

ABSTRACT
Cell identity involves both selective gene activity and specialization of cytoplasmic architecture and protein machinery. Similarly, reprogramming differentiated cells requires both genetic program alterations and remodeling of the cellular architecture. While changes in genetic and epigenetic programs have been well documented in dedifferentiating cells, the pathways responsible for remodeling the cellular architecture and eliminating specialized protein complexes are not as well understood. Here, we utilize a zebrafish model of adult muscle regeneration to study cytoplasmic remodeling during cell dedifferentiation. We describe activation of autophagy early in the regenerative response to muscle injury, while blocking autophagy using chloroquine or Atg5 and Becn1 knockdown reduced the rate of regeneration with accumulation of sarcomeric and nuclear debris. We further identify Casp3/caspase 3 as a candidate mediator of cellular reprogramming and Fgf signaling as an important activator of autophagy in dedifferentiating myocytes. We conclude that autophagy plays a critical role in cell reprogramming by regulating cytoplasmic remodeling, facilitating the transition to a less differentiated cell identity.
KEYWORDS: Atg5, autolysosome, dedifferentiation, electron microscopy, extraocular muscle, regeneration, reprogramming, stem cell, strabismus, zebrafish
Introduction
In multicellular organisms, cellular differentiation from a multipotent progenitor involves genetic programs that lead to increasing specialization, with formation of organs and tissues. Losing specialization through somatic cell reprogramming requires turning off the genetic program for specialization, activating the genetic program of the more pluripotent progenitor cell, and converting the cell components (cytoskeleton, organelles, plasma membrane receptors, etc.) from a differentiated to a more potent state. Much effort has been made to unlock the genetics of cellular reprogramming that underlies dedifferentiation. However, the mechanisms that govern disassembly or removal of the cytoplasmic apparatus during cellular reprogramming have received less attention.
Autophagy (“self-eating”) is a normal cellular process that degrades unnecessary or dysfunctional components. During macroautophagy (hereafter autophagy), intracellular components, such as long-lived proteins and damaged or superfluous organelles, are engulfed within double-membrane compartments termed phagophores that mature into autophagosomes; the latter fuse with lysosomes, generating autolysosomes, resulting in degradation of the autophagosomal cargo.1 Defects in autophagy cause embryonic lethality2 and are associated with a range of pathophysiologies including degenerative diseases3 and cancer.4
Zebrafish is a powerful model for vertebrate embryogenesis and regenerative medicine due to its innate high capacity to regenerate adult tissues. These tissues include retina,5 muscle,6,7 heart8,9 and fin.10,11 Zebrafish regenerates adult damaged tissues through either formation of a blastema or, alternatively, through cellular reprogramming of differentiated cells from the damaged tissue.7,12-14 The ability of zebrafish to reprogram differentiated cells into multipotent progenitor cells may be a particularly important regenerative pathway. Research on regeneration through cellular reprogramming and dedifferentiation, a mechanism that does not rely on stem cells, has mainly focused on the genetic mechanisms that underlie the reprogramming process. This leaves unanswered a biological dilemma: muscles are a highly specialized tissue composed mostly of sarcomeres, the contractile protein machinery; therefore, muscle reprogramming would require active disassembly of the sarcomeres, a process likely requiring substantial degradation.
We hypothesized that autophagy plays a key role in cell reprogramming during muscle regeneration by participating in sarcomere disassembly. To test our hypothesis, we took advantage of our recently described adult zebrafish extraocular muscle (EOM) regeneration model.7 We discovered that after a large injury (50% myectomy), myocytes activated autophagy to disassemble the sarcomeres, facilitating nuclear reprogramming and cellular proliferation. In this model, autophagy was mainly regulated at the protein level and Fgf (fibroblast growth factor) played a role in this regulation. We conclude that cellular dedifferentiation involves both nuclear and cytoplasmic reprogramming, giving autophagy a key role in the process.
Results
Autophagy is upregulated during zebrafish EOM regeneration
As previously determined, reprogramming occurs within 18 h postinjury (hpi).7 Therefore, our studies focused on this time period. To determine whether zebrafish EOMs activate autophagy following a large myectomy (50% of the muscle), several approaches were followed. First, wild-type fish underwent lateral rectus myectomy, and were stained with LysoTracker Red and processed for cryosectioning 16 hpi. LysoTracker Red is a vital dye that nonspecifically accumulates in acidic compartments such as autolysosomes. LysoTracker Red staining in the injured muscle was evident at lower (Fig. 1A, B) and high (Fig. 1C, D) magnification, suggesting autophagy activation. Map1lc3a/b (microtubule-associated protein 1 light chain 3 α/β; Lc3 hereafter) is a protein that is ubiquitously distributed in the cytoplasm. During the induction of autophagy the soluble form (Lc3-I) is conjugated to a form (Lc3-II) that localizes to, and accumulates within, the autophagosome membrane.15 Transgenic fish expressing GFP-tagged Lc3 (GFP-Lc3)16 underwent LR myectomy. Next, they were stained with LysoTracker Red and visualized by whole-mount craniectomy (Fig. 1E to I ). Both LysoTracker Red (Fig. 1J) and GFP-Lc3 (Fig. 1K, M) clearly accumulated in the injured muscle (Fig. 1L), suggesting an increase in autophagy. Last, autophagy activation was tested by Lc3 western blot hybridization. As expected, Lc3-II accumulated in the injured muscle (Fig. 1N) supporting the conclusion that there was activation of autophagy.
Figure 1.
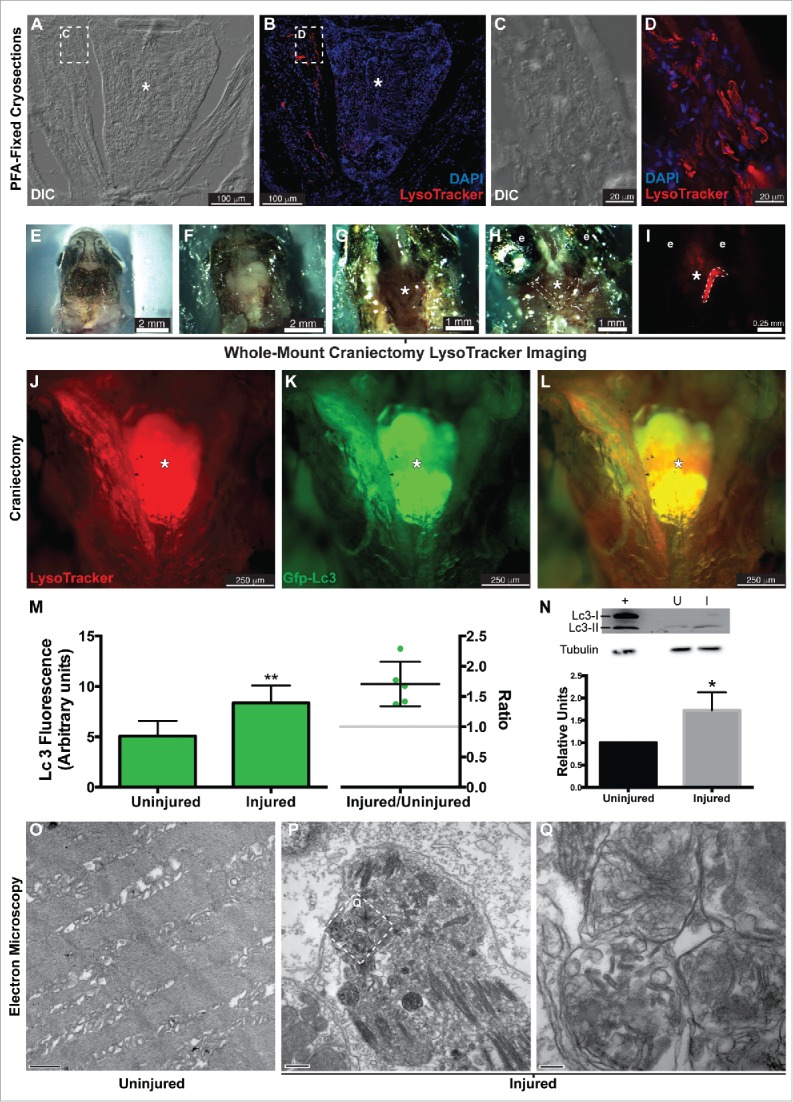
Autophagy in the regenerating EOMs. (A to L) LysoTracker Red was used to label autophagy in the regenerating LR (left side), after staining fish heads were processed for cryosectioning ((A to D). (A, B) Sections containing the regenerating LR were imaged at lower magnification. (C, D) Higher-resolution detail of the boxes in (A)and (B), respectively. ((E)to L) Craniectomy was also used to visualize LysoTracker Red labeling in situ. At 16 hpi, zebrafish heads were mounted in agarose (E), and the top of the skulls was removed (F). Brain was extracted to expose the skull base (*) where the pituitary is located and both LR muscles attach to the bone (G). Then the lateral bones of the skull were removed to allow complete visualization of the LR muscles (H). (I) Fluorescent visualization of H. LysoTracker Red (J) and GFP-Lc3 (K) clearly accumulate and colocalize (L) in the regenerating LR. (M) Fluorescence intensity of GFP signal was therefore higher in the injured muscle; in fact, the ratio of the intensities of injured vs control was higher than 1 in every fish, meaning a net increase of GFP-Lc3 content of the injured muscle (Student t test, **, P < 0.01, n = 5). (N) Western blot of Lc3 showed an increase of Lc3-II in the injured muscle, indicating again autophagy activation (Mann-Whitney test, *, P < 0.05, n = 4). Protein loading was assayed with an anti-Tubg1/tubulin antibody. +, positive control (rat protein); U, uninjured; I, injured. (O to Q) Electron microscopy showed that double-membrane organelles were easily detected in the injured muscle (P, scale bar: 500 nm; Q, scale bar: 100 nm) while virtually undetectable in the control muscle (O, scale bar: 500 nm). Check Figure 3O for a diagram of a craniectomyzed zebrafish head.
The data presented so far support the hypothesis that autophagy is activated in the regenerating muscle within the critical 16 hpi window. However, Lc3-II accumulation and LysoTracker Red staining are also found in other biological processes. Therefore, we next analyzed the injured muscle at 16 hpi, when LysoTracker Red (Fig. 1J) and GFP-Lc3 (Fig. 1K, M) were detected, by transmission electron microscopy (TEM) to determine the presence or absence of autophagosomes (double-membrane organelles unique to autophagy). TEM revealed that the injured muscle was replete with autophagosomes (Fig. 1P, Q), whereas uninjured control muscles were generally devoid of autophagosomes (Fig. 1O), confirming that autophagy is activated during myocyte reprogramming in the injured LR muscle.
Next, expression of genes involved in the autophagy process was measured by qRT-PCR at 14 hpi (Fig. 2). Among the analyzed genes, atg5/autophagy-related 5 was upregulated in the injured muscle, while the other tested genes were not. We interpret these collective data to indicate that activation of autophagy in our model is primarily regulated at the protein level.
Figure 2.
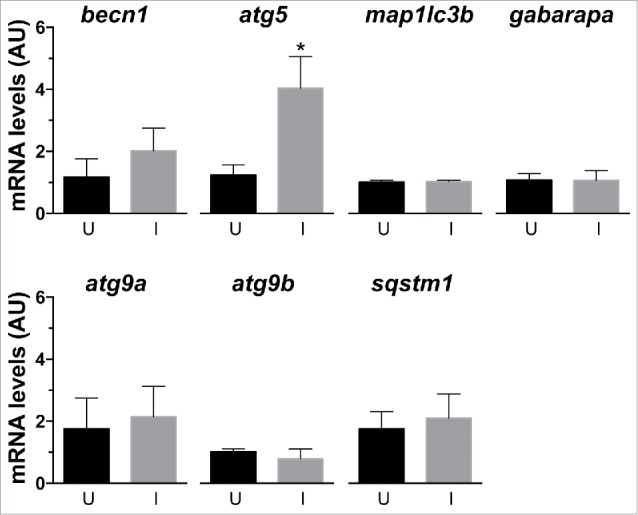
Analysis of expression of genes involved in autophagy. Gene expression in uninjured (U) and injured (I) LR muscles was assessed using qRT-PCR. Among the studied genes only atg5 was differentially expressed in the injured muscles, at levels 4-fold higher than control muscles (*, P < 0.05, n = 5). Gene mRNA levels (arbitrary units [AU]) were normalized to 18s rRNA using the ΔΔ C(t) method.
Autophagy is required for EOMs regeneration
Activation of autophagy does not necessarily imply a key role for this process in EOM regeneration. Therefore, we decided to block autophagy and evaluate regeneration. First, we confirmed that CQ treatment was effective blocking autophagy by Sqstm1/Sequestosome 1-Lc3 western blot (Fig. 3A, S1) and by LysoTracker Red staining and our craniectomy approach to visualize the injured muscle (Fig. 3B to G). Next, we took advantage of the craniectomy technique to measure LR regeneration as previously described.7 We found a significant decrease of LR regeneration in fish treated with 1 mM CQ compared with untreated control fish at 3 d (Fig. 3H to J). To confirm this phenotype, we treated fish with different CQ doses (0, 0.5, 1 and 2 mM CQ) and extended the length of the regenerating period to 5 d. The length of the muscle beyond the myectomy site reflects the length of newly regenerated muscle. Since myectomy left approximately 50% of the muscle remaining, the measurements are shown for the entire muscle with a gray box indicating the approximate location of the myectomy site (gray area in Fig. 3J to M). Our measurements reveal that muscle regeneration in control fish was significantly higher than in the CQ treated groups (Fig. 3K).
Figure 3.
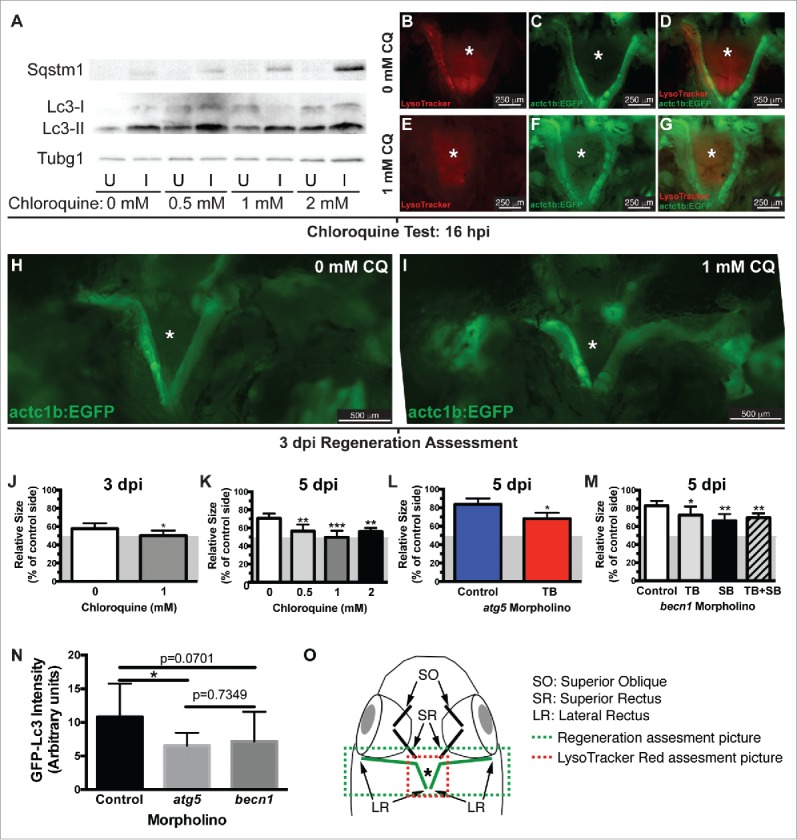
Autophagy is required for EOM regeneration. (A) Western blot of fish treated with increasing CQ concentrations showed a dose-dependent increase of Sqstm1 in the injured muscle and Lc3-II in both injured and uninjured muscles (see Figure S1), indicating real autophagy flux. Protein loading was assayed with an anti-Tubg1 antibody. U, uninjured; I, injured. (B o G) The lack of LysoTracker Red staining in injured LR muscles of transgenic actc1b/α-actin:EGFP zebrafish (to visualize the muscles) using our craniectomy technique confirmed that CQ effectively blocked autophagy. (H to J) Craniectomy of an untreated (H) and CQ-treated fish (I) at 3 dpi; note the difference in length of the injured muscle between both groups (pictures are representative examples of 5 fish per group). (J) Quantification of LR muscle regeneration of fish shown in H and I; values are averages ± SD (Student t test, *, P < 0.05, n = 5). (K) Quantification of LR muscle regeneration at 5 dpi treated with different CQ concentrations; values are averages ± SD (One-way ANOVA, P < 0.0001, n=5; **, P < 0.01; ***, P < 0.001; Newman-Keuls multiple comparisons test). (L) Regeneration of LR muscles injected with control or atg5 translation-blocking (TB) MO was measured at 5 dpi; values are averages ± SD (Student t test, *, P < 0.05, n = 5). (M) Regeneration of LR muscles injected with control or becn1 MOs (translation-blocking, TB; splicing-blocking, SB; combination of both, TB+SB) was measured at 5 dpi; values are averages ± SD (Student t test; *, P < 0.05; **, P < 0.01, n = 5). (J to M) Since myectomy left approximately 50% of the muscle remaining, the measurements are shown for the entire regenerating muscle with a gray box indicating the approximate location of the myectomy site. (N) Fluorescence intensity of LR muscles of GFP-Lc3 fish injected with control, atg5 TB or becn1 TB+SB MOs was measured at 18 hpi; values are averages ± SD (Student t test; *, P < 0.05, n = 8). (O) Diagram of a craniectomized zebrafish head; muscles visualized by this technique are shown, and LR muscles are highlighted in green. Green and red boxes show approximately the picture used for regeneration or LysoTracker Red (or GFP-Lc3) assessment, respectively. * Skull base where the pituitary is located.
To further test the role of autophagy in EOM regeneration, Atg5 expression was knocked down using previously described translation-blocking antisense MOs,17,18 which were delivered via microinjection followed by electroporation prior to myectomy. LR regeneration at 5 dpi was measured as before, showing that knockdown of Atg5 effectively reduced LR regeneration (Fig. 3L). To confirm these results, becn1/beclin 1 translation-blocking and splicing-blocking antisense MOs were designed. Knockdown of becn1 expression effectively reduced LR regeneration (Fig. 3M). The efficacy of atg5 and becn1 MOs inhibiting autophagy was confirmed by injecting them into LR muscles of GFP-Lc3 fish and measuring the GFP fluorescence intensity of the injured muscle (Fig. 3N). Hence, knockdown of either Atg5 or Becn1 expression further confirmed the key role of autophagy in LR regeneration.
Autophagy is not required for nutrient or energy recycling in regenerating muscle
We next turned our attention to the potential role of autophagy in regeneration. One of the biological functions of autophagy is to recycle cellular contents into nutrients and energy during periods of stress and starvation. To test this idea, fish were fasted (20% weight loss) to decrease the circulating levels of nutrient supplies. At this point, fed and fasted fish underwent LR myectomy and were treated with (1 mM CQ) or without CQ, to generate 4 different comparison groups. Fish were injected with EdU at 20 hpi and sacrificed at 24 hpi to analyze cell proliferation (Fig. 4A to H). Both fasting treatment (“fed 0 mM” versus “fasted 0 mM;” Fig. 4I) and CQ treatment (“fed 0 mM” vs. “fed 1 mM;” Fig. 4I) reduced cell proliferation. Cell proliferation in fasted fish was further reduced by CQ treatment (“fasted 0 mM” versus “fasted 1 mM;” Fig. 4I), and the effect was similar to the effect of CQ in fed fish (Fig. 4J). The interaction between feeding condition and CQ treatment was further analyzed by 2-way ANOVA, showing that although both fasting (P=0.0176) and CQ treatment (P=0.0018) have a statistically significant effect, the interaction between them was not statistically significant (P=0.2909). We interpret our results to mean that there is no synergy between CQ inhibition and nutrient deprivation in regulating EOM regeneration, and therefore the main role of autophagy in EOM regeneration is not to provide energy or recycle nutrients.
Figure 4.
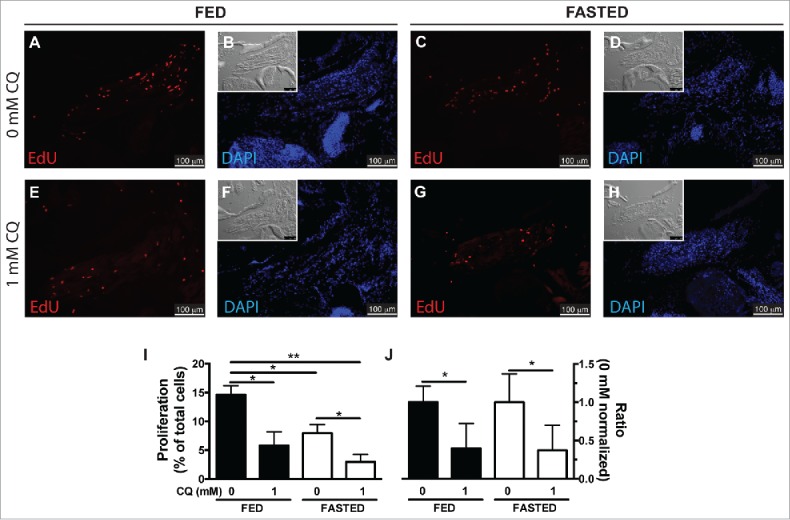
Autophagy as an energy-providing and recycling process. (A to H) Fed and fasted fish were myectomized and treated with or without CQ. Proliferation was visualized at 24 hpi staining for EdU; the inset shows the differential interference contrast image. (I) Quantification of proliferation showed that both fasting and CQ treatment reduced proliferation in the regenerating LR muscle; values are averages ± SEM (Student t test, *, P < 0.05; **, P < 0.01; n = 4). (J) To better illustrate the interaction of feeding condition and CQ treatment, proliferation values were normalized to the 0 mM CQ value of each group showing that CQ treatment reduced proliferation similarly in both fed and fasted fish. Values are averages ± SEM (Student t test; *, P < 0.05, n = 4).
Autophagy is required for the proper cytoplasmic organization of the regenerated EOMs
Histological assessment using light microscopy at 5 dpi of regenerating LR muscles of fish treated with CQ did not show drastic changes at the microscopic level when compared to regenerating muscles of untreated fish (data not shown). However, they were clearly different when their ultrastructure was investigated by TEM. Five days after injury, the muscle of control fish was composed of the typical sarcomeric structure (Fig. 5A to C) and multiple vesicular structures that could be autophagosomes and/or lipid droplets (Fig. 5A and B). In contrast, the regenerating muscle of CQ-treated fish showed a clear accumulation of unresolved autophagosomes (Fig. 5D to G) and failed to properly organize the sarcomeres (Fig. 5H and I). Importantly, sarcomeric remnants appeared to be present within accumulated autophagosomes.
Figure 5.
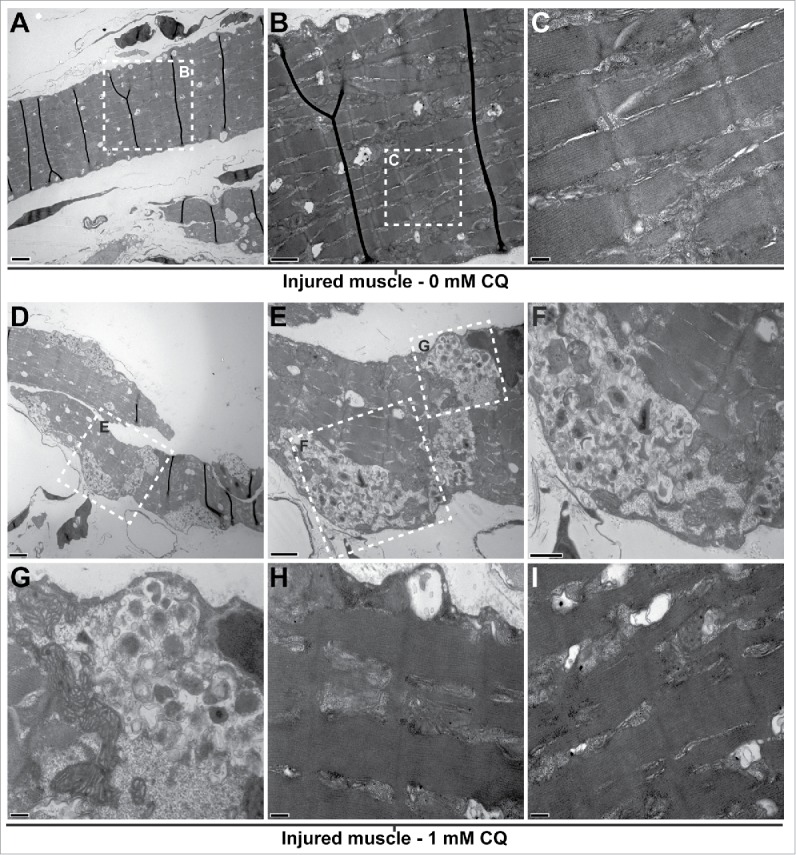
Blocking autophagy affects cytoplasmic organization of the regenerating muscle. Fish were myectomized and treated with or without CQ, 5 d later the regenerating muscle was extracted and imaged by electron microscopy. (A) Electron micrography of regenerating muscles without CQ treatment, scale bar: 2 μm. (B, C) High-magnification images of the boxes in (A)and (B)showing the typical sarcomeric structure; scale bars represent 1 μm and 200 nm, respectively. (D) Overview of the regenerating muscle of treated fish, note the high amount of unresolved autophagosomes, scale bar: 2 μm. (E) View of the box in (D)showing an example of unresolved autophagosomes, scale bar: 1 μm. (F, G) High-magnification views of the boxes in (E)revealed that the autophagosomes were filled with undigested cellular debris; scale bars represent 600 nm and 200 nm, respectively. (H, I) In addition to the unresolved autophagosomes, the analysis of the CQ-treated fish revealed that the regenerating muscle contained a high proportion of disorganized sarcomeres (compare to C); scale bars: 200 nm. This was not found in the injured muscles of untreated fish.
Autophagy inhibition does not induce apoptosis in regenerating muscle
Since inhibition of autophagy triggers apoptosis in some models,19,20 we tested the possibility that blocking autophagy reduced LR regeneration via apoptosis of dedifferentiating muscle fibers. First, levels of active Casp3 were analyzed by western blot in fish treated with CQ and compared to those of untreated fish. Surprisingly, we found higher levels of active Casp3 in the injured muscle of untreated fish than in the uninjured muscle. Treatment with CQ clearly reduced the levels of active Casp3 in the injured muscle while mildly increasing its levels in the uninjured muscle (Figs. 6A, S1). Next, we analyzed cell death in the regenerating muscle by TUNEL assay. The injured muscle of untreated fish did not show TUNEL staining (Fig. 6B, C). By contrast, the injured muscle of CQ treated fish showed high levels of TUNEL staining (Fig. 6D, E). Interestingly, high-magnification images reveal clear cytoplasmic TUNEL staining (Fig. 6F) instead of nuclear staining that represents apoptosis. Cytoplasmic TUNEL staining most likely represents labeled DNA fragments in unresolved autophagosomes that accumulate in the CQ treated fish, likely remnants of degraded nuclei from syncytial multinucleated myocytes as they reprogram into myoblasts (Fig. 5F, G). In conclusion, we find no evidence of apoptosis in the regenerating muscle of either CQ-treated or untreated fish.
Figure 6.
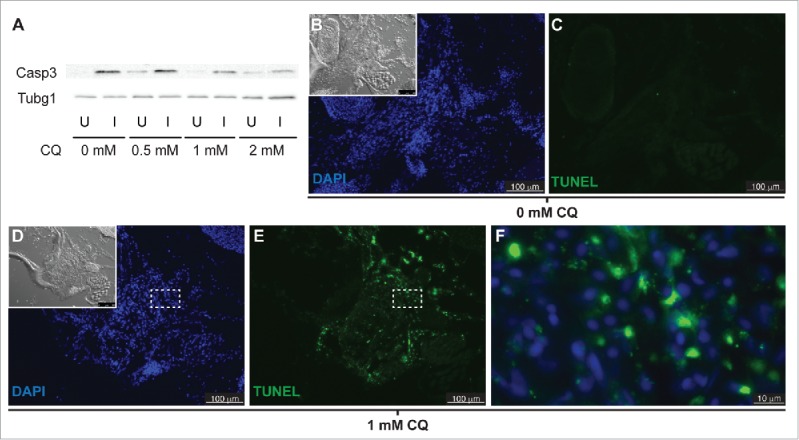
Apoptosis is not activated in response to autophagy inhibition. (A) Western blot of fish treated with increasing CQ concentrations showed a dose-dependent decrease of active Caspase 3 (Casp3) in the injured muscle (see Figure S1), suggesting a role of Casp3 in muscle regeneration. Protein loading was assayed with an anti-Tubg1/tubulin antibody. U, uninjured; I, injured. (B to F) Fish were treated with or without CQ. Apoptosis was visualized by TUNEL staining; the inset shows the differential interference contrast image (pictures are representative examples of 5 fish per group). (F) High-magnification view of the box in (D) and (E)shows cytoplasmic localization of TUNEL signal in fish treated with CQ.
Fgf promotes autophagy activation in EOMs regeneration
As a first step to understand the regulation of autophagy in muscle regeneration, we tested whether Fgf signaling plays a role in autophagy activation during EOM regeneration, as has been described in tail fin regeneration.18 Fgf signaling was blocked using the hsp70l:dnfgfr1a-EGFP fish line, which expresses a dominant-negative (dn) Fgf receptor following heat shock treatment, and autophagy was visualized using LysoTracker Red and whole-mount craniectomy (Fig. 7A, B). Ectopic expression of dnFgfr1a-EGFP significantly reduced LysoTracker Red fluorescence in the regenerating muscle (Fig. 7C), indicating a decrease in autophagy activation. To further confirm this result, LR muscles of actc1b/α-actin:EGFP and hsp70l:dnfgfr1a-EGFP fish were assessed by western blot (Fig. 7D) showing higher levels of Sqstm1 and lower levels of Lc3-II when the dnFgfr1a-EGFP protein was ectopically expressed (Fig. 7E). Therefore we conclude that Fgf signaling is necessary for the full activation of autophagy during EOM regeneration.
Figure 7.
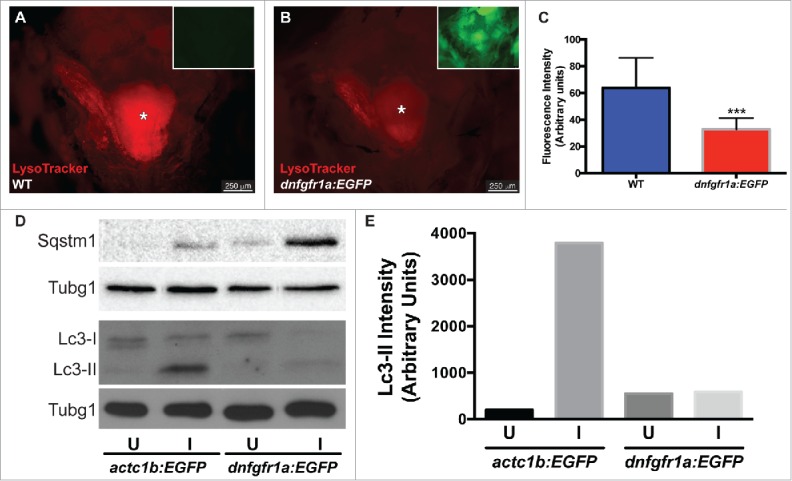
Autophagy is regulated by Fgf during EOM regeneration. (A, B) Craniectomy was again used to visualize LysoTracker Red-labeling in (A) wild-type (WT) fish, and (B) hsp70l:dnfgfr1a-EGFP; the inset shows an image of the GFP channel to confirm transgene expression and, consequently, Fgfr inhibition (pictures shown are representative examples of the 12 fish per group quantified). (C) Quantification of LysoTracker Red fluorescence intensity shows a clear decrease of LysoTracker Red labeling in hsp70l:dnfgfr1a-EGFP fish, indicating a lower autophagic flux. Values are averages ± SD (Student t test; ***, P < 0.001, n = 12). (D) Western blot of Sqstm1 and Lc3. The injured muscle of hsp70l:dnfgfr1a-EGFP fish showed higher levels of Sqstm1 and lower levels of Lc3-II (E), indicating again a lower autophagic flux. Protein loading was assayed with an anti-Tubg1/tubulin antibody. (D, E) Show a representative example of 3 independent experiments. U, uninjured; I, injured. * Skull base, where the pituitary is located.
Discussion
Cellular dedifferentiation is a highly regulated process that resets differentiated cells to a more potent progenitor state, requiring nuclear reprogramming that includes changes in chromatin and gene expression. At the same time, differentiated cells contain specialized cytoplasmic machinery that must be disassembled to truly reprogram the cell.21 The importance of cellular reprogramming in many biological processes cannot be overemphasized, with examples that include the zygote-to-embryo transition,22 tissue regeneration23 and oncogenesis.24 While the genetic regulation of cellular dedifferentiation has been heavily studied and somatic cells can now be reprogrammed into induced pluripotent stem cells overexpressing a defined set of transcription factors,25 the cytoplasmic events are still poorly understood, although their importance is already clear in the classical Xenopus nuclear transfer experiments.22
In order to obtain a better mechanistic understanding of cellular reprogramming and dedifferentiation, we investigated cytoplasmic reorganization by asking how sarcomeres are disassembled using our robust zebrafish model of adult muscle regeneration because we observed that dedifferentiated myocytes lost their sarcomeric machinery to form proliferating myoblasts during the dedifferentiation process.7 Using a multidisciplinary approach, we discovered that autophagy is induced rapidly and early in the regeneration process. Consistent with a role in sarcomere disassembly, autophagy was not involved in nutrient recycling. Furthermore, we show that inhibition of autophagy resulted in DNA accumulation in unresolved autophagosomes in the regenerating muscle and reduced proliferation of dedifferentiating myocytes, suggesting that cytoplasmic remodeling and degradation of excess nuclei are important for reprogramming syncytial multinucleated myocyte into myoblasts and for cell-cycle reentry of dedifferentiated cells. Finally, we show that inhibition of Fgf signaling was associated with reduced autophagy, providing a mechanistic framework for cytoplasmic remodeling through induction of autophagy.
Autophagy has mostly been considered a waste management and recycling process that is important during embryogenesis,2 degenerative conditions3 and cancer.4 Interestingly, recent studies have shown the role of autophagy in stem cell biology26 and cell reprogramming.21 And it has been shown that the proteome changes drastically during cellular reprogramming.27 In agreement with this, our data support the hypothesis that autophagy is critical in the regulation of cellular reprogramming, and would act in concert with genetic and epigenetic mechanisms to prepare and remodel the cell.
Eukaryotic cells have 2 primary systems for degrading cellular components: the proteasome is responsible for degrading short-lived proteins marked by covalent modifications, whereas autophagy functions on a larger scale, degrading long-lived proteins, large protein complexes, and entire organelles28 (including the nucleus29,30). Therefore, when considering the dramatic reprogramming that was observed in regenerating zebrafish EOMs, we hypothesized that autophagy would be activated to degrade the sarcomeric protein machinery and some of the nuclei of the syncytial multinucleated myocyte, to facilitate its dedifferentiation. Accordingly, TEM of regenerating zebrafish muscle revealed disorganized sarcomeres, as well as clear accumulation of unresolved DNA containing autophagosomes when autophagy was blocked. A similar phenotype of vacuolar accumulation has been reported in myopathies with autophagy defects.31 Our results indicate that autophagy is rapidly activated, presumably in concert with genetic and epigenetic mechanisms, to eliminate unnecessary cytoplasmic elements and prepare the cell to assume a new identity.
Caspase enzymes (aspartate-specific, cysteine proteases) function as key mediators of apoptosis.32 However, regenerating muscles activated Casp3 (Fig. 6) with no detectable levels of apoptosis (Figs. 6, S2). Therefore, Casp3 appears to play a different role in myocyte reprogramming during muscle regeneration, which may be related to its roles in myocyte homeostasis (reviewed by Bell et al.33). Hundreds of caspase targets have been reported34 including several key cell cycle regulators35-37 and cytoskeletal components;38 and because caspase proteolytic activity involves irreversible or long-lasting signal transduction,39 caspases are ideal candidates as regulators of cellular processes such as cell reprogramming. That might explain why active Casp3 levels decreased when myocyte cell reprogramming was impaired with CQ.
Fgf signaling is important for regeneration in both zebrafish and mammalian models,40,41 as well as in the biology of cancer stem cells,42 yet its functions in cellular reprogramming are not fully understood. Our data reveal that Fgf signaling promotes autophagy activation to remodel the cellular architecture during EOM regeneration since (1) blocking Fgf signaling reduced autophagy, and (2) blocking autophagy reduced proliferation of dedifferentiated myoblasts and also reduced the efficiency of muscle regeneration to a similar extent as seen with blocking Fgf signaling.43 The role of Fgf signaling, presumably through the Mapk-Erk pathway,44-46 suggests potential targets in the regulation of muscle disorders and repair. A recent report on the activation of autophagy by Fgf and its signaling pathway during zebrafish caudal fin regeneration18 supports our hypothesis.
In summary, we show that cytoplasmic remodeling through activation of autophagy plays a key role in zebrafish muscle regeneration. Further studies of the mechanisms underlying cytoplasmic remodeling in cells undergoing reprogramming stand to enhance our understanding of degenerative disease processes as well as facilitate development of novels therapies to promote tissue repair and block pathological cell reprogramming in cancer.
Materials and methods
Zebrafish (Danio rerio) rearing and surgeries
All animal work was performed in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and approved by the University of Michigan Committee on the Use and Care of Animals, protocol 06034. Sexually mature adult (4- to 18-mo old) zebrafish were spawned in our fish facility and raised per standard protocol at 28°C with a 14-h light/10-h dark alternating cycle.
Adult zebrafish were anesthetized (0.05% tricaine methanosulfate, Western Chemical, Tricaine-S) and approximately 50% of the lateral rectus (LR) muscle was surgically removed.7 The remaining amount of muscle left after surgery (48.42 ± 4.9%, average ± SD) is represented in Figure 3J to M as a gray area and was quantified by craniectomy as described previously.7
Specimen processing
Zebrafish were fixed in 4% paraformaldehyde (Sigma-Aldrich, 441244) and evaluated microscopically using coronal frozen sections (12 μm). Decalcification was performed using either Morse solution (45% Formic Acid, ACROS, 42375–0025; 20% Sodium Citrate, R&D Systems 3161500G) or 10% ethylenediamine-tetraacetic acid (EDTA, Fisher, BP118) in phosphate-buffered saline (pH 7.2 to 7.4; Fisher BioReagents, BP665).
Staining and biochemical analysis
Adult zebrafish were incubated in LysoTracker Red DND-99 (Thermo Fisher Scientific, L-7528) for one h and then washed in fresh fish water (3 × 20 min). Then fish were processed for cryosectioning (250 μM LysoTracker Red) or craniectomy (500 nM LysoTracker Red, Figure 1E to I) as described. LysoTracker Red staining was quantified measuring the fluorescence intensity of the area corresponding to the regenerating muscle in pictures taken from craniectomized fish with ImageJ software (http://rsbweb.nih.gov/ij/). Pictures were taken at the same time and with the same microscope settings to minimize experimental error.
Western blots were performed following standard protocols. Injured or uninjured LR from 10 to 15 fish were pooled and homogenized in lysis buffer containing protease (Roche Diagnostics Corporation, cOmplete) and phosphatase (Roche Diagnostics Corporation, PhosSTOP) inhibitors. The transgenic actc1b/α-actin:GFP fish47 were used to visualize the muscles. Anti-Tubg1/-tubulin (tubulin, gamma 1) antibody (1:10,000, T5326) was obtained from Sigma-Aldrich, anti-Map1lc3a/b (1:3,000, NB100–2331) was purchased from Novus Biologicals, anti-active Casp3/caspase 3 antibody (1:500, ab13847) was purchased from abcam and anti-Sqstm1/p62 (sequestosome 1) antibody (1:1000, 5114) was purchased from Cell Signaling Technology. Quantification of the bands was done with ImageJ software.
Electron microscopy
Thin sections (70 nm) were prepared and examined on a transmission electron microscope (JEOL JEM-1400 Plus, Peabody, MA, USA) at 80 kV as described previously.7 Briefly, fish were perfused with 4% paraformaldehyde + 1.5% glutaraldehyde (Sigma-Aldrich, 49626) in 0.1 M cacodylate buffer (pH 7.4; Hampton Research, HR2575), fixed overnight in 2.5% glutaraldehyde in 0.1 M cacodylate buffer and decalcified in 2.5% glutaraldehyde in 7.5% disodium EDTA for 2 d. Then, the LR muscles were dissected and further fixed for 2 h in 1% OsO4 in 0.1 M sodium cacodylate buffer.
RNA extraction and qRT-PCR
RNA for qRT-PCR analysis was prepared from frozen tissue sections using Laser Microdissection (LMD7000, Leica Microsystems Inc., Buffalo Grove, IL) and the Reliaprep RNA tissue Miniprep System (Promega, Z6110), following the manufacturer's protocols and recovered in 15 µl RNase-free H2O. Preparation and amplification of cDNA was made with the Ovation Pico WTA System V2 (NuGen, 3302); then it was purified using the QIAquick PCR Purification Kit (QIAGEN, 28104) and eluted in 30 µl nuclease-free H2O. Quality of amplified cDNA was assessed with the D1k ScreenTape System on an Agilent 2200 TapeStation (Agilent, Santa Clara, CA).
Gene expression was measured using CFX384 and CFX96 Real-Time PCR detection systems (Bio-Rad Laboratories, Hercules, CA). Diluted cDNA (1ng/reaction), SsoFast EvaGreen supermix (Bio-Rad Laboratories, 1725200) and specific primers (Table S1) were used in 20 µl PCR reactions following the manufacturer's suggestions; reactions were performed in triplicate. The efficiency of PCR reactions for target and reference genes varied between 86% and 105%. The dynamic range of standard curves (serial dilutions of 30 h postfertilization whole embryo cDNA) spanned 5 orders of magnitude. The specificity of reaction was verified by analysis of melting curves and by electrophoresis of PCR-amplified products. Gene expression was calculated by the ΔΔ C(t) method48 using 18S ρρνΑ as the reference gene.
Drug treatments
Chloroquine (CQ, Sigma-Aldrich, C6628), an inhibitor of lysosomal acidification, was dissolved directly in fish water. Up to 8 fish were treated in 1 L of water, and tanks were maintained at 28.5°C with CQ solutions replaced every 24 h. The experimental concentration of CQ was determined in a preliminary experiment using LysoTracker Red to label autolysosomes. Although CQ was very effective blocking autophagy in a wide range of concentrations (0.5 to 2 mM), fish mortality varied among experiments with no clear relationship to fish or CQ batches. Therefore, unless stated otherwise, several CQ concentrations were used in each experiment and the result shown represents the highest CQ concentration group with no significant mortality.
Morpholino oligonucleotide injection and electroporation
Microinjection of morpholino oligonucleotides (MOs; Gene-Tools, LLC, Philomath, OR), a widely used technique to perform knockdown experiments in adult zebrafish,49-51 was used. Briefly,s lissamine-tagged MOs were directly microinjected into the LR muscle followed by electroporation (6 to 10 pulses at 48 V cm, BTX ECM830 electroporator; Harvard Apparatus, Holliston, MA). The MO sequence for the atg5 gene was previously validated in zebrafish embryos17 and in adult zebrafish tail regeneration.18 The sequences for becn1 translation-blocking and splicing-blocking MOs were CTAGAAAACCTCAAAGTCTCCATGC and TCATCCTGCAAAACACAAATGGCTT, respectively.
Fish fasting
Wild-type fish (n=56) were initially weighted and measured to select fish with similar body mass and condition factor (data not shown). Twenty of the selected fish were randomly divided in 2 groups (289.5 ± 12.4 vs 289.9 ± 10.87 mg) and one group was fasted for 2 wk (20% of body mass loss). At this point, fish were myectomized and treated with CQ as described (Fed-0 mM, Fed-1 mM, Fasted-0 mM, Fasted-1 mM).
EdU incorporation assays
Cellular proliferation was assessed by intraperitoneal injections of 5-ethynyl-2′-deoxyuridine (EdU; Invitrogen, 10187).52 Fish were injected with EdU(20 µl, 10 mM in phosphate-buffered saline) at 20 h postinjury and sacrificed 4 h later. The injured muscle of 4 fish per experimental group was analyzed. EdU+ and total (DAPI) nuclei were counted from 2 to 6 sections per muscle (more than 70 sections were analyzed), representing approximately 1800 total nuclei (range 1007 to 3016) per muscle. Cell proliferation is represented as the percentage of EdU+ nuclei in the injured muscle.
Cell death analysis
Terminal Transferase dUTP Nick End Labeling (TUNEL) assay was performed on frozen sections using the TUNEL Apo-Green Detection Kit (Biotool, B31112), with some minor modifications. The tissue was permeabilized in 0.5% Triton X-100 PBS. TdT reaction was performed at 37°C for 1 h per manufacturer's suggestion. Positive controls were generated by treating tissue sections with DNase I (3000 U/ml in 50 mM TRIS, 10 mM MgCl2, 1 mg/ml BSA buffer) for 10 minutes at room temperature before performing the TdT reaction. Negative controls were performed with no enzyme in the TdT reaction mix (Fig. S2).
Adult heat induction experiments
Heat shocks were performed to hsp70l:dnfgfr1a-EGFP fish40 and wild-type fish directly in the housing racks using our customized system.53 Before cutting the muscles, fish were exposed to hot water for 5 d gradually increasing the time from 1 h to 3 h 30 min per day. This protocol was developed in a preliminary experiment where fish were heat shocked daily until GFP fluorescence was clearly present in the LR muscle (data not shown) to ensure the effective blockage of Fgf signaling in the regenerating muscle.
Statistics
Data were analyzed by Student t test (Mann-Whitney test when data did not met the requirements of parametric statistics) or one-way analysis of variance (ANOVA) followed by the Newman-Keuls multiple comparisons test, and the interaction between feeding condition and CQ treatment was further analyzed by 2-way ANOVA (P < 0.05), using the statistical software Prism 6.03 for Mac OSX (GraphPad Software, Inc.).
Supplementary Material
Abbreviations
- Atg
autophagy related
- Becn1
Beclin 1, autophagy related
- Casp3
caspase 3, apoptosis-related cysteine peptidase
- CQ
chloroquine
- dnFgfr1a-EGFP
EGFP-tagged dominant-negative fibroblast growth factor receptor 1a
- EdU
5-ethynyl-2′-deoxyuridine
- EOM
extraocular muscle
- Fgf
fibroblast growth factor
- GFP
green fluorescent protein
- hpi
hours post injury
- Map1lc3a/b
microtubule-associated protein 1 light chain 3 α/β
- LR
lateral rectus
- MOs
morpholino oligonucleotides
- Sqstm1
Sequestosome 1
- TEM
transmission electron microscopy
- TUNEL
terminal transferase dUTP nick end labeling
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank David Zacks for helpful discussions and suggestions regarding the fasting experiments, Amy Stevenson and Amy Robbins for their expertise in zebrafish husbandry, and Dotty Sorenson for electron microscopy technical support.
Funding
This study was supported by a Physician-Scientist Award from RPB and grant R01 EY022633 from the NEI of the NIH (AK). This research utilized the Vision Research Core (P30 EY007003) and the Cancer Center Research Core (P30 CA046592) at the University of Michigan. AK is supported by the Mrs. William Davidson Emerging Scholar Award from the A. Alfred Taubman Medical Research Institute, and DJK by NIH grant GM053396. The Zebrafish International Resource Center is supported by grant P40 RR012546 from the NIH NCRR. Funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
References
- [1].Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/ 10.1038/ncb2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/ 10.1038/ncb0910-823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 2010; 13:805-11; PMID:20581817; http://dx.doi.org/ 10.1038/nn.2575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Eng CH, Abraham RT. The autophagy conundrum in cancer: influence of tumorigenic metabolic reprogramming. Oncogene 2011; 30:4687-96; PMID:21666712; http://dx.doi.org/ 10.1038/onc.2011.220 [DOI] [PubMed] [Google Scholar]
- [5].Hitchcock PF, Raymond PA. The teleost retina as a model for developmental and regeneration biology. Zebrafish 2004; 1:257-71; PMID:18248236; http://dx.doi.org/ 10.1089/zeb.2004.1.257 [DOI] [PubMed] [Google Scholar]
- [6].Rodrigues AM, Christen B, Marti M, Belmonte JC. Skeletal muscle regeneration in Xenopus tadpoles and zebrafish larvae. Bmc Dev Biol 2012; 12:9; PMID:22369050; http://dx.doi.org/ 10.1186/1471-213X-12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Saera-Vila A, Kasprick DS, Junttila TL, Grzegorski SJ, Louie KW, Chiari EF, Kish PE, Kahana A. Myocyte dedifferentiation drives extraocular muscle regeneration in adult zebrafish. Invest Ophth Vis Sci 2015; 56:4977-93; http://dx.doi.org/ 10.1167/iovs.14-16103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science 2002; 298:2188-90; PMID:12481136; http://dx.doi.org/ 10.1126/science.1077857 [DOI] [PubMed] [Google Scholar]
- [9].Raya A, Koth CM, Buscher D, Kawakami Y, Itoh T, Raya RM, Sternik G, Tsai HJ, Rodríguez-Esteban C, Izpisúa-Belmonte JC. Activation of Notch signaling pathway precedes heart regeneration in zebrafish. P Natl Acad Sci USA 2003; 100:11889-95; http://dx.doi.org/ 10.1073/pnas.1834204100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Poss FD, Shen JX, Nechiporuk A, McMahon G, Thisse B, Thisse C, Thisse C, Keating MT. Roles for Fgf signaling during zebrafish fin regeneration. Dev Biol 2000; 222:347-58; PMID:10837124; http://dx.doi.org/ 10.1006/dbio.2000.9722 [DOI] [PubMed] [Google Scholar]
- [11].Pfefferli C, Jaźwińska A. The art of fin regeneration in zebrafish. Regeneration 2015; 2:72-83; http://dx.doi.org/ 10.1002/reg2.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Goldman D. Muller glial cell reprogramming and retina regeneration. Nat Rev Neurosci 2014; 15:431-42; PMID:24894585; http://dx.doi.org/ 10.1038/nrn3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lenkowski JR, Raymond PA. Muller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Prog Retin Eye Res 2014; 40:94-123; PMID:24412518; http://dx.doi.org/ 10.1016/j.preteyeres.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang RL, Han PD, Yang HB, Ouyang KF, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, et al.. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature 2013; 498:497-501; PMID:23783515; http://dx.doi.org/ 10.1038/nature12322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol 2008; 445:77-88; PMID:18425443; http://dx.doi.org/ 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- [16].He CC, Bartholomew CR, Zhou WB, Klionsky DJ. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 2009; 5:520-6; PMID:19221467; http://dx.doi.org/ 10.4161/auto.5.4.7768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hu ZY, Zhang JP, Zhang QY. Expression pattern and functions of autophagy-related gene atg5 in zebrafish organogenesis. Autophagy 2011; 7:1514-27; PMID:22082871; http://dx.doi.org/ 10.4161/auto.7.12.18040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Varga M, Sass M, Papp D, Takacs-Vellai K, Kobolak J, Dinnyes A, Klionsky DJ, Vellai T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ 2014; 21:547-56; PMID:24317199; http://dx.doi.org/ 10.1038/cdd.2013.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Amaravadi RK, Yu D, Lum JJ, Bui T, Christophorou MA, Evan GI, Thomas-Tikhonenko A, Thompson CB. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J Clin Invest 2007; 117:326-36; PMID:17235397; http://dx.doi.org/ 10.1172/JCI28833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al.. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/ 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang S, Xia PY, Rehm M, Fan Z. Autophagy and cell reprogramming. Cell Mol Life Sci 2015; 72:1699-713; PMID:25572296; http://dx.doi.org/ 10.1007/s00018-014-1829-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gurdon J. Nuclear reprogramming in eggs. Nat Med 2009; 15:1141-4; PMID:19812574; http://dx.doi.org/ 10.1038/nm1009-1141 [DOI] [PubMed] [Google Scholar]
- [23].Jopling C, Boue S, Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nat Rev Mol Cell Bio 2011; 12:79-89; http://dx.doi.org/ 10.1038/nrm3043 [DOI] [PubMed] [Google Scholar]
- [24].Semi K, Matsuda Y, Ohnishi K, Yamada Y. Cellular reprogramming and cancer development. Int J Cancer 2013; 132:1240-8; PMID:23180619; http://dx.doi.org/ 10.1002/ijc.27963 [DOI] [PubMed] [Google Scholar]
- [25].Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126:663-76; PMID:16904174; http://dx.doi.org/ 10.1016/j.cell.2006.07.024 [DOI] [PubMed] [Google Scholar]
- [26].Pan HZ, Cai N, Li M, Liu GH, Belmonte JC. Autophagic control of cell “stemness'. Embo Mol Med 2013; 5:327-31; PMID:23495139; http://dx.doi.org/ 10.1002/emmm.201201999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hansson J, Rafiee MR, Reiland S, Polo JM, Gehring J, Okawa S, Huber W, Hochedlinger K, Krijgsveld J. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Rep 2012; 2:1579-92; PMID:23260666; http://dx.doi.org/ 10.1016/j.celrep.2012.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mizushima N, Klionsky DJ. Protein turnover via autophagy: Implications for metabolism. Annu Rev Nutr 2007; 27:19-40; PMID:17311494; http://dx.doi.org/ 10.1146/annurev.nutr.27.061406.093749 [DOI] [PubMed] [Google Scholar]
- [29].Dou ZX, Xu CY, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al.. Autophagy mediates degradation of nuclear lamina. Nature 2015; 527:105-9; PMID:26524528; http://dx.doi.org/ 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015; 522:359-62; PMID:26040717; http://dx.doi.org/ 10.1038/nature14506 [DOI] [PubMed] [Google Scholar]
- [31].Cho A, Noguchi S. Autophagy in GNE Myopathy In: Bailly Y, ed. Autophagy - A Double-Edged Sword - Cell Survival or Death?, 2013 [Google Scholar]
- [32].Shalini S, Dorstyn L, Dawar S, Kumar S. Old, new and emerging functions of caspases. Cell Death Differ 2015; 22:526-39; PMID:25526085; http://dx.doi.org/ 10.1038/cdd.2014.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bell RA, Al-Khalaf M, Megeney LA. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet Muscle 2016; 6:16; PMID:27054028; http://dx.doi.org/ 10.1186/s13395-016-0086-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Luthi AU, Martin SJ. The CASBAH: a searchable database of caspase substrates. Cell Death Differ 2007; 14:641-50; PMID:17273173; http://dx.doi.org/ 10.1038/sj.cdd.4402103 [DOI] [PubMed] [Google Scholar]
- [35].Hashimoto T, Kikkawa U, Kamada S. Contribution of caspase(s) to the cell cycle regulation at Mitotic Phase. Plos One 2011; 6:e18449; PMID:21479177; http://dx.doi.org/ 10.1371/journal.pone.0018449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang J, Kabra NH, Cado D, Kang C, Winoto A. FADD-deficient T cells exhibit a disaccord in regulation of the cell cycle machinery. J Biol Chem 2001; 276:29815-8; PMID:11390402; http://dx.doi.org/ 10.1074/jbc.M103838200 [DOI] [PubMed] [Google Scholar]
- [37].Frost V, Al-Mehairi S, Sinclair AJ. Exploitation of a non-apoptotic caspase to regulate the abundance of the cdkI p27(KIP1) in transformed lymphoid cells. Oncogene 2001; 20:2737-48; PMID:11420686; http://dx.doi.org/ 10.1038/sj.onc.1204367 [DOI] [PubMed] [Google Scholar]
- [38].Helfer B, Boswell BC, Finlay D, Cipres A, Vuori K, Kang TB, Wallach D, Dorfleutner A, Lahti JM, Flynn DC, et al.. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res 2006; 66:4273-8; PMID:16618751; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4183 [DOI] [PubMed] [Google Scholar]
- [39].Kuranaga E. Beyond apoptosis: caspase regulatory mechanisms and functions in vivo. Genes Cells 2012; 17:83-97; PMID:22244258; http://dx.doi.org/ 10.1111/j.1365-2443.2011.01579.x [DOI] [PubMed] [Google Scholar]
- [40].Lee Y, Grill S, Sanchez A, Murphy-Ryan M, Poss KD. Fgf signaling instructs position-dependent growth rate during zebrafish fin regeneration. Development 2005; 132:5173-83; PMID:16251209; http://dx.doi.org/ 10.1242/dev.02101 [DOI] [PubMed] [Google Scholar]
- [41].Floss T, Arnold HH, Braun T. A role for FGF-6 in skeletal muscle regeneration. Gene Dev 1997; 11:2040-51; PMID:9284044; http://dx.doi.org/ 10.1101/gad.11.16.2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dvorak P, Dvorakova D, Hampl A. Fibroblast growth factor signaling in embryonic and cancer stem cells. Febs Lett 2006; 580:2869-74; PMID:16516203; http://dx.doi.org/ 10.1016/j.febslet.2006.01.095 [DOI] [PubMed] [Google Scholar]
- [43].Saera-Vila A, Kish PE, Kahana A. Fgf regulates dedifferentiation during skeletal muscle regeneration in adult zebrafish. Cell Signal 2016; 28(9):1196-1204; PMID:27267062; http://dx.doi.org/ 10.1016/j.cellsig.2016.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth F R 2005; 16:233-47; http://dx.doi.org/ 10.1016/j.cytogfr.2005.01.007 [DOI] [PubMed] [Google Scholar]
- [45].Corcelle E, Dierbi N, Mari M, Nebout M, Fiorini C, Fenichel P, Hofman P, Poujeol P, Mograbi B. Control of the autophagy maturation step by the MAPK ERK and p38 - Lessons from environmental carcinogens. Autophagy 2007; 3:57-9; PMID:17102581; http://dx.doi.org/ 10.4161/auto.3424 [DOI] [PubMed] [Google Scholar]
- [46].Kang C, You YJ, Avery L. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Gene Dev 2007; 21:2161-71; PMID:17785524; http://dx.doi.org/ 10.1101/gad.1573107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Higashijima S, Okamoto H, Ueno N, Hotta Y, Eguchi G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev Biol 1997; 192:289-99; PMID:9441668; http://dx.doi.org/ 10.1006/dbio.1997.8779 [DOI] [PubMed] [Google Scholar]
- [48].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- [49].Craig SE, Thummel R, Ahmed H, Vasta GR, Hyde DR, Hitchcock PF. The Zebrafish Galectin Drgal1-L2 is expressed by proliferating muller glia and photoreceptor progenitors and regulates the regeneration of rod photoreceptors. Invest Ophth Vis Sci 2010; 51:3244-52; http://dx.doi.org/ 10.1167/iovs.09-4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ramachandran R, Fausett BV, Goldman D. Ascl1a regulates Muller glia dedifferentiation and retinal regeneration through a Lin-28-dependent, let-7 microRNA signalling pathway. Nat Cell Biol 2010; 12:1101-U106; PMID:20935637; http://dx.doi.org/ 10.1038/ncb2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thummel R, Bai S, Sarras MP, Song PZ, McDermott J, Brewer J, Perry M, Zhang X, Hyde DR, Godwin AR. Inhibition of zebrafish fin regeneration using in vivo electroporation of morpholinos against fgfr1 and msxb. Dev Dynam 2006; 235:336-46; http://dx.doi.org/ 10.1002/dvdy.20630 [DOI] [PubMed] [Google Scholar]
- [52].Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. P Natl Acad Sci USA 2008; 105:2415-20; http://dx.doi.org/ 10.1073/pnas.0712168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Saera-Vila A, Kish PE, Kahana A. Automated scalable heat shock modification for standard aquatic housing systems. Zebrafish 2015; 12:312-4; PMID:25942613; http://dx.doi.org/ 10.1089/zeb.2015.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.