

Abstract
Environmental exposure to silica nanoparticles (SiNPs) is inevitable due to their widespread application in industrial, commercial, and biomedical fields. In recent years, most investigators focus on the evaluation of cardiovascular effects of SiNPs in vivo and in vitro. Endothelial injury and dysfunction is now hypothesized to be a dominant mechanism in the development of cardiovascular diseases. This study aimed to explore interaction of SiNPs with endothelial cells, and extensively investigate the exact effects of reactive oxygen species (ROS) on the signaling molecules and cytotoxicity involved in SiNPs-induced endothelial injury. Significant induction of cytotoxicity as well as oxidative stress, apoptosis, and autophagy was observed in human umbilical vein endothelial cells following the SiNPs exposure (P<0.05). The oxidative stress was induced by ROS generation, leading to redox imbalance and lipid peroxidation. SiNPs induced mitochondrial dysfunction, characterized by membrane potential collapse, and elevated Bax and declined bcl-2 expression, ultimately leading to apoptosis, and also increased number of autophagosomes and autophagy marker proteins, such as LC3 and p62. Phosphorylated ERK, PI3K, Akt, and mTOR were significantly decreased, but phosphorylated JNK and p38 MAPK were increased in SiNPs-exposed endothelial cells. In contrast, all of these stimulation phenomena were effectively inhibited by N-acetylcysteine. The N-acetylcysteine supplement attenuated SiNPs-induced endothelial toxicity through inhibition of apoptosis and autophagy via MAPK/Bcl-2 and PI3K/Akt/mTOR signaling, as well as suppression of intracellular ROS property via activating antioxidant enzyme and Nrf2 signaling. In summary, the results demonstrated that SiNPs triggered autophagy and apoptosis via ROS-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling in endothelial cells, and subsequently disturbed the endothelial homeostasis and impaired endothelium. Our findings may provide experimental evidence and explanation for cardiovascular diseases triggered by SiNPs. Furthermore, results hint that the application of antioxidant may provide a novel way for safer use of nanomaterials.
Keywords: nanoparticle, cytotoxicity, mechanism, oxidative stress
Introduction
Engineered nanoparticles (NPs) have been mass produced and widely applied with the development of nanotechnology and materials science. Silica nanoparticles (SiNPs) are among the top five commonly used NPs, and are on the priority lists for toxicity evaluation by the Organization for Economic Cooperation and Development. They have found broad applications in many fields. Specifically, due to many advantages, such as high compatibility, easy surface functionalization, and resistance to biodegradation in the cellular environment, SiNPs are currently intensively studied for potential uses in medical and biomedical fields, including image, diagnosis, and treatment of diseases. Recently, the cancer-targeted diagnostic probes, “C-dots” composed of SiNPs and approved by the US Food and Drug Administration, have been allowed for the stage I human clinical trials.1 However, both in vitro and in vivo studies demonstrated that SiNPs induced toxicological effects, and the excessive production of reactive oxygen species (ROS) mediated oxidative stress and contributed to SiNPs-induced cellular damage.2
In recent years, the adverse effects of SiNPs on cardiovascular toxicity and atherosclerosis development have gained attention, and studies demonstrated a clear cardiovascular health risk associated with SiNPs exposure.3,4 Vascular endothelia line the inner surface of blood vessels, and provide an essential structural and biological barrier to maintain vascular function and homeostasis. Endothelial dysfunction is now hypothesized to be a dominant mechanism in the development of cardiovascular diseases, including atherosclerosis and ischemic heart disease.5 Currently, study on the biological behavior of engineered nanomaterials in endothelial cells is an essential part of their safety evaluation. Endothelial cells could direct contact with the NPs, making NPs–endothelial interactions potentially pathogenically relevant.6 Our previous studies showed that intratracheal-instilled SiNPs could pass through the alveolar–capillary barrier into systemic circulation,7 and SiNPs triggered endothelial dysfunction.8 However, knowledge on mechanisms of endothelial dysfunction is still limited.
In our previous study, we found that SiNPs could increase ROS generation.8 However, the exact effects of ROS on the signaling molecules and cytotoxicity involved in SiNPs-induced endothelial injury have not been extensively studied. N-acetylcysteine (NAC) is known as an “antioxidant” in experimental models. It has been extensively used as an antioxidant and free radical scavenger for the treatment of a range of clinical diseases, including paracetamol overdose, acute heavy metal poisoning, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, and hepatic encephalopathy.9–11 It has been reported to reduce cell damage induced by NPs including nano-titanium dioxide12 and multi-walled carbon nanotubes.13 However, the mechanisms underlying its beneficial effects are poorly defined. This study is an extension of our previous study, which aimed at fully understanding the effects and interaction mechanisms of SiNPs-induced disturbance of endothelial cell homeostasis, and also determining the therapeutic effects and possible mechanisms of NAC on attenuating the SiNPs-triggered ROS signaling in vascular endothelial cells. The research work may provide persuasive evidence for safety evaluation and risk management of SiNPs, and also for NAC as a chemopreventive agent.
Materials and methods
Chemicals and reagents
NAC, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylben-zimidazolylcarbocyanine iodide (JC-1), acridine orange (AO), and ethidium bromide (EB) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum, Dulbecco’s Modified Eagle’s Medium (DMEM), and MitoSOX™ were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA) and dihydroethidium (DHE) were acquired from Beyotime (Nantong, People’s Republic of China). A MiniBest universal RNA extraction kit, PrimeScript™ RT Master Mix (Perfect Real Time), and SYBR Premix Ex Taq™ II were obtained from Takara (Tokyo, Japan). All the chemicals and reagents used were of analytical purity and obtained from commercial sources.
Preparation and characterization of amorphous SiNPs
Amorphous SiNPs were prepared using the Stöber method and characterized as described in our previous study.8 Briefly, 2.5 mL of tetraethylorthosilicate was added to a premixed ethanol solution (50 mL) containing ammonia (2 mL) and water (1 mL). The reaction mixture was stirred (150 rpm) at 40°C for 12 hours. The particles were isolated by centrifugation (12,000 rpm) for 15 minutes and rinsed with deionized water for three times; they were then dispersed in deionized water (50 mL). The size and distribution of SiNPs were assessed by transmission electron microscopy (TEM; JEM2100; JEOL, Tokyo, Japan) and ImageJ software. In addition, the zeta potential, hydrodynamic sizes, and polydispersity index of SiNPs were detected with a Zetasizer (Nano ZS90; Malvern Instruments, Malvern, UK). Prior to the experimental use, the stock suspensions of SiNPs were sonicated for 5 minutes through a sonicator (160 W, 20 kHz, 5 minutes; Bioruptor UCD-200; Diagenode, Liege, Belgium) in order to minimize their aggregation. Meanwhile, inductively coupled plasma atomic emission spectrometry (ICP-AES) (Agilent 720; Agilent Technologies, Santa Clara, CA, USA) was used for purity detection, and gel-clot limulus amebocyte lysate assay kit for endotoxin measurement in SiNP suspensions was purchased from Zhanjiang Bokang Marine Biological Co. Ltd (Zhanjiang, People’s Republic of China).
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were purchased from the Cell Resource Center, Shanghai Institutes for Biological Sciences, People’s Republic of China. Cells were cultured in DMEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 humidified incubator. Cells were incubated up to ~24 hours and grown to ~80% confluence before experiments. SiNPs were suspended in DMEM and diluted to appropriate concentrations. NAC was dissolved in phosphate-buffered saline (PBS; 500 mM) and added to DMEM to treat cells for 2 hours. After pretreatment, NAC was removed, and cells were washed with PBS, replaced with either fresh culture medium or SiNPs, and cultured for another 24 or 48 hours. Lastly, cells or supernatants were collected for a series of analyses according to the experiment schedule.
Cell viability assay
Viability of HUVECs was determined using the MTT assay as previously described.14 Briefly, 1×104 HUVECs were placed in each of 96 wells. After treatment, the cell supernatant was discarded to avoid the reaction between NAC and MTT, and 100 μL fresh medium plus 10 μL of 5 mg/mL MTT was added, and finally, the solution was incubated at 37°C for 4 hours. The conversion of the tetrazolium salt MTT to a colored formazan by mitochondrial dehydrogenase was used to assess cell viability. Ultimately, absorbance was measured at 490 nm using a microplate reader (SpectraMax M5; Molecular Devices LLC, Sunnyvale, CA, USA).
Lactate dehydrogenase release assay
The release of intracellular lactate dehydrogenase (LDH) into culture medium is an indicator of irreversible cell death due to cell-membrane damage. To determine the effect of SiNPs on the cell membrane, LDH release was assessed using a commercial LDH kit (Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China) according to the manufacturer’s protocols. Briefly, cells were cultured in 24-well plates, with ~8×104 cells/well. After 12 hours of growth, cells were treated with 5 mM NAC for 2 hours and/or SiNPs with the concentration of 50 μg/mL at 37°C for 24 hours. The supernatants were collected, and LDH activity was measured by an ultraviolet–visible spectrophotometer (Beckman DU-640B; Beckman Coulter, Brea, CA, USA) at 440 nm. The amount of LDH released is expressed as LDH activity (U/L) in culture media.
SiNPs uptake
ICP-AES is a spectral technique that can determine the presence and concentrations of metal samples. Nowadays, it is widely applied for the quantification of NPs uptake.15 Thus, the SiNPs uptake was assayed by ICP-AES. Briefly, HUVECs were washed with PBS, and trypsinized after treatment. Cell density was determined with a hemocytometer. The cell suspensions were centrifuged at 1,500 rpm for 5 minutes. In total, 5 mL cell lysate containing HNO3 and H2O2 (3:1) was added in cell pellets. Then, the silicon content in the lysis solution was detected using ICP-AES. The number of internalized SiNPs was calculated via the silicon content in the lysis solution divided by the number of cells, providing a quantitative measurement of SiNPs uptake.
Intracellular ROS measurement
The production of intracellular ROS was measured using DCFH-DA, DHE, and MitoSOX™. DCFH-DA, an oxidation-sensitive probe, has been widely used for the detection of intracellular H2O2 level. After SiNPs plus NAC exposure, cells were incubated with PBS containing 10 mM DCFH-DA for 30 minutes in a CO2 incubator. After incubation, the cells were washed with PBS. DCF fluorescence was observed using a flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) and a laser scanning confocal microscope (LSM 710; Carl Zeiss Meditec AG, Jena, Germany) by capturing images at ×400 or ×1,200 magnification. Meanwhile, DHE, an indicator of superoxide anion radical (O2−2), and MitoSOX™, a mitochondrial superoxide indicator, were used to measure the production of O2− and mitochondrial ROS. After treatment, cells were incubated with either DHE or MitoSOX™ working solution at 37°C for 10 minutes. Then, cells were harvested and washed with PBS buffer for three times. Ultimately, the DHE or MitoSOX™ red fluorescence intensity was determined through flow cytometry (FCM).
Assessment of oxidative damage
Malondialdehyde (MDA) content was measured as an end product of lipid peroxidation, which was carried out using commercially available kits (Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China) according to the manufacturer’s instructions. Furthermore, the defense systems against free radical attack were assessed by the measurement of superoxide dismutase (SOD) activity and glutathione (GSH) level, using the corresponding commercial kit from Nanjing Jiancheng Bioengineering Institute (Jiancheng, People’s Republic of China).
Quantification of mRNA expression using real-time polymerase chain reaction
The relative amount of redox-related factors, including Nrf2 and its downstream genes, such as SOD2, NQO1, TXN-1, and TXNRD-1, GCLC, and also hsp70 were quantified using real-time polymerase chain reaction according to our previous study.8 All quantifications were performed with β-actin as an internal standard, and the relative amount of mRNA was calculated using the 2−ΔΔCT method.
Cell apoptosis analysis
Morphological evidence of apoptosis was obtained using AO/EB staining. Briefly, cells were seeded in 24-well plates at a density of 5×104 cells/well. After treatment, one droplet of mixture solution, containing 100 μg/mL each of AO and EB, was added to stain cells, which were then immediately observed under inverted fluorescence microscope (BX-60; Olympus Corporation, Tokyo, Japan) and photographed at ×200 magnification. Moreover, the apoptotic cells were quantified and analyzed by FCM using an Annexin V-FITC apoptosis detection kit (KeyGen, Nanjing, People’s Republic of China) according to the manufacturer’s instructions.
Evaluation of mitochondrial membrane potential
Mitochondrial membrane potential (MMP) of the cells was monitored using JC-1 probe, which can selectively enter into mitochondria and reversibly change color from red to green as the membrane potential decreased. The ratio of red to green expresses the change of MMP. After treatment, cells were washed with PBS for three times, and then incubated with 10 mg/mL working solution of JC-1 for 20 minutes. Ultimately, cells were washed with PBS twice and observed using laser scanning confocal microscope by capturing images at ×1,200 magnification. Meanwhile, the red/green ratio was analyzed by Leica QWin image analysis software (Leica Microsystems, Wetzlar, Germany). At least 20 fields were picked in each group for the measurement of mean values of red and green fluorescence intensities, and then the mean value of red/green ratio was calculated.
Autophagy measurement
Firstly, the autophagy induced by SiNPs in HUVECs was measured by TEM. Briefly, after treatment, the cell samples were collected and immediately fixed overnight in 3% glutaraldehyde. Then, the samples were rinsed three times with 0.1 M phosphate buffer and postfixed with 1% osmic acid for 2 hours. After being rinsed three times with 0.1 M phosphate buffer and serially dehydrated with 50%, 70%, 80%, 90%, and 100% alcohol and 100% acetone, the samples were embedded in epoxy resin for making the blocks of cells. Ultrathin sections (50 nm) obtained with an ultramicrotome were stained with 5% aqueous uranyl acetate and 2% aqueous lead citrate, air dried, and imaged under TEM. Besides TEM observation, MAP1LC3β or LC3B, the autophagy protein biomarker, was detected by immunofluorescent staining. Intracellular LC3B intensity was quantified by Leica QWin image analysis software. At least 20 fields were picked in each group for the calculation of mean value of fluorescence intensity. Meanwhile, both mRNA and protein expressions of LC3B and p62 were determined by real-time polymerase chain reaction and Western blot, respectively. The primary antibodies for LC3B and p62 were purchased from Cell Signaling Technology (Danvers, MA, USA).
Western blot analysis
The total and phosphorylated ERK, p38 MAPK, JNK, PI3K, Akt, and mTOR protein in HUVECs was detected by Western blot analysis, besides the protein levels of bcl-2 and Bax. As an internal control, GAPDH was detected. The primary antibodies for these proteins were all acquired from Cell Signaling Technology. Densitometric analysis of the Western blots was performed using Image Lab™ Software (Bio-Rad Version 3.0; Bio-Rad Laboratories Inc., Hercules, CA, USA). The relative values of the samples were measured by normalizing all data to the respective control samples of each experiment.
Statistical analysis
Data were expressed as mean ± SD and examined for their statistical significance of differences by Student’s t-test. All comparisons were two-tailed, and P-values <0.05 was considered to be statistically significant.
Results
Characterization of SiNPs
The TEM images of SiNPs exhibited near-spherical shape with relatively favorable dispersibility (Figure 1). The SiNPs was fully characterized in our previous studies:8 the average diameter of the SiNPs was ~57.66±7.30 nm. When the SiNPs were added in DMEM for 24 hours, their hydrodynamic size, zeta potential, and polydispersity index were 107.94±1.47 nm, −30.33±1.35 mV, and 0.11±0.02, respectively, reflecting that SiNPs displayed good monodispersity in the culture medium. Furthermore, the purity of the SiNPs used in our study was better than 99.9%, and SiNPs were endotoxin-free. Our results confirmed that the SiNPs possessed favorable dispersibility and stability in the culture medium.
Figure 1.
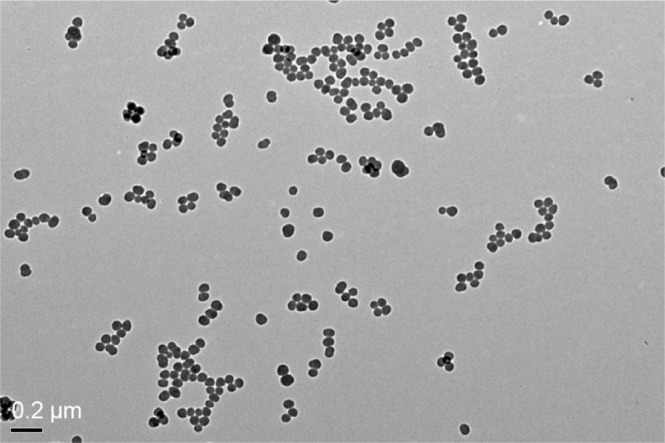
TEM of tested SiNPs.
Notes: Representative transmission electron micrograph of SiNPs. All particles were near-spherical in shape, and no stable aggregates were visible (scale bar =0.2 μm).
Abbreviations: TEM, transmission electron microscopy; SiNPs, silica nanoparticles.
SiNPs declined cell viability, while NAC restored it
Firstly, in order to find the optimum concentration of NAC to be administered in HUVECs, an NAC dose–response curve was created, and 5 mM NAC was found to be the optimum dose, which was used in all the experiments (Figure 2A). The exposure of HUVECs to SiNPs led to a significant loss of cell viability. However, NAC pretreatment showed significant protective effect on viability of HUVECs exposed to SiNPs (Figure 2B).
Figure 2.

The declined cell viability of HUVECs induced by SiNPs, and restored by NAC.
Notes: (A) The cytotoxicity of NAC on HUVECs assessed by MTT assay. Cells were treated with NAC for 24 hours at various concentrations, which were 0.125, 0.25, 0.5, 1, 2, 4, 5, 8, and 10 mM, respectively. Control received culture medium only. Finally, 5 mM was chosen as the dose of NAC for the following study. (B) Cell viability of HUVECs measured by MTT assay. Cells were pretreated with 5 mM NAC for 2 hours followed by exposure of SiNPs (12.5, 25, 50, and 100 μg/mL, respectively) for either 24 or 48 hours. All values are mean ± SD of three identical experiments made in triplicate. *P<0.05 vs control and #P<0.05 for SiNPs vs SiNPs plus NAC.
Abbreviations: HUVECs, human umbilical vein endothelial cells; SiNPs, silica nanoparticles; NAC, N-acetylcysteine; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide; SD, standard deviation.
SiNPs induced LDH release, which was attenuated by NAC
According to IC50 of SiNPs calculated from the MTT results, the dose of 50 μg/mL was chosen for the following study. After exposure to 50 μg/mL SiNPs for 24 hours, the LDH activity (U/L) in cell media increased from 117.81±38.62 (control) to 686.76±21.32 (P<0.05), indicating a statistically significant LDH release induced by SiNPs. NAC pretreatment was found to be effective in decreasing LDH release to 567.58±37.48 U/L compared to SiNPs only (P<0.05), although not to its normal state. The treatment with NAC alone (104.16±15.88 U/L) had no significant effect on LDH release as compared with control (Table 1).
Table 1.
LDH activity and silicon content after treatment with SiNPs and NAC
| Group | LDH activity (U/L) | Silicon content (μg/106 cells) |
|---|---|---|
| Control | 117.81±38.62 | 0.02±0.01 |
| NAC | 104.16±15.88 | 0.03±0.01 |
| SiNPs | 686.76±21.32* | 24.46±4.52* |
| SiNPs + NAC | 567.58±37.48*,# | 20.93±2.21* |
Notes: Data are expressed as mean ± SD from three independent experiments performed in triplicate.
P<0.05 vs control;
P<0.05 for SiNPs vs SiNPs plus NAC.
Abbreviations: LDH, lactate dehydrogenase; SiNPs, silica nanoparticles; NAC, N-acetylcysteine; SD, standard deviation.
No influence on SiNPs uptake by NAC
Results showed that after incubation with 50 μg/mL SiNPs for 24 hours, the average silicon content internalized by each 106 cells increased from 0.02±0.01 (control) to 24.46±4.52 μg (P<0.05), suggesting a statistically significant increase in the uptake of the SiNPs by HUVECs. NAC pretreatment had no significant effect on SiNPs uptake, which was 20.93±2.21 μg as compared to SiNPs only. Meanwhile, treatment with NAC alone (0.03±0.01 μg) also manifested no significant effect as compared with control (Table 1).
SiNPs increased ROS production, which was attenuated by NAC
To examine the intracellular ROS production induced by SiNPs, intracellular H2O2 and O2− levels were measured using DCFH-DA and DHE probe, respectively. As shown in Figure 3A, the DCF fluorescence intensity in HUVECs treated with SiNPs significantly increased as compared with the control group, while it was relatively weak after pretreated with NAC compared to that treated with SiNPs only. Quantification of the fluorescence was done by FCM (Figure 3B). The generation of both H2O2 and O2− in HUVECs increased nearly by 75% (H2O2) and 50% (O2−) under the SiNPs exposure compared with the control group (P<0.05). However, NAC pretreatment declined the elevated H2O2 levels and O2− mediated by SiNPs (P<0.05). It is assumed that mitochondria serve as the major intracellular source of ROS. The mitochondrial ROS was measured by monitoring MitoSOX™ fluorescence intensity. Similarly, the production of mitochondrial ROS also manifested such a trend of change (Figure 3C and D). All these results suggested that NAC pretreatment could effectively inhibit the ROS production induced by SiNPs.
Figure 3.

ROS production induced by SiNPs, which was attenuated by NAC.
Notes: (A) Fluorescence image of HUVECs under laser confocal microscope observed through DCFH-DA probe at a magnification of ×400. Cells were pretreated with 5 mM NAC for 2 hours, followed by 50 μg/mL SiNPs exposure for 24 hours. Control received culture medium only. (B) Quantification of the fluorescence by FCM using DCFH-DA and DHE probes for the detection of intracellular H2O2 and O2−, respectively. (C) MitoSOX™ via FCM for the determination of mitochondrial ROS. (D) The representative FCM scatter plots of intracellular H2O2, O2−, and mitochondrial ROS, respectively. Data are expressed as mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: ROS, reactive oxygen species; SiNPs, silica nanoparticles; NAC, N-acetylcysteine; HUVECs, human umbilical vein endothelial cells; DCFH-DA, 2′,7′-dichlorofluorescein diacetate; FCM, flow cytometry; DHE, dihydroethidium; SD, standard deviation; MFI, mean fluorescence intensity.
SiNPs induced oxidative injury, while NAC reduced it
As shown in Figure 4A, SiNPs exposure increased the MDA content in HUVECs when compared to control group (P<0.05). But pretreatment with NAC remarkably reduced MDA content induced by SiNPs (P<0.05). On the other hand, both the GSH content and SOD activity in SiNPs exposure group increased significantly in comparison to that in the control group (P<0.05), and that in the group pretreated with NAC followed by SiNPs exposure had an obvious increase compared to SiNPs exposure group (P<0.05), indicating that NAC pretreatment further increased the elevated GSH content and SOD activity mediated by SiNPs in HUVECs (Figure 4B and C).
Figure 4.

Oxidative injury induced by SiNPs and reduced by NAC.
Notes: After exposure to 50 μg/mL SiNPs for 24 hours plus pretreatment with 5 mM NAC for 2 hours, the (A) production of MDA, (B) GSH content, and (C) SOD activity were measured. Data are expressed as mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: SiNPs, silica nanoparticles; NAC, N-acetylcysteine; MDA, malondialdehyde; GSH, glutathione; SOD, superoxide dismutase; SD, standard deviation; prot, protein.
SiNPs promoted the activation of Nrf2 signaling, and NAC further enhanced it
Nrf2 has been recognized as a transcription factor that controls mechanisms of cellular defense response against oxidative stress. As shown in Figure 5, the mRNA expressions of Nrf2 and its key downstream genes tested were upregulated with SiNP exposure in HUVECs, and NAC pretreatment further promoted the activation of Nrf2 signaling as evidenced by enhanced expressions of Nrf2 and its downstream genes.
Figure 5.

The activation of redox-mediated Nrf2 signaling pathway.
Notes: The mRNA expressions of Nrf2 and its key downstream genes, including SOD2, NQO1, TXN-1, TXNRD-1, GCLC, and hsp70, were measured through quantitative real-time PCR. Data are expressed as mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: PCR, polymerase chain reaction; SD, standard deviation; SiNPs, silica nanoparticles; NAC, N-acetylcysteine.
SiNPs promoted endothelial cell apoptosis, while NAC inhibited it
To investigate the impact of SiNPs on endothelial apoptosis, the cellular morphology was assessed by AO/EB staining (Figure 6A). Cells in the control group were uniformly green colored with a normal morphology, and many cells in SiNPs exposure group manifested apoptotic morphological changes, while cells under apoptosis became less with NAC pretreatment. Furthermore, the quantification data on apoptosis acquired by FCM analysis showed that the apoptotic rate increased from 5.44% (control) to 23.99% at 50 μg/mL SiNPs exposure for 24 hours; on the other hand, pretreatment with NAC resulted in protection against SiNPs-induced cell apoptosis, with the apoptotic rate declined to 14.32% (Figure 6B), suggesting that SiNPs promoted apoptosis in HUVECs, while NAC pretreatment prevented HUVECs from apoptosis induced by SiNPs.
Figure 6.

Apoptosis promoted by SiNPs, and the prevention by NAC.
Notes: (A) AO/EB staining and flow cytometry using (B) Annexin V-FITC double-stain were used to detect the apoptosis of HUVECs after treatment with 50 μg/mL SiNPs for 24 hours plus NAC pretreatment (5 mM, 2 hours). Scale bar =50 μm, with the magnification of ×200. Data are expressed as mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: SiNPs, silica nanoparticles; NAC, N-acetylcysteine; AO, acridine orange; EB, ethidium bromide; HUVECs, human umbilical vein endothelial cells; SD, standard deviation.
SiNPs induced MMP collapse, which was suppressed by NAC
An increase in ROS and a consequent loss of MMP were reported to be typical phenomena during mitochondria-dependent apoptosis. The MMP was determined with JC-1 probe by confocal microscopy. In live cells, JC-1 existed either as a green-fluorescent JC-1 monomer at depolarized membrane potentials or as a red-fluorescent JC-1 aggregate at hyperpolarized membrane potentials. As shown in Figure 7A, the HUVECs stained with JC-1 showed the enhanced cytoplasmic diffusion of green monomer fluorescence following exposure to SiNPs. The loss of the MMP was reflected by a decrease in the ratio of red (JC-1 aggregates) to green (JC-1 monomers) fluorescence. As shown in Figure 7B, SiNPs induced the decrease of MMP as compared to control (P<0.05), while NAC pretreatment increased MMP compared to SiNPs-only group (P<0.05), suggesting that NAC effectively suppressed the SiNPs-induced loss of MMP.
Figure 7.

The loss of MMP induced by SiNPs and suppressed by NAC.
Notes: HUVECs were treated with 50 μg/mL SiNPs for 24 hours plus NAC pretreatment for 2 hours. The MMP was determined using the fluorescent probe, JC-1. (A) Confocal images of MMP at a magnification of ×1,200. (B) The JC-1 red/green fluorescence intensity ratio was used to express the changes of MMP, and the decreased ratio indicates decrease of MMP. Data are expressed as mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: MMP, mitochondrial membrane potential; SiNPs, silica nanoparticles; NAC, N-acetylcysteine; HUVECs, human umbilical vein endothelial cells; SD, standard deviation; DAPI, 4′,6-diamidino-2-phenylindole.
SiNPs induced autophagy, which was reversed by NAC
TEM is considered as the gold standard method to determine double-membrane vacuole structures. Firstly, autophagy activation in HUVECs treated with SiNPs was measured by ultrastructural analysis via TEM. As shown in Figure 8A, HUVECs exposed to SiNPs exhibited typical autophagic vacuoles with degraded cytoplasmic contents and highly electron-dense SiNPs when compared to the control cells. By inspecting the TEM image carefully, the whole process of autophagy was observed in HUVECs exposed to SiNPs, including double-membranous phagophores, autophagosomes with cytoplasmic materials inside, and autolysosomes. Furthermore, swollen or cristae-rupturing mitochondria and damaged lysosome existed in the cytoplasm of HUVECs exposed to SiNPs. Results from the ultrastructural analysis clearly indicated that SiNPs triggered the autophagy process in vascular endothelial cells.
Figure 8.

Induction of autophagy by SiNPs and reversed by NAC.
Notes: (A) Induction of autophagy in HUVECs by 50 μg/mL SiNPs observed under TEM. Control shows typical distribution of organelles within vascular endothelial cells. SiNPs-treated cell shows appearance of numerous perinuclear vacuoles typical of an autophagic cell. The white and black arrows indicate damaged mitochondrial and impaired lysosome, respectively, whereas the red arrow indicates a double-membranous phagophore developing into an autophagosome; (a and b) early autophagic vacuoles (AVi) containing membrane-bound cytoplasmic material, (c and d) and late autophagic vacuoles (AVs) with partially degraded cytoplasmic materials. LC3B expression was determined by immunofluorescence. Images demonstrate enhanced LC3-positive dots in HUVECs under 50 μg/mL SiNPs exposure, (B and C) while NAC pretreatment reversed the elevated LC3 level to nearly normal state. (D) Quantification of LC3 and p62 expressions was done by quantitative real-time PCR at mRNA level, and by Western blot at corresponding protein level. Data represented are the mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: SiNPs, silica nanoparticles; NAC, N-acetylcysteine; HUVECs, human umbilical vein endothelial cells; TEM, transmission electron microscopy; PCR, polymerase chain reaction; SD, standard deviation; DAPI, 4′,6-diamidino-2-phenylindole.
Autophagy activated by SiNPs was further confirmed by assessing the autophagy protein biomarker, LC3. Immuno-fluorescence observation and the corresponding fluorescent intensity analysis demonstrated enhanced LC3-positive dots in HUVECs under the exposure of SiNPs, while NAC pretreatment reversed the elevated LC3 level to normal state (Figure 8B and C). The quantification of mRNA and protein expressions also revealed the enhanced LC3 and p62 levels in SiNPs-treated HUVECs, while NAC pretreatment significantly lowered it down to almost normal level (Figure 8D). All these results indicated that SiNPs could activate the autophagy process but with impaired autophagy degradation in vascular endothelial cells, while NAC pretreatment could effectively reverse it.
SiNPs regulated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling, which was retroregulated by NAC
As shown in Figure 9A and B, SiNPs significantly promoted the phosphorylation of both JNK and p38 MAPK, but reduced that of ERK (P<0.05). NAC pretreatment reversed such phosphorylation (P<0.05). We also analyzed expressions of the proapoptotic protein, Bax, and antiapoptotic protein, bcl-2, by Western blot. The bcl-2/Bax ratio is called as “molecular switch” in apoptosis initiation. SiNPs significantly declined the bcl-2/Bax ratio as compared to control (P<0.05), while NAC pretreatment alleviated the elevated Bax levels and increased the bcl-2/Bax ratio when compared to SiNPs only (P<0.05). Furthermore, as PI3K/Akt/mTOR signaling is a key negative regulator of autophagy, we examined p-PI3K, p-Akt, and p-mTOR upon different treatments. Results revealed that SiNPs significantly inhibited the phosphorylation of PI3K, Akt, and mTOR in HUVECs (P<0.05). Furthermore, NAC pretreatment markedly attenuated the dephosphorylation in PI3K/Akt/mTOR signaling pathway (P<0.05).
Figure 9.

Regulation of MAPK/Bcl-2 and PI3K/Akt/mTOR signaling.
Notes: The expressions of ERK, p-ERK, p38 MAPK, p-p38 MAPK, JNK, p-JNK, PI3K, p-PI3K, Akt, p-Akt, mTOR, p-mTOR, bcl-2, and Bax were measured by Western blot assay after 24 hours of exposure to SiNPs plus NAC in HUVECs. (A) All blots shown are representative of three independent experiments. (B) Relative densitometric analysis of the protein bands was performed and presented. Data are expressed as the mean ± SD from three independent experiments. *P<0.05 vs control; #P<0.05 for SiNPs vs SiNPs + NAC.
Abbreviations: SiNPs, silica nanoparticles; NAC, N-acetylcysteine; HUVECs, human umbilical vein endothelial cells; SD, standard deviation.
Discussion
Oxidative stress generated by an imbalance between ROS and antioxidants has been implicated in the causation of many human disorders, such as cardiovascular, liver, and respiratory diseases. In general, ROS-mediated oxidative stress has been suggested as one among the key events that triggers the NPs-induced cytotoxicity.16–18 We observed a dose-dependent ROS production either in HUVECs or in HepG2 cells when exposed to SiNPs at 25, 50, and 100 μg/mL for 24 hours.8,19 However, ROS-independent mechanisms were also involved in NPs’ toxicity.20,21 Recently, SiNPs have been reported to protect pea seedings against Cr(VI) phytotoxicity via reducing oxidative stress, and enhancing antioxidant defense system.22 These data of agreement and discordance with regard to NPs’ toxicity and its mechanisms indicate that hazards and exact mechanisms have not been clearly defined. We previously demonstrated that SiNPs exposure induced inhibition of cell viability, an increase of cytokines, as well as apoptosis in vascular endothelial cells, HUVECs.8 In vivo study also provided support for disturbance of endothelial cell homeostasis, and induction of autophagy mediated by SiNPs.23 In recent years, the pharmaceutical application potential of NAC supplement has attracted much interest from researchers in exploring strategies against NPs’ toxicity. Thus, in order to certify the critical role of ROS in SiNPs-induced endothelial cell injury and also explore a mean for preserving cell function, the cellular antioxidant NAC was used in the present study.
NAC has already been approved for use in human clinical practice for several years, and it appears to be safe, especially when administered via ingestion.10 It is a source of GSH and sulfhydryl groups, and its antioxidant activity results from its abilities to scavenge free radicals by interacting with ROS and raise levels of cellular GSH. To be noted, some reports showed an effective decline in cellular uptake of NPs in the presence of NAC,24,25 whereas our results demonstrated that NAC pretreatment had no effect on SiNPs uptake, which might be associated with the characterization and concentration of NPs used in experiments according to the study by Davda and Labhasetwar.26 Long et al demonstrated that ROS generation is an initial cellular response to NP internalization.27 As a result, increased ROS production has been associated with the internalization of SiNPs within cells, while NAC pretreatment could scavenge the ROS generated by internalized SiNPs as evidenced by the reduction of intracellular H2O2, superoxide anion (O2−), and also mitochondrial ROS with pretreatment of NAC in HUVECs. Our results confirmed the involvement of ROS in the process of toxicity and is in agreement with previous studies.28 Elevated levels of MDA implicated an oxidative attack leading to cellular damage and failure of antioxidant defense mechanisms. It has been reported that NAC could promote the antioxidant defense system, including the enhancement of antioxidant activity (ie, SOD, GSH-Px, and CAT)29 and GSH level, and also the activation of the Nrf2 signaling.30,31 Our study confirmed that NAC pretreatment significantly decreased the MDA level induced by SiNPs, and also enhanced antioxidant defense system, including upregulated GSH content, SOD activity, and also Nrf2 antioxidant signaling, resulting in restoration of the decreased viability of HUVECs induced by SiNPs. All these indicated that the protective effects of NAC on SiNPs-induced endothelial cell injury were related to the alleviation of ROS-mediated oxidative stress and also the augmentation of antioxidant defense system. Meanwhile, results confirmed that the SiNPs-induced endothelial injury was ROS-dependent. Cell death, including apoptosis, necrosis, and autophagy, is a highly regulated process and critical to the maintenance of tissue homeostasis in multicellular organisms. ROS production and oxidative stress have received much attention in NPs-induced apoptosis.32 ROS have been suggested to be signaling molecules for the initiation and execution of the apoptotic death program.33 The scavenger of ROS, NAC, was proved to attenuate the SiNPs-induced apoptosis of endothelial cells in this study. Mitochondria are the main source of intracellular ROS, and are also vulnerable under oxidative stress. ROS can induce mitochondrial dysfunction, leading to ATP depletion, opening of the mitochondrial permeability transition pore, MMP collapse, and ultimately cell death.34 Consistent with previous studies,28 an increase of mitochondrial superoxide and a decline of MMP induced by SiNPs were observed. NAC could upregulate the decreased MMP induced by SiNPs, and also significantly alleviate the generation of mitochondrial superoxide. We speculated that the decline of ROS, especially mitochondrial ROS level, by NAC alleviated the MMP collapse, and then apoptosis of HUVECs exposed to SiNPs.
It is known that NPs-induced mitochondrial disruption leads to the subsequent cytochrome c release from the mitochondria which initiates the process of apoptosis. The proapoptotic members of Bcl-2 family such as Bax, Bad, and Bak initiate the release of cytochrome c, but antiapoptotic members of this family such as bcl-2 and bcl-xl antagonize it. In the present study, SiNPs regulated Bcl-2 family proteins, a process characterized by the upregulated Bax and down-regulated bcl-2 (Figure 9A and B). The crucial role of bcl-2/Bax in mitochondrial permeability and MMP loss has been well established. The decreased MMP with a concomitant decrease in the bcl-2/Bax ratio suggested that SiNPs-induced apoptosis was mitochondria dependent. Simultaneously, SiNPs also activated the MAPK signaling, as shown by the increased expressions of phosphorylated JNK and p38 MAPK, but inactivated ERK. It has been reported that NPs-induced ROS production and oxidative stress through the MAPK pathway regulates apoptosis.35–37 MAPK plays an essential role in modulating the functions of pro- and anti-apoptotic proteins located in mitochondria. For example, JNK has been shown to be critically required for the release of cytochrome c from mitochondria.38 Following activation by apoptotic stimuli, JNK readily translocates to mitochondria. The activated JNK and p38 MAPK could not only phosphorylate bcl-2 or bcl-xl to suppress their antiapoptotic activity39,40 but also phosphorylate Bax leading to its mitochondrial translocation prior to apoptosis.41 Meanwhile, the ROS-mediated ERK inactivation could downregulate bcl-xl expression via suppressing ERK-mediated c-Jun phosphorylation,42 resulting in mitochondria-dependent apoptosis. These findings suggested that SiNPs induced endothelial cell apoptosis through MAPK/Bcl-2 signaling. However, NAC pretreatment could reverse SiNPs-induced alteration of MAPK/Bcl-2 signaling, indicating that the MAPK/Bcl-2 signaling is ROS-dependent. ROS-mediated oxidative stress activates MAPK signaling; for example, JNK is activated by ROS through upstream kinases, primarily ASK1. Conversely, research demonstrated that JNK signaling contributes to the amplification of ROS generation.43 Therefore, we speculated that the suppressive effect of NAC pretreatment on SiNPs-induced JNK activation may contribute to the decline of elevated ROS level in HUVECs exposed to SiNPs.
Autophagy, also known as type II cell death, is an evolutionarily conserved process for bulk degradation, including the elimination of long-lived proteins, macromolecular aggregates, and nonessential or dysfunctional organelles in living cells, such as mitochondria, Golgi apparatus, endoplasmic reticulum, and peroxisomes.44 Recently, NPs are also considered to cause toxic effects through autophagy and lysosomal dysfunction.45–47 Fullerene NPs were shown to induce autophagic vacuolization of HUVECs.48 In our study, the appearance of cellular ultrastructures observed by TEM presented the characteristics of an autophagic response of the endothelial cells following exposure to SiNPs, including generalized high vacuolization, autophagosomes, and autolysosomes with some of them containing NPs (Figure 8A). However, detailed mechanisms of NPs-induced autophagy have not been fully investigated. Several lines of evidence indicate that ROS are the upstream modulators of autophagy and that oxidative stress coupled with defective autophagy in endothelial cells causes endothelial dysfunction and disruption of vascular homeostasis, further leading to the pathogenesis of a range of diseases, including cardiovascular dysfunction, atherosclerosis, and cerebrovascular disorders.49–51 ROS could activate autophagy, probably representing a feedback mechanism to limit ROS-mediated cell activation by removing oxidatively damaged molecules and cell structure.52 Currently, it remains unclear whether or how oxidative stress is linked to the autophagy in the context of SiNPs-induced endothelial cell death.
During our evaluation of the effects of NAC on autophagy in SiNPs-exposed endothelial cells, we detected LC3 and p62 expressions. LC3, a key autophagy protein marker, converts from the cytoplasmic LC3-I form to the lipidated LC3-II form during autophagy. Conversion of LC3-I into LC3-II is commonly used to monitor autophagy.53 Recently, a few studies demonstrated that LC3-II/LC3-I increased significantly under SiNPs exposure.23,54,55 We demonstrated SiNPs-specific induction of the formation of LC3 puncta viewed as aggregate dot-like structures in HUVECs (Figure 8B), which is another alternative hallmark of autophagy.56 Consistent with the enhanced LC3 puncta, an elevated LC3 mRNA expression and conversion of LC3-I to LC3-II in HUVECs exposed to SiNPs were also observed (Figure 8D), suggesting the autophagosome formation in endothelial cells induced by SiNPs exposure. These results fully supported our TEM observations. As for NPs-mediated autophagy, it might be an adaptive cellular response to the degradation and clearance of NPs initially. Recent research reported that the autophagic activity might be a key mechanism leading to cell survival under 20 nm SiNPs exposure.57 However, prolonged and uncontrolled autophagy results in harmful cellular dysfunction and also cell death. Lysosome is one of the major intracellular storage sites for SiNPs that enter into cells.58 SiNPs accumulation in lysosomes has been suggested as a mechanism of cytotoxicity in lung epithelial cells, and its overload leading to delayed clearance and particle persistence can negatively impact lysosome function.59 It has been reported that internalized SiNPs could cause destabilization of lysosomes and permeabilization of lysosome membranes,60 resulting in ROS generation, cytosolic acidification, and eventually cellular death.61,62 Additionally, lysosome is vulnerable to ROS generated by NPs.63 Accordingly, although the lysosome plays an integral part in the degradation stage of autophagy by fusing with the autophagosome to generate the autolysosome, SiNPs-mediated lysosome impairment would cause a continued accumulation of autophagosomes, and ultimately disturbance of autophagy.64–66 We did observe lysosome impairment after SiNPs exposure, which may be responsible for the autophagy disturbance (Figure S1). Furthermore, the intracellular p62 protein (SQSTM1) is a selective substrate of autophagic degradation, and has been used to monitor autophagic flux, and been proposed to be an indicator of autophagy disturbance.53 An induction of the autophagic flux leads to increased degradation of p62, whereas autophagy disturbance, that is, an impaired autophagic flux or inhibition of autophagosome degradation, would result in p62 accumulation. Besides the increase in LC3 level, we further detected an increase of p62 level, suggesting an impaired autophagic degradation induced by SiNPs. It has been reported that accumulation of p62 could trigger a positive amplifying loop for ROS generation, enhanced oxidative stress, and aggravated genomic instability.67,68 Notably, SiNPs exposure affected autophagy of endothelial cells in two ways, including the induction of autophagosome synthesis via increasing LC3-II expression, and reduction of autophagosome degradation as shown by the lysosome impairment and p62 accumulation. As for the influence of NAC on autophagy, up to now, several studies have revealed that NAC could decrease the conversion of LC3-I to LC3-II in HepG2 cells exposed to SiNPs,55 and in ZnO NPs-exposed macrophage.69 This was in line with our results that NAC pretreatment reversed the enhanced LC3 and p62 expressions in HUVECs induced by SiNPs, which also indicated that SiNPs could induce ROS-mediated autophagy on endothelial cell.
A large number of studies reported ROS as early inducers of autophagy, but many unknowns still exist regarding how ROS-mediated oxidative stress exactly drives the autophagy process. The data gathered so far undoubtedly show that autophagy is negatively regulated by the serine–threonine protein kinase mTOR. PI3K/Akt signaling is one well-characterized signaling pathway that contributes to mTOR activation. In this work, we demonstrated that the crucial upstream autophagy regulator PI3K was inhibited by SiNPs, and also the phosphorylated Akt and mTOR, suggesting that inhibition of PI3K/Akt/mTOR signaling pathway contributed to SiNPs-induced autophagy, in consistent with our previous studies.70 It has been reported that the downregulation of phosphorylated PI3K/Akt influences endothelial cell survival, migration, and vascular permeability. Furthermore, NAC could attenuate these inhibition effects, indicating that the PI3K/Akt/mTOR pathway may be ROS dependent. Taken together, our studies also indicate that SiNPs induced autophagy via ROS-mediated PI3K/Akt/mTOR signaling; meanwhile, the inhibition of autophagy via ROS-mediated PI3K/Akt/mTOR signaling pathway may be a new strategy of NAC supplement against SiNPs-induced endothelial injury. Additionally, MAPK/ERK signaling is well known as a major negative regulator of autophagy, which could lead to activation of mTOR. The observed suppression of ERK phosphorylation induced by SiNPs might be correlated with induction of autophagy. It is reported that ERK and JNK could contribute to autophagic cell death via phosphorylation of bcl-2, further leading to bcl-2 separation from Beclin-1, and thereby alleviation of the inhibition effect on Beclin-1.71–74 Recently, NAC was reported to attenuate ischemia–reperfusion-induced autophagy via ROS/JNK/bcl-2 pathway.75 Therefore, we speculated that ROS-mediated MAPK/Bcl-2 may be a novel mechanism in the protective effects of NAC on SiNPs-induced autophagy in endothelial cells. Studies revealed that mTOR inhibition could lead to and even promote apoptosis,76,77 suggesting that the detected downregulation of mTOR may also be responsible for SiNPs-induced apoptosis. Importantly, this indicated cross talk between the SiNPs-mediated apoptosis and autophagy in vascular endothelial cells. Mitochondria play an essential role in the process of apoptosis and autophagy. Autophagy can, to some extent, eliminate the dysfunctional mitochondria (Figure S2A–F). However, SiNPs exposure could not only trigger autophagy activation but also impair autophagic degradation and disturb the proper autophagy flux, characterized by p62 accumulation, leading to the accumulation of sequestered but undigested defective mitochondria and ultimately cell death. Thus, we do believe that our observed autophagy could coordinate with and further augment SiNPs-induced apoptosis, which is in line with ZnO NPs-induced apoptosis by enhancement of autophagy.69 The cross talk and the exact signaling molecules involved in the interplay between apoptosis and autophagy need to be extensively investigated. A schematic model of the amorphous SiNPs that triggers vascular endothelial cell injury through apoptosis and autophagy via ROS-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling is presented in Figure 10.
Figure 10.

Schematic model of ROS-dependent MAPK/Bcl-2 and PI3K/Akt/mTOR signaling mediated cross talk between autophagy and apoptosis involved in endothelial injury triggered by SiNPs.
Abbreviations: ROS, reactive oxygen species; SiNPs, silica nanoparticles; NAC, N-acetylcysteine.
Conclusion
In summary, the present study indicated that SiNPs can disrupt endothelial cell homeostasis, including the induction of apoptosis and autophagy disturbance as a result of SiNPs-induced ROS generation and oxidative stress. NAC has been reported to decrease cell oxidative stress directly, inhibit activation of the nuclear transcription factor NF-κB,78 suppress cytokine elaboration,79 or indirectly restore the GSH content.80 Our results demonstrated that NAC pretreatment had the protective effects on SiNPs-induced endothelial dysfunction by its antioxidant properties, including facilitating GSH biosynthesis, enhancing SOD activity, scavenging ROS via activating Nrf2 signaling pathway, and also by suppressing apoptosis and autophagy. ROS-dependent MAPK/Bcl-2 and PI3K/Akt/mTOR signaling pathways were involved in SiNPs-induced apoptosis and autophagy. Moreover, our findings speculated cross talk between apoptosis and autophagy via MAPK/Bcl-2 and PI3K/Akt/mTOR signaling pathway. These results provide laboratory evidence not only for the safety evaluation and biomedical application of NPs but also for the exploration of NAC as a chemopreventive agent.
Supplementary materials
Lysosome impairment of HUVECs exposed to the SiNPs (50 μg/mL) for 24 hours.
Notes: LysoTracker Red (Beyotime) was used for the staining of lysosome in HUVECs. Compared with the control group, the significant decrease of fluorescence intensity in the SiNPs group indicated that SiNPs induced lysosome impairment in HUVECs.
Abbreviations: HUVECs, human umbilical vein endothelial cells; SiNPs, silica nanoparticles.
TEM images of HUVECs exposed to the SiNPs (50 μg/mL) for 24 hours.
Notes: (A) Control group. (B) The magnification of selected area of control showed evidently intact mitochondria. (C) The induction of mitochondrial swelling and cristae rupturing and disappearance after SiNPs exposure (black arrows), and also SiNPs deposition in mitochondria. (D and E) Severe mitochondrial swelling in SiNPs-treated endothelial cells (black arrows), and (E and F) autophagosome including cytoplasmic material, especially impaired mitochondria (white arrow).
Abbreviations: TEM, transmission electron microscopy; HUVECs, human umbilical vein endothelial cells; SiNPs, silica nanoparticles.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No 81102095, No 81202242, No 81573176, No 81230065), General Program of Beijing Natural Science Foundation (7162021, 7162022), Science and Technology Development Program of the Beijing Municipal Commission of Education (KM201510025005), and Opening Project of Beijing Key Laboratory of Metabolic Disorders Related Cardiovascular Diseases (2015DXWL01).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Benezra M, Penate-Medina O, Zanzonico PB, et al. Multimodal silica nanoparticles are effective cancer-targeted probes in a model of human melanoma. J Clin Invest. 2011;121(7):2768–2780. doi: 10.1172/JCI45600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park EJ, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009;184(1):18–25. doi: 10.1016/j.toxlet.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Duan J, Yu Y, Li Y, et al. Low-dose exposure of silica nanoparticles induces cardiac dysfunction via neutrophil-mediated inflammation and cardiac contraction in zebrafish embryos. Nanotoxicology. 2016;10(5):575–585. doi: 10.3109/17435390.2015.1102981. [DOI] [PubMed] [Google Scholar]
- 4.Petrick L, Rosenblat M, Paland N, Aviram M. Silicon dioxide nanoparticles increase macrophage atherogenicity: stimulation of cellular cytotoxicity, oxidative stress, and triglycerides accumulation. Environ Toxicol. 2016;31(6):713–723. doi: 10.1002/tox.22084. [DOI] [PubMed] [Google Scholar]
- 5.Park KH, Park WJ. Endothelial dysfunction: clinical implications in cardiovascular disease and therapeutic approaches. J Korean Med Sci. 2015;30(9):1213–1225. doi: 10.3346/jkms.2015.30.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peters K, Unger RE, Kirkpatrick CJ, Gatti AM, Monari E. Effects of nano-scaled particles on endothelial cell function in vitro: studies on viability, proliferation and inflammation. J Mater Sci Mater Med. 2004;15(4):321–325. doi: 10.1023/b:jmsm.0000021095.36878.1b. [DOI] [PubMed] [Google Scholar]
- 7.Du ZJ, Zhao DL, Jing L, et al. Cardiovascular toxicity of different sizes amorphous silica nanoparticles in rats after intratracheal instillation. Cardiovasc Toxicol. 2013;13(3):194–207. doi: 10.1007/s12012-013-9198-y. [DOI] [PubMed] [Google Scholar]
- 8.Guo C, Xia Y, Niu P, et al. Silica nanoparticles induce oxidative stress, inflammation, and endothelial dysfunction in vitro via activation of the MAPK/Nrf2 pathway and nuclear factor-kappaB signaling. Int J Nanomedicine. 2015;10:1463–1477. doi: 10.2147/IJN.S76114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cazzola M, Calzetta L, Page C, et al. Influence of N-acetylcysteine on chronic bronchitis or COPD exacerbations: a meta-analysis. Eur Respir Rev. 2015;24(137):451–461. doi: 10.1183/16000617.00002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8(12):1955–1962. doi: 10.1517/14728220802517901. [DOI] [PubMed] [Google Scholar]
- 11.Rushworth GF, Megson IL. Existing and potential therapeutic uses for N-acetylcysteine: the need for conversion to intracellular glutathione for antioxidant benefits. Pharmacol Ther. 2014;141(2):150–159. doi: 10.1016/j.pharmthera.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Xue C, Liu W, Wu J, Yang X, Xu H. Chemoprotective effect of N-acetylcysteine (NAC) on cellular oxidative damages and apoptosis induced by nano titanium dioxide under UVA irradiation. Toxicol In Vitro. 2011;25(1):110–116. doi: 10.1016/j.tiv.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Srivastava RK, Rahman Q, Kashyap MP, Lohani M, Pant AB. Ameliorative effects of dimetylthiourea and N-acetylcysteine on nanoparticles induced cyto-genotoxicity in human lung cancer cells-A549. PLoS One. 2011;6(9):e25767. doi: 10.1371/journal.pone.0025767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li YB, Zhang HX, Guo CX, et al. Cytotoxicity and DNA damage effect of TGA-capped CdTe quantum dots. Chem Res Chin Univ. 2012;28(2):276–281. [Google Scholar]
- 15.Chithrani BD, Ghazani AA, Chan WC. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6(4):662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 16.Alarifi S, Ali D, Alakhtani S, Al Suhaibani ES, Al-Qahtani AA. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol Trace Elem Res. 2014;157(1):84–93. doi: 10.1007/s12011-013-9871-9. [DOI] [PubMed] [Google Scholar]
- 17.Sadeghnia HR, Zoljalali N, Hanafi-Bojd MY, Nikoofal-Sahlabadi S, Malaekeh-Nikouei B. Effect of mesoporous silica nanoparticles on cell viability and markers of oxidative stress. Toxicol Mech Methods. 2015;25(6):433–439. [PubMed] [Google Scholar]
- 18.Mateo D, Morales P, Avalos A, Haza AI. Oxidative stress contributes to gold nanoparticle-induced cytotoxicity in human tumor cells. Toxicol Mech Methods. 2014;24(3):161–172. doi: 10.3109/15376516.2013.869783. [DOI] [PubMed] [Google Scholar]
- 19.Sun L, Li Y, Liu XM, et al. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicology In Vitro. 2011;25(8):1619–1629. doi: 10.1016/j.tiv.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 20.Goncalves DM, Girard D. Zinc oxide nanoparticles delay human neutrophil apoptosis by a de novo protein synthesis-dependent and reactive oxygen species-independent mechanism. Toxicol In Vitro. 2014;28(5):926–931. doi: 10.1016/j.tiv.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 21.Chairuangkitti P, Lawanprasert S, Roytrakul S, et al. Silver nanoparticles induce toxicity in A549 cells via ROS-dependent and ROS-independent pathways. Toxicol In Vitro. 2013;27(1):330–338. doi: 10.1016/j.tiv.2012.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Tripathi DK, Singh VP, Prasad SM, Chauhan DK, Dubey NK. Silicon nanoparticles (SiNp) alleviate chromium (VI) phytotoxicity in Pisum sativum (L.) seedlings. Plant Physiol Biochem. 2015;96:189–198. doi: 10.1016/j.plaphy.2015.07.026. [DOI] [PubMed] [Google Scholar]
- 23.Duan J, Yu Y, Li Y, et al. Silica nanoparticles enhance autophagic activity, disturb endothelial cell homeostasis and impair angiogenesis. Part Fibre Toxicol. 2014;11:50. doi: 10.1186/s12989-014-0050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi AO, Cho SJ, Desbarats J, Lovric J, Maysinger D. Quantum dot-induced cell death involves Fas upregulation and lipid peroxidation in human neuroblastoma cells. J Nanobiotechnology. 2007;5:1. doi: 10.1186/1477-3155-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Sun X, Lin Y, et al. Endothelial cell injury and dysfunction induced by silver nanoparticles through oxidative stress via IKK/NF-kappaB pathways. Biomaterials. 2014;35(24):6657–6666. doi: 10.1016/j.biomaterials.2014.04.093. [DOI] [PubMed] [Google Scholar]
- 26.Davda J, Labhasetwar V. Characterization of nanoparticle uptake by endothelial cells. Int J Pharm. 2002;233(1–2):51–59. doi: 10.1016/s0378-5173(01)00923-1. [DOI] [PubMed] [Google Scholar]
- 27.Long JF, Waldman WJ, Kristovich R, Williams M, Knight D, Dutta PK. Comparison of ultrastructural cytotoxic effects of carbon and carbon/iron particulates on human monocyte-derived macrophages. Environ Health Perspect. 2005;113(2):170–174. doi: 10.1289/ehp.7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Sun JA. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-kappa B pathways. Biomaterials. 2010;31(32):8198–8209. doi: 10.1016/j.biomaterials.2010.07.069. [DOI] [PubMed] [Google Scholar]
- 29.Cheng G, Guo W, Han L, et al. Cerium oxide nanoparticles induce cytotoxicity in human hepatoma SMMC-7721 cells via oxidative stress and the activation of MAPK signaling pathways. Toxicol In Vitro. 2013;27(3):1082–1088. doi: 10.1016/j.tiv.2013.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Cai Z, Lou Q, Wang F, et al. N-acetylcysteine protects against liver injure induced by carbon tetrachloride via activation of the Nrf2/HO-1 pathway. Int J Clin Exp Pathol. 2015;8(7):8655–8662. [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Zhu Z, Liu J, Hu Z. Protective effect of N-acetylcysteine (NAC) on renal ischemia/reperfusion injury through Nrf2 signaling pathway. J Recept Signal Transduct Res. 2014;34(5):396–400. doi: 10.3109/10799893.2014.908916. [DOI] [PubMed] [Google Scholar]
- 32.Ahamed M, Akhtar MJ, Siddiqui MA, et al. Oxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cells. Toxicology. 2011;283(2–3):101–108. doi: 10.1016/j.tox.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Stone V, Donaldson K. Nanotoxicology: signs of stress. Nat Nanotechnol. 2006;1(1):23–24. doi: 10.1038/nnano.2006.69. [DOI] [PubMed] [Google Scholar]
- 34.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10(3):579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 35.Verma G, Datta M. The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. J Cell Physiol. 2012;227(5):1791–1795. doi: 10.1002/jcp.22903. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Deng X, Zhang F, Chen D, Ding W. ZnO nanoparticle-induced oxidative stress triggers apoptosis by activating JNK signaling pathway in cultured primary astrocytes. Nanoscale Res Lett. 2014;9(1):117. doi: 10.1186/1556-276X-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen KC, Willmore WG, Tayabali AF. Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology. 2013;306:114–123. doi: 10.1016/j.tox.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 38.Schroeter H, Boyd CS, Ahmed R, et al. c-Jun N-terminal kinase (JNK)-mediated modulation of brain mitochondria function: new target proteins for JNK signalling in mitochondrion-dependent apoptosis. Biochem J. 2003;372(Pt 2):359–369. doi: 10.1042/BJ20030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markou T, Dowling AA, Kelly T, Lazou A. Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic Res. 2009;43(9):809–816. doi: 10.1080/10715760903071649. [DOI] [PubMed] [Google Scholar]
- 41.Kim BJ, Ryu SW, Song BJ. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J Biol Chem. 2006;281(30):21256–21265. doi: 10.1074/jbc.M510644200. [DOI] [PubMed] [Google Scholar]
- 42.Changchien JJ, Chen YJ, Huang CH, Cheng TL, Lin SR, Chang LS. Quinacrine induces apoptosis in human leukemia K562 cells via p38 MAPK-elicited BCL2 down-regulation and suppression of ERK/c-Jun-mediated BCL2L1 expression. Toxicol Appl Pharmacol. 2015;284(1):33–41. doi: 10.1016/j.taap.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Chambers JW, LoGrasso PV. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem. 2011;286(18):16052–16062. doi: 10.1074/jbc.M111.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajawat YS, Bossis I. Autophagy in aging and in neurodegenerative disorders. Hormones (Athens) 2008;7(1):46–61. doi: 10.14310/horm.2002.1111037. [DOI] [PubMed] [Google Scholar]
- 45.Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9:20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li JJ, Hartono D, Ong CN, Bay BH, Yung LY. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31(23):5996–6003. doi: 10.1016/j.biomaterials.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Ha SW, Weitzmann MN, Beck GR. Bioactive silica nanoparticles promote osteoblast differentiation through stimulation of autophagy and direct association with LC3 and p62. ACS Nano. 2014;8(6):5898–5910. doi: 10.1021/nn5009879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamawaki H, Iwai N. Cytotoxicity of water-soluble fullerene in vascular endothelial cells. Am J Physiol Cell Physiol. 2006;290(6):C1495–C1502. doi: 10.1152/ajpcell.00481.2005. [DOI] [PubMed] [Google Scholar]
- 49.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 50.Schrijvers DM, De Meyer GR, Martinet W. Autophagy in atherosclerosis: a potential drug target for plaque stabilization. Arterioscler Thromb Vasc Biol. 2011;31(12):2787–2791. doi: 10.1161/ATVBAHA.111.224899. [DOI] [PubMed] [Google Scholar]
- 51.Perrotta I, Aquila S. The role of oxidative stress and autophagy in atherosclerosis. Oxid Med Cell Longev. 2015;2015:130315. doi: 10.1155/2015/130315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009;16(7):1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 53.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun T, Yan Y, Zhao Y, Guo F, Jiang C. Copper oxide nanoparticles induce autophagic cell death in A549 cells. PLoS One. 2012;7(8):e43442. doi: 10.1371/journal.pone.0043442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Duan J, Li Y, et al. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater. 2014;270:176–186. doi: 10.1016/j.jhazmat.2014.01.028. [DOI] [PubMed] [Google Scholar]
- 56.Kabeya Y, Mizushima N, Uero T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nowak JS, Mehn D, Nativo P, et al. Silica nanoparticle uptake induces survival mechanism in A549 cells by the activation of autophagy but not apoptosis. Toxicol Lett. 2014;224(1):84–92. doi: 10.1016/j.toxlet.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Schutz I, Lopez-Hernandez T, Gao Q, et al. Lysosomal dysfunction caused by cellular accumulation of silica nanoparticles. J Biol Chem. 2016;291(27):14170–14184. doi: 10.1074/jbc.M115.710947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shapero K, Fenaroli F, Lynch I, Cottell DC, Salvati A, Dawson KA. Time and space resolved uptake study of silica nanoparticles by human cells. Mol Biosyst. 2011;7(2):371–378. doi: 10.1039/c0mb00109k. [DOI] [PubMed] [Google Scholar]
- 60.Kusaka T, Nakayama M, Nakamura K, Ishimiya M, Furusawa E, Ogasawara K. Effect of silica particle size on macrophage inflammatory responses. PLoS One. 2014;9(3):e92634. doi: 10.1371/journal.pone.0092634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miyayama T, Matsuoka M. Involvement of lysosomal dysfunction in silver nanoparticle-induced cellular damage in A549 human lung alveolar epithelial cells. J Occup Med Toxicol. 2016;11:1. doi: 10.1186/s12995-016-0090-0. eCollection 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5(11):886–897. doi: 10.1038/nrc1738. [DOI] [PubMed] [Google Scholar]
- 63.Petters C, Thiel K, Dringen R. Lysosomal iron liberation is responsible for the vulnerability of brain microglial cells to iron oxide nanoparticles: comparison with neurons and astrocytes. Nanotoxicology. 2016;10(3):332–342. doi: 10.3109/17435390.2015.1071445. [DOI] [PubMed] [Google Scholar]
- 64.Wan B, Wang ZX, Lv QY, et al. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol Lett. 2013;221(2):118–127. doi: 10.1016/j.toxlet.2013.06.208. [DOI] [PubMed] [Google Scholar]
- 65.Ma XW, Wu YY, Jin SB, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. doi: 10.1021/nn202155y. [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Zhang H, Liang X, et al. Iron oxide nanoparticles induce autophagosome accumulation through multiple mechanisms: lysosome impairment, mitochondrial damage, and ER stress. Mol Pharm. 2016;13(7):2578–2587. doi: 10.1021/acs.molpharmaceut.6b00405. [DOI] [PubMed] [Google Scholar]
- 67.Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17(9):422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 68.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roy R, Singh SK, Chauhan LK, Das M, Tripathi A, Dwivedi PD. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol Lett. 2014;227(1):29–40. doi: 10.1016/j.toxlet.2014.02.024. [DOI] [PubMed] [Google Scholar]
- 70.Duan J, Yu Y, Li Y, et al. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int J Nanomedicine. 2014;9:5131–5141. doi: 10.2147/IJN.S71074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xiao D, Choi S, Johnson DE, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23(33):5594–5606. doi: 10.1038/sj.onc.1207747. [DOI] [PubMed] [Google Scholar]
- 72.Li C, Xing G, Dong M, et al. Beta-asarone protection against beta-amyloid-induced neurotoxicity in PC12 cells via JNK signaling and modulation of Bcl-2 family proteins. Eur J Pharmacol. 2010;635(1–3):96–102. doi: 10.1016/j.ejphar.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 73.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 74.Liu L, Fang YQ, Xue ZF, He YP, Fang RM, Li L. Beta-asarone attenuates ischemia-reperfusion-induced autophagy in rat brains via modulating JNK, p-JNK, Bcl-2 and Beclin 1. Eur J Pharmacol. 2012;680(1–3):34–40. doi: 10.1016/j.ejphar.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 75.Wang C, Chen K, Xia Y, et al. N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS One. 2014;9(9):e108855. doi: 10.1371/journal.pone.0108855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Werzowa J, Koehrer S, Strommer S, et al. Vertical inhibition of the mTORC1/mTORC2/PI3K pathway shows synergistic effects against melanoma in vitro and in vivo. J Invest Dermatol. 2011;131(2):495–503. doi: 10.1038/jid.2010.327. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Frankel A, Radvanyi LG, Penn LZ, Miller RG, Mills GB. Rapamycin enhances apoptosis and increases sensitivity to cisplatin in vitro. Cancer Res. 1995;55(9):1982–1988. [PubMed] [Google Scholar]
- 78.Hur GM, Ryu YS, Yun HY, et al. Hepatic ischemia/reperfusion in rats induces iNOS gene transcription by activation of NF-kappaB. Biochem Biophys Res Commun. 1999;261(3):917–922. doi: 10.1006/bbrc.1999.1143. [DOI] [PubMed] [Google Scholar]
- 79.Weigand MA, Plachky J, Thies JC, et al. N-acetylcysteine attenuates the increase in alpha-glutathione S-transferase and circulating ICAM-1 and VCAM-1 after reperfusion in humans undergoing liver transplantation. Transplantation. 2001;72(4):694–698. doi: 10.1097/00007890-200108270-00023. [DOI] [PubMed] [Google Scholar]
- 80.Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free Radic Biol Med. 2005;38(1):136–145. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Lysosome impairment of HUVECs exposed to the SiNPs (50 μg/mL) for 24 hours.
Notes: LysoTracker Red (Beyotime) was used for the staining of lysosome in HUVECs. Compared with the control group, the significant decrease of fluorescence intensity in the SiNPs group indicated that SiNPs induced lysosome impairment in HUVECs.
Abbreviations: HUVECs, human umbilical vein endothelial cells; SiNPs, silica nanoparticles.
TEM images of HUVECs exposed to the SiNPs (50 μg/mL) for 24 hours.
Notes: (A) Control group. (B) The magnification of selected area of control showed evidently intact mitochondria. (C) The induction of mitochondrial swelling and cristae rupturing and disappearance after SiNPs exposure (black arrows), and also SiNPs deposition in mitochondria. (D and E) Severe mitochondrial swelling in SiNPs-treated endothelial cells (black arrows), and (E and F) autophagosome including cytoplasmic material, especially impaired mitochondria (white arrow).
Abbreviations: TEM, transmission electron microscopy; HUVECs, human umbilical vein endothelial cells; SiNPs, silica nanoparticles.