

Abstract
Food nutrients and metabolic supply-demand dynamics constitute environmental factors that interact with our genome influencing health and disease states. These gene–environment interactions converge at the metabolic-epigenome-genome axis to regulate gene expression and phenotypic outcomes. Mounting evidence indicates that nutrients and lifestyle strongly influence genome-metabolic functional interactions determining disease via altered epigenetic regulation. The mitochondrial network is a central player of the metabolic-epigenome-genome axis, regulating the level of key metabolites (NAD+, AcCoA, ATP) acting as substrates/cofactors for acetyl transferases, kinases (e.g., protein kinase A), deacetylases (e.g., sirtuins). The chromatin, an assembly of DNA and nucleoproteins, regulates the transcriptional process, acting at the epigenomic interface between metabolism and the genome. Within this framework, we review existing evidence showing that preservation of mitochondrial network function is directly involved in decreasing the rate of damage accumulation thus slowing aging and improving healthspan.
Keywords: epigenetic modification, histones, chromatin, acetylation, mitochondrial fusion-fission, biogenesis, autophagy, mitophagy, diet, caloric restriction, cardiovascular disease, aging
Introduction
Aerobic metabolism constitutes the energetic foundation of the great majority of living systems on Earth. By processing oxygen to harness the essential redox and phosphorylation energy required for cellular function, mitochondria have become central players in aerobic life, and their function pivotal for health, disease, and aging [1–4]. Despite the highly efficient chemical reduction of O2 through cytochrome oxidase [5–7], mitochondria still generate significant levels of reactive oxygen species (ROS) during this essential function. Cellular and mitochondrial physiological levels of ROS are attained when production and scavenging are balanced [8, 9], and controlled emission of the freely diffusible H2O2 can act as a rather specific signaling molecule [10, 11].
The functional organization of mitochondria into networks is an emerging paradigm [12–14]. A diverse array of mechanisms underlie the dynamic function of mitochondrial networks such as chemical communication [15–19], physical contact via partial or complete fusion enabling complementation [20–23], intermitochondrial junctions [24], and nanotunneling [25]. Functional organization within localized mitochondrial clusters also represents an additional important organizational principle of mitochondria in cells and organs [17, 26–28]. Examples of the functional importance of localized clusters are given by subsarcolemmal and interfibrillar mitochondrial subpopulations in human and rodent cardiac cells [29–31] and in skeletal muscle [20]. Evidence in brain [16, 32, 33] and in cardiac and skeletal muscle excitation-contraction coupling [20, 34], arrhythmias [35–38] and sudden cardiac death [39] underscores the importance of mitochondrial networks function.
The network organization of mitochondrial structure-function can be characterized from at least four different perspectives [13, 40]: (i) architectural (structural morphology, e.g. punctuated, fused), (ii) topological (spatial connectivity, e.g. lattice, reticular), (iii) molecular (components, e.g., enzymes, carriers, transporters, DNA); and (iv) dynamical (spatio-temporal dynamics, e.g., fission-fusion, respiration, mitophagy, oscillations). Various mitochondrial network functions emerge from a dynamic interplay between all these aspects. For example, the predominance of fission over fusion in glucose and lipid excess happens concomitantly with higher expression of fission proteins [41], or the occurrence of mitochondrial membrane potential (ΔΨm) and NAD(P)H oscillations occurring with intense oxidative stress and overwhelming of the antioxidant systems [15, 19, 42].
Mitochondria function as hubs in the overall cellular network formed by metabolic, signaling, genetic and epigenetic pathways [13, 43]. Genetic, signaling, metabolic and transport networks underlie the morphological-topological appearance of the mitochondrial network (Fig. 1). The contribution of mitochondria to cell function results from their collective cluster dynamics in different cellular locations (perinuclear, subsarcolemmal, intermyofibrillar) connected via local signaling events [17, 19, 26, 27]. Mitochondria drive catabolic and anabolic reactions supplying energy (e.g. ATP, electrochemical gradients) and metabolites with biosynthetic (e.g. citrate, AcCoA) and signaling (e.g., AcCoA, cytochrome c, H2O2, O2·−) roles [13, 44]. They influence the cellular redox status introducing ROS, namely H2O2) [3, 32, 45–47], and are reciprocally influenced by the cytoplasmic redox environment (e.g., glutathione, H2O2) in a dynamic way [46, 48]. Likewise, mitochondria respond to signaling molecules (e.g., Ca2+, O2·−, ADP) released from mitochondrial neighbors, other organelles (e.g., endoplasmic or sarcoplasmic reticulum, peroxisomes) or contractile proteins (e.g., myosin ATPase). Mitochondria also maintain an anterograde and retrograde signaling cross-talk with the nucleus that generates reciprocal activation-repression patterns of gene expression [4, 44, 49]. Mitochondria can determine apoptotic and necrotic cell death mediated by Ca2+ overload and opening of the permeability transition pore [19, 50, 51], and their turnover is modulated by the mitochondrial quality control machinery including the fission-fusion, mitophagy and biogenesis processes [41, 52–54].
Figure 1.
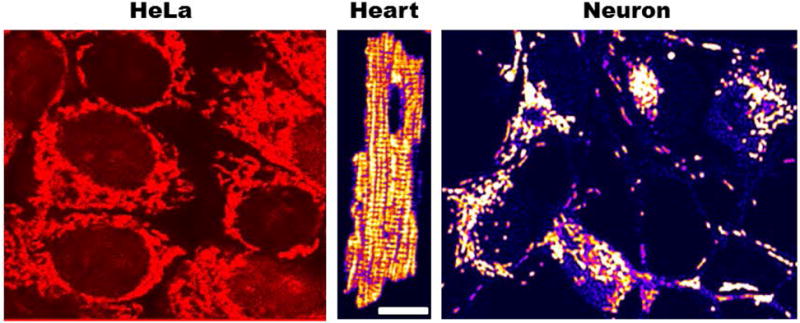
Morphological appearance of intracellular mitochondrial networks labeled with the cationic potentiometric dye tetramethylrhodamine ethyl ester (TMRE) used to monitor mitochondrial membrane potential. Notice the different architecture exhibited by mitochondrial networks in HeLa, cardiomyocyte and cortical neuron cells ranging from reticular (Hela and neuron) to lattice-like (ventricular cardiomyocyte).
The index of aging-relatedness (π): human healthspan and mitochondria
In this review, we adopt the definition of healthspan by Levy and Levin (2014) in the context of an evolutionary view of human aging. These authors introduced the index of aging-relatedness, π, as a measure of “living life to the fullest” or “the maintenance of full function as nearly as possible to the end of life” when applied to intrinsic mortality data [55].
Levy and Levin (2014) postulated a mixture model encompassing the Gompertz law and the Weibull hazard function. Based on empirical human mortality observations, the Gompertz law describes an exponential relation between age-specific mortality rates and age of the form: hG (x) = λe θx, where hG (x) is the hazard function (or instantaneous death rate, or force of mortality); λ > 0 is a parameter denoting the initial mortality rate (at birth or another arbitrary age; λ is also called vulnerability parameter), and θ is an exponential rate parameter (rate of aging or Gompertz parameter) [55]. The relationship between age-specific mortality (or failure) rates and age according to the Weibull hazard function (a model of the strength of materials) is described by a power function of the form: hw (x) = α γ xγ−1 where α > 0 and γ > 0 are parameters (γ, shape parameter; α−1, scale parameter) [55]. (Weibull plays a prominent role in reliability theory which applies to the failure of mechanical devices similar to the role played by the Gompertz law in demography.
The model of Levy and Levin [55] implies that if all deaths or cases of a specific disease in a population were caused by one of a large number of sufficient causes from Late Onset Genetic Effects (LOGE), the residual time to event after age 10y would follow an approximate Gompertz distribution, while if all deaths or cases of a specific disease in a population were caused by one of a large number of sufficient causes from Evolutionarily Recent Environmental Factors (EREF), the residual time to event after age 10y would follow an approximate Weibull distribution [55] (Fig. 2).
Figure 2. Survival curves according to Gompertz, Weibull and the mixture model of Levy and Levin.
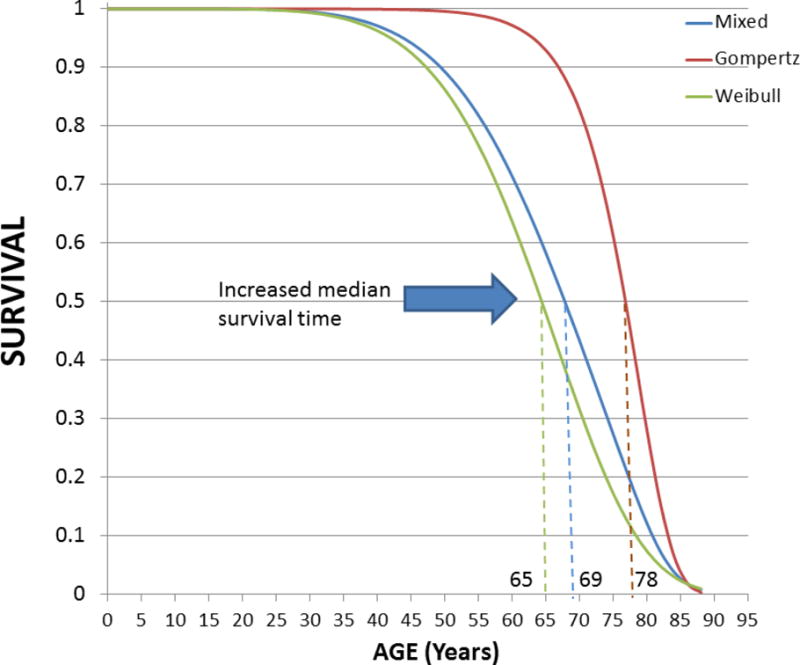
The evolutionary biology-based mixture model of Levy and Levin encompasses Gompertz and Weibull components [55]. Notice the increase in the median survival time from the Weibull, to the mixed and the Gompertz distributions (dashed lines). A visible decline in the survival function starts at 35–40y for the Weibull and mixed distributions whereas the decline is only noticeable at about 60–65y for the Gompertz component, accelerating at ~75y, and then showing a steep decline between ~80–90y while approaching an overall rectangular form. At 80y, ~10% and ~35% of the population is alive according to the mixed and Gompertz components, respectively. Prevention of premature deaths (e.g., accidental, neonatal and infant) would bring population survival closer to the Gompertz component of the mixture model. The index of aging-relatedness applied to intrinsic mortality data is a measure of “living life to the fullest” (rectangularization of the survival curve) thus representing a quantitative characterization of health span.
Aging is a function of multiple genetic, environmental factors and their interactions. In the Levy and Levin formulation, biological aging is by definition genetic in the sense of related to sufficient causes containing LOGE in the absence of EREF, and the π index may be said to represent the contribution of biological aging to the overall (genetic and non-genetic) aging in the population [55]. Otherwise stated, π indicates the proportion of deaths or cases of a specific disease in the population due to sufficient causes of LOGE in the absence of EREF. For example, π of 20% implies that 20% of the intrinsic deaths in the population are due to sufficient causes containing LOGE in the absence of EREF (i.e., 80% of the intrinsic deaths are due to sufficient causes containing EREF). Thus, as a caveat, population biological aging, defined as an exponential increase in age-specific mortality rates, is a more restrictive and hence, specific, definition than chronological aging or the “mere passage of time”.
In the evolutionary view, what makes modern life exposures “abnormal” is the lack of human adaptation to the effects of, e.g., smoking, air pollution, excess calorie intake, or the mismatch between the human genetic makeup and modern lifestyle. Epigenetics, viewed as the inheritable response to environmental factors, can help identify the mechanisms by which intrinsic as well as extrinsic (environmental) factors derail human health provoking dysfunction and, ultimately disease. In this context, mitochondrial functionality is centrally involved in human healthspan because as main energy suppliers they participate in energy availability and allocation to maintenance and repair processes [56, 57]. Preservation of mitochondrial network function is critically important for limiting the rate of damage accumulation and thus a way of slowing the rate of senescence and of increasing life expectancy as well as of life quality.
Increasing evidence shows that the health status of the mitochondrial network in mammalian tissues is highly sensitive to the cellular energy/redox capacity [45, 58], fusion-fission dynamics [41], and mitochondrial turnover as a result of the dynamic balance between mitochondrial loss vs. scavenging and replacement, e.g., by mitophagy [52] and biogenesis [54, 59]. Alterations of mitochondrial quality control due to altered fusion-fission dynamism and mitophagy can explain differences in cumulative mitochondrial damage. The response of these processes to physiological conditions imposed by, e.g., diet, exercise, via the epigenome exert a direct impact on mitochondrial health and health-span.
Depending upon the nutrient composition of the diet, certain substrates, cofactors and effectors (ATP, AcCoA, NADH, α-ketoglutarate) become available and their metabolism render intermediates that in turn generate patterns of epigenetic modifications. Among the latter with gene expression-modulating capacity are methylation, acetylation, or oxidation (as in demethylation catalyzed by iron-containing jumonji-domain demethylases that consumes α-ketoglutarate and O2 [60] or in 5-methylcytosine [an epigenetic mark on DNA] to render 5-hydroxymethylcytosine catalyzed by the ten-eleven translocation, TET, family of dioxygenases [61, 62]) (Fig. 3). Mitochondrial sensitivity to the body’s balance of substrate/energy supply-demand determined by lifestyle decisions (e.g. sedentarism vs. physical activity, over- vs. balanced-nutrition) will favor, or avoid altogether, the effects of metabolic disorder upon the gene-environment interaction and their impact on healthspan.
Figure 3. The fluxome and the metabolic-epigenetic-genomic axis.
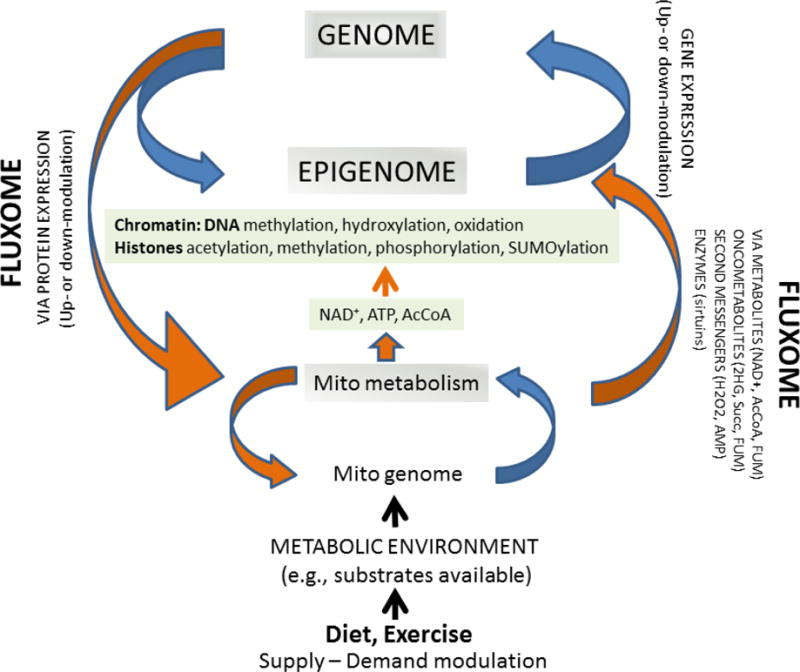
The fluxome corresponds to the fluxes of metabolite transformations from a substrate precursor and the fluxes distribution among different pathways. Lifestyle via diet and exercise or physical activity affect (peri- and postnatally) the balance of energy supply-demand promoting, e.g. over-nutrition or calorie restriction. These interventions have an impact on the fluxome and the levels of metabolites, second messengers, that in turn modify the epigenome via chromatin by, e.g. methylation, acetylation, of the DNA and/or histones. NAD+, ATP and AcCoA are key metabolic intermediates in major carbon catabolic pathways and in fuel metabolism, including glucose and pyruvate oxidation, as well as in β-oxidation of fatty acids (FAs). In mammalian cells, AcCoA can be generated both in the mitochondrial and nucleocytoplasmic compartments. In the mitochondria, AcCoA is synthesized from pyruvate by the pyruvate dehydrogenase complex or by β-oxidation, and then feeds into the TCA cycle.
A key component of the epigenomic interphase between metabolism and the genome is the assembly of DNA and nucleoproteins that constitutes the chromatin. The chromatin regulates the transcriptional machinery by conformational changes and genome compartmentalization, thus a key component of the epigenomic interphase between metabolism and the genome. Epigenetic modifications regulate the transcriptional and gene expression machinery by conformational changes and genome compartmentalization. Stable patterns of epigenetic modifications may represent a mechanism of cell memory, important for interpreting the long-lasting footprints of hyperglycemic episodes [230] or developmental and nutritional milieus associated with predisposition and progression of disease [62, 80]. The feedback from the genome to the epigenome, exemplified by posttranslational modifications (PTMs) occurring on histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases, and demethylating enzymes, suggests a highly ordered interactive network comprising many components capable of adding and removing modifications at both the chromatin template as well as each other.
Key to symbols: fluxome, orange arrows; gene expression, blue arrows.
The role and the meaning of mitochondrial dysfunction
Maintaining efficient energy supply, redox environment and signaling are three of the main mitochondrial functions. In aerobic metabolism mitochondria harness phosphorylation and redox energy from oxygen and oxidizable substrates, and thus they are constantly challenged to maintain energy supply while keeping ROS within physiological levels compatible with signaling [8, 45, 46, 48].
Under disease conditions mitochondria can become dysfunctional exhibiting in general (at least) three main impairments: uncoupling of oxidative phosphorylation (OxPhos), excess ROS emission [63–66] and abnormal Ca2+ uptake [67–69]. These defects impair function by altering energy availability, the cellular redox environment and cell viability. As a matter of fact, in the heart impairment of mitochondrial respiration and antioxidant systems, and alterations in structure and respiratory chain complexes, have been extensively documented in both type 1 and type 2 (T2DM) diabetes animal models (see Table 1 from [45], for a review). Mitochondrial Ca2+ uptake impairment has been described in diabetes [45, 67] and heart failure [68, 69] producing defective ATP supply that in concert with ROS can lead to cell death by necrosis/apoptosis [70, 71] mediated by sensitization of the permeability transition pore [51, 72].
In the heart, the respiratory complex I appears as a relevant and common site of defects across chronic T1DM and T2DM conditions, although mitochondrial energetic dysfunction in diabetes is a manifold failure, involving the other electron carriers as well [73]. Contractile failure will ensue since mitochondria provide more than 90% of the energy required for function and the contractile machinery depends upon a proper redox status to develop force. Oxidizing conditions in diabetes readily affect contractility and Ca2+ handling [63, 66, 74, 75]. Energy/redox stressful conditions produced evident heart contractile impairment in T2DM diabetic mice as compared to controls [66]. Because a balanced cellular/mitochondrial redox environment is vital for optimal energy supply and excitation-contraction coupling, key mitochondrial functions are apparently protected [1, 45]. In heart trabeculae from T2DM Zucker rats, maintained redox balance via increased glutathione- and thioredoxin-antioxidant activities, apparently to resist oxidative stress, is an essential protective response of the diabetic heart to keep contractile function [75].
One of the main theses of this review is that the proportion of energy-redox defective mitochondria increases due to impairments in their turnover (by e.g., mitophagy, biogenesis) which depends on the mitochondrial quality control axis [53, 76] (see below). Degradation of the mitochondrial network’s quality, as can be judged from energy-redox impairment [41, 45], alters the dynamic balance of fusion-fission processes [53]. It has been hypothesized that a predominance of mitochondrial fission over fusion may increase the frequency of mutations in the mitochondrial DNA (mtDNA) [58]. Deleterious mtDNA mutations can create an intracellular mixture of mutant and normal mtDNAs, a state known as “heteroplasmy” [77]. When a threshold level of heteroplasmy level is crossed, extensive reprogramming of nuclear DNA (nDNA) gene expression can happen mediated by retrograde signaling [78]. Mechanisms mediated by mtDNA damage may be relevant for diabetic cardiomyopathy because they connect basic mitochondrial biology such as fusion/fission [41] that impinge on apoptosis [79] and are closely integrated with the mitophagy quality control pathway [23, 52–54, 76, 77].
mtDNA mutations and retrograde signaling to the nucleus
Increasing appreciation for the role that mitochondria play in metabolic dysfunction as well as other disorders argues in favor of a role for aberrant mtDNA modification in disease development and progression [62, 80]. Since the vast majority of mitochondrial proteins are encoded in the nuclear genome, appropriate communication between the nuclear, cytoplasmic and mitochondrial compartments is essential for maintaining proper mitochondrial function. Mitochondria signals to the nucleus (via retrograde signaling) reprogram gene expression underscoring the relevance of understanding the mechanisms through which mitochondria and the nucleus communicate.
Several studies have established a direct cause-and-effect relationship between mitochondrial dysfunction and disease. The contributory role of mitochondrial dysfunction in various metabolic and degenerative diseases, and aging has being demonstrated in mice harboring mitochondrial gene mutations [81]. mtDNA has a higher intrinsic mutation rate than nDNA [82] thus generating heteroplasmy [83] while there are indications that high mutation rates might be a major driving force for the population prevalence of heteroplasmy [84]. The accumulation of somatic mtDNA mutations probably affects healthspan, and may influence lifespan, contributing to the onset and progression of complex diseases. In mice, mtDNA mutations shorten lifespan and lead to age-related phenotypes, which include reduced body mass and an increase in the heart:body weight ratio, consistent with age-related cardiomyopathy [85]. In longevity, an alternative view of heteroplasmy as a reservoir of genetic variability to cope with stress during life has also been put forward [86]. Reported evidence indicates that low level heteroplasmies in blood are detectable, transmitted and maintained within families until extreme age [86, 87] with an apparent unique profile for each family [87]. Low level mtDNA heteroplasmy in humans also plays a role in mitochondrial-related diseases such as myopathies, lactic acidosis, stroke-like episodes and complex disorders, including T2DM, aging, cancer, and late-onset neurodegenerative diseases [4, 78, 81, 83, 85, 88–91]. In healthy aged humans, the overall tissue burden of mtDNA mutations is low (1%–2%), but in single cells the percentage can reach mutation threshold levels known to cause a defect in OxPhos [85]. In a study by Ye and colleagues [84], 4,342 heteroplasmies were identified in 1,085 individuals, of which 973 individuals (~90%) had at least one heteroplasmy. These heteroplasmies were observed at 2,531 mtDNA sites across different mtDNA regions, and 1,757 (~70%) of these sites were heteroplasmic in only one individual suggesting that the pattern of heteroplasmies can be quite varied among individuals. Among the 4,342 detected heteroplasmies, 301 (~7%) were reported to be disease-associated and 210 individuals (~20% of 1,085) carried at least one disease-associated heteroplasmy of which mitochondrial myopathy and encephalomyopathy were overrepresented [84]. Mitochondrial heteroplasmy tends to show high pathogenicity, and is significantly overrepresented in disease-associated loci [88]. Maternally transmitted mtDNA mutations have been shown to underlie mild aging phenotypes in mice. Importantly, mtDNA mutator mice (PolgAmut/mut) heterozygous for the allele (PolgAwt/mut) aggravate premature aging phenotypes [88]. An increase in the proportion of mutant mtDNAs might drive the cell to an energetic threshold beyond which its energy output capacity declines, leading to insufficient energy supply to sustain cellular function. Thus, such DNA damage can impair organ metabolic functions by causing cell dysfunction, death or senescence and can also induce tissue inflammation perturbing systemic metabolism [92].
The mitochondrial genome consists of roughly 1500 genes distributed across the maternal mtDNA and the nDNA [81, 93]. The genome of mtDNA is organized in circular double-stranded molecules, packed in compact particles termed ‘nucleoids’ tethered to mitochondrial membranes [94, 95]. Although nucleoids do not contain histones, mtDNA is packaged with structural proteins and, like its nuclear counterpart, is subject to remodeling events [62, 96]). Human mtDNA is maternally inherited and contains 37 genes encoding for 13 subunits of complexes I, III, IV, and V; 2 ribosomal RNAs, and 22 tRNAs [97]. Thus, it is not possible to build a mitochondrion based on nDNA alone, despite the majority of about 1000 – 2000 mitochondrial proteins being encoded in the nDNA. mtDNA mutations may cause bioenergetic failure due to the essential OxPhos components encoded by the mtDNA [78]. The importance of this topic has been further underscored by recent work showing that relatively subtle changes in mtDNA heteroplasmic levels can have dramatic phenotypic effects [78, 84].
Most mitochondrial proteins are synthesized in the cytosol and are subsequently imported into the organelle by protein translocases, ensuring proper targeting to the different mitochondrial subcompartments [93, 98, 99]. Several nuclear transcription factors are imported to directly facilitate mitochondrial gene expression [100]. Transcriptional regulation of a small number of human mitochondrial genes associated with respiration implicates metabolic-driven epigenetic changes [62]. Several proteins have emerged as major regulators of mitochondrial gene expression, capable of increasing transcription of mitochondrial genes in response to the physiological demands of the cell. For the purpose of this review we consider effectors of mitochondrial function such as the biogenesis regulator PGC-1α, the lysine deacetylase SIRT1, the metabolic sensor adenosine monophosphate (AMP)-activated protein kinase (AMPK), and the tumor suppressor protein P53, a transcription factor that acts in response to cellular stress signals (e.g., DNA damage, hypoxia, oxidative, and nitrosative stress) [101]. The mammalian target of rapamycin (mTOR) is a serine-threonine kinase that functions as an intracellular energy sensor in mammalian cells, existing in two distinct protein complexes: mTORC1 and mTORC2) which affect metabolic regulation during aging [56] and the response to calorie restriction [102]. P53 is also able to inhibit mTORC1 by activation of AMPK, which is subsequently followed by induction of autophagy [103–105], a function likely to contribute to the role of p53 in tumor suppression [105].
The metabolism-epigenome-genome axis and the fluxome
The metabolism-epigenome-genome axis is comprised by a dynamically reciprocal feedback loop between the epigenome and the genome driven by metabolism via, e.g., histones acetylation and deacetylation, DNA methylation (Figs. 3, 4). Reciprocally, the feedback from the genome to the epigenome can be exemplified by the expression level of histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases, demethylating enzymes, and single nucleotide polymorphisms that impact epigenetic modifications, in particular DNA cytosine methylation [62, 106]. This suggests a highly ordered interactive network comprising many components capable of adding and removing modifications at both the chromatin template as well as upon each other.
Figure 4. Substrates fuel interrelationships between main body organs, mitochondria and the epigenome.
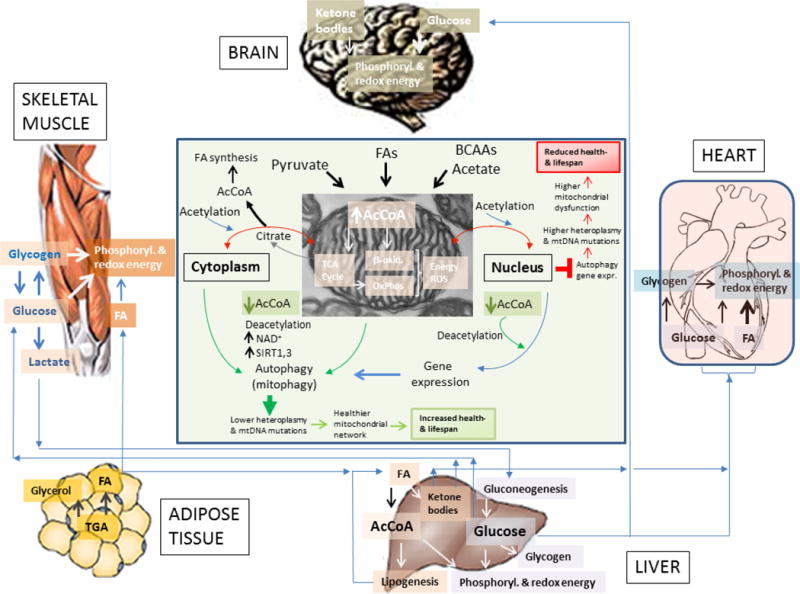
Depicted are the differential uses of glucose and fatty acids (FAs) by the brain, muscle (cardiac and skeletal), liver and adipose tissue. As shown, the liver appears as a main provider of glucose and FAs for muscle and brain. Adipose tissue provides FAs to the liver; the skeletal muscle can also supply lactate to the liver where it is utilized for gluconeogenesis, especially in the early fed state, when the liver continues in ketogenic and gluconeogenic modes.
Displayed is the mitochondrial supply of AcCoA, the main acetyl donor that via its nucleo-cytoplasmic pools determine the autophagic response in mammals [168] and, with a few difference, in yeast as well [162]. The scheme at the center emphasizes that AcCoA excess (red right) or decrease (green bottom) will preclude or stimulate autophagy and gene expression, respectively; decreased autophagy will favor higher mtDNA mutations and heteroplasmy driving accumulation of dysfunctional mitochondria that, in the long-term, will reduce health- and lifespan (red box, top right). Opposite changes will occur under a more balanced AcCoA supply (green box, bottom). Hypothetically, healthy and nutritionally well-adjusted lifestyle, including physical activity or exercise training, would favor balanced energy supply and demand in concert with mitochondrial fusion/fission-driven morphological changes, turnover (mitophagy, biogenesis) and proper energy-redox function [201]. The interplay between these processes would determine not only the overall functionality of the mitochondrial network but also the integrity of the mitochondrial genome [4, 78].
From a systems perspective, the epigenome represents an interface between metabolism and the gene expression machinery of nDNA and mtDNA whereas the fluxome is the biochemical readout of the combined metabolic activities within the cell, which subsequently alter the epigenome (Fig. 3). Specifically, the fluxome corresponds to the fluxes of metabolite transformations (in moles per unit time) from a substrate precursor (e.g., glucose, lipids), and their distribution among different pathways (e.g., glycolysis, pentose phosphate, etc). The ensemble of metabolic fluxes that the fluxome embodies is the final result from genes expressed and proteins translated including their posttranslational modifications (PTMs). The fluxome represents a dynamic picture of the phenotype encompassing the metabolome and its functional interactions with the epigenome and signaling networks. The fluxome dynamics are expected to change during physical activity and vital cellular functions such as cell contraction, growth, division, differentiation, autophagy, apoptosis/necrosis, or the response to key environmental signals such as starvation, hypoxia, etc [107–109]. For example, in exercise short (~ 2min), high intensity activities including, for example, power lift, baseball pitch, 200-meter swimming, are fueled by the ATP-creatine phosphate and the lactic acid system which can proceed anaerobically, e.g., from stored ATP-creatine-phosphate and glycogen (Fig. 4). In contrast, aerobic activities like prolonged endurance or ultra-endurance exercise (e.g., running, cycling, marathon) depend on carbohydrate and fat, more of the latter than the former during rest (~ 60/40 %) and greater carbohydrate use during exercise [110]. Muscle glycogen provides most substrate fuel (> 80%) during high intensity exercise depending on training status, intensity and duration, diet, and circulating levels of insulin [110, 111]. Adrenaline mobilizes fats from adipocytes that circulate as free FAs and are taken up by skeletal and cardiac muscles for use and storage as intramyocellular triacylglycerol in lipid droplets (reviewed in [112] and [1]). In addition to providing substrate for the TCA cycle, FAs are converted to ketone bodies for export as a glucose- sparing energy source to metabolically active tissues (Fig. 4). Under normal conditions, ketones are produced at relatively low rates in well-nourished individuals. However, during extended caloric restriction, starvation, or intense exercise, depletion of TCA cycle intermediates by gluconeogenesis in the liver, coupled with increased mobilization of FAs, divert AcCoA to ketone production. This allows continued β-oxidation and energy transport to extra-hepatic tissue when AcCoA accumulates beyond the capacity of the TCA cycle.
The fluxome influences the epigenome via metabolite and second messenger levels, which together represent a unique phenotypic signature (Fig. 3). Mitochondrial metabolism and glycolysis convert carbohydrates and fats into readily available metabolites ATP, AcCoA, NADH, ROS that together with the main methyl donor S-adenosylmethionine (SAM) generated in the cytoplasm, drive epigenetic modifications (acetylation, methylation, phosphorylation, oxidation). ATP drives phosphorylation of nuclear and cytoplasmic signal transduction proteins, and histone tails. AcCoA is the main acetyl donor of acetyl transferases in the acetylation of chromatin and signal transduction proteins modifying nDNA transcription and replication [4, 62, 106] thus linking cellular energetics with chromatin dynamics and transcription [62] while acting as a phylogenetically conserved inhibitor of starvation and age-associated autophagy [4, 113, 114]. Sirtuins use NAD+ as a cofactor for deacetylating proteins. DNA is methylated via SAM, the active methyl donor group utilized by most methyltransferase enzymes.
The fluxome can be characterized at the steady state [115–117] and in time-dependent phenomena [109, 118]. The control and regulation of the fluxome can be known, including fluxes and metabolite concentrations, when accounting for regulatory information [9, 109, 115, 119–121]. Knowledge about the control and regulation of metabolites and second messenger concentrations as well as their temporal evolution is crucial to understand the control of phenomena such as the cellular response to hypoxia, or autophagy in response to starvation that can happen within minutes to hours, respectively. As a matter of fact, recent compelling evidence supports the central role of AcCoA metabolism in autophagy regulation [113, 114]. In the cytoplasm, AcCoA can be generated via ATP citrate lyase (ACL) from mitochondrial citrate or via acyl-CoA synthase short-chain (ACSS2) from acetate. Knocking-down these enzymes results in very low levels of global histone acetylation [122]. Compartmentation of these pathways plays a role because the nuclear and cytoplasmic pools of AcCoA, rather than the mitochondrial, appear to modulate the autophagic flux [113]. In the mitochondria, AcCoA is generated by pyruvate decarboxylation catalyzed by pyruvate dehydrogenase (PDH) and from FA oxidation. It has been proposed that efficient utilization of AcCoA by mitochondrial tricarboxylic acid (TCA) cycle activity and OxPhos would prevent AcCoA overflow into protein acetylation and/or histone acetylation pathways that would negatively influence autophagy and other pro survival processes during aging and age-associated disease [113]. Indeed, higher acetylation of histones and cytoplasmic proteins decreases the rate of autophagy in yeast and mammalian cells, despite differences in the routes of AcCoA synthesis [114]. Mitochondrial AcCoA can also be synthesized by oxidative decarboxylation of the branched chain amino acids (BCAAs) valine, isoleucine, lysine, via branched-chain alpha-ketoacid dehydrogenase complex (BCKDH). First, BCAAs are transaminated to branched-chain a-ketoacids (e.g., α-ketoisocaproic acid from leucine), which subsequently undergo oxidative decarboxylation, an irreversible reaction catalyzed by BCKDH yielding AcCoA as the end product.
Effectors of the epigenome
Different effectors of the epigenome spanning from metabolite intermediates to proteins and transcriptional factors have been reported. Genetic-metabolic responses of the body converge at the epigenome reciprocally influencing each other. Thus, altered metabolism may contribute to disease by distorting epigenetic regulation. The stability and patterns of epigenetic modifications with gene expression-modulating abilities may confer a “footprint” of cell memory. For example, altered chromatin methylation patterns involving methyltransferases competing for bioavailable S-adenosyl methionine and posttranslational methylations are extensively implicated in metabolic gene activation and repression [123–125].
Sirtuin (SIRT)
The immense interest surrounding the SIRTs enzyme functions in metabolism stems from their close homology to yeast Sir289 and similar proteins associated with longevity in flies [126] and worms [127]. SIRTs catalyze deacetylation processes of the chromatin, opposite to protein acetylation by acetyltransferases. Seven mammalian SIRTs are differentially distributed throughout the cell, with SIRT1, SIRT6, and SIRT7 mainly localized for nuclear functions. Important studies have identified roles for SIRT1 and SIRT6 in the regulation of mammalian life span and several lines of evidence strongly implicate SIRTs in the life-extending effects of calorie restriction; however, the precise molecular mechanisms are unclear [128]. SIRT3 serves to induce the catalytic activity of the enzyme ACCS2 that converts acetate into AcCoA in mitochondria by removing an inhibitory acetyl group. Nuclear SIRT1 serves to deacetylate the transcriptional coactivator PGC1α, thereby enabling the expression of genes whose products are required in energy-depleted cells [129].
Nicotinamide adenine dinucleotide (NAD+)
This is an essential cofactor for reactions catalyzed by the highly conserved SIRT HDAC family [130]. Other NAD+ consuming enzymes such as ADP-ribosyltransferases have also been shown to covalently ADP-ribosylate core histones [62, 131]. PAR polymerases (PARPs) utilize NAD+ to catalyze Poly(ADP-ribose) synthesis and are involved in the cellular stress response [132]. PARP-1 activation leads to cytosolic NAD+ depletion and mitochondrial-mediated apoptosis and cell death [133]. Continual re-synthesis of NAD+ through salvage/recycling pathways maintains the functions of cytosolic and nuclear enzymes. Nicotinamide phosphoribosyltransferase and nicotinamide mononucleotide adenylyltransferase 1 constitute the major NAD+ salvage pathway in the mammalian nucleus, whereas the respective localization of nicotinamide mononucleotide adenylyl transferases 2 and 3 isoforms to the Golgi and mitochondria further indicates the importance of subcellular metabolite compartmentalization [62, 134].
S-adenosylmethionine (SAM)
SAM is produced by the condensation of methionine and ATP during the first of nine steps required for the conversion of methionine to succinyl-CoA, a predominantly cytoplasmic pathway that ends up in the mitochondria [124, 129, 135]. It contains the active methyl donor group utilized by most methyltransferase enzymes. Histone methylation is a dynamic, reversible process. For example, LSD1 demethylase (also called KDM1A or AOF2) is a nuclear FAD-dependent enzyme capable of demethylating methylated H3K4 both in vitro and in vivo, in agreement with the dynamically reversible nature of histone methylation [136]. LSD1 demethylase activity acts on H3K4me1/2 and H3K9me1/2 but cannot demethylate trimethylated groups because it requires protonated nitrogen in the substrates thus restricting its activity to mono- or dimethylated peptides [136]. Similar to SIRTs, LSD1 activity is regulated by redox state and is stimulated when FAD is in its oxidized form but not when it is reduced to FADH2 as it occurs in the TCA cycle [137]. LSD1, in turn, regulates mitochondrial respiration and energy expenditure. Specifically, LSD1 binds directly to genes such as PPARγ coactivator-1alpha (PGC1α), PDK4, FATP1, and adipose triglyceride lipase (ATGL), and represses their transcription associated with loss of H3K4 methylation [138].
Flavin adenine dinucleotide (FAD)
Derived from the vitamin riboflavin (vitamin B2), mitochondrial-generated FAD functions as the prosthetic group for certain oxidation-reduction enzymes. Riboflavin is phosphorylated by riboflavin kinase to generate riboflavin 50-phosphate (sometimes called flavin monucleotide or FMN), which is then converted to FAD by FAD synthase (also called FMN adenyltransferase). Instead of NAD+, the nuclear-located LSD1 uses FAD, which is composed of riboflavin (vitamin B2) and ADP. It has been suggested that the nuclear location of LSD1 might render it particularly sensitive to changes in FAD availability (and the ratio of FAD to FADH2) arising from the activities of other flavin-linked dehydrogenases and oxidases, including those associated with fatty acid β-oxidation and the TCA cycle in mitochondria.
Beta-hydroxybutyrate (β-HB)
This metabolite modulates several signaling pathways leading to epigenetic changes. The ketone body β-HB inhibits the activity of many NAD+-independent HDACs [139], and is able to not only fuel metabolic adaptation to starvation but also to help sustain a protective epigenetic state by inhibiting the activities of NAD+-independent HDACs.
Alpha-ketoglutarate (α-KG)
The α-KG-malate carrier regulates mitochondria-cytosol transport of this metabolite. The ten-eleven translocation (TET) family of dioxygenases mediate the oxidation of 5-methylcytosine [5mC]. The potential for the TET family (TET1/2/3) to regulate diverse physiological functions including metabolic signaling requires the TCA cycle metabolite α-ketoglutarate, and this activity is inhibited by 2-hydroxyglutarate. Jumonji C domain containing histone demethylases are α-KG-dependent [140, 141]. While studies are yet to determine the TET-metabolism connection, mutations in isocitrate dehydrogenase genes are associated with reduced α-KG and elevated 2HG levels leading to genome-wide changes in histone and DNA methylation patterns [62, 142].
Succinate, Fumarate and 2-hydroxyglutarate (2HG)
Succinate dehydrogenase (SDH), fumarate hydratase (FH), and isocitrate dehydrogenase (IDH) mutations cause the accumulation of succinate, fumarate, and R-2HG (R enantiomer of 2-hydroxyglutarate), respectively. These metabolites appear to cause cancer by affecting the behavior of various α-KG-dependent dioxygenases, including dioxygenases linked to DNA and histone methylation, and therefore have the potential to alter the epigenome [143]. IDH mutations can impair differentiation in non-transformed cells from multiple cells of origin, and this impairment is linked to 2HG-mediated epigenetic dysregulation. In human cells proliferating in hypoxia, α-KG can accumulate and be metabolized through an enhanced reductive activity of wild-type IDH2 in the mitochondria, leading to 2HG accumulation in the absence of IDH mutation [144]. The ability of 2HG to alter epigenetics may reflect its evolutionarily ancient status as a signal for elevated glutamine/glutamate metabolism and/or oxygen deficiency [143].
Mitochondrial function and the epigenome
After the notion of “epigenetic landscape” introduced early on by Waddington (1957) to describe cellular differentiation beyond genetic inheritance [145], the concept of epigenetics has evolved into “modifications of the DNA or associated proteins, other than DNA sequence variation that carry information content during cell division”, e.g., DNA cytosine methylation within cytosine–guanidine (CpG) dinucleotides converting cytosine to 5-methylcytosine [146, 147], hydroxylation and oxidation [61], acetylation, methylation, phosphorylation, ubiquitination, Small Ubiquitin-related MOdifier (SUMO)ylation [49], and succinylation and malonylation [148] of the lysine and/or arginine residues of histones. These modifications are thought to determine the genome accessibility to transcriptional machinery [149]. More recently, epigenetics is acquiring wider connotations because we increasingly recognize the relevant role played by reciprocal feedbacks between the metabolic state (e.g., metabolites, second messengers) and the regulatory state (transcription factors, gene expression modification through, e.g. histones acetylation, DNA methylation) and vice versa via the epigenome [49, 107, 150, 151]. As an example, acetylation from AcCoA of the FOXO transcription factors, PGC-1α and β transcriptional co-activators depresses OxPhos and redirects catabolism toward glycolysis in the fed state [4]. This subject is relevant for communication and temporal coupling of circadian rhythm and metabolic cycles by epigenetic modifications.
The reciprocal influence between mitochondrial function and the epigenome depends upon substrate availability, and is mediated by energy and redox intermediates [4, 152]. More specifically, the epigenome is affected by the metabolic status in turn determined by the balance of substrate supply (calories availability) and demand (e.g., physical activity or over-nutrition) that directly influence mitochondrial supply of intermediates (Fig. 4). Mitochondria and glycolysis convert carbohydrates and fats into readily available AcCoA, SAM, high energy transfer groups as ATP, and redox equivalents as NADH [4]. These metabolites drive the modification of the epigenome via phosphorylation, acetylation and methylation that regulate signal transduction pathways.
In response to changes in nutrient intake and metabolism, many enzymes that regulate epigenetic modifications are potentially susceptible to changes in the levels of ATP, AcCoA, SAM, NAD, FAD, and α-KG [129, 137]. Oncogene-, tumor suppressor- or oncometabolite (i.e., metabolites with potential tumorigenic effect)-mediated alterations in signaling pathways and cellular differentiation may lead to metabolic reprogramming consisting of increased nutrient uptake and biosynthesis, involving the shift of mitochondrial metabolism from a catabolic to a predominant anabolic mode to support high proliferation rates [153]. As a matter of fact, recent evidence shows that mitochondria are not defective in most tumors and that, instead, their metabolism is actively reprogrammed, challenging Warburg’s proposal that damaged mitochondria are at the root of the aerobic glycolysis exhibited by tumors. In place of oxidative metabolism of both glucose and glutamine, some cancers preferentially perform reductive and carboxylating biosynthetic reactions from glutamine carbon [144, 154, 155]. The potential tumorigenic effects of the oncometabolites succinate and fumarate arising from loss-of-function mutations in the TCA cycle enzymes SDH and FH, respectively, are likely due to their increased levels [153]. The tumorigenesis promoting role of elevated succinate and fumarate can be in part due to epigenetic modulation, e.g., fumarate modification of cysteine residues to inhibit a negative regulator of the Nrf2 transcription factor that leads to the up-regulation of antioxidant response genes [156]. Loss-of-function mutations in the cytosolic NADP+-dependent isocitrate dehydrogenase 1 gene (IDH1) or in the mitochondrial IDH2, both found in tumors, not only hamper isocitrate and α-KG interconversion but also acquire a novel reductive activity to convert α-KG to the rare metabolite 2-hydroxyglutarate. The fact that mutations in both IDHs result in 2-hydroxyglutarate generation suggests its potential oncometabolite role since it appears that this metabolite is a primary feature being selected in tumors [153].
Impact of diet on metabolism and the epigenome
Macroautophagy (or autophagy) is a mechanism through which organelles or cytoplasmic molecules are sequestered in autophagosomes (double-membrane vesicles) that subsequently fuse with lysosomes for bulk digestion and recycling of the autophagic cargo [157, 158]. When extracellular nutrients are scarce, cells meet their energetic demand via autophagy that mobilizes endogenous macromolecules, maintaining organelle turnover and avoiding proteotoxic stress, thereby attenuating or precluding age-associated processes while mediating cytoprotection [159]. Consequently, as a result of autophagy impairment, hallmarks of aging and disease such as certain kinds of molecular damage, malfunctioning organelles, defective enzymes, protein aggregates, and DNA mutations, accumulate.
Chronic carbohydrate and lipid excess elevates AcCoA suppressing autophagy thereby accelerating the manifestation of age-associated pathologies [157]. Hyperglycemic conditions mediate chromatin changes that underlie altered gene transcription in vascular and inflammatory cells likely due to the epigenome sensitivity to glycemic levels [160, 161]. AcCoA accumulation increases protein acetylation; in the nucleus, acetylation of lysines (Lys) on DNA-binding proteins neutralizes their positive charge, reducing protein affinity for DNA thus favoring their detachment and stimulating transcription, replication, and cell proliferation. However, the cytosolic accumulation of acetate, an AcCoA precursor, also causes histone hyperacetylation, reducing the autophagic flux by repressing the transcription of the autophagy essential gene ATG7 [162]. Therefore, agents that favor protein deacetylation may extend longevity of model organisms, either by stimulating deacetylases (such as resveratrol) [163] or by inhibiting acetyltransferases (such as spermidine) [157].
Among the cytoprotective effects of autophagy are: (a) buffering of cellular stress in conditions of fluctuating nutrient availability by enhanced provision of substrates for bioenergetic metabolism and anabolic reactions; (b) removal of dysfunctional and harmful organelles, including uncoupled mitochondria; and (c) clearance of aggregate-prone, potentially toxic proteins [157]. Consequently, autophagy-stimulating conditions elicited by nutritional, pharmacologic, and genetic interventions may also contribute to longevity. Mitochondrial dysfunction provoked by lack of autophagy elicits cellular stress, accumulation of protein aggregates and excessive ROS driving perturbations of the intracellular redox environment which result in accumulation of misfolded proteins within the endoplasmic reticulum [164, 165] or protein oxidation of, e.g., the contractile machinery of the heart, negatively affecting its contractile performance [45, 166, 167].
Under carbohydrate and fat limitation (e.g. fasting), AcCoA levels decrease, acetylation decreases, chromatin condenses, and cellular gene expression, replication and proliferation are suppressed [4]. Autophagy is stimulated by AcCoA depletion [168] provoked by decreased levels of intracellular amino acids, ATP, and NADH that in turn elicit inhibition of mTORC1, AMPK stimulation, and SIRT1 activation, respectively [169]. Accordingly, interruption of glycolytic and lypolytic AcCoA generation in mitochondria via inhibition of the mitochondrial pyruvate carrier complex or carnitine palmitoyl transferase 1 (CPT1) can induce cytoprotective autophagy [168].
Together, the evidence presented suggests that dietary and pharmacological manipulations causing a decrease in AcCoA might promote a healthier outcome by stimulating autophagy. Although the broad applicability of this concept remains to be explored, it offers nonetheless a promising research direction worth exploring.
Mitochondria as a hub of the nutritional impact
The direct influence of autophagy upon the turnover of molecular and cellular components, including mitochondria, offers a mechanistic link between diet-life style interventions with health- and lifespan. High autophagic flux is associated with extended life and/or healthspan. The contribution of nutrient oversupply and physical inactivity to overall health and the pathogenesis of cardiovascular disease, metabolic syndrome and insulin resistance have been extensively documented [1, 45, 170–172]. Emerging evidence also indicates that mitochondrial quality control via mitophagy and mitochondrial fusion-fission dynamism and biogenesis are altered by both nutrient excess and physical inactivity. The liver [173] and cardiac and skeletal muscle [174–176] are among the most affected organs.
Sedentarism combined with the dietary abundance of fructose, ethanol, BCAAs and trans-fats have been proposed to underlie the obesity and type 2 diabetes epidemics in the United States [173, 177]. Dietary fat, favored as one of the main culprits of metabolic syndrome, has not changed over the past 30 years in terms of the absolute consumption of total dietary fat in the US; in fact, the percentage of calories ingested from saturated fats has decreased from 40% to 30% [173]. Cardiometabolic risk relates more to the balance of saturated vs. unsaturated fats than to the total amount ingested [178]. Similarities among the abundant aforementioned four dietary foodstuffs are (i) their primary utilization for energy in the liver, (ii) lack of insulin regulation and of mechanisms leading to glycogen storage, and (iii) all the intermediates resulting from their metabolism being delivered directly to the mitochondria (Fig. 4). Their excess provokes the accumulation of metabolic intermediates (e.g., AcCoA) driving epigenomic changes, excessive ROS and de novo lipogenesis, impaired β-oxidation, driving insulin resistance and the downstream comorbidities of the metabolic syndrome [45, 173]. In liver, the insulin-independent degradation of fructose or ethanol results in their conversion to AcCoA, bypassing the pathway leading to storage as glycogen, provoking de novo lipogenesis, inflammation and insulin resistance [173].
The so-called “hedonic pathway of food reward” from the brain limbic areas responds to excessive stimulation by fructose and ethanol intake. The neural circuit from the lateral hypothalamic to the ventral tegmental area of the brain was shown to process reward by reducing compulsive sucrose seeking but not food consumption in hungry mice [179]. An important question is whether excess dietary fat can promote epigenetic regulation in the liver since mitochondria are the main sites of β-oxidation and sources of precursors for lipogenesis and FAs for the body. Recent data links high fat diets with widespread chromatin remodeling likely involving regulatory regions of genes encoding for histone modifying enzymes in mouse liver [62, 180].
Glucose produces a lower hepatic substrate burden compared to fructose or ethanol. Comparatively, after ingestion of 120 kcal in the form of glucose or ethanol, or sucrose (a molecule composed of 50% glucose/50% fructose) 24, 96 or 72 kcal reach the liver, respectively [177]. Thus, in the presence of fructose or ethanol the liver processes much higher levels of substrate as compared to glucose alone. In the case of fructose, after its conversion to fructose-1-P, further degradation results in an overload of the mitochondrial AcCoA pool that, in part, exits to the cytoplasm as citrate where it can initiate lipogenesis [135, 177, 181]. Excessive FA export from the liver increases delivery to the skeletal muscle where, in the absence of energy demand, may promote insulin resistance. Ethanol bypasses glycolysis, being converted, first, to acetaldehyde by alcohol dehydrogenase, and then to acetate by aldehyde dehydrogenase 2. Acetate can generate AcCoA in the mitochondria, but due to the increase in NADH/NAD+ ratio and subsequent inhibition of the TCA cycle and gluconeogenesis provoked by ethanol degradation, it is likely no further processed in the liver and thus exported for further metabolism in other organs [135].
BCAAs are essential amino acids thus provided by the diet, >20% of the dietary protein [182]. Normally used for protein biosynthesis and cell growth, they are diverted away toward energy utilization when in excess [183]. Apart from increasing the cellular pool of AcCoA, dietary excess of BCAAs also provoke their increase in the systemic circulation. In the liver, the level of activity of the branched-chain aminotransferase is relatively low. The increased peripheral delivery of BCAAs may elicit insulin resistance via accumulation of lipid-derived metabolites such as diacylglycerols and ceramides originating from saturation of mitochondrial capacity for β-oxidation due to over accumulation of C3 and C5 acylcarnitines [184].
Impact of lifestyle on the balance of energy supply-demand and the status of mitochondrial function and turnover
Changes in mitochondrial morphology driven by fission-fusion processes reflect energetic-redox adaptation along the mitochondrial life cycle [23, 53]. The changes in mitochondrial morphology through fusion and fission events render more or less connected or separated mitochondria enabling the reorganization of mitochondrial components and the elimination of damaged material [53, 54]. The dynamism of fission-fusion occurs with different kinetics depending on cell type, e.g., faster in β-cells, hepatocytes, slower in cardiomyocytes [22, 53]. Although fusion and fission proteins are abundantly expressed in adult hearts, mitochondrial fission-fusion processes are slow [22], a cycle of fusion-fission roughly estimated to occur within two weeks in adult mice [185, 186]. Notwithstanding, genetic disruption of fission and/or fusion in the heart negatively affects mitochondrial quality control [22, 186].
Healthy mitochondria replicate by symmetrical fission whereas asymmetrical fission represents the initial step of mitophagy, a process that sequesters and eliminates damaged organelles or their components [23, 76]. Fission is a key process in mitochondrial quality control [53]. The involvement of mitochondrial fission in sequestering damaged mitochondria, and of mitochondrial fusion in reintroducing healthy mitochondria, along with biogenesis, are critical for keeping the overall health status of the mitochondrial network. Consequently, turnover requires both fusion events and the segregation of damaged components by fission. In the heart and fibroblasts, comparative studies of mitochondrial fission- and fusion-deficiency showed that Dynamin related protein (Drp)-1-mediated mitochondrial fission is essential to properly target mitochondrial autophagy and to restrain cell necrosis via permeability transition [186].
Mitochondria targeted for mitophagy have a relatively depolarized membrane potential [23], remaining solitary due to reduced likelihood of being involved in fusion events, as OPA1 fusion proteins are either cleaved or degraded [54, 187]. These organelles constitute a transient (1 to 3h) pool of pre-autophagic mitochondria [54, 76]. The energetic-redox impairment of mitochondrial function can be explained by accumulation of irreversibly damaged mitochondria that cannot be segregated due to failure of the fission process [23]. The non-removal of damaged mitochondria also interrupts normal mitophagy.
For the purpose of this review, we recognize the importance of the cellular sensitive response to the nutritional environment [188] exhibited by the overall dynamism of mitochondrial fusion-fission and autophagy-biogenesis processes [53, 58]. While nutrient excess is associated with a pro-fission phenotype of mitochondria leading to a more fragmented network, mitochondrial elongation prevails under nutrient depletion. Nutrient starvation inhibits mitochondrial fission contributing to a pro-fusion phenotype resulting in elongated mitochondria that maintain function and are spared from autophagic degradation ([189, 190]; reviewed in [41]). Nutrient excess can also impair autophagic flux by disruption of lysosomal function, which is required for autophagic degradation [191]. Short and small mitochondria form within minutes in response to acute hyperglycemia or metabolic stressful conditions leading to ROS elevation [192]. Chronic hyperglycemia results in more severe fragmentation of mitochondria and apoptotic cell death (reviewed in [41]). Mitochondrial fragmentation as a short-term protective response to nutrient overload has deleterious consequences on mitochondrial quality control when nutrient excess becomes chronic [76, 193]. A tight regulation of mitochondria number and quality is essential for cellular fitness and survival [194]. Mitochondria are main players in redox regulation of autophagy; removal of damaged mitochondria and oxidized proteins, in most cases, supports cell survival [195].
Evidence of decreased mitochondrial biogenesis has been reported in situations involving both the loss of mitochondrial fission and fusion [186]. Apparently, an intact fission-fusion cycle is important for normal biogenesis [196, 197]. Still unclear is whether de novo complete mitochondrial biogenesis or specific replacement of damaged mitochondrial parts happens [54]. On the other hand, mitochondrial biogenesis requires a sophisticated transcriptional program capable of responding to the energetic demands of the cell by coordinating expression of both nuclear and mitochondrial encoded genes [59].
The ensemble of the aforementioned evidence shows that alterations of mitochondrial quality control due to altered fusion-fission dynamism and mitophagy can play critical roles in the cumulative mitochondrial damage. Degradation of mitochondrial quality results in the overall energetic-redox impairment of the mitochondrial network, as shown by mitochondrial populations isolated from organs affected by metabolic disease [45, 194, 198] or by ablation of fission proteins [41, 186]. Importantly, the mitochondrial reserve capacity – operationally defined as the maximal respiratory capacity after subtracting baseline respiration – can be used as an index of mitochondrial health [194, 198, 199]. The closer a cell is operating to its bioenergetic limit the more stringent its response would be toward stressful conditions [194]. Additionally, the bioenergetic reserve capacity can be sensed by the mitophagic machinery [200] and has been implicated in immunity, cell proliferation, and neurodegeneration (see [194] for a review). We also infer that a healthy and nutritionally well-adjusted lifestyle should favor balanced energy supply and demand in concert with mitochondrial fission-fusion and mitophagy-biogenesis processes leading to proper organelle turnover and energy-redox function of the mitochondrial population [53, 201]. The balanced interplay between the mentioned processes determines not only the overall functionality of the mitochondrial network but also the integrity of the mitochondrial genome and, ultimately, health-span.
Eating patterns, exercise and mitochondrial health and health-span
We now know that calorie intake is linked to health and lifespan. Calorie restriction (e.g., at the magnitude of 40% reduction in caloric intake) is considered to be the most powerful non-genetic intervention to delay the onset and progression of most chronic diseases and extend lifespan [202, 203]. The increase in lifespan and slower rate of aging elicited by calorie restriction has been reported in multiple organisms across a wide range of species from yeast to non-human primates [137, 204–206], although the data on non-human primates is mixed [204, 206] and the apparent contradictions await reconciliation.
The incidence of cardiovascular disease (CVD), cancer, and the degree of longevity, depends on the amount of calories ingested and diet quality (e.g., the ratio saturated/insaturated FAs, the presence of antioxidants). In the case of mitochondria, and as described herein, the mechanisms that participate in the deterioration of their function and in health span have been dissected. The biochemical impact of diet on cell and organ function underscores the crucial role played by eating patterns on general health. All main substrates derived from the dietary intake (e.g., lipids, amino acids, glucose/pyruvate) converge on mitochondrial metabolism (Fig. 4; for a specific discussion on the essential BCAAs see section: Mitochondria as a hub of the nutritional impact) influencing the metabolism-epigenome-genome axis (Fig. 3). Rising levels of generational exposure of vulnerable populations to over- or under-nutrition combined with imbalanced eating patterns and/or sedentarism, significantly impact the epigenome throughout development and adulthood [125]. This view is supported by emerging evidence showing that stable epigenetic memories are established by developmental and adult nutritional milieus, and are strongly associated with disease predisposition and progression (reviewed in [62]). For example, influenced by maternal diet, cholesterol, and stress, atherosclerosis may begin before birth, with accumulating lipids, monocytes, and local oxidative damage, while leading later to progression of arterial degeneration [205, 207, 208]. More recently, a test for permanent phenotypic consequences in the offspring through epigenetic mechanisms related to the establishment of DNA methylation during the periconceptional period (i.e., from before conception to early pregnancy) was performed on rural Gambian women subjected to pronounced naturally occurring seasonal differences in their diet [209]. It was found that DNA methylation exhibited significant variations in response to mothers’ periconceptional dietary intakes and plasma concentrations of key methyl-donors (e.g., methionine, choline, betaine) and related cofactors (e.g., folate) and intermediary metabolites (e.g., S-adenosylmethionine, SAM). DNA methyltransferases utilize SAM, a product of 1-carbon cycle metabolism that involves zinc, methionine, and vitamin B family members including folate, choline, and vitamin B12 [137, 210]. Mitochondrial ATP and folate syntheses modulate SAM generation [80, 211]. Folate cycle reactions are duplicated in cytosol and mitochondria, linked through exchange of serine and glycine which are interconverted by the mitochondrial and cytosolic serine hydroxymethyltransferase through methylenetetrahydrofolate [80]. High fetal demands for folate and choline are needed for neural tube closure and normal development [210]. The level of several of these maternal biomarkers predicted increased/decreased methylation at metastable epialleles (MEs, i.e., DNA loci whose epigenetic state is independent of cell differentiation and which exhibit dynamic stochasticity [210]) in DNA extracted from lymphocytes and hair follicles in infants postnatally [209]. Although this data represent the first-in-human confirmation that the maternal blood biomarker status of substrates and cofactors required for methyl-donor pathways, measured around the time of conception, can predict methylation patterns in offspring, the consequences of these variations in methylation are not yet known. However, they are known in mice where maternal dietary changes affecting methyl-donor availability alter epigenetic development at MEs causing permanent phenotypic variation in the isogenic offspring of yellow agouti mice [212]. The change in the offspring’s coat color in these mice can be easily visualized, ranging from yellow through increasing amounts of agouti (brown) mottling to completely agouti, depending on the extent of DNA methylation in response to moderate diet supplementation of dams with folate, vitamin B12, choline and betaine [212]. Also known in animals is that short-term maternal voluntary exercise prior to and during healthy pregnancy and nursing in mice, or exercise during pregnancy in rats, can enhance long-term glucose homeostasis [213] or insulin sensitivity and improved glucose homeostasis [214] in their offspring, probably decreasing the susceptibility to insulin resistant-related diseases such as T2DM.
In the human body, the heart handles several-fold higher amounts of O2 on a specific basis than other organs such as skeletal muscle, brain and the lung. Thus, the heart faces the highest degree of exposure to possible oxidative damage [45]. In heart function, mitochondria harness essential redox and phosphorylation energy while generating ROS. In doing so, mitochondria have to accomplish reliable energy supply, while keeping ROS within physiological limits compatible with signaling [9, 45]. Potentially, the higher vulnerability of the heart to redox damage accounts for the prevalence of morbidity and mortality from cardiovascular complications in diverse populations responding to eating habits [215], metabolic syndrome [175, 176, 216], physical activity and exercise training [174, 217]. In diabetic patients, CVD (including both arterial and myocardial dysfunction) is the leading cause of mortality. Extensive and compelling evidence shows that exercise training helps prevent and treat obesity and T2DM. Exercise amends systemic metabolic changes associated with diabetes/obesity such as insulin resistance, glycemic control, plasma lipid levels, and inflammatory status [217], and beneficial cardiac effects after exercise training [174].
The concept that the intrinsic aerobic capacity of an organism can play a role in determining longevity is the foundation of the observation that low aerobic exercise capacity is a powerful predictor of premature morbidity and mortality for healthy adults as well as those with CVD [218]. The maximal metabolic rate (MMR), expressed as maximal respiratory rate capacity (VO2max), represents the functional parameter defining the limiting flow rate through all steps of the respiratory system, from lung to muscle mitochondria. Consequently, MMR underscores the crucial role played by mitochondria. Moreover, mitochondrial respiration scales as a function of body size both under rest and maximal metabolic performance. The aerobic capacity of locomotor muscle scales with MMR, and the latter scales with the surface area of inner mitochondrial membranes, where OxPhos takes place [219]. Interestingly, the active mitochondrial surface, i.e., the inner membrane, shows invariant density both with respect to body mass and aerobic capacity such that for each ml of O2 consumed per minute at VO2max the muscle contains 7m2 of active membrane [219, 220].
Relevant to CVD are recent evaluations of long-term studies on diet and eating patterns. Under the main hypothesis that prevalence, incidence and mortality from coronary heart disease (CHD) and other CVDs in different populations can be explained by eating habits, the longitudinal Seven Countries Study of Cardiovascular Diseases was started at the end of the 1950s [215]. Sixteen cohorts for a total of 12,763 middle aged men (40–59 years old) were enrolled, one in the USA, two in Finland, one in the Netherlands, three in Italy, two in Croatia, three in Serbia, two in Greece and two in Japan [215]. This international study allowed comparisons both among cohorts and individuals of the different cohorts that represented different cultures with respect to their agriculture and traditional eating patterns. Findings from these studies, produced and published over the last 50 years, indicate the protective role of dietary habits characterized by a predominance of plant foods and fish over animal foods and sugar that were typical of the Mediterranean areas. This conclusion holds valid under different techniques of dietary measurements and analyses based on ecological or individual approach, the use of all cohorts or subgroups of them, different lengths of follow up and end points [215]. Mediterranean diet is defined as the eating habits that were typical of some populations on the Mediterranean shores in the mid-20th century. The virtue of these eating habits was their documented association with low incidence, prevalence and mortality rates from CHD, other CVDs, and low all-cause mortality including cancer.
Dietary patterns rather than single nutrients or food groups or their combinations, were utilized to better define healthy eating habits. The factor score [221] and the Mediterranean Adequacy Index (MAI) [222] as indicators of intake food patterns gave similar results. For example, the MAI – calculated from the ratio of the sum of the percentage of total energy from ‘typical’ Mediterranean foods (bread, cereals, legumes, potatoes, vegetables, fruit, fish, wine and vegetable oils) over the sum of total energy from non-typical Mediterranean foods (milk, cheese, meat, eggs, animal fat, margarine, pastries, sugar and sweet beverages) – was high in the Mediterranean cohorts and Japan (usually ≥ 4), and very low in the US, Finland and the Netherlands (< 1). Applied to the 16 cohorts of the Seven Countries Study, the MAI index was inversely correlated (r= −0.84) to 25-year CHD death rates [222], confirming findings obtained with factor analysis [221] as well as other health indicators [215]. Similar conclusions were reached when other end points such as cancer were evaluated suggesting that dietary habits can help prevent other maladies than CVD or CHD [215].
We have seen that nutrient intake shapes metabolism, altering the metabolic-epigenomic-genomic axis which results in fluxome modifications (section, The metabolism-epigenome-genome axis and the fluxome). On the one hand, the bidirectional influence between mitochondrial function and the epigenome is mediated by energy and redox intermediates which levels depend upon substrate availability and demand (section, Mitochondrial function and the epigenome). On the other hand, age-associated pathologies (e.g., CVD or CHD) associated with metabolic disorders involving chronic carbohydrates and lipids excess affect mitochondrial turnover (see sections, Impact of diet on metabolism and the epigenome, and Mitochondria as a hub of the nutritional impact) determining aerobic capacity, healthspan, and, likely, longevity (see above). In this regard, it came as a surprise that the presumed absence of modern triggers of atherosclerosis such as sedentarism and calorie excess did not preclude the presence of arterial disease in our human ancestors. The incidence of atherosclerosis in preindustrial, including preagricultural hunter-gatherer, human populations, across a wide span of human history and eating patterns, suggest that the occurrence of arterial disease per se was common and, probably, not characteristic of any specific diet or lifestyle [223]. As a matter of fact, evidence of arterial disease manifestation in the form of atherosclerosis and calcification of large arteries (carotid, aorta, iliac) has been reported for Egyptian mummies from 3,500 years ago, earlier by [224, 225] and more recently confirmed and extended using X-ray computed tomography by [223, 226]. One of the oldest cases of arterial disease described, showing calcification of both carotid arteries and portions of aorta and iliac artery, corresponds to the Tyrolean iceman from 5,300 years ago, found in present Italy and who died from traumatic wounding and hemorrage at about age 45 [205, 227]. Although, taken together, this evidence does not indicate a high prevalence of arterial disease or its significant contribution to mortality of humans from ancient populations in Egypt, China, Alaskan Inuit, Peru, amongst others [223, 226], it can be argued that advanced atherosclerosis is not an exclusive modern condition [223, 226]. This leads to the question of why should our ancestors have developed such circulatory lesions in the likelihood of absent modern triggers of atherosclerosis such as smoking, sedentarism, hypertension, calorie excess, in their daily living? It has been proposed that infection and inflammation may have played an important role. High levels of chronic infection leading to inflammatory load could have promoted cardiovascular lesion development which might have become fatal for many with the passage of time [228]. We now know that inflammation participates in atherosclerosis from its inception and development to its ultimate endpoint, thrombotic complications [229]. The link of inflammation with atherosclerosis increases with advanced age, although not as an inevitable component since lifestyle, diet, and appropriate medication can help prevent or attenuate inflammatory processes [229].
Concluding remarks
The bulk of the scientific evidence available clearly supports a direct link between eating habits, physical activity with human healthspan and the quality of aging, and with the incidence as well as management of metabolic disorder, and major neurodegenerative, cancer, and cardiovascular diseases. Successful aging understood as “adding life to the years as well as years to life” involves extrinsic factors such as diet, exercise, personal habits and psychosocial aspects that largely explain non-disease aging with respect to physiological and cognitive impairments [55]. The recent exciting advances surveyed herein show that this knowledge is underlain by a major and complex cell biology phenomenon dubbed the mitochondrial quality control axis [76], comprising mitochondrial turnover through biogenesis, fusion-fission dynamism, and mitophagy. Being a convergent path of substrate degradation, recycling and signaling, mitochondrial healthy function, as revealed by aerobic capacity, represents a critical probe and index of health- and lifespan.
From an evolutionary perspective, the metabolic-epigenome-genome axis sets the stage of human healthspan. In this axis, the epigenome embodies the inheritable response to environmental factors represented by the metabolic response to, e.g., disease, nutrition, lifestyle, ultimately translated into the nuclear and mitochondrial genomes. The resulting gene expression-mediated changes of the metabolome-fluxome will feed-back into the epigenome signaling pathways thus closing the loop of healthspan that may be virtuous or vicious depending upon the delay in the onset of diseases related to genetic origin/predisposition and our nutritional, lifestyle and other factors.
Interpretatively, the index of aging relatedness (π) in the mixture model of Levy and Levin represents the proportion of the intrinsic mortality or disease incidence experience of the population. An increasing π would reflect a positive development in population health, mainly achieved through the prevention of premature deaths and delayed disease onset, the latter as a result of late age-specific genetic effects. In the context of a public health effort aimed at increasing human healthspan and a hopeful picture of aging for the 21st century, π is a measure of “living life to the fullest or to the limits established by the evolutionary process”.
Acknowledgments
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Abbreviations
- 2-HG
2-hydroxyglutarate
- 5-mC
5-methylcytosine
- 5-meOHC
5-hydroxymethylcytosine
- α-KG
alpha-ketoglutarate
- AlkB
alkane hydroxylase
- AcCoA
acetyl CoA
- ACSS
acyl-CoA synthase short-chain
- AMPK
AMP-activated protein kinase
- ACL
ATP citrate lyase
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- ATGL
adipose triglyceride lipase
- BCCA
branched chain amino acid
- BCKDH
branched-chain alpha-ketoacid dehydrogenase complex
- β-HB
Beta-hydroxybutyrate
- CHD
coronary heart disease
- MAI
Mediterranean Adequacy Index
- CPT1
carnitine palmitoyl transferase 1
- CpG
cytosine-guanidine dinucleotide
- CR
caloric restriction
- CVD
cardiovascular disease
- Drp1
Dynamin related protein-1
- EREF
Evolutionarily Recent Environmental Factors
- FAD
Flavin adenine dinucleotide
- FA
fatty acid
- FH
fumarate hydratase
- FMN
Flavin mononucleotide
- FUM
fumarate
- GSH
glutathione
- GSSG
glutathione disulfide
- H2O2
hydrogen peroxide
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HMT
histone methyltransferase
- IDH
isocitrate dehydrogenase
- jmjC
jumonji-domain containing
- LOGE
Late Onset Genetic Effects
- ME
metastable epiallele
- mTOR
mammalian target of rapamycin
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- NAD+
nicotinamide adenine dinucleotide
- O2.−
superoxide anion
- OxPhos
oxidative phosphorylation
- PDH
pyruvate dehydrogenase
- PGC-1α
peroxisome proliferator-activated receptor
- PMT
posttranslational modifications
- R-2HG
R-enantiomer of 2-hydroxyglutarate
- ROS
reactive oxygen species
- SAH
S-adenosyl homocysteine
- SAM
S-adenosyl methionine
- SDH
succinate dehydrogenase
- SIRT
sirtuin
- Succ
succinate
- Succ
succinate
- SUMO
Small Ubiquitin-related Modifier
- T2DM
type 2 diabetes mellitus
- TCA
tricarboxylic acid cycle
- TET
ten-eleven translocation family of dioxygenases
- VO2max
maximal respiratory rate capacity
Footnotes
All Authors declare no conflict of interest.
Author contributions
M.A.A. researched the literature and wrote the manuscript. S.C., M.J., S.J.S. researched the literature and edited the manuscript.
References
- 1.Aon MA, Bhatt N, Cortassa S. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014;5:1–13. doi: 10.3389/fphys.2014.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finkel T. Radical medicine: treating ageing to cure disease. Nat Rev Mol Cell Biol. 2005;6:971–976. doi: 10.1038/nrm1763. [DOI] [PubMed] [Google Scholar]
- 3.Kembro JM, Cortassa S, Aon MA. In: Mitochondrial ROS and arrhythmias In Systems Biology of free radicals and anti-oxidants. Laher I, editor. Springer-Verlag; 2014. [Google Scholar]
- 4.Wallace DC, Fan W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31. doi: 10.1016/j.mito.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stowe DF, Camara AK. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxid Redox Signal. 2009;11:1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 2003;552:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: A unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–877. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortassa S, O’Rourke B, Aon MA. Redox-optimized ROS balance and the relationship between mitochondrial respiration and ROS. Biochim Biophys Acta. 2014;1837:287–295. doi: 10.1016/j.bbabio.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Autreaux B, Toledano MB. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol. 2007;8:813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 11.Pourova J, Kottova M, Voprsalova M, Pour M. Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiol (Oxf) 2010;198:15–35. doi: 10.1111/j.1748-1716.2009.02039.x. [DOI] [PubMed] [Google Scholar]
- 12.Aon MA. From isolated to networked: a paradigmatic shift in mitochondrial physiology. Frontiers in Physiology. 2010;1 doi: 10.3389/fphys.2010.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aon MA, Cortassa S, O’Rourke B. On the network properties of mitochondria. Wiley-VCH; 2007. [Google Scholar]
- 14.Weiss JN, Yang L, Qu Z. Systems biology approaches to metabolic and cardiovascular disorders: network perspectives of cardiovascular metabolism. J Lipid Res. 2006;47:2355–2366. doi: 10.1194/jlr.R600023-JLR200. [DOI] [PubMed] [Google Scholar]
- 15.Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44744. doi: 10.1074/jbc.M302673200. [DOI] [PubMed] [Google Scholar]
- 16.Breckwoldt MO, Armoundas AA, Aon MA, Bendszus M, O’Rourke B, Schwarzlander M, Dick TP, Kurz FT. Mitochondrial redox and pH signaling occurs in axonal and synaptic organelle clusters. Sci Rep. 2016;6:23251. doi: 10.1038/srep23251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurz FT, Aon MA, O’Rourke B, Armoundas AA. Spatio-temporal oscillations of individual mitochondria in cardiac myocytes reveal modulation of synchronized mitochondrial clusters. Proc Natl Acad Sci U S A. 2010;107:14315–14320. doi: 10.1073/pnas.1007562107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A Reaction-Diffusion Model of ROS-Induced ROS Release in a Mitochondrial Network. PLoS computational biology. 2010;6:e1000657. doi: 10.1371/journal.pcbi.1000657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eisner V, Lenaers G, Hajnoczky G. Mitochondrial fusion is frequent in skeletal muscle and supports excitation-contraction coupling. J Cell Biol. 2014;205:179–195. doi: 10.1083/jcb.201312066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato A, Nakada K, Hayashi J. Mitochondrial dynamics and aging: Mitochondrial interaction preventing individuals from expression of respiratory deficiency caused by mutant mtDNA. Biochim Biophys Acta. 2006;1763:473–481. doi: 10.1016/j.bbamcr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Song M, Dorn GW., 2nd Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab. 2015;21:195–205. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008a;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picard M, McManus MJ, Csordas G, Varnai P, Dorn GW, 2nd, Williams D, Hajnoczky G, Wallace DC. Trans-mitochondrial coordination of cristae at regulated membrane junctions. Nat Commun. 2015;6:6259. doi: 10.1038/ncomms7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci U S A. 2013;110:2846–2851. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurz FT, Aon MA, O’Rourke B, Armoundas AA. Cardiac mitochondria exhibit dynamic functional clustering. Front Physiol. 2014;5:329. doi: 10.3389/fphys.2014.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurz FT, Derungs T, Aon MA, O’Rourke B, Armoundas AA. Mitochondrial networks in cardiac myocytes reveal dynamic coupling behavior. Biophys J. 2015;108:1922–1933. doi: 10.1016/j.bpj.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lloyd D, Williams CF. New tunes from the heart. Biophys J. 2015;108:1841–1842. doi: 10.1016/j.bpj.2015.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, Nichols CE, Long DM, Olfert IM, Jagannathan R, Hollander JM. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol. 2014;307:H54–65. doi: 10.1152/ajpheart.00845.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–540. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol. 2009;296:H359–369. doi: 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Breckwoldt MO, Pfister FM, Bradley PM, Marinkovic P, Williams PR, Brill MS, Plomer B, Schmalz A, St Clair DK, Naumann R, Griesbeck O, Schwarzlander M, Godinho L, Bareyre FM, Dick TP, Kerschensteiner M, Misgeld T. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat Med. 2014;20:555–560. doi: 10.1038/nm.3520. [DOI] [PubMed] [Google Scholar]
- 33.Picard M. Mitochondrial synapses: intracellular communication and signal integration. Trends Neurosci. 2015;38:468–474. doi: 10.1016/j.tins.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Zhou L, Cortassa S, Wei AC, Aon MA, Winslow RL, O’Rourke B. Modeling cardiac action potential shortening driven by oxidative stress-induced mitochondrial oscillations in guinea pig cardiomyocytes. Biophys J. 2009;97:1843–1852. doi: 10.1016/j.bpj.2009.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Invest. 2005;115:3527–3535. doi: 10.1172/JCI25371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brown DA, Aon MA, Frasier CR, Sloan RC, Maloney AH, Anderson EJ, O’Rourke B. Cardiac arrhythmias induced by glutathione oxidation can be inhibited by preventing mitochondrial depolarization. J Mol Cell Cardiol. 2010;48:673–679. doi: 10.1016/j.yjmcc.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solhjoo S, O’Rourke B. Mitochondrial instability during regional ischemia-reperfusion underlies arrhythmias in monolayers of cardiomyocytes. Journal of Molecular and Cellular Cardiology. 2015;78:90–99. doi: 10.1016/j.yjmcc.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, Trayanova N, O’Rourke B. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circ Arrhythm Electrophysiol. 2014;7:143–151. doi: 10.1161/CIRCEP.113.000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang KC, Kyle JW, Makielski JC, Dudley SC., Jr Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. 2015;116:1937–1955. doi: 10.1161/CIRCRESAHA.116.304691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aon MA, Cortassa S. Mitochondrial network energetics in the heart. Wiley Interdiscip Rev Syst Biol Med. 2012;4:599–613. doi: 10.1002/wsbm.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galloway CA, Yoon Y. Mitochondrial dynamics in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1545–1562. doi: 10.1089/ars.2015.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aon MA, Cortassa S, O’Rourke B. Percolation and criticality in a mitochondrial network. Proc Natl Acad Sci U S A. 2004;101:4447–4452. doi: 10.1073/pnas.0307156101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin F, Cadenas E. Mitochondria: the cellular hub of the dynamic coordinated network. Antioxid Redox Signal. 2015;22:961–964. doi: 10.1089/ars.2015.6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aon MA, Tocchetti CG, Bhatt N, Paolocci N, Cortassa S. Protective mechanisms of mitochondria and heart function in diabetes. Antioxid Redox Signal. 2015;22:1563–1586. doi: 10.1089/ars.2014.6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kembro JM, Cortassa S, Aon MA. Complex oscillatory redox dynamics with signaling potential at the edge between normal and pathological mitochondrial function. Front Physiol. 2014;5:257. doi: 10.3389/fphys.2014.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stanley BA, Sivakumaran V, Shi S, McDonald I, Lloyd D, Watson WH, Aon MA, Paolocci N. Thioredoxin reductase-2 is essential for keeping low levels of H(2)O(2) emission from isolated heart mitochondria. J Biol Chem. 2011;286:33669–33677. doi: 10.1074/jbc.M111.284612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kembro JM, Aon MA, Winslow RL, O’Rourke B, Cortassa S. Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys J. 2013;104:332–343. doi: 10.1016/j.bpj.2012.11.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 2010;584:2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubli DA, Gustafsson AB. Unbreak my heart: targeting mitochondrial autophagy in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1527–1544. doi: 10.1089/ars.2015.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shirihai OS, Song M, Dorn GW., 2nd How mitochondrial dynamism orchestrates mitophagy. Circ Res. 2015;116:1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levy G, Levin B. The biostatistics of aging: from Gompertzian mortality to an index of aging relatedness. John Wiley & Sons, Inc; Hoboken, New Jersey: 2014. [Google Scholar]
- 56.Finkel T. The metabolic regulation of aging. Nat Med. 2015;21:1416–1423. doi: 10.1038/nm.3998. [DOI] [PubMed] [Google Scholar]
- 57.Riera CE, Dillin A. Tipping the metabolic scales towards increased longevity in mammals. Nat Cell Biol. 2015;17:196–203. doi: 10.1038/ncb3107. [DOI] [PubMed] [Google Scholar]
- 58.Picard M, Turnbull DM. Linking the metabolic state and mitochondrial DNA in chronic disease, health, and aging. Diabetes. 2013;62:672–678. doi: 10.2337/db12-1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene. 2002;286:81–89. doi: 10.1016/s0378-1119(01)00809-5. [DOI] [PubMed] [Google Scholar]
- 60.Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15:551–589. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. 2014;33:1198–1211. doi: 10.15252/embj.201488290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keating ST, El-Osta A. Epigenetics and metabolism. Circ Res. 2015;116:715–736. doi: 10.1161/CIRCRESAHA.116.303936. [DOI] [PubMed] [Google Scholar]
- 63.Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW, 3rd, Kang L, Rabinovitch PS, Szeto HH, Houmard JA, Cortright RN, Wasserman DH, Neufer PD. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009b;119:573–581. doi: 10.1172/JCI37048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 65.Bugger H, Abel ED. Molecular mechanisms for myocardial mitochondrial dysfunction in the metabolic syndrome. Clin Sci (Lond) 2008;114:195–210. doi: 10.1042/CS20070166. [DOI] [PubMed] [Google Scholar]
- 66.Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O’Rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, Aon MA. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes. 2012;61:3094–3105. doi: 10.2337/db12-0072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Physiol. 1996;271:H192–202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 68.Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288. doi: 10.1161/CIRCRESAHA.108.175919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nickel A, Loffler J, Maack C. Myocardial energetics in heart failure. Basic Res Cardiol. 2013;108:358. doi: 10.1007/s00395-013-0358-9. [DOI] [PubMed] [Google Scholar]
- 70.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 71.Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta. 2009;1787:1402–1415. doi: 10.1016/j.bbabio.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 72.Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res. 2005;66:233–244. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 73.Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–671. doi: 10.1007/s00125-014-3171-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009a;54:1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bhatt NM, Aon MA, Tocchetti CG, Shen X, Dey S, Ramirez-Correa G, O’Rourke B, Gao WD, Cortassa S. Restoring redox balance enhances contractility in heart trabeculae from type 2 diabetic rats exposed to high glucose. Am J Physiol Heart Circ Physiol. 2015;308:H291–302. doi: 10.1152/ajpheart.00378.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008b;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Picard M, Zhang J, Hancock S, Derbeneva O, Golhar R, Golik P, O’Hearn S, Levy S, Potluri P, Lvova M, Davila A, Lin CS, Perin JC, Rappaport EF, Hakonarson H, Trounce IA, Procaccio V, Wallace DC. Progressive increase in mtDNA 3243A>G heteroplasmy causes abrupt transcriptional reprogramming. Proc Natl Acad Sci U S A. 2014;111:E4033–4042. doi: 10.1073/pnas.1414028111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martinou JC, Youle RJ. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev Cell. 2011;21:92–101. doi: 10.1016/j.devcel.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. 2013;110:25–34. doi: 10.1016/j.ymgme.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 81.Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev. 2009;23:1714–1736. doi: 10.1101/gad.1784909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sato A, Endo H, Umetsu K, Sone H, Yanagisawa Y, Saigusa A, Aita S, Kagawa Y. Polymorphism, heteroplasmy, mitochondrial fusion and diabetes. Biosci Rep. 2003;23:313–337. doi: 10.1023/b:bire.0000019189.35983.b9. [DOI] [PubMed] [Google Scholar]
- 83.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci U S A. 2014;111:10654–10659. doi: 10.1073/pnas.1403521111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Keogh M, Chinnery PF. Hereditary mtDNA heteroplasmy: a baseline for aging? Cell Metab. 2013;18:463–464. doi: 10.1016/j.cmet.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 86.Rose G, Passarino G, Scornaienchi V, Romeo G, Dato S, Bellizzi D, Mari V, Feraco E, Maletta R, Bruni A, Franceschi C, De Benedictis G. The mitochondrial DNA control region shows genetically correlated levels of heteroplasmy in leukocytes of centenarians and their offspring. BMC Genomics. 2007;8:293. doi: 10.1186/1471-2164-8-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Giuliani C, Barbieri C, Li M, Bucci L, Monti D, Passarino G, Luiselli D, Franceschi C, Stoneking M, Garagnani P. Transmission from centenarians to their offspring of mtDNA heteroplasmy revealed by ultra-deep sequencing. Aging (Albany NY) 2014;6:454–467. doi: 10.18632/aging.100661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ross JM, Stewart JB, Hagstrom E, Brene S, Mourier A, Coppotelli G, Freyer C, Lagouge M, Hoffer BJ, Olson L, Larsson NG. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501:412–415. doi: 10.1038/nature12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2007;76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955. [DOI] [PubMed] [Google Scholar]
- 91.Wallace DC. Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen. 2010;51:440–450. doi: 10.1002/em.20586. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu I, Yoshida Y, Suda M, Minamino T. DNA damage response and metabolic disease. Cell Metab. 2014;20:967–977. doi: 10.1016/j.cmet.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 93.Muller M, Lu K, Reichert AS. Mitophagy and mitochondrial dynamics in Saccharomyces cerevisiae. Biochim Biophys Acta. 2015 doi: 10.1016/j.bbamcr.2015.02.024. [DOI] [PubMed] [Google Scholar]
- 94.Al Rawi S, Louvet-Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science. 2011;334:1144–1147. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 95.Birky CW., Jr Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc Natl Acad Sci U S A. 1995;92:11331–11338. doi: 10.1073/pnas.92.25.11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kucej M, Kucejova B, Subramanian R, Chen XJ, Butow RA. Mitochondrial nucleoids undergo remodeling in response to metabolic cues. J Cell Sci. 2008;121:1861–1868. doi: 10.1242/jcs.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 99.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 100.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 101.Vurusaner B, Poli G, Basaga H. Tumor suppressor genes and ROS: complex networks of interactions. Free Radic Biol Med. 2012;52:7–18. doi: 10.1016/j.freeradbiomed.2011.09.035. [DOI] [PubMed] [Google Scholar]
- 102.Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol. 2015;33:55–66. doi: 10.1016/j.ceb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 103.Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal. 2013;25:50–65. doi: 10.1016/j.cellsig.2012.09.020. [DOI] [PubMed] [Google Scholar]
- 104.Melnik BC. Leucine signaling in the pathogenesis of type 2 diabetes and obesity. World J Diabetes. 2012;3:38–53. doi: 10.4239/wjd.v3.i3.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Poels J, Spasic MR, Callaerts P, Norga KK. Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. Bioessays. 2009;31:944–952. doi: 10.1002/bies.200900003. [DOI] [PubMed] [Google Scholar]
- 106.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123:2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Aon MA. Complex systems biology of networks: The riddle and the challenge. In: Aon MA, Saks V, Schlattner U, editors. Systems Biology of Metabolic and Signaling Networks. Energy, Mass and Information transfer. Springer-Verlag Berlin Heidelberg; Heidelberg, New York, Dordrecht, London: 2013. pp. 19–35. [Google Scholar]
- 108.Cascante M, Marin S. Metabolomics and fluxomics approaches. Essays in biochemistry. 2008;45:67–81. doi: 10.1042/BSE0450067. [DOI] [PubMed] [Google Scholar]
- 109.Cortassa S, Caceres V, Bell LN, O’Rourke B, Paolocci N, Aon MA. From metabolomics to fluxomics: a computational procedure to translate metabolite profiles into metabolic fluxes. Biophys J. 2015;108:163–172. doi: 10.1016/j.bpj.2014.11.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Colberg SR. Diabetic athlete’s handbook. Human Kinetics; Champaign, IL: 2009. [Google Scholar]
- 111.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 112.Kienesberger PC, Pulinilkunnil T, Nagendran J, Dyck JR. Myocardial triacylglycerol metabolism. J Mol Cell Cardiol. 2013;55:101–110. doi: 10.1016/j.yjmcc.2012.06.018. [DOI] [PubMed] [Google Scholar]
- 113.Eisenberg T, Schroeder S, Andryushkova A, Pendl T, Kuttner V, Bhukel A, Marino G, Pietrocola F, Harger A, Zimmermann A, Moustafa T, Sprenger A, Jany E, Buttner S, Carmona-Gutierrez D, Ruckenstuhl C, Ring J, Reichelt W, Schimmel K, Leeb T, Moser C, Schatz S, Kamolz LP, Magnes C, Sinner F, Sedej S, Frohlich KU, Juhasz G, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F. Nucleocytosolic depletion of the energy metabolite acetyl-coenzyme a stimulates autophagy and prolongs lifespan. Cell Metab. 2014;19:431–444. doi: 10.1016/j.cmet.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schroeder S, Pendl T, Zimmermann A, Eisenberg T, Carmona-Gutierrez D, Ruckenstuhl C, Marino G, Pietrocola F, Harger A, Magnes C, Sinner F, Pieber TR, Dengjel J, Sigrist SJ, Kroemer G, Madeo F. Acetyl-coenzyme A: a metabolic master regulator of autophagy and longevity. Autophagy. 2014;10:1335–1337. doi: 10.4161/auto.28919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cortassa S, Aon MA, Iglesias AA, Aon JC, Lloyd D. An Introduction to Metabolic and Cellular Engineering. World Scientific Publishers; Singapore: 2012. [Google Scholar]
- 116.Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Winter G, Kromer JO. Fluxomics – connecting ’omics analysis and phenotypes. Environ Microbiol. 2013;15:1901–1916. doi: 10.1111/1462-2920.12064. [DOI] [PubMed] [Google Scholar]
- 118.Aon MA, Cortassa S. Systems biology of the fluxome. Processes. 2015;3:607–618. [Google Scholar]
- 119.Cortassa S, O’Rourke B, Winslow RL, Aon MA. Control and regulation of mitochondrial energetics in an integrated model of cardiomyocyte function. Biophys J. 2009a;96:2466–2478. doi: 10.1016/j.bpj.2008.12.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cortassa S, O’Rourke B, Winslow RL, Aon MA. Control and regulation of integrated mitochondrial function in metabolic and transport networks. Int J Mol Sci. 2009b;10:1500–1513. doi: 10.3390/ijms10041500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fell DA. Understanding the Control of Metabolism. Portland Press; London: 1996. [Google Scholar]
- 122.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Burdge GC, Lillycrop KA. Fatty acids and epigenetics. Curr Opin Clin Nutr Metab Care. 2014;17:156–161. doi: 10.1097/MCO.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 124.Chiacchiera F, Piunti A, Pasini D. Epigenetic methylations and their connections with metabolism. Cell Mol Life Sci. 2013;70:1495–1508. doi: 10.1007/s00018-013-1293-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lillycrop KA, Burdge GC. Environmental challenge, epigenetic plasticity and the induction of altered phenotypes in mammals. Epigenomics. 2014;6:623–636. doi: 10.2217/epi.14.51. [DOI] [PubMed] [Google Scholar]
- 126.Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guarente L. Calorie restriction and sirtuins revisited. Genes Dev. 2013;27:2072–2085. doi: 10.1101/gad.227439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cosentino C, Mostoslavsky R. Metabolism, longevity and epigenetics. Cell Mol Life Sci. 2013;70:1525–1541. doi: 10.1007/s00018-013-1295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, Rutishauser D, Huang D, Caflisch A, Hottiger MO. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010;38:6350–6362. doi: 10.1093/nar/gkq463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 135.Salway JG. Metabolism at a glance. Blackwell Publishing Ltd; Malden, MA, USA; Oxford, UK; Carlton, Australia: 2004. [Google Scholar]
- 136.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 137.Donohoe DR, Bultman SJ. Metaboloepigenetics: interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. 2012;227:3169–3177. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai K, Nakao M. FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat Commun. 2012;3:758. doi: 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. 2014a;26:1598–1603. doi: 10.1016/j.cellsig.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 141.Salminen A, Kauppinen A, Hiltunen M, Kaarniranta K. Krebs cycle intermediates regulate DNA and histone methylation: epigenetic impact on the aging process. Ageing Res Rev. 2014b;16:45–65. doi: 10.1016/j.arr.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 142.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ward PS, Cross JR, Lu C, Weigert O, Abel-Wahab O, Levine RL, Weinstock DM, Sharp KA, Thompson CB. Identification of additional IDH mutations associated with oncometabolite R(-)-2-hydroxyglutarate production. Oncogene. 2012;31:2491–2498. doi: 10.1038/onc.2011.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Waddington CH. Strategy of the genes. Allen & Unwin; London: 1957. [Google Scholar]
- 146.Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–1350. doi: 10.1001/jama.299.11.1345. [DOI] [PubMed] [Google Scholar]
- 147.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 148.Xie Z, Dai J, Dai L, Tan M, Cheng Z, Wu Y, Boeke JD, Zhao Y. Lysine succinylation and lysine malonylation in histones. Mol Cell Proteomics. 2012;11:100–107. doi: 10.1074/mcp.M111.015875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 150.Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–28. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 151.McKnight SL. On getting there from here. Science. 2010;330:1338–1339. doi: 10.1126/science.1199908. [DOI] [PubMed] [Google Scholar]
- 152.Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 159.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 160.Miao F, Smith DD, Zhang L, Min A, Feng W, Natarajan R. Lymphocytes from patients with type 1 diabetes display a distinct profile of chromatin histone H3 lysine 9 dimethylation: an epigenetic study in diabetes. Diabetes. 2008;57:3189–3198. doi: 10.2337/db08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Okabe J, Orlowski C, Balcerczyk A, Tikellis C, Thomas MC, Cooper ME, El-Osta A. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ Res. 2012;110:1067–1076. doi: 10.1161/CIRCRESAHA.112.266171. [DOI] [PubMed] [Google Scholar]
- 162.Eisenberg T, Schroeder S, Buttner S, Carmona-Gutierrez D, Pendl T, Andryushkova A, Marino G, Pietrocola F, Harger A, Zimmermann A, Magnes C, Sinner F, Sedej S, Pieber TR, Dengjel J, Sigrist S, Kroemer G, Madeo F. A histone point mutation that switches on autophagy. Autophagy. 2014;10:1143–1145. doi: 10.4161/auto.28767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Novelle MG, Wahl D, Dieguez C, Bernier M, de Cabo R. Resveratrol supplementation: Where are we now and where should we go? Ageing Res Rev. 2015;21:1–15. doi: 10.1016/j.arr.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kapoor A, Sanyal AJ. Endoplasmic reticulum stress and the unfolded protein response. Clin Liver Dis. 2009;13:581–590. doi: 10.1016/j.cld.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 165.Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–1106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 167.Christians ES, Benjamin IJ. Proteostasis and REDOX state in the heart. Am J Physiol Heart Circ Physiol. 2012;302:H24–37. doi: 10.1152/ajpheart.00903.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, Schroeder S, Pendl T, Harger A, Niso-Santano M, Zamzami N, Scoazec M, Durand S, Enot DP, Fernandez AF, Martins I, Kepp O, Senovilla L, Bauvy C, Morselli E, Vacchelli E, Bennetzen M, Magnes C, Sinner F, Pieber T, Lopez-Otin C, Maiuri MC, Codogno P, Andersen JS, Hill JA, Madeo F, Kroemer G. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710–725. doi: 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 169.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fontana L, Partridge L. Promoting health and longevity through diet: from model organisms to humans. Cell. 2015;161:106–118. doi: 10.1016/j.cell.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Schwab U, Uusitupa M. Diet heart controversies–Quality of fat matters. Nutr Metab Cardiovasc Dis. 2015;25:617–622. doi: 10.1016/j.numecd.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 172.Widmer RJ, Flammer AJ, Lerman LO, Lerman A. The Mediterranean diet, its components, and cardiovascular disease. Am J Med. 2015;128:229–238. doi: 10.1016/j.amjmed.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bremer AA, Mietus-Snyder M, Lustig RH. Toward a unifying hypothesis of metabolic syndrome. Pediatrics. 2012;129:557–570. doi: 10.1542/peds.2011-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hafstad AD, Boardman N, Aasum E. How exercise may amend metabolic disturbances in diabetic cardiomyopathy. Antioxid Redox Signal. 2015;22:1587–1605. doi: 10.1089/ars.2015.6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Schrauwen-Hinderling VB, Kooi ME, Schrauwen P. Mitochondrial Function and Diabetes: Consequences for Skeletal and Cardiac Muscle Metabolism. Antioxid Redox Signal. 2016;24:39–51. doi: 10.1089/ars.2015.6291. [DOI] [PubMed] [Google Scholar]
- 176.Sung MM, Hamza SM, Dyck JR. Myocardial metabolism in diabetic cardiomyopathy: potential therapeutic targets. Antioxid Redox Signal. 2015;22:1606–1630. doi: 10.1089/ars.2015.6305. [DOI] [PubMed] [Google Scholar]
- 177.Lustig RH. Fructose: metabolic, hedonic, and societal parallels with ethanol. J Am Diet Assoc. 2010;110:1307–1321. doi: 10.1016/j.jada.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 178.Willett WC. The great fat debate: total fat and health. J Am Diet Assoc. 2011;111:660–662. doi: 10.1016/j.jada.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 179.Nieh EH, Matthews GA, Allsop SA, Presbrey KN, Leppla CA, Wichmann R, Neve R, Wildes CP, Tye KM. Decoding neural circuits that control compulsive sucrose seeking. Cell. 2015;160:528–541. doi: 10.1016/j.cell.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Leung A, Parks BW, Du J, Trac C, Setten R, Chen Y, Brown K, Lusis AJ, Natarajan R, Schones DE. Open chromatin profiling in mice livers reveals unique chromatin variations induced by high fat diet. J Biol Chem. 2014;289:23557–23567. doi: 10.1074/jbc.M114.581439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15:536–550. doi: 10.1038/nrm3841. [DOI] [PubMed] [Google Scholar]
- 182.Layman DK, Walker DA. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J Nutr. 2006;136:319S–323S. doi: 10.1093/jn/136.1.319S. [DOI] [PubMed] [Google Scholar]
- 183.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15:606–614. doi: 10.1016/j.cmet.2012.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 185.Chen Y, Liu Y, Dorn GW., 2nd Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011;109:1327–1331. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW., 2nd Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab. 2015;21:273–285. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol. 2010;191:1367–1380. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 189.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci U S A. 2011;108:10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Las G, Serada SB, Wikstrom JD, Twig G, Shirihai OS. Fatty acids suppress autophagic turnover in beta-cells. J Biol Chem. 2011;286:42534–42544. doi: 10.1074/jbc.M111.242412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mouli PK, Twig G, Shirihai OS. Frequency and selectivity of mitochondrial fusion are key to its quality maintenance function. Biophys J. 2009;96:3509–3518. doi: 10.1016/j.bpj.2008.12.3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hill BG, Benavides GA, Lancaster JR, Jr, Ballinger S, Dell’Italia L, Jianhua Z, Darley-Usmar VM. Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem. 2012;393:1485–1512. doi: 10.1515/hsz-2012-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 196.Kim B, Kim JS, Yoon Y, Santiago MC, Brown MD, Park JY. Inhibition of Drp1-dependent mitochondrial division impairs myogenic differentiation. Am J Physiol Regul Integr Comp Physiol. 2013;305:R927–938. doi: 10.1152/ajpregu.00502.2012. [DOI] [PubMed] [Google Scholar]
- 197.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res. 2012;111:1012–1026. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Hill BG, Dranka BP, Zou L, Chatham JC, Darley-Usmar VM. Importance of the bioenergetic reserve capacity in response to cardiomyocyte stress induced by 4-hydroxynonenal. Biochem J. 2009;424:99–107. doi: 10.1042/BJ20090934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Higdon AN, Dranka BP, Hill BG, Oh JY, Johnson MS, Landar A, Darley-Usmar VM. Methods for imaging and detecting modification of proteins by reactive lipid species. Free Radic Biol Med. 2009;47:201–212. doi: 10.1016/j.freeradbiomed.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Aon MA, Foster DB. Diabetic cardiomyopathy and the role of mitochondrial dysfunction: novel insights, mechanisms, and therapeutic strategies. Antioxid Redox Signal. 2015;22:1499–1501. doi: 10.1089/ars.2015.6349. [DOI] [PubMed] [Google Scholar]
- 202.Armanios M, de Cabo R, Mannick J, Partridge L, van Deursen J, Villeda S. Translational strategies in aging and age-related disease. Nat Med. 2015;21:1395–1399. doi: 10.1038/nm.4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Brunner S, Herndler-Brandstetter D, Weinberger B, Grubeck-Loebenstein B. Persistent viral infections and immune aging. Ageing Res Rev. 2011;10:362–369. doi: 10.1016/j.arr.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 204.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–204. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Finch CE. Evolution in health and medicine Sackler colloquium: Evolution of the human lifespan and diseases of aging: roles of infection, inflammation, and nutrition. Proc Natl Acad Sci U S A. 2010;107(Suppl 1):1718–1724. doi: 10.1073/pnas.0909606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, Barnard D, Ward WF, Qi W, Ingram DK, de Cabo R. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature. 2012;489:318–321. doi: 10.1038/nature11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Palinski W, Nicolaides E, Liguori A, Napoli C. Influence of maternal dysmetabolic conditions during pregnancy on cardiovascular disease. J Cardiovasc Transl Res. 2009;2:277–285. doi: 10.1007/s12265-009-9108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mazumder B, Almond D, Park K, Crimmins EM, Finch CE. Lingering prenatal effects of the 1918 influenza pandemic on cardiovascular disease. J Devel Origin Health Disease. 2009;1:1–9. doi: 10.1017/S2040174409990031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Dominguez-Salas P, Moore SE, Baker MS, Bergen AW, Cox SE, Dyer RA, Fulford AJ, Guan Y, Laritsky E, Silver MJ, Swan GE, Zeisel SH, Innis SM, Waterland RA, Prentice AM, Hennig BJ. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat Commun. 2014;5:3746. doi: 10.1038/ncomms4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Dominguez-Salas P, Cox SE, Prentice AM, Hennig BJ, Moore SE. Maternal nutritional status, C(1) metabolism and offspring DNA methylation: a review of current evidence in human subjects. Proc Nutr Soc. 2012;71:154–165. doi: 10.1017/S0029665111003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ghosh S, Singh KK, Sengupta S, Scaria V. Mitoepigenetics: The different shades of grey. Mitochondrion. 2015;25:60–66. doi: 10.1016/j.mito.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 212.Waterland RA, Jirtle RL. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol Cell Biol. 2003;23:5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Carter LG, Lewis KN, Wilkerson DC, Tobia CM, Ngo Tenlep SY, Shridas P, Garcia-Cazarin ML, Wolff G, Andrade FH, Charnigo RJ, Esser KA, Egan JM, de Cabo R, Pearson KJ. Perinatal exercise improves glucose homeostasis in adult offspring. Am J Physiol Endocrinol Metab. 2012;303:E1061–1068. doi: 10.1152/ajpendo.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Carter LG, Qi NR, De Cabo R, Pearson KJ. Maternal exercise improves insulin sensitivity in mature rat offspring. Med Sci Sports Exerc. 2013;45:832–840. doi: 10.1249/MSS.0b013e31827de953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Menotti A, Puddu PE. How the Seven Countries Study contributed to the definition and development of the Mediterranean diet concept: a 50-year journey. Nutr Metab Cardiovasc Dis. 2015;25:245–252. doi: 10.1016/j.numecd.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 216.Schilling JD. The mitochondria in diabetic heart failure: from pathogenesis to therapeutic promise. Antioxid Redox Signal. 2015;22 doi: 10.1089/ars.2015.6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Colberg SR. Exercise and diabetes. A clinician’s guide to prescribing physical activity. American Diabetes Association; Alexandria, Virginia: 2013. [Google Scholar]
- 218.Koch LG, Kemi OJ, Qi N, Leng SX, Bijma P, Gilligan LJ, Wilkinson JE, Wisloff H, Hoydal MA, Rolim N, Abadir PM, van Grevenhof EM, Smith GL, Burant CF, Ellingsen O, Britton SL, Wisloff U. Intrinsic aerobic capacity sets a divide for aging and longevity. Circ Res. 2011;109:1162–1172. doi: 10.1161/CIRCRESAHA.111.253807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Weibel ER, Hoppeler H. Exercise-induced maximal metabolic rate scales with muscle aerobic capacity. J Exp Biol. 2005;208:1635–1644. doi: 10.1242/jeb.01548. [DOI] [PubMed] [Google Scholar]
- 220.Hoppeler H, Weibel ER. Scaling functions to body size: theories and facts. J Exp Biol. 2005;208:1573–1574. doi: 10.1242/jeb.01630. [DOI] [PubMed] [Google Scholar]
- 221.Menotti A, Kromhout D, Blackburn H, Fidanza F, Buzina R, Nissinen A. Food intake patterns and 25-year mortality from coronary heart disease: cross-cultural correlations in the Seven Countries Study. The Seven Countries Study Research Group. Eur J Epidemiol. 1999;15:507–515. doi: 10.1023/a:1007529206050. [DOI] [PubMed] [Google Scholar]
- 222.Fidanza F, Alberti A, Lanti M, Menotti A. Mediterranean Adequacy Index: correlation with 25-year mortality from coronary heart disease in the Seven Countries Study. Nutr Metab Cardiovasc Dis. 2004;14:254–258. doi: 10.1016/s0939-4753(04)80052-8. [DOI] [PubMed] [Google Scholar]
- 223.Thompson RC, Allam AH, Lombardi GP, Wann LS, Sutherland ML, Sutherland JD, Soliman MA, Frohlich B, Mininberg DT, Monge JM, Vallodolid CM, Cox SL, Abd el-Maksoud G, Badr I, Miyamoto MI, el-Halim Nur el-Din A, Narula J, Finch CE, Thomas GS. Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations. Lancet. 2013;381:1211–1222. doi: 10.1016/S0140-6736(13)60598-X. [DOI] [PubMed] [Google Scholar]
- 224.Bruetsch WL. The earliest record of sudden death possibly due to atherosclerotic coronary occlusion. Circulation. 1959;20:438–441. doi: 10.1161/01.cir.20.3.438. [DOI] [PubMed] [Google Scholar]
- 225.Ruffer M. On arterial lesions found in Egyptian mummies. J Pathol Bacteriol. 1911;15:453–462. [Google Scholar]
- 226.Allam AH, Thompson RC, Wann LS, Miyamoto MI, Nur El-Din Ael H, El-Maksoud GA, Al-Tohamy Soliman M, Badr I, El-Rahman Amer HA, Sutherland ML, Sutherland JD, Thomas GS. Atherosclerosis in ancient Egyptian mummies: the Horus study. JACC Cardiovasc Imaging. 2011;4:315–327. doi: 10.1016/j.jcmg.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 227.Murphy WA, Jr, Nedden Dz D, Gostner P, Knapp R, Recheis W, Seidler H. The iceman: discovery and imaging. Radiology. 2003;226:614–629. doi: 10.1148/radiol.2263020338. [DOI] [PubMed] [Google Scholar]
- 228.Heagerty AM. Scanning ancient history for evidence of modern diseases. Lancet. 2013;381:1165–1166. doi: 10.1016/S0140-6736(13)60639-X. [DOI] [PubMed] [Google Scholar]
- 229.Libby P. Inflammation and cardiovascular disease mechanisms. Am J Clin Nutr. 2006;83:456S–460S. doi: 10.1093/ajcn/83.2.456S. [DOI] [PubMed] [Google Scholar]
- 230.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]