are far more likely to be misinformative. Further, specific characteristics of nonadherent subjects can inform enrichment strategies that may be employed in subsequent trials to reduce misinformative data, such as using an adherence‐monitoring system during a placebo lead‐in period and censoring data for those subjects with poor adherence during the lead‐in.8 By gathering detailed dosing information and patterns of patient behavior, clinical researchers may be able to stratify subjects according to their levels of adherence and better understand the dose‐response rate and “forgiveness” of an investigational product.7, 36 New types of analyses could further correlate actual dosing patterns to determine when the medication might be most effective in a real‐world setting. Adherence monitoring can complicate trial design and add to trial costs, but the increased cost should be weighed against the information it may provide about the true safety and efficacy of an investigational product.11, 12, 26 However, it may also provide reduced costs as a result of improved power, lower enrollment, and shorter trial duration.
Regulatory Considerations
This article has discussed the negative impact of artificial nonadherence on clinical trial outcomes, various methods to detect such nonadherence, and interventions that may be implemented during a trial in order to mitigate its effects. Pharmaceutical companies, subject to regulatory oversight, are understandably interested in how regulatory agencies might view such interventions.
Regulatory agencies have an interest in improving the quality and output of registration trials. For example, part of the FDA's mission statement is “The FDA is responsible for advancing the public health by helping to speed innovations that make medicines more effective, safer. . . .”53 Improving the precision of clinical trials is therefore one approach to advancing the FDA's mission, and that would include interventions to improve adherence and to detect nonadherence.
The FDA has been supportive of adding the types of interventions suggested in this article to registration trials. More problematic, from a regulatory standpoint, is the question of when one can intervene in the conduct of a trial and the impact such an intervention has on trial integrity. Trial design and patient selection methods intended to have an impact on which patients are randomized generally do not represent a trial integrity issue for regulators, nor do various screening procedures, eg, adjudication of diagnosis by nonsite interviewers or checking on adherence during a run‐in period. It is even acceptable to decide to exclude certain patients from the primary analysis, as long as this rule is prespecified in the statistical analysis plan (SAP) and is based on information collected prior to randomization. For example, out of concern for rater inflation of scores prior to randomization, some depression protocols have dropped threshold levels on standard ratings, such as the Hamilton Depression Rating Scale (HAMD); however, unknown to the investigators, the SAP may contain a rule excluding patients falling below a certain HAMD threshold on the baseline rating from the analysis, even though they are continued in the study.54, 55
However, from a regulatory standpoint, it is problematic to exclude from the analysis patients who are determined, based on postrandomization observations, to have been nonadherent.54 Because the decision to become nonadherent could be a result of treatment assignment—eg, a patient may stop taking assigned treatment either because of a side effect (generally based on assignment to active drug) or because of a perceived lack of effectiveness (often, but not always, based on assignment to placebo)— such exclusions from the analysis data set could introduce bias and thereby compromise the randomization. Theoretically, it should be possible to exclude from the analysis a patient who made the decision to be nonadherent prior to taking any of the assignment treatment. In fact, the FDA's usual definition of a mITT analysis includes only patients who are known (or believed) to have taken at least 1 dose of assignment treatment and to have both baseline and at least 1 postbaseline efficacy assessment.56 The challenge is in determining whether, in fact, no assigned treatment was ever taken.
A recent report of a study with a drug to treat opioid‐induced constipation revealed that 15 patients had been excluded from the ITT analysis because they were all determined (presumably postrandomization) to be participating at more than 1 study center.51 Although the paper did not comment on whether or not their level of treatment adherence had been assessed, they were clearly not adherent with the protocol exclusion of dual enrollment, and they would be receiving (although probably not compliant with) assigned treatment from multiple sites. The exclusion of these patients from the ITT analysis does not mean that the FDA will agree with this view. Nevertheless, this example raises the question of whether subjects who decide to be nonadherent even prior to taking their first dose of assigned treatment might be excluded from the ITT analysis.
A related question is what to do with patients who, during the course of a study (postrandomization), are determined to be significantly nonadherent. As noted above, there are many reasons for such nonadherence, including side effects and a perceived lack of efficacy; patients are in fact, free to leave for any reason. However, what about a patient who has been discovered to be participating at multiple centers or who is determined to be completely nonadherent with assigned treatment? He or she may admit to not taking assigned treatment and to have never had any intention to take the assigned treatment. An investigator should have the option of dropping such patients, but it would be helpful to have discussions with regulatory agencies on these types of questions so that standard policies and approaches could be developed, especially now that better methods are being developed to detect such patients. Another question is whether or not to permit interventions during the conduct of the trial to improve adherence of patients who may be sincere in trying to be active participants in the trial but need help in being more adherent. Finally, it is generally understood that there are no regulatory contraindications to a variety of post hoc analyses that might be done to inform future decisions about a program and are not intended to be viewed as primary analyses for that trial.
Conclusion
Recommendations of the Working Group
At site selection, eliminate requirements that most phase 2 to 4 subjects should come from internal databases and set limits on the number of previous studies a phase 2 to 4 subject has participated in over a specified time period (eg, no more than x studies during the past 24 months).
Provide PK and treatment assignment information from previous studies in a timely manner to investigators.
Recognize that increased complexity of phase 2 to 4 protocols, particularly of eligibility criteria, may give professionals an advantage over appropriate subjects.
Limit phase 2 to 4 subject stipends to a reasonable (and appropriate to the local cost of living) reimbursement for time and travel.
Encourage use of recruitment techniques that draw appropriate and novel sources of patients into clinical trials.
Utilize an outside source, such as a study partner/relative or medical/pharmacy records to confirm history.
Utilize an available subject registry to identify and eliminate duplicate and professional subjects.
Eliminate overly restrictive screen‐fail ratios, which adversely incentivize investigators.
Monitor ratings consistency, diary compliance, and subject adherence and consider an outside adjudication process at screen to improve the patient sample.
Consider performing PK sampling on background treatments and consider a biomarker or medication adherence technology during run‐in.
Prespecify who will be included in the final analysis based on information available on subjects prior to randomization.
Monitor individual subject adherence with a medication adherence technology, not pill counts alone; when appropriate, provide subjects and investigators with prompt feedback when nonadherence is detected.
Prompt discontinuation of subjects who are deceptive, duplicate, or egregiously nonadherent may be desirable in order to minimize the impact of the subjects’ data (MMRM).
Consider stratification of subpopulations based on adherence and behavior.
Utilize adherence data to inform protocol design and go/no‐go decisions in later studies.
Acknowledgments
Special thanks to Rachel Oseas, BS, for her contribution in preparing and editing the manuscript. The authors would also like to thank their fellow participants in the ISCTM working group: Josephine Cucchiaro, PhD, David Walling, PhD, Louise Thurman, MD, PhD, Marlene Zarrow, EdM, Sabrina Schoneberg, BA, Paul Greene, PhD, Anjana Bose, PhD, Herbert Noack, PhD, Ibrahim Turkoz, PhD, Judith Quinian, MSc, Timothy Hsu, MD, and Lori Davis, MD.
Disclosure
Thomas M. Shiovitz is president of CTSdatabase, LLC, a clinical trial subject registry discussed in the review, and Adam Hanina is CEO of AiCure, one of the medication adherence technologies mentioned. Phil Skolnick and David J. McCann are full‐time employees of NIDA. There are no financial disclosures to report for any authors.
References
- 1. World Health Organization . World Health Report 2003: Shaping the Future. Geneva, Switzerland: World Health Organization; 2003:17–19. [Google Scholar]
- 2. National Institute for Health and Care Excellence. Medicines adherence: involving patients in decisions about prescribed medicines and supporting adherence. NICE clinical guideline 76. Available at www. nice.org.uk/guidance/cg76. Accessed October 2, 2015. [PubMed]
- 3. Osterberg L, Blaschke T. Drug therapy: adherence to mediation. N Engl J Med. 2005;353:487–497. [DOI] [PubMed] [Google Scholar]
- 4. Gossec L, Tuback F, Dougados M, Ravaud P. Reporting of adherence to medication in recent randomized controlled trials of 6 chronic diseases: a systematic literature review. Am J Med Sci. 2007;334(4):248–254. [DOI] [PubMed] [Google Scholar]
- 5. Shiovitz TM, Wilcox CS, Gevorgyan L, et al. CNS sites cooperate to detect duplicate subjects with a clinical trial registry. Innov Clin Neurosci. 2013;10(2):17–21. [PMC free article] [PubMed] [Google Scholar]
- 6. Mitchell AJ, Selmes T. Why don't patients take their medicine? Reasons and solutions in psychiatry. Adv Psychiatr Treat. 2007;13:336–346. [Google Scholar]
- 7. Vrijens B, Urquhart J. Methods of measuring, enhancing, and accounting for medication adherence in clinical trials. Clin Pharmacol Ther. 2014;95(6);617–626. [DOI] [PubMed] [Google Scholar]
- 8. Czobor P, Skolnick P. The secrets of a successful clinical trial: compliance, compliance, and compliance. Mol Interv. 2011;11(2):107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Devine EG, Waters ME, Putnam M, et al. Concealment and fabrication by experienced research subjects. Clin Trials. 2013;10:935–948. [DOI] [PubMed] [Google Scholar]
- 10. Stichele RV. Measurement of patient compliance and the interpretation of randomized clinical trials. Eur J Clin Pharmacol. 1991;41(1):27–35. [DOI] [PubMed] [Google Scholar]
- 11. McCann D, Petry NM, Bresell AI, et al. Medication nonadherence, “professional subjects,” and apparent placebo responders: overlapping challenges for medications development. J Clin Psychopharmacol. 2015;35(1):566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vitolins MZ, Rand CS, Rapp SR, Ribisl PM, Sevick MA. Measuring adherence to behavioral and medical interventions. Control Clin Trials. 2000;21(5):188–194. [DOI] [PubMed] [Google Scholar]
- 13. World Health Organization . Adherence to Long‐Term Therapies: Evidence for Action. Geneva, Switzerland: World Health Organization Report; 2003. [Google Scholar]
- 14. Zanini GM, Marone C. A new job: research volunteer? Swiss Med Wkly. 2005;135(21‐22):315–317. [DOI] [PubMed] [Google Scholar]
- 15. Brody B, Leon AC, Kocsis JH. Antidepressant clinical trials and subject recruitment: just who are symptomatic volunteers? Am J Psychiatry. 2011;168(12):1245–1247. [DOI] [PubMed] [Google Scholar]
- 16. Liu KS, Snavely DB, Ball WA, Lines CR, Reines SA, Potter WZ. Is bigger better for depression trials? J Psychiatr Res. 2008;42:622–630. [DOI] [PubMed] [Google Scholar]
- 17. Shiovitz TM, Gevorgyan L, Shiovitz ZW, Zarrow ME. How far are duplicate subjects willing to go? Poster presented at ACNP 52nd annual meeting; December 2013; Hollywood, FL.
- 18. Shiovitz TM, Zarrow ME, Schoneberg SH, Seikh LM. Catch me if you can: how a subject registry combines voluntary, investigator‐based use at prescreen and sponsor‐mandated use at screen to reduce duplicate enrollment. Poster presented at ACNP 53rd Annual Meeting; December 2014; Hollywood, FL.
- 19. National Research Council . Transforming Clinical Research in the United States: Challenges and Opportunities: Workshop Summary. Washington, DC: The National Academies Press, 2010. [PubMed] [Google Scholar]
- 20. Weingard K, Efros M. The impact of implementing a national research subject database to prevent dual enrollment in early and late phase central nervous system trials. Poster presented at CNS Summit; November 2014; Hollywood, FL.
- 21. Resnik DB, Koski G. A national registry for healthy volunteers in phase 1 clinical trials. JAMA. 2011;305(12):1236–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Servick K. “Nonadherence”: A bitter pill for drug trials. Science. 2014;346(6207):288–289. [DOI] [PubMed] [Google Scholar]
- 23. Department of Health and Human Services . Recruiting human subjects: pressures in industry‐sponsored clinical research. http://oig.hhs.gov/oei/reports/oei‐01‐97‐00195.pdf. June, 2000. Accessed October 2, 2015. [Google Scholar]
- 24. Anderson AL, Li SH, Biswas K, et al. Modafinil for the treatment of methamphetamine dependence. Drug Alcohol Depend. 2012;120(1‐3):135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson AL, Li SH, Markova D, et al. Bupropion for the treatment of methamphetamine dependence in non‐daily users: a randomized, double‐blind, placebo‐controlled trial. Drug Alcohol Depend. 2015;150:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vrijens B, Vincze G, Kristanto P, Urquhart J, Burnier M. Adherence to prescribed antihypertensive drug treatments: longitudinal study of electronically compiled dosing histories. BMJ. 2008;336(7653):1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baros AM, Latham PK, Moak DH, Voronin K, Anton RF. What role does measuring medication compliance play in evaluating the efficacy of naltrexone? Alcohol Clin Exp Res. 2007;31(4):596–603. [DOI] [PubMed] [Google Scholar]
- 28. Hampson A, Babalonis S, Lofwall MR, Nuzzo PA, Walsh SL. Acetazolamide, a new adherence marker for clinical trials? J Alcohol Drug Depend. 2015;146:e133–e144. [Google Scholar]
- 29. McDonald HP, Garg AX, Haynes RB. Interventions to enhance patient adherence to medication prescriptions: scientific review. JAMA. 2002;288(22):2868–2879. [DOI] [PubMed] [Google Scholar]
- 30. Blaschke T, Osterberg L, B Vrijens, Urquhart J. Adherence to medications: insights arising from studies on the unreliable link between prescribed and actual drug dosing histories. Annu Rev Pharmacol Toxicol. 2012;52:275–301. [DOI] [PubMed] [Google Scholar]
- 31. Pledger GW. Compliance in clinical trials: impact on design, analysis and interpretation. Epilepsy Res Suppl. 1988;1:125–133. [PubMed] [Google Scholar]
- 32. Fimińska Z. Why drugs fail to get approved. http://social.eyeforpharma.com/research‐development/why‐drugs‐fail‐get‐approved. Accessed September 2015.
- 33. Farmer KC. Methods for measuring and monitoring medication regimen adherence in clinical trials and clinical practice. Clin Ther. 1999;21(6):1074–1090. [DOI] [PubMed] [Google Scholar]
- 34. Ho PM, Bryson CL, Rumsfeld JS. Medication adherence: its importance in cardiovascular outcomes. Circulation. 2009;119:3028–3035. [DOI] [PubMed] [Google Scholar]
- 35. Robiner WN, Flaherty M, Fossum TA, Nevins TE. Desirability and feasibility of wireless electronic monitoring of medications in clinical trials. Transl Behav Med. 2015;5(3):285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Demonceau J, Ruppar T, Kristanto P, et al. Identification and assessment of adherence‐enhancing interventions in studies assessing medication adherence through electronically completed drug‐dosing histories: a systematic literature review and meta‐analysis. Drugs. 2013;73(6):545–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaillon P. Healthy volunteers data bank: where and how? Fundam Clin Pharmacol. 1990;4(Suppl 2):177s–181s. [PubMed] [Google Scholar]
- 38. The Over Volunteering Prevention System (TOPS) . Web site. http://www.tops.org.uk/site/cms/contentChapterView.asp?chapter=1. Accessed September, 2012.
- 39. Boyer D, Goldfarb NM. Preventing overlapping enrollment in clinical studies. J Clin Res Best Pract. 2010;6(4):1–4. [Google Scholar]
- 40. Hanson E, Hudgins B, Tyler D, et al. A method to identify subjects who attempt to enroll in more than 1 trial within a drug development program and preliminary review of subject characteristics. Poster presented at CNS Summit; November 2011; Boca Raton, FL.
- 41. Shiovitz TM, Redundancy or integration: the future of subject registries. Talk presented at: Clinical Research Subject Registries: Past Experience and Future Directions, ASCP Conference; June 2015.
- 42. Harriette GC, Toren A, Kiss A, Fowler RA. Eligibility criteria of randomized controlled trials published in high‐impact general medical journals. JAMA. 2007;297(11):1233–1240. [DOI] [PubMed] [Google Scholar]
- 43. Shiovitz TM, Zarrow ME, Shiovitz AM, Bystritsky AM. Failure rate and “professional subjects” in clinical trials of major depressive disorder [letter]. J Clin Psychiatry. 2011;72(9):1284. [DOI] [PubMed] [Google Scholar]
- 44. Ariely D. The Honest Truth About Dishonesty: How We Lie to Everyone—Especially Ourselves. New York: Harper Collins; 2005. [Google Scholar]
- 45. Wyse RKH. Accelerating patient recruitment in clinical trials: in‐depth report from the SMI 2nd Annual Conference. July 2006.
- 46. Fava M, Evins AE, Dorer DJ, Shoenfeld DA. The problem of the placebo response in clinical trials for psychiatric disorders: culprits, possible remedies, and a novel study design approach. Psychother Psychosom. 2003;72:115–127. [DOI] [PubMed] [Google Scholar]
- 47. Kobak KA, Leuchter A, DeBrota D, et al. Site versus centralized raters in clinical depression trial. J Clin Psychopharmacol. 2010;30(2):193–197. [DOI] [PubMed] [Google Scholar]
- 48. Faeries DE, Heiligenstein JH, Tollefson GD, Potter WZ. The double‐blind variable placebo lead‐in period: results from two antidepressant clinical trials. J Clin Psychopharmacol. 2001;21(6):561–568. [DOI] [PubMed] [Google Scholar]
- 49. Agot K, Taylor D, Corneli AL, et al. Accuracy of self‐report and pill‐count measures of adherence in the FEM‐PrEP clinical trial: implications for future HIV‐prevention trials. AIDS Behav. 2015;19:743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fossler MJ. Patient adherence: clinical pharmacology's embarrassing relative. J Clin Pharmacol. 2015;55(4):365–367. [DOI] [PubMed] [Google Scholar]
- 51. Chey WD, Webster LW, Sostek M, et al. Naloxegol for opioid‐induced constipation in patients with noncancer pain. N Engl J Med. 2014;370:2387–2396. [DOI] [PubMed] [Google Scholar]
- 52. AiCure, Phase II study: subject data, 2012–2014, provided by AiCure; New York, NY.
- 53. Food and Drug Administration Statement of FDA Mission . http://www.fda.gov/downloads/AboutFDA/ReportsManualsForms/Reports/BudgetReports/UCM298331.pdf. Accessed September 2015.
- 54. Lee S, Walker JR, Jakul L, Sexton K. Does elimination of placebo responders in a placebo run‐in increase the treatment effect in randomized clinical trials? A meta‐analytical evaluation. Depress Anxiety. 2004;19(1):10–19. [DOI] [PubMed] [Google Scholar]
- 55. Trivedi MH, Rush J. Does a placebo run‐in or a placebo treatment cell affect the efficacy of antidepressant medications? Neuropsychopharmacology. 1994;11:33–43. [DOI] [PubMed] [Google Scholar]
- 56. Food and Drug Administration . Guideline for the format and content of the clinical and statistical sections of applications. http://www.fda.gov/downloads/Drugs/…./UCM071665.pdf. Accessed October 2015.