

Abstract
Aim
The aim of this study was to investigate the etiological factors, clinical features, and prognostic factors in children with hemophagocytic lymphohistiocytosis (HLH).
Methods
Fifty-six children with HLH in Wuhan Union Hospital, People’s Republic of China, were retrospectively analyzed in recent years. We reviewed the medical records of 56 HLH children hospitalized from 2000 to 2013 to identify the possible prognostic factors.
Results
In more than half of the cases (64.29%), the etiological factor was found to be infection. Clinical characteristics such as prolonged fever (100.00%), hepatosplenomegaly (95.24%), and pancytopenia (100.00%) were observed. Characteristic laboratory values presented with increased ferritin (64.29%), triglycerides (78.57%), transaminases (80.95%), bilirubin (67.54%), lactate dehydrogenase (95.23%), and decreased fibrinogen (61.90%), natrium (40.48%), and potassium (30.94%). Bone marrow aspiration showed hemophagocytosis in 48 cases (85.71%). Forty-two patients were treated according to HLH-2004 protocol. Out of the 42 patients, 19 cases acquired remission and 13 cases died. Ten cases lost follow-up. High lactate dehydrogenase (>2,000 U/L), high bilirubin (>2 mg/mL), and younger age (<2 years) at the time of diagnosis were adverse prognostic factors.
Conclusion
HLH is a life-threatening syndrome caused by complicated etiology. The level of lactate dehydrogenase and bilirubin accompanied with younger age were adverse factors.
Keywords: HLH, clinical features, children, prognostic factors
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare disease characterized by a rapidly fatal hematological disorder usually associated with malignancies and severe infections. It presents with prolonged high fever that could not be controlled by antibiotics; hepatosplenomegaly; cytopenia; widespread infiltration with lymphocytes; and benign-looking histiocytes with hemophagocytosis in the bone marrow, liver, spleen, or lymph nodes. A steadily increasing attention has been paid to HLH in the past decade. Early diagnosis and combined treatment can effectively control this life-threatening disease. In this study, we have described the progression of 56 children with HLH who underwent treatment in Wuhan Union Hospital, People’s Republic of China, and have emphasized the prognostic factors in an effort to improve the outcome of patients with HLH.
Methods
Diagnosis and patients
A retrospective analysis on HLH was carried out from January 1, 2000 to November 30, 2013, at the Pediatric Department of Wuhan Union Hospital. The criteria for diagnosis of HLH were based on the guideline of the HLH-2004 protocol of Henter et al.1 Fifty-six pediatric patients who received a discharge diagnosis of HLH during the period were enrolled in our study. Clinical and laboratory data were recorded and collected by a single investigator. This study was approved by the Ethics Committee of Wuhan Union Hospital, People’s Republic of China. Written informed consent was obtained from the guardians of the patients.
Statistical analysis
Data are reported as mean ± standard deviation (SD). Median values were reported for nonnormally distributed data. Regression logistic was used to identify prognostic factors. All statistical analyses were performed with SPSS Software Version 15.0 (SPSS, Chicago, IL, USA). Differences were considered statistically significant if P<0.05.
Results
Clinical features
Of the 56 pediatric patients, 35 were boys and 21 were girls with a median age of 4.5 years (range: 4 months to 12 years) at the onset of HLH. The median duration before diagnosis of HLH was 1.5 weeks (range: 1 week to 6 weeks). The most common clinical symptoms observed were fever (100.00%), hepatomegaly or splenomegaly (95.24%), and pancytopenia (100.0%). The clinical features observed in the patients are shown in Figure 1.
Figure 1.
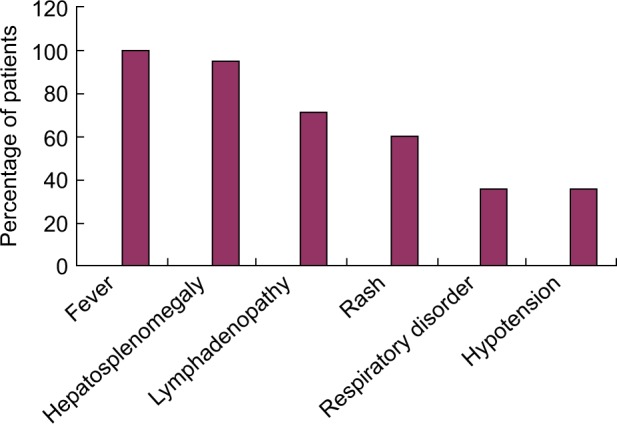
Clinical features of 56 patients with HLH.
Abbreviation: HLH, hemophagocytic lymphohistiocytosis.
Laboratory findings
The laboratory results obtained for all pediatric patients with HLH are given in Table 1. Anemia was observed in 39 patients (five cases with hemoglobin level <90 g/L, 27 cases <60–89 g/L, seven cases <30–59 g/L), neutropenia (neutrophil count <1.5×109/L) in 28 patients (one case <1.0–1.5×109/L, 22 cases <0.5–1.0×109/L, and five cases <0.5×109/L) and thrombocytopenia (platelet count <100×109/L) in 35 patients (33 cases with platelet count <20–99×109/L and two cases with platelet count <20×109/L).
Table 1.
Laboratory findings of 56 patients with HLH
| Variables | Data median(range) |
|---|---|
| Hemoglobin (g/L) | 80(30–118) |
| White blood cell count (×109/L) | 3.0(0.3–11.5) |
| Neutrophil count (×109/L) | 0.6(0.1–5.8) |
| Platelet count (×109/L) | 49.0(11–450) |
| Alanine aminotransferase (U/L) | 115(15–1,187) |
| Aspartate aminotransferase (U/L) | 175(17–2,928) |
| Triglycerides (mmol/L) | 5.0(1.5–10.8) |
| Fibrinogen (g/L) | 2.7(0.3–6.0) |
| Ferritin (μg/L) | 6,900(45–87,800) |
| Total bilirubin (μmol/L) | 28.7(3.5–269.0) |
| Lactate dehydrogenase(U/L) | 2,300(1,123–5,062) |
| APTT (seconds) | 31.0 (22.0–120) |
| PT (seconds) | 14.0 (10.0–85) |
Abbreviations: HLH, hemophagocytic lymphohistiocytosis; PT, prothrombin time; APTT, activeated partial thromboplastin time.
Increased level of serum ferritin was observed in 36 (64.29%) patients. Elevated level of serum lactate dehydrogenase (LDH) was found in the majority (95.24%) of our patients. The highest LDH level observed was 5,062 U/L. Increased triglycerides, transaminases, and bilirubin were observed in 44 (78.57%), 45 (80.95%), and 38(67.86%) patients, respectively. Hyponatremia and kaliopenia were observed in 23 (41.07%) and 17 (30.36%) cases, respectively. Decreased fibrinogen was observed in 34 cases (60.71%). Prolonged prothrombin time and partial thromboplastin time were observed in 38 (67.86%) cases. Examination of bone marrow aspiration showed hemophagocytosis in 48 (85.71%) patients. Unidentified cells were observed in the bone marrow aspiration of three patients and normal cells in three patients.
Associated underlying diseases
Infectious agents were identified in 35 (61.90%) patients. Non-infectious agents of HLH were identified in six (10.70%) patients. No agents were identified in the remaining 15 cases (26.78%). Analysis of etiology is shown in Figure 2.
Figure 2.

Etiology analysis of HLH.
Abbreviation: HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein–Barr virus; CMV, cytomegalovirus; HSV, herpes simplex virus; MP, mycoplasma; CP,chlamydia.
Treatment and outcome
Forty-two patients were treated according to the HLH-94 or HLH-2004 guidelines; 14 patients refused treatment and lost follow-up because of rapid fatality or financial difficulty and belief of HLH incurability. The common supportive treatments included packed red cell, fresh plasma, and platelet transfusions. Those patients who underwent treatment received steroid and intravenous immunoglobulin. Of the 42 treated patients, 25 acquired remission and 17 died of relapse.
Prognostic factors analysis
Among the clinical and laboratory features analyzed, high LDH (>2,000 U/L), high bilirubin (>2 mg/mL), and younger age (<2 years old) at the time of diagnosis indicated worst outcome (Table 2).
Table 2.
Analysis of prognostic factors of HLH
| Risk factors | β | Standard error | χ2 | OR | 95% Confidence interval | P-value |
|---|---|---|---|---|---|---|
| Age <2 years | 2.905 | 8.515 | 7.636 | 18.661 | 1.562–197.781 | 0.007 |
| N <1.0×109/L | −0.836 | 0.202 | 1.244 | 0.424 | 0.021–4.881 | 0.288 |
| Hb <60 g/L | 1.775 | 2.266 | 0.715 | 5.891 | 0.612–60.271 | 0.395 |
| Plt <50 g/L | 0.911 | 1.164 | 2.060 | 2.491 | 0.214–23.115 | 0.142 |
| LDH >2,000 U/L | 3.335 | 13.881 | 8.891 | 29.771 | 2.681–317.114 | 0.003 |
| AST >400 U/L | 2.603 | 6.337 | 2.249 | 13.771 | 1.298–133.141 | 0.133 |
| ALT >400 U/L | 2.515 | 5.661 | 1.441 | 12.361 | 1.017–130.712 | 0.230 |
| EBV infection | 2.055 | 3.660 | 1.958 | 10.123 | 0.612–71.124 | 0.163 |
| CMV infection | 1.061 | 1.335 | 0.615 | 7.821 | 0.216–38.218 | 0.411 |
| Bilirubin >2 mg/mL | 3.066 | 11.315 | 10.331 | 19.145 | 1.225–113.512 | 0.013 |
Abbreviations: HLH, hemophagocytic lymphohistiocytosis; N, neutrophil; Hb, hemoglobin; Plt, platelet; LDH, lactate dehydrogenase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; EBV, Epstein–Barr virus; CMV, cytomegalovirus; OR, odds ratio.
Discussion
HLH is classified into genetic (primary) HLH and acquired (secondary) HLH. The pathogenesis and etiology of HLH are still unknown. Primary HLH is thought to be associated with impaired natural killer cell function. Secondary HLH may be acquired under a variety of conditions such as infections2 (including infections caused by bacteria, viruses, and fungi), malignancies,3 autoinflammatory and autoimmune diseases,4 and acquired immune deficiency states.5–7 Thirty-one patients were found to have secondary HLH. Epstein–Barr virus is the main viral infectious agent, which accounted for 19.05% of the infections. In our study, hematological malignancies were the second conditions (9.52%) associated with HLH, in lower proportion than previous reports.8,9
The onset characteristic of HLH is complicated by common clinical and laboratory findings that may present in other infectious disease, such as pneumonia or sepsis. The patients’ condition became worse rapidly. Early recognition of HLH and immediate appropriate therapy to HLH patients have been identified to improve or to relieve the disease progression. The time from the onset of symptoms to the diagnosis varied from 1 to 6 weeks (median 1.5 weeks) in our patients. The delay of diagnosis or prolonged duration of diagnosis (1.5 weeks) reflected the difficulties in diagnosing HLH. Persistent fever, hepatosplenomegaly, cytopenia, hypertriglyceridemia, and/or hypofibrinogenemia, together with the progressive course, suggest suspicion of an HLH diagnosis and the pediatricians should be aware of the hemophagocytosis in bone marrow.10 Gupta et al11 reported that the sensitivity is 60% for hemophagocytosis in initial bone marrow aspirate in patients diagnosed with HLH. Of the 56 patients with HLH, hemophagocytosis was not found in six patients. So repeated bone marrow aspiration was recommended to these patients. Physicians should note the fact that cytological characteristic of hemophagocytosis is not the gold standard for the diagnosis of HLH. Some potential prognostic factors, such as cytokine levels, may reveal a consistent elevation in HLH patients. It was reported that a cytokine storm occurred in most of the HLH patients, which was responsible for the clinical manifestations and abnormal laboratory data of HLH.12–14 High levels of LDH and bilirubin accompanied with younger age were observed in our patients which may predict the severity of HLH. Further investigations should focus on identify the result.
Previous studies of HLH indicated that older age (in adults), multidrug chemotherapy, and Epstein–Barr virus reactivation were related to poor treatment outcomes in these patients.15 According to the guideline of HLH (HLH-94) in children, the drugs such as dexamethasone, etoposide, cyclosporine A were employed (after 8 weeks) in combination with antithymocyte globulin/antilymphocyte globulin and high-dose intravenous immunoglobulin as systemic therapy.16 Mild cases may require remission on condition that corticosteroids are used properly. Patients with primary HLH required Hematopoietic Stem Cell Transplantation17 and they were treated with the aforementioned drugs until a transplant could be performed.18,19 Etoposide can destroy infected antigen-presenting cells. Dexamethasone can penetrate into the cerebrospinal fluid. Cyclosporine A inhibits T-cell activation and is considered as valuable cytolytic corticosteroids. HLH-94 and HLH-2004 enrolled 26 cases in our study, one patient was presumed primary HLH and treated with maintenance treatment, followed by Hematopoietic Stem Cell Transplantation. The event-free survival of the patient till the last follow-up was 5 years.
Conclusion
In conclusion, the etiology of HLH is complicated and early diagnosis is difficult. The level of LDH and bilirubin may predict the severity of HLH. Depending on the clinical presentation, individualized therapy is needed for different patients.
Footnotes
Author contributions
Yan-Rong Wang, Yi-Ning Qiu, and Yan Bai collected clinical data, and analyzed and wrote the manuscript. Yi-Ning Qiu and Jia-Wei Xu collected and reviewed the laboratory data. Yan-Rong Wang and Xian-Feng Wang reviewed the final manuscript and approved the version to be submitted and all the authors contributed to the final preparation of the article. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Henter JI, Horne A, Aricó M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 2.Endo R, Yoshioka M, Ebihara T, Ishiguro N, Kikuta H, Kobayashi K. Clonal expansion of multiphenotypic Epstein-Barr virus-infected lymphocytes in chronic active Epstein-Barr virus infection. Med Hypotheses. 2004;63(4):582–587. doi: 10.1016/j.mehy.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 3.Machaczka M, Vaktnäs J, Klimkowska M, Hägglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–619. doi: 10.3109/10428194.2010.551153. [DOI] [PubMed] [Google Scholar]
- 4.Pérard L, Costedoat-Chalumeau N, Limal N, et al. Hemophagocytic syndrome in a pregnant patient with systemic lupus erythematosus, complicated with preeclampsia and cerebral hemorrhage. Ann Hematol. 2007;86(7):541–544. doi: 10.1007/s00277-007-0277-7. [DOI] [PubMed] [Google Scholar]
- 5.Henter JI, Andersson B, Elinder G, Jakobson A, Lubeck PO, Soder O. Elevated circulating levels of interleukin-1 receptor antagonist but not IL-1 agonists in hemophagocytic lymphohistiocytosis. Med Pediatr Oncol. 1996;27(1):21–25. doi: 10.1002/(SICI)1096-911X(199607)27:1<21::AID-MPO5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 6.Tiab M, Mechinaud F, Harousseau JL. Haemophagocytic syndrome associated with infections. Baillieres Best Pract Res Clin Haematol. 2000;13(2):163–178. doi: 10.1053/beha.2000.0066. [DOI] [PubMed] [Google Scholar]
- 7.Re A, Facchetti F, Borlenghi E, et al. Fatal hemophagocytic syndrome related to active human herpesvirus-8/Kaposi sarcoma-associated herpesvirus infection in human immunodeficiency virus-negative, non-transplant patients without related malignancies. Eur J Haematol. 2007;78(4):361–364. doi: 10.1111/j.1600-0609.2007.00828.x. [DOI] [PubMed] [Google Scholar]
- 8.Sailler L, Duchayne E, Marchou B, et al. Etiological aspects of reactive hemophagocytoses: retrospective study in 99 patients. Rev Med Interne. 1997;18(11):855–864. doi: 10.1016/s0248-8663(97)81959-5. [DOI] [PubMed] [Google Scholar]
- 9.Shabbir M, Lucas J, Lazarchick J, Shirai K. Secondary hemophagocytic syndrome in adults: a case series of 18 patients in a single institution and a review of literature. Hematol Oncol. 2011;29(2):100–106. doi: 10.1002/hon.960. [DOI] [PubMed] [Google Scholar]
- 10.Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(2):192–194. doi: 10.1002/pbc.21441. [DOI] [PubMed] [Google Scholar]
- 11.Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(3):402–404. doi: 10.1002/pbc.21564. [DOI] [PubMed] [Google Scholar]
- 12.My LT, Lien le B, Hsieh WC, et al. Comprehensive analyses and characterization of haemophagocytic lymphohistiocytosis in Vietnamese children. Br J Haematol. 2010;148(2):301–310. doi: 10.1111/j.1365-2141.2009.07957.x. [DOI] [PubMed] [Google Scholar]
- 13.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 14.Pachlopnik Schmid J, Ho CH, Chretien F, et al. Neutralization of IFN gamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. 2009;1(2):112–124. doi: 10.1002/emmm.200900009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishii E, Ohga S, Imashuku S, et al. Nationwide survey of hemophagocytic lymphohistiocytosis in Japan. Int J Hematol. 2007;86(1):58–65. doi: 10.1532/IJH97.07012. [DOI] [PubMed] [Google Scholar]
- 16.Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–e628. doi: 10.1542/peds.2006-3164. [DOI] [PubMed] [Google Scholar]
- 17.Ohga S, Kudo K, Ishii E, et al. Hematopoietic stem cell transplantation for familial hemophagocytic lymphohistiocytosis and Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Pediatr Blood Cancer. 2010;54(2):299–306. doi: 10.1002/pbc.22310. [DOI] [PubMed] [Google Scholar]
- 18.Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622–630. doi: 10.1111/j.1365-2141.2005.05501.x. [DOI] [PubMed] [Google Scholar]
- 19.Filipovich AH. Life-threatening hemophagocytic syndromes: current outcomes with hematopoietic stem cell transplantation. Pediatr Transplant. 2005;9(Suppl 7):87–91. doi: 10.1111/j.1399-3046.2005.00449.x. [DOI] [PubMed] [Google Scholar]