

Abstract
Fetal alcohol exposure may impair growth, development, and function of multiple organ systems, and is encompassed by the term Fetal Alcohol Spectrum Disorders (FASD). Research has so far focused on the mechanisms, prevention, and diagnosis of FASD, while the risk for adult-onset chronic diseases in individuals exposed to alcohol in utero is not well explored. David Barker’s hypothesis on Developmental Origins of Health and Disease (DOHaD) suggests that insults to the milieu of the developing fetus program it for adult-development of chronic diseases. In the 25 years since the introduction of this hypothesis, epidemiological and animal model studies have made significant advancements in identifying in utero developmental origins of chronic adult-onset diseases affecting cardiovascular, endocrine, musculoskeletal, and psycho-behavioral systems. Teratogen exposure is an established programming agent for adult diseases, and recent studies suggest that prenatal alcohol exposure correlates with adult-onset of neuro-behavioral deficits, cardiovascular disease, endocrine dysfunction, nutrient homeostasis instability, warranting additional investigation of alcohol-induced DOHaD, as well as patient follow-up well into adulthood for affected individuals. In utero epigenetic alterations during critical periods of methylation is a key potential mechanism for programming and susceptibility of adult-onset chronic diseases, with imprinted genes affecting metabolism being critical targets. Additional studies in epidemiology, phenotypic characterization in response to timing, dose and duration of exposure, as well as elucidation of mechanisms underlying FASD-DOHaD inter-relation are thus needed to clinically define chronic disease associated with prenatal alcohol exposure. These studies are critical to establish interventional strategies that decrease incidence of these adult-onset diseases and promote healthier aging among individuals affected with FASD.
Keywords: DOHaD, FASD, Alcohol, Barker hypothesis, Epigenetics
INTRODUCTION
Alcohol consumption during pregnancy may produce deleterious effects on multiple fetal organ systems; the best studied are its effects on the developing brain and the resulting behavioral deficits (Riley and McGee, 2005, Riley et al., 2011). These developmental anomalies are collectively termed “fetal alcohol spectrum disorders” or FASD (Sokol et al., 2003). The targets of developmental alcohol exposure include cardiac (Burd et al., 2007, Ren et al., 2002), vascular (Ramadoss and Magness, 2012, Parnell et al., 2007, Bake et al., 2012), endocrine (Zhang et al., 2005, Wilcoxon et al., 2005), neurobehavioral (Warren et al., 2011, Riley and McGee, 2005, Lewis et al., 2015, Gautam et al., 2015), and uteroplacental systems (Ramadoss and Magness, 2012, Sawant et al., 2014, Gareri et al., 2009, Gundogan et al., 2008), demonstrating that alcohol can target virtually any fetal organ system. The severity and the specific regional deficits following prenatal alcohol exposure is a function of the amount of alcohol consumed, the timing of exposure during pregnancy, the duration of exposure, and the pattern of alcohol consumption in both humans (Feldman et al., 2012, O'Leary et al., 2010, May et al., 2013) as well as animal models of FASD (Maier et al., 1999, Livy et al., 2003, Sawant et al., 2013, Ramadoss et al., 2007a, Ramadoss et al., 2007b, Bonthius and West, 1988, Thomas et al., 1996). Current estimates of FASD are at least 2–5% among young school children in the United States (May et al., 2009, May et al., 2014).
Around 25 years ago, David Barker proposed that adverse events during development may permanently alter fetal development and physiologic function, leading to a higher risk for adult-onset diseases, an idea termed “Developmental Origins of Adult Health and Disease (DOHaD)” or “Barker’s Hypothesis” (Barker, 2004a, Barker, 2004b, Barker, 2012, Barker, 2007). It is suggested that patterns of fetal growth program blood pressure, insulin responsivity, glucose metabolism, and cardiovascular function later in life (Barker et al., 1993, Hales et al., 1991). Adverse events affecting fetal development may be initiated or influenced by factors altering the in utero environment. Examples include maternal nutrition, endocrine disruptors, maternal stress, gestational diabetes, obesity, and substance abuse, etc. (Gabory et al., 2013, Adair and Dahly, 2005, Barker, 2006, Moore and Riley, 2015). Prior to the proposal of Barker’s hypothesis, recognition of an environmental trigger followed by a latent period and then disease manifestation was widely accepted as, but limited to, the etiology for various cancers. Since the proposal of Barker’s hypothesis, evidence across disciplines supports recognition of the causative role these environmental triggers during development play in the etiology of adult chronic disease (Gluckman et al., 2008). Investigation of fetal integrative physiology, along with its impact on adult-onset diseases, has since gained increasing momentum in a time when genetic programming and life style contribution were considered the primary determining factors of causation (Hanson, 2015).
The current review article summarizes (1) major work performed so far in the DOHaD field, (2) specific studies on DOHaD and alcohol, (3) epigenetic pathways underlying DOHaD in relation to in utero alcohol exposure, and (4) future directions for the FASD-DOHaD field.
ADVANCEMENTS IN THE DOHaD FIELD
Studies performed to date utilizing animal models and human epidemiologic methods suggest adverse influences on the intrauterine environment during time sensitive periods of organ development result in intrauterine growth restriction (IUGR), altered structure, physiology and metabolism of developing organs, low birth weight, variations in patterns of hormone release and response, and altered postnatal growth rate; all of which may influence fetal programming for chronic diseases. Furthermore, these insults affect the developing fetus in such a way that disrupts gene expression and epigenetic mechanisms, resulting in very early programming of chronic adult diseases
Human reports on DOHaD
We will first review reports containing human study observations correlating prenatal insults and later onset of diseases in adult life. We have classified these findings based on the adult-organ system whose function is impaired. Cardiovascular, metabolic, and respiratory: Epidemiological studies have shown low birth weight and poor nutrition throughout pregnancy strongly associate with increased risk of adult onset of cardiovascular disease, obesity, and type 2 diabetes (Barker, 2012). These individuals are also at higher risk for adult onset of chronic diseases like hypertension, stroke, and hyperlipidemia (Barker, 1995). Reduced fetal birth weight and infant growth rate correlate with increased adult incidence of death from ischemic heart disease, as at age 1, infants weighing ≤ 18 pounds are three times more likely than infants weighing ≥ 27 pounds to die from ischemic heart disease in adulthood (Barker et al., 1989). In a study of 286 South Indian men and women, adult mean forced expiratory volume (FEV1) and forced vital capacity (FVC) fell with decreasing birth weight, and small head circumference was associated with decreased FEV1/FVC ratio in males, suggesting adult lung function is programmed in utero (Stein et al., 1997). Prenatal exposure to the Dutch Famine, a 5-month period of extreme food shortage during the winter of 1944–45, resulted in offspring, who as adults, exhibited a predisposition to chronic adult diseases, with increased incidence and early onset of coronary artery disease (Painter et al., 2006). Those exposed to the famine in early gestation had an increased atherogenic lipid profile, higher plasma fibrinogen and reduced plasma factor VII, increased incidence of adult obesity, and increased incidence of coronary heart disease; mid gestation exposure to famine also had a higher incidence of obstructive airway disease, and both mid and late gestation famine exposure produced decreased glucose tolerance in adults (Roseboom et al., 2001). Brain and behaviour: Evidence suggests that prenatal exposure to maternal smoking, alcohol, and drug use can compromise brain development in utero and may increase the risk for compromised mental health, and other psychopathologies later in life (Schlotz and Phillips, 2009). Skeletal system: Birth weight, length, and placental weight are also positively associated with bone mass. Birth weight is a predetermining factor in adult bone mineral content, basal activity of growth hormone/insulin-like growth factor (IGF)-1, and hypothalamic-pituitary-adrenal axes; maternal smoking and nutrition have been shown to directly affect skeletal mineralization, suggesting that adult onset of osteoporosis may have fetal programmed origins (Cooper et al., 2002). Antenatal steroids: Since the first controlled clinical trial in 1972, antenatal steroid administration has proven effective in promoting lung maturation in infants at risk for pre-term birth, and it is estimated to have reduced preterm infant mortality by 53% and morbidity by 37% (Liggins and Howie, 1972, Mwansa-Kambafwile et al., 2010). It is generally regarded that benefits outweigh the risks, and a single course of antenatal steroid administration is standard care in pre-term patients in North America and Western Europe (Consensus Statement NIH, 2000). This has allowed for a unique cohort of individuals to be studied following a specifically dosed and timed antenatal treatment. One study following a cohort of 210 preterm survivors concluded antenatal corticosteroid therapy is associated with higher systolic and diastolic blood pressures in adolescence (Doyle et al., 2000). Interestingly, administration of multiple doses of exogenous glucocorticoids during pregnancy in animal model studies correlates with low birthweight and hypertension later in life (Edwards et al., 1993, Wood and Keller-Wood, 2016). Additional animal studies indicate that excess glucocorticoid exposure early in life may alter central nervous system development and have long term physiologic consequences, and these may not become apparent until later in life (Matthews, 2000).
Animal studies
Animal model experiments have effectively replicated examples of fetal programming of adult diseases. These models are advantageous as they allow exquisite control of both study conditions while excluding multiple effect-modifying exposures that may alter outcomes. The latter frequently confounds alcohol related human studies, i.e. polysubstance abuse or malnutrition combined with drinking while pregnant may have additive negative effects that are almost impossible to exclusively differentiate. The findings from animal model DOHaD studies currently provide vital clues on developmental alcohol effects on adult-onset disease states and we hope that this review will encourage future DOHaD human studies in the alcohol field. We herein will classify select major findings based on the in utero environmental insult. Altering blood flow in utero-placental compartment: A CD-1 mouse model demonstrated gestational crowding of pups into one uterine horn decreased placenta blood flow up to 4-fold and IUGR occurred; pups produced in both the bottom and top 5th percentiles of birth weight developed obesity as adults. (Coe et al., 2008). Prenatal caffeine exposure:. Prenatal caffeine (120 mg/kg) administration in rats on gestational days 11–20 resulted in low birth weight and increased susceptibility to non-alcoholic fatty liver disease as adults when introduced to a high fat diet (Wang et al., 2014). Prenatal benzodiazepine exposure: Prenatal exposure to benzodiazepine ligands over gestational days 14–20 resulted in increased levels of thiobarbituric acid-reactive material, decreased intracellular pH, and significant change in phosphocreatine utilization in the brain when compared with controls; these changes were undetectable until early adulthood. This long term effect of late gestational exposure to benzodiazepines suggests selective binding of receptor ligands during development can program for altered adult cerebral metabolism (Miranda et al., 1989, Miranda et al., 1990a, Miranda et al., 1990b). Low-protein diet: After an isocaloric low-protein diet fed during gestational days 3–5, 28 week old offspring had elevated angiotensin converting enzyme activity in the serum in females and in the lung in males, and these offspring had elevated systolic blood pressure as adults (Watkins et al., 2010). High fat diet: A maternal high saturated fat diet in mice produced adult offspring with insulin resistance, hyperglycemia, obesity, and hypertension, despite being fed a standard caloric diet postnatally (Liang et al., 2009). These offspring had decreased average bone mineral density in the femoral epiphysis at 6 months of age and dysregulation of distal femoral trabecular architecture at 12 months of age, indicating a prenatal origin for osteoporosis (Liang et al., 2009). Interestingly, recent evidence indicates that parental diet can affect cholesterol and lipid metabolism in offspring, and suggest preconception factors also influence the incidence of adult pathologies (Carone et al., 2010).
Intervention strategies
Combined data from epidemiologic, experimental, and clinical studies indicate early life events in utero may influence and initiate pathogenesis of chronic adult diseases such as hypertension, heart disease, stroke, type 2 diabetes mellitus, obesity, metabolic syndrome, and osteoporosis. By determining the factors that program long term health, interventional strategies combined with screening for epigenetic markers may be used to prevent onset of these chronic diseases and to customize targeted treatment regimens for susceptible individuals. Recent studies indicate deviant phenotypes induced by insults during early development in utero exhibit potential for reversibility and recovery (Gluckman et al., 2008). One example of successful intervention utilizing a mouse model is the finding that perinatal exercise resulted in improved glucose clearance in both male and female adult offspring in response to oral glucose challenges (Carter et al., 2012). Supplementing a maternal high fat diet with the antioxidant quercetin improved offspring birth size and resulted in a protective effect on adult rat diseases, decreasing incidence adult osteoporotic changes and metabolic dysfunction, indicating an oxidative stress mediated role in pathogenesis (Liang et al., 2009). Additionally, glycine supplementation when fed concurrently with a low-protein diet throughout pregnancy mitigated vascular dysfunction, decreased systolic blood pressure, and improved mesenteric artery nitric oxide release in adult offspring (Jackson et al., 2002, Brawley et al., 2004).
PRENATAL ALCOHOL-INDUCED ADULT ONSET DISEASES
Surprisingly, although 1 in 10 pregnant women and 1 in 2 non-pregnant women consume alcohol, and 1 in 33 pregnant women report binge drinking in the past 30 days (Tan et al., 2015), limited work has been performed on alcohol-induced adult-onset disease vulnerability. What has been established is that health problems of adults with FASD is largely different from diagnostic criteria applied to infants and children. At birth, defining characteristics of individuals with FASD include low birth weight and/or growth deficiency, dysmorphic facial structures, and neurologic impairment (Jones and Smith, 1973, Hoyme et al., 2005). Conversely, aging in FASD individuals is associated with attenuation of facial dysmorphology (Streissguth et al., 1991), increased rates of obesity (Fuglestad et al., 2014), endocrine dysfunction (Hellemans et al., 2008), as well as learning and memory impairments (Olson et al., 1998) and increased incidence of mental illnesses (Streissguth, 1996). These aging-related problems are complex, requiring high-level of care, and consequential expense.
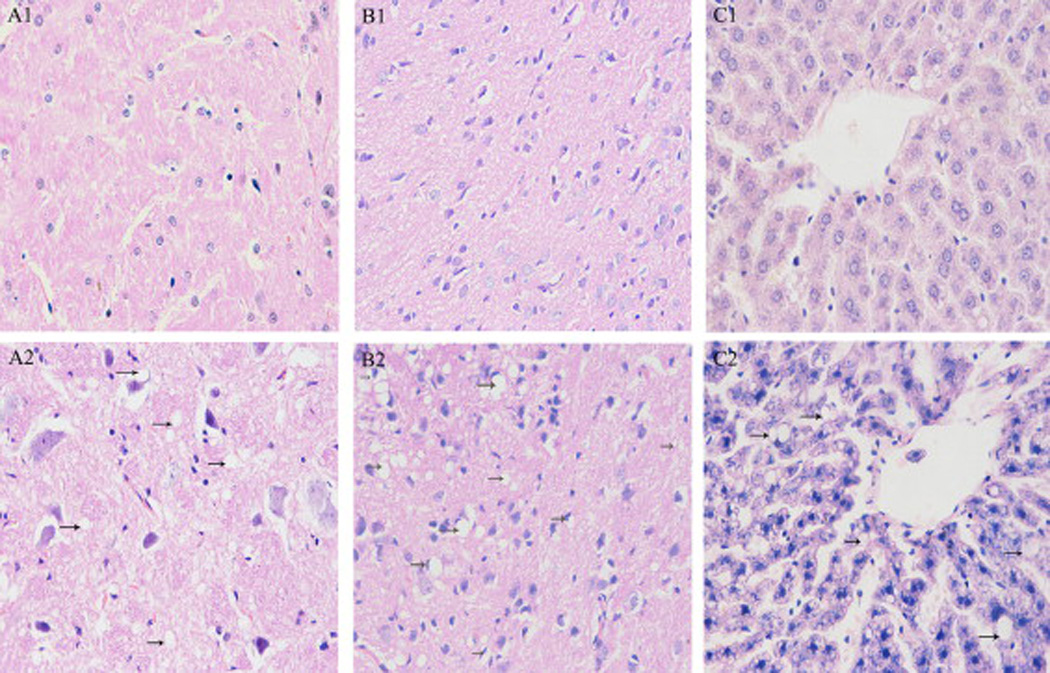
The following is a composite of examples of DOHaD studies previously established in the alcohol field. We have not described studies herein that measure FASD phenotypes following prenatal alcohol exposure, exclusively in the fetal and in the neonatal period, as these studies do not fit under the DOHaD definition. Behavioral: Questionnaires were used to collect data on the number of days mothers had consumed the equivalent of 4 units of alcohol in the past month during pregnancy at 18 and 32 weeks gestation, and reported that prenatal alcohol significantly associated with border-line personality disorder in children between 11 and 12 years of age; specifically, with those exposed to 4 or more units of alcohol at 32 weeks gestation after controlling for birth weight and sex, alcohol was positively associated with border-line personality disorder (Winsper et al., 2015). Previous studies in mice showed one bout of alcohol (5.8 g/kg) on gestational day 9 during the first trimester-equivalent resulted in long-term retention deficits in adulthood at ages 12 and 24 months, although such deficits were barely detectable at 3 months of age (Dumas and Rabe, 1994). In another study, rats administered ad libitum liquid alcohol diet (36% alcohol-derived calories) throughout pregnancy, followed by 10 days of chronic mild stress procedure on offspring at 60–78 days of age, had different effects in males and females. Alcohol-exposed males showed impaired hedonic responsivity, locomotor hyperactivity, and alterations in affiliative and non-affiliative behaviors compared to control males. Alcohol-exposed females showed greater levels of behavioral despair in the forced swim test and altered behavior in the final 5 minutes of the social interaction test (Hellemans et al., 2010). Growth: Sprague-Dawley rats were fed a liquid diet with or without 6% v/v alcohol throughout gestation, and growth patterns were studied in the offspring up to 1 year; slower growth was detected in males from alcohol-fed dams between 7 and 12 months of age, whereas no differences were found in females (Probyn et al., 2012). Cardiac: Using a low level of 6% v/v alcohol during pregnancy, it was discovered that gene expression of a number of cardiac growth factors in the rat fetus (harvested on gestation day 20) and cardiomyocyte number in weanling offspring (postnatal day 30) were not altered by alcohol, whereas at 8 months age, there were significant increases in left ventricle anterior and posterior wall thickness during diastole in alcohol-exposed offspring (Nguyen et al., 2014). Thyroid: Prenatal alcohol effects on adult thyroid function have been studied in a rat model after 5% (w/v) alcohol was fed during pregnancy. At 90–100 postnatal days, basal thyroid stimulating hormone (TSH) was significantly increased in males compared to the females except in the fetal alcohol exposed animals (Wilcoxon and Redei, 2004). This prenatal alcohol exposure resulted in adult hypothyroidism; total T3 thyroid hormone was decreased in both males and females, whereas T4 was not altered, and T4 replacement during pregnancy could not normalize adult thyroid deficiency (Wilcoxon and Redei, 2004). Recently, the same group demonstrated that glucose tolerance and insulin aberrations could be observed in second generation (F2) offspring following prenatal alcohol exposure (~5% w/v) between gestational days 8–20 (Harper et al., 2014); sex-specific hypoglycemic and hyperinsulinemic glucose-tolerance test response patterns were observed in F2 offspring, and administration of thyroid hormone (T4) to the alcohol-consuming grandmother prevented deficits in glucose tolerance test responses in F2 progeny alone, suggesting T4 prevented, reversed, or interfered with the effects of alcohol on F1 germ cells (Harper et al., 2014). This study exemplifies an important feature of alcohol exposure during pregnancy, that is in addition to impacting the mother and fetus, the fetal germline is vulnerable. Neurobiological: Rats were administered alcohol (6 mg/kg) and were assessed for auditory brain stem response at 22 days, 7 months, and 17 months after birth; in accordance with the Barker hypothesis, the authors reported developmental delay that dissipated at 7 months, and enhanced deficits at 17 months, thus recommending long-term follow up of prenatally alcohol-exposed kids (Church et al., 2012). Nutrient homeostasis: Alcohol-exposed rats were studied up to 14 months after birth, and male rats specifically demonstrated significantly higher fasting serum triglyceride levels. These disturbances were replicated in females by treating them with testosterone, and the hypertriglyceridemia could be inhibited in males by castration (Pennington et al., 2002). These effects show the significance of studying alcohol-induced testosterone programming of adult-onset diseases. Glucose homeostasis was investigated in another study where rats received a liquid diet containing 6% (v/v) alcohol (15% alcohol-derived calories) throughout gestation, and offspring were studied up to 8 months of age (Probyn et al., 2013); longitudinal assessments of fasting basal glucose and insulin over these 8 months did not show any differences, whereas four month old male rats displayed elevated first phase insulin secretion in a glucose challenge, showing vulnerability to adult-onset insulin resistance due to prenatal alcohol exposure (Probyn et al., 2013). In another study, 2 g/kg alcohol was administered twice daily via intra-gastric gavage to rats throughout gestation. Glucose and insulin levels with fasting blood glucose concentrations were higher by >7.0 mmol/l in alcohol-treated offspring, resulting in insulin resistance despite hyperinsulinemia in 3 month old rats (Yao and Gregoire Nyomba, 2007). Pregnant guinea pigs were administered 4g/kg alcohol for five days a week throughout gestation followed by magnetic resonance imaging between postnatal days 100–140. Results demonstrated increased visceral and subcutaneous adiposity, and between postnatal day 150–200, increased pancreatic adipocyte area and decreased β-cell insulin-like immunopositive area, concluding that prenatal alcohol exposure induced adult-onset metabolic syndrome (Figure 1) (Dobson et al., 2012). Recently, the same group utilized a similar paradigm, demonstrating prenatal alcohol exposure resulted in sex-specific alteration in adult offspring mRNA expression of several insulin and insulin-like growth factor-related genes in the liver and prefrontal cortex, and concluded prenatal alcohol exposure induced metabolic dysregulation in adult offspring (Dobson et al., 2014). Liver: In rats, it was demonstrated that prenatal alcohol exposure (4 g/kg gavage) from gestational day 11until birth followed by high fat diet post-weaning in female offspring resulted in down-regulation of hepatic IGF-1 expression and lipid output, whereas lipid synthesis was significantly increased (Shen et al., 2014); the authors concluded that prenatal alcohol-exposed female offspring exhibited increased susceptibility to a high fat diet-induced non-alcoholic fatty liver disease in adulthood (Shen et al., 2014). Adrenal axis: Pregnant Wistar rats were administered 4g/kg alcohol intragastrically from gestational day 11 until birth, and male offspring were fed a high-fat diet from postnatal week 4 at weaning until postnatal week 20 (Figure 2) (Xia et al., 2014). At postnatal week 17, offspring were subjected to an unpredictable chronic stress procedure for 21 days (Xia et al., 2014). Male alcohol-exposed offspring exhibited lower birth weight, later catch-up growth, lower basal activity of hypothalamic-pituitary-adrenal (HPA) axis, and enhanced sensitivity to stress. This resulted in increased serum glucose, insulin, insulin resistance index, and cholesterol, concluding prenatal alcohol exposure followed by high fat postnatal diet results in increased susceptibility to metabolic syndrome in adulthood (Xia et al., 2014). Paternal alcohol exposure: Current studies are limited in considering the effect of paternal alcohol exposure on offspring. Just as alcohol crosses the placenta, it also crosses the blood-testis barrier, and its potential effects on male germ cells should not be discredited. Neither animal nor human studies have directly connected paternal alcohol consumption with lower birthweight (Windham et al., 1995, Abel and Lee, 1988), the most replicated predictor for future risk of chronic adult diseases within multiple organ systems (Barker, 1998). However, more recent studies have linked paternal alcohol consumption with growth, developmental and behavioral abnormalities. A compelling study by Abel’s group in 2002 determined paternal alcohol consumption reduced cytosine methyltransferase mRNA in sperm, which may reflect alteration in genomic imprinting by reducing DNA methylation and potentially silencing paternal alleles (Bielawski et al., 2002). This finding may discern a mechanistic role in gene expression associated with both paternal alcohol consumption and adult chronic disease.
Figure 1.
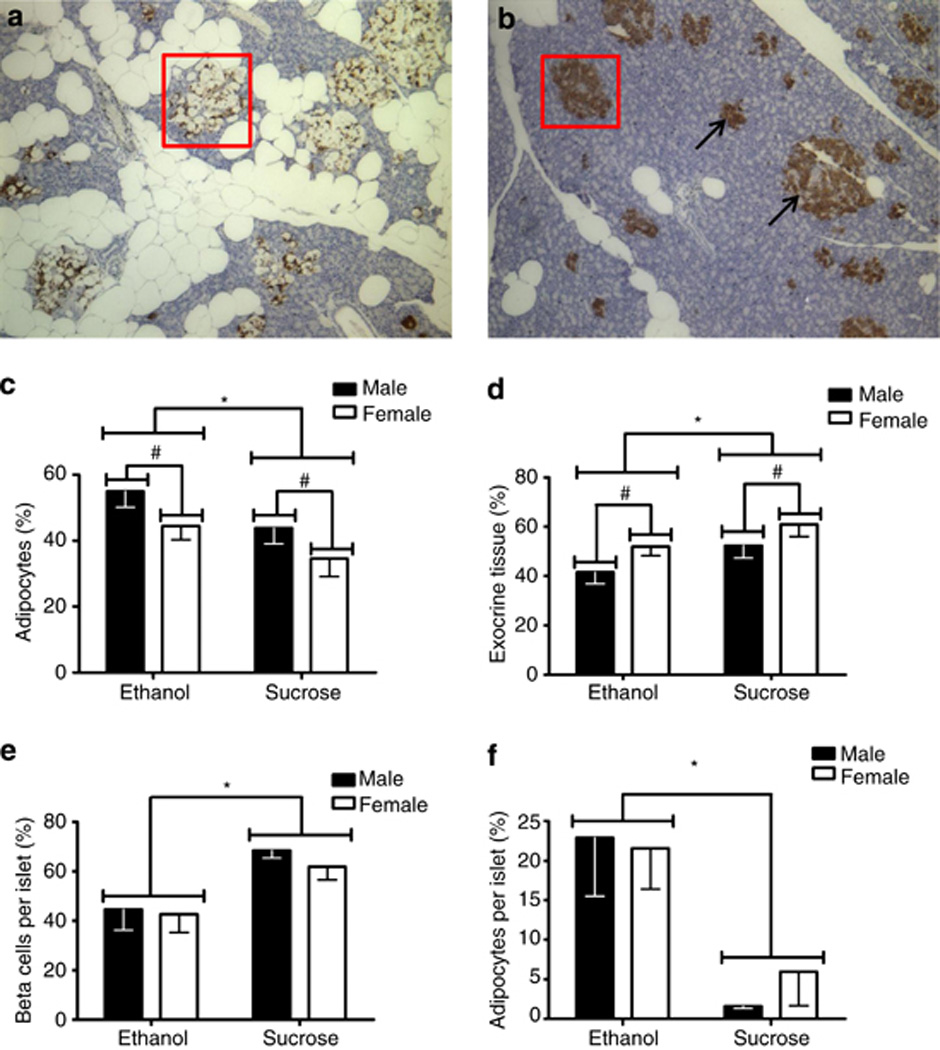
Figure 2.
EPIGENETIC MECHANISMS UNDERLYING DOHaD
From the studies discussed above, we have come to suspect that in utero exposures to alcohol have the capacity to alter the developmental program and impart a predisposition for the adult-onset of chronic disease. As these in utero encounters manifest their effects well beyond the window of exposure, deciphering the molecular mechanisms is incredibly challenging. Similar to many other agents, alcohol is suspected to have the capacity to alter the dynamic regulation of chromatin structure during critical developmental transitions, and therefore transmit a lasting signature of exposure on into later states (Feil and Fraga, 2011, Ungerer et al., 2013). However, the fundamental mechanisms by which alcohol-induced alterations in chromatin structure lead to increased vulnerability to adult-onset disease states following in utero insults remains largely unexplored.
Epigenetics is a term that refers to a series of biochemical processes through which heritable changes in gene transcription are achieved throughout the lifecycle of an organism, without a change in DNA sequence (Jaenisch and Bird, 2003). Within this context, the term heritable can be used to denote the transmission of transcriptional states through cell division on into later stages of development/post-natal life (cellular inheritance), or the marking of genes in the germ-line with the potential to impact gene expression in the offspring (D'Urso and Brickner, 2014). Collectively, epigenetic changes in chromatin structure have been associated with a memory of environmentally induced transcriptional change, alterations in animal phenotype, and an increased susceptibility (or lack of adaptability) to challenges later in life. For example, studies examining the interferon gamma-induced transcriptional response have shown that cells previously exposed to a stimulant remodel their chromatin in anticipation of the need for a more robust response, and that this priming can persist or be inherited for at least four mitotic generations (Gialitakis et al., 2010). Similarly, prenatal and postnatal stress has been shown to alter DNA methylation within the regulatory regions of key genes modulating behavior, and therefore pass a transcriptional memory of early life (with a clear impact on long-term health) on into later stages (Stankiewicz et al., 2013). Therefore alterations in chromatin structure represent a viable mechanism by which early exposures can alter the transcriptional program and potentially sensitize, or in some way limit the developmental capacity of physiological systems, resulting in pathology later in life. Interestingly, each exposure episode for the fetus can actually be considered a trigenerational exposure, as the mother has the potential to be impacted, as well as the fetus and the gametes developing within the fetal germline. While compelling evidence has begun to emerge suggesting alcohol can modulate chromatin structure, the extent to which these changes influence development and disease remains very poorly explored (Ungerer et al., 2013).
Studies in embryonic stem cells and other in vitro developmental models have revealed a multitude of large-scale changes in chromatin architecture induced by the processes of differentiation. In animal models, correlation of acute alcohol exposures with major periods of organ growth indicate that different tissues are largely susceptible to alcohol-induced teratogenesis during very specific developmental windows (Becker et al., 1996). It is therefore tempting to speculate that the processes of chromatin remodeling inherent to differentiation make specific cell populations vulnerable to the teratogenic actions of alcohol, and that in select instances, the impact of alcohol-induced changes in chromatin structure do not manifest until much later in life. In support of this notion, using the Agouti viable yellow (Avy) mouse model, Kaminen-Ahola and colleagues demonstrated maternal alcohol exposure between gestational days 0.5–8.5 induced changes in DNA methylation, which led to altered coat color in the adolescent offspring (Kaminen-Ahola et al., 2010). While several studies have suggested that alcohol exposure can induce acute changes in chromatin structure, these observations were among the first to suggest that at least some loci can inherit alcohol-induced epigenetic changes through into postnatal life. Since this initial work, other studies have identified heritable alterations in chromatin structure arising from early gestational exposures, suggesting epigenetic mechanisms are relevant to the development of FASDs (Chen et al., 2013, Veazey et al., 2015, Zhang et al., 2015, Marjonen et al., 2015). As an example, studies examining blood samples derived from FAS children have demonstrated altered DNA methylation within the regulatory regions of select imprinted genes, suggesting that alcohol-induced alterations in chromatin structure may persist through development and into early adulthood (Laufer et al., 2015, Masemola et al., 2015). Interestingly, recent work suggests some of these epigenetic lesions may transmit through the male germline into the next generation (Govorko et al., 2012).
Collectively, these individual observations support the hypothesis that several adult pathologies may be linked to epigenetic lesions inherited from a prenatal exposure. Emerging evidence is now beginning to suggest the impact of these encounters may last into postnatal life, and alter key homeostatic processes. For instance, the imprinted gene Insulin-like Growth Factor 2 (IGF-2), is maternally silenced / paternally expressed through complex epigenetic means, and plays a major role in both directing fetal growth and regulating adult metabolic pathways (Waterland and Michels, 2007). Downing and colleagues recently reported that when mice were fed 5.8g/kg alcohol on gestational day 9, alterations in the methylation of the IGF-2 regulatory regions were observed on gestational day 18, and these changes in DNA methylation correlated with a concomitant 1.5 fold decrease in the transcript levels (Downing et al., 2011). Given the prominent role IGF-2 plays in programming basic metabolism and growth, these alterations could have a lasting impact upon energy balance and cardiovascular health (Burton and Fowden, 2012, Loke et al., 2013). Although many tantalizing correlations exist, further long-term studies are required to follow alcohol-induced epigenetic lesions through into adult life before we can definitively link inherited changes in chromatin structure to adult disease. It is also important to remember that for the vast majority histone modifications, no known mechanism of inheritance exist, and researchers need to be careful not to confuse transient alterations in chromatin biology with inherited lesions, as they are not necessarily one and the same (Ptashne, 2007).
For the past 60 years, human genetic research has focused exclusively on the nucleic acid sequence of DNA as being the sole heritable molecule responsible for transmitting information controlling phenotype from parent to offspring. We are now beginning to understand that select loci within the sperm and the egg also possess some epigenetic information transmitted through to the offspring. For example, studies examining chronically exposed humans and rats have demonstrated that alcohol alters DNA methylation levels in sperm, and that these epigenetic errors are associated with reductions in offspring weight (Abel, 1991, Gabrielli and Mednick, 1983, Bielawski and Abel, 1997, Jamerson et al., 2004, Ouko et al., 2009). Moreover, studies by Govorko and colleagues in rodents have begun to suggest that some epigenetic lesions may be heritable transgenerationally (Govorko et al., 2012). If these observations hold true for humans, this could have a profound impact upon our understanding of the origins and inheritance of substance abuse disorders, as well as the origins of chronic disease. However, evidence to support this assertion is still very limited and more work exploring this aspect of FASD needs to be conducted.
DIRECTIONS FOR FASD-DOHaD FIELD
This is the first systematic review of prenatal alcohol exposure and DOHaD (Figure 3). It is clear that the studies are limited in the field and there are a dauntingly large number of unknowns. The following studies are most needed to fill the gap and advance the science in the DOHaD field.
Epidemiological studies: There is an absolutely essential need for human epidemiological studies that systematically assess the impact of alcohol exposure on adult-onset diseases. These studies are critical to establish the clinical significance of this area of scientific exploration. These studies come with limitations, as they can at best provide associations. Moreover, causal relationships between prenatal alcohol exposure and outcomes are difficult to establish, as women who drink while pregnant may also be subject to suboptimal nutrition and prenatal care, and may employ poly substance abuse, etc. (May et al., 2014, Viljoen et al., 2002). As these effects overlap and potentially interact, careful studies utilizing both animal models for FASD which control for confounding variables as well as relevant longitudinal comparison studies of human populations subject to similar risk factors yet abstain from alcohol are imperative to identify chronic disease risk factors associated with prenatal alcohol exposure.
Characterization of phenotypes: It is necessary to conduct animal studies under controlled conditions to identify the effects of dose of alcohol during gestation, duration of alcohol exposure, and timing of insult on development of adult-onset diseases. It is possible that higher doses of alcohol would lead to birth defects as well as adult-onset diseases, whereas lower doses of alcohol may result in subtle alterations that may not show any visible phenotypes at birth, but would nevertheless result in increased vulnerability to adult-onset disease states (Nguyen et al., 2014). Moreover, early gestation alcohol exposure is likely to produce adverse effects on diverse organs systems, whereas late gestation alcohol exposure is likely to result in tissue- and cell type-specific effects (Waterland and Michels, 2007) and the consequent adult disease outcomes are likely to be different.
Delineating mechanisms and interventions: The direct cause and effect is hard to establish between a substance of abuse and a specific adult-onset disease state. Better characterization of epigenetic mechanisms of inheritance and their impact upon pathology are needed before we can hypothesize potential mechanisms that may contribute to specific adult-onset functional deficits. Delineating connection between prenatal alcohol exposure and adult disease may help strengthen and broaden prevention strategies for drinking during pregnancy that rely solely on preventing FASD as their primary message. Limited work has been performed, even in non-alcohol literature, on interventions or prevention strategies for DOHaD, and it is essential to delineate ameliorative strategies following prenatal alcohol exposure. It is also important to address whether or not current research strategies for FASD will help further understanding of the mechanisms responsible for alcohol-induced DOHaD.
Figure 3.
Acknowledgments
Grants: NIH AA19446, AA23035, AA23520 (JR).
Footnotes
Conflict of Interest: None
REFERENCES
- Abel EL. Alcohol consumption does not affect fathers but does affect their offspring in the forced swimming test. Pharmacol Toxicol. 1991;68:68–69. doi: 10.1111/j.1600-0773.1991.tb01211.x. [DOI] [PubMed] [Google Scholar]
- Abel EL, Lee JA. Paternal alcohol exposure affects offspring behavior but not body or organ weights in mice. Alcohol Clin Exp Res. 1988;12:349–355. doi: 10.1111/j.1530-0277.1988.tb00205.x. [DOI] [PubMed] [Google Scholar]
- Adair L, Dahly D. Developmental determinants of blood pressure in adults. Annu Rev Nutr. 2005;25:407–434. doi: 10.1146/annurev.nutr.25.050304.092538. [DOI] [PubMed] [Google Scholar]
- Bake S, Tingling JD, Miranda RC. Ethanol exposure during pregnancy persistently attenuates cranially directed blood flow in the developing fetus: evidence from ultrasound imaging in a murine second trimester equivalent model. Alcohol Clin Exp Res. 2012;36:748–758. doi: 10.1111/j.1530-0277.2011.01676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Fetal origins of coronary heart disease. BMJ. 1995;311:171–174. doi: 10.1136/bmj.311.6998.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. In utero programming of chronic disease. Clin Sci (Lond) 1998;95:115–128. [PubMed] [Google Scholar]
- Barker DJ. Developmental origins of adult health and disease. J Epidemiol Community Health. 2004a;58:114–115. doi: 10.1136/jech.58.2.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. The developmental origins of well-being. Philos Trans R Soc Lond B Biol Sci. 2004b;359:1359–1366. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Adult consequences of fetal growth restriction. Clin Obstet Gynecol. 2006;49:270–283. doi: 10.1097/00003081-200606000-00009. [DOI] [PubMed] [Google Scholar]
- Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–417. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- Barker DJ. Sir Richard Doll Lecture. Developmental origins of chronic disease. Public Health. 2012;126:185–189. doi: 10.1016/j.puhe.2011.11.014. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Osmond C, Simmonds SJ, Wield GA. The relation of small head circumference and thinness at birth to death from cardiovascular disease in adult life. BMJ. 1993;306:422–426. doi: 10.1136/bmj.306.6875.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–580. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- Becker HC, Diaz-Granados JL, Randall CL. Teratogenic actions of ethanol in the mouse: a minireview. Pharmacol Biochem Behav. 1996;55:501–513. doi: 10.1016/s0091-3057(96)00255-9. [DOI] [PubMed] [Google Scholar]
- Bielawski DM, Abel EL. Acute treatment of paternal alcohol exposure produces malformations in offspring. Alcohol. 1997;14:397–401. doi: 10.1016/s0741-8329(97)87951-7. [DOI] [PubMed] [Google Scholar]
- Bielawski DM, Zaher FM, Svinarich DM, Abel EL. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcohol Clin Exp Res. 2002;26:347–351. [PubMed] [Google Scholar]
- Bonthius DJ, West JR. Blood alcohol concentration and microencephaly: a dose-response study in the neonatal rat. Teratology. 1988;37:223–231. doi: 10.1002/tera.1420370307. [DOI] [PubMed] [Google Scholar]
- Brawley L, Torrens C, Anthony FW, Itoh S, Wheeler T, Jackson AA, Clough GF, Poston L, Hanson MA. Glycine rectifies vascular dysfunction induced by dietary protein imbalance during pregnancy. J Physiol. 2004;554:497–504. doi: 10.1113/jphysiol.2003.052068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd L, Deal E, RIOS R, Adickes E, Wynne J, Klug MG. Congenital heart defects and fetal alcohol spectrum disorders. Congenit Heart Dis. 2007;2:250–255. doi: 10.1111/j.1747-0803.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- Burton GJ, Fowden AL. Review: The placenta and developmental programming: balancing fetal nutrient demands with maternal resource allocation. Placenta. 2012;33(Suppl):S23–S27. doi: 10.1016/j.placenta.2011.11.013. [DOI] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE, LI R, Bock C, LI C, GU H, Zamore PD, Meissner A, Weng Z, Hofmann HA, Friedman N, Rando OJ. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell. 2010;143:1084–1096. doi: 10.1016/j.cell.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LG, Lewis KN, Wilkerson DC, Tobia CM, Ngo Tenlep SY, Shridas P, Garcia-Cazarin ML, Wolff G, Andrade FH, Charnigo RJ, Esser KA, Egan JM, De Cabo R, Pearson KJ. Perinatal exercise improves glucose homeostasis in adult offspring. Am J Physiol Endocrinol Metab. 2012;303:E1061–E1068. doi: 10.1152/ajpendo.00213.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Ozturk NC, ZHOU FC. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS One. 2013;8:e60503. doi: 10.1371/journal.pone.0060503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church MW, Hotra JW, Holmes PA, Anumba JI, Jackson DA, Adams BR. Auditory brainstem response (ABR) abnormalities across the life span of rats prenatally exposed to alcohol. Alcohol Clin Exp Res. 2012;36:83–96. doi: 10.1111/j.1530-0277.2011.01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BL, Kirkpatrick JR, Taylor JA, VOM SAAL FS. A new 'crowded uterine horn' mouse model for examining the relationship between foetal growth and adult obesity. Basic Clin Pharmacol Toxicol. 2008;102:162–167. doi: 10.1111/j.1742-7843.2007.00195.x. [DOI] [PubMed] [Google Scholar]
- CONSENSUS STATEMENT NIH. Antenatal Corticosteroids Revisited: Repeat Courses. NIH Consensus Statement. 2000 [PubMed] [Google Scholar]
- Cooper C, Javaid MK, Taylor P, Walker-Bone K, Dennison E, Arden N. The fetal origins of osteoporotic fracture. Calcif Tissue Int. 2002;70:391–394. doi: 10.1007/s00223-001-0044-z. [DOI] [PubMed] [Google Scholar]
- D'Urso A, Brickner JH. Mechanisms of epigenetic memory. Trends Genet. 2014;30:230–236. doi: 10.1016/j.tig.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CC, Mongillo DL, Brien DC, Stepita R, Poklewska-Koziell M, Winterborn A, Holloway AC, Brien JF, Reynolds JN. Chronic prenatal ethanol exposure increases adiposity and disrupts pancreatic morphology in adult guinea pig offspring. Nutr Diabetes. 2012;2:e57. doi: 10.1038/nutd.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CC, Thevasundaram K, Mongillo DL, Winterborn A, Holloway AC, Brien JF, Reynolds JN. Chronic prenatal ethanol exposure alters expression of central and peripheral insulin signaling molecules in adult guinea pig offspring. Alcohol. 2014;48:687–693. doi: 10.1016/j.alcohol.2014.09.001. [DOI] [PubMed] [Google Scholar]
- Downing C, Johnson TE, Larson C, Leakey TI, Siegfried RN, Rafferty TM, Cooney CA. Subtle decreases in DNA methylation and gene expression at the mouse Igf2 locus following prenatal alcohol exposure: effects of a methyl-supplemented diet. Alcohol. 2011;45:65–71. doi: 10.1016/j.alcohol.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle LW, Ford GW, Davis NM, Callanan C. Antenatal corticosteroid therapy and blood pressure at 14 years of age in preterm children. Clin Sci (Lond) 2000;98:137–142. [PubMed] [Google Scholar]
- Dumas RM, RABE A. Augmented memory loss in aging mice after one embryonic exposure to alcohol. Neurotoxicol Teratol. 1994;16:605–612. doi: 10.1016/0892-0362(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Edwards CR, Benediktsson R, Lindsay RS, Seckl JR. Dysfunction of placental glucocorticoid barrier: link between fetal environment and adult hypertension? Lancet. 1993;341:355–357. doi: 10.1016/0140-6736(93)90148-a. [DOI] [PubMed] [Google Scholar]
- Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2011;13:97–109. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- Feldman HS, Jones KL, Lindsay S, Slymen D, Klonoff-Cohen H, Kao K, Rao S, Chambers C. Prenatal alcohol exposure patterns and alcohol-related birth defects and growth deficiencies: a prospective study. Alcohol Clin Exp Res. 2012;36:670–676. doi: 10.1111/j.1530-0277.2011.01664.x. [DOI] [PubMed] [Google Scholar]
- Fuglestad AJ, Boys CJ, Chang PN, Miller BS, Eckerle JK, Deling L, Fink BA, Hoecker HL, Hickey MK, Jimenez-Vega JM, Wozniak JR. Overweight and obesity among children and adolescents with fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2014;38:2502–2508. doi: 10.1111/acer.12516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabory A, Roseboom TJ, Moore T, Moore LG, Junien C. Placental contribution to the origins of sexual dimorphism in health and diseases: sex chromosomes and epigenetics. Biol Sex Differ. 2013;4:5. doi: 10.1186/2042-6410-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GabriellI WF, Jr, Mednick SA. Intellectual performance in children of alcoholics. J Nerv Ment Dis. 1983;171:444–447. doi: 10.1097/00005053-198307000-00009. [DOI] [PubMed] [Google Scholar]
- Gareri J, Brien J, Reynolds J, Koren G. Potential role of the placenta in fetal alcohol spectrum disorder. Paediatr Drugs. 2009;11:26–29. doi: 10.2165/0148581-200911010-00010. [DOI] [PubMed] [Google Scholar]
- Gautam P, Lebel C, Narr KL, Mattson SN, May PA, Adnams CM, Riley EP, Jones KL, Kan EC, Sowell ER. Volume changes and brain-behavior relationships in white matter and subcortical gray matter in children with prenatal alcohol exposure. Hum Brain Mapp. 2015;36:2318–2329. doi: 10.1002/hbm.22772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gialitakis M, Arampatzi P, Makatounakis T, Papamatheakis J. Gamma interferon-dependent transcriptional memory via relocalization of a gene locus to PML nuclear bodies. Mol Cell Biol. 2010;30:2046–2056. doi: 10.1128/MCB.00906-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359:61–73. doi: 10.1056/NEJMra0708473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorko D, Bekdash RA, Zhang C, Sarkar DK. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry. 2012;72:378–388. doi: 10.1016/j.biopsych.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogan F, Elwood G, Longato L, Tong M, Feijoo A, Carlson RI, Wands JR, De la Monte SM. Impaired placentation in fetal alcohol syndrome. Placenta. 2008;29:148–157. doi: 10.1016/j.placenta.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson M. The birth and future health of DOHaD. J Dev Orig Health Dis. 2015:1–4. doi: 10.1017/S2040174415001129. [DOI] [PubMed] [Google Scholar]
- Harper KM, Tunc-Ozcan E, Graf EN, Redei EE. Intergenerational effects of prenatal ethanol on glucose tolerance and insulin response. Physiol Genomics. 2014;46:159–168. doi: 10.1152/physiolgenomics.00181.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KG, Verma P, Yoon E, Yu W, Weinberg J. Prenatal alcohol exposure increases vulnerability to stress and anxiety-like disorders in adulthood. Ann N Y Acad Sci. 2008;1144:154–175. doi: 10.1196/annals.1418.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellemans KG, Verma P, Yoon E, Yu WK, Young AH, Weinberg J. Prenatal alcohol exposure and chronic mild stress differentially alter depressive- and anxiety-like behaviors in male and female offspring. Alcohol Clin Exp Res. 2010;34:633–645. doi: 10.1111/j.1530-0277.2009.01132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyme HE, May PA, Kalberg WO, Kodituwakku P, Gossage JP, Trujillo PM, Buckley DG, Miller JH, Aragon AS, Khaole N, Viljoen DL, Jones KL, Robinson LK. A practical clinical approach to diagnosis of fetal alcohol spectrum disorders: clarification of the 1996 institute of medicine criteria. Pediatrics. 2005;115:39–47. doi: 10.1542/peds.2004-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AA, Dunn RL, Marchand MC, Langley-Evans SC. Increased systolic blood pressure in rats induced by a maternal low-protein diet is reversed by dietary supplementation with glycine. Clin Sci (Lond) 2002;103:633–639. doi: 10.1042/cs1030633. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jamerson PA, Wulser MJ, Kimler BF. Neurobehavioral effects in rat pups whose sires were exposed to alcohol. Brain Res Dev Brain Res. 2004;149:103–111. doi: 10.1016/j.devbrainres.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Jones KL, Smith DW. Recognition of the fetal alcohol syndrome in early infancy. Lancet. 1973;302:999–1001. doi: 10.1016/s0140-6736(73)91092-1. [DOI] [PubMed] [Google Scholar]
- Kaminen-Ahola N, Ahola A, Maga M, Mallitt KA, Fahey P, Cox TC, Whitelaw E, Chong S. Maternal ethanol consumption alters the epigenotype and the phenotype of offspring in a mouse model. PLoS Genet. 2010;6:e1000811. doi: 10.1371/journal.pgen.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufer BI, Kapalanga J, Castellani CA, Diehl EJ, Yan L, Singh SM. Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics. 2015:1–16. doi: 10.2217/epi.15.60. [DOI] [PubMed] [Google Scholar]
- Lewis CE, Thomas KG, Dodge NC, Molteno CD, Meintjes EM, Jacobson JL, Jacobson SW. Verbal learning and memory impairment in children with fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2015;39:724–732. doi: 10.1111/acer.12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C, Oest ME, Prater MR. Intrauterine exposure to high saturated fat diet elevates risk of adult-onset chronic diseases in C57BL/6 mice. Birth Defects Res B Dev Reprod Toxicol. 2009;86:377–384. doi: 10.1002/bdrb.20206. [DOI] [PubMed] [Google Scholar]
- Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics. 1972;50:515–525. [PubMed] [Google Scholar]
- Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25:447–458. doi: 10.1016/s0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- Loke YJ, Galati JC, Morley R, Joo EJ, Novakovic B, Li X, Weinrich B, Carson N, Ollikainen M, Ng HK, Andronikos R, Aziz NK, Saffery R, Craig JM. Association of maternal and nutrient supply line factors with DNA methylation at the imprinted IGF2/H19 locus in multiple tissues of newborn twins. Epigenetics. 2013;8:1069–1079. doi: 10.4161/epi.25908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SE, Miller JA, Blackwell JM, West JR. Fetal alcohol exposure and temporal vulnerability: regional differences in cell loss as a function of the timing of binge-like alcohol exposure during brain development. Alcohol Clin Exp Res. 1999;23:726–734. doi: 10.1111/j.1530-0277.1999.tb04176.x. [DOI] [PubMed] [Google Scholar]
- Marjonen H, Sierra A, Nyman A, Rogojin V, Grohn O, Linden AM, Hautaniemi S, Kaminen-Ahola N. Early maternal alcohol consumption alters hippocampal DNA methylation, gene expression and volume in a mouse model. PLoS One. 2015;10:e0124931. doi: 10.1371/journal.pone.0124931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masemola ML, Van der Merwe L, Lombard Z, Viljoen D, Ramsay M. Reduced DNA methylation at the PEG3 DMR and KvDMR1 loci in children exposed to alcohol in utero: a South African Fetal Alcohol Syndrome cohort study. Front Genet. 2015;6:85. doi: 10.3389/fgene.2015.00085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews SG. Antenatal glucocorticoids and programming of the developing CNS. Pediatr Res. 2000;47:291–300. doi: 10.1203/00006450-200003000-00003. [DOI] [PubMed] [Google Scholar]
- May PA, Baete A, Russo J, Elliott AJ, Blankenship J, Kalberg WO, Buckley D, Brooks M, Hasken J, Abdul-Rahman O, Adam MP, Robinson LK, Manning M, Hoyme HE. Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics. 2014;134:855–866. doi: 10.1542/peds.2013-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Blankenship J, Marais AS, Gossage JP, Kalberg WO, Joubert B, Cloete M, Barnard R, De Vries M, Hasken J, Robinson LK, Adnams CM, Buckley D, Manning M, Parry CD, Hoyme HE, Tabachnick B, Seedat S. Maternal alcohol consumption producing fetal alcohol spectrum disorders (FASD): quantity, frequency, and timing of drinking. Drug Alcohol Depend. 2013;133:502–512. doi: 10.1016/j.drugalcdep.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Gossage JP, Kalberg WO, Robinson LK, Buckley D, Manning M, Hoyme HE. Prevalence and epidemiologic characteristics of FASD from various research methods with an emphasis on recent in-school studies. Dev Disabil Res Rev. 2009;15:176–192. doi: 10.1002/ddrr.68. [DOI] [PubMed] [Google Scholar]
- Miranda R, Ceckler T, Guillet R, Kellogg C. Early developmental exposure to benzodiazepine ligands alters brain 31P–NMR spectra in young adult rats. Brain Res. 1990a;506:85–92. doi: 10.1016/0006-8993(90)91202-r. [DOI] [PubMed] [Google Scholar]
- Miranda R, Ceckler T, Guillet R, Kellogg CK. Aging-related changes in brain metabolism are altered by early developmental exposure to diazepam. Neurobiol Aging. 1990b;11:117–122. doi: 10.1016/0197-4580(90)90044-z. [DOI] [PubMed] [Google Scholar]
- Miranda RC, Wagner JP, Kellogg CK. Early developmental exposure to benzodiazepine ligands alters brain levels of thiobarbituric acid-reactive products in young adult rats. Neurochem Res. 1989;14:1119–1127. doi: 10.1007/BF00965618. [DOI] [PubMed] [Google Scholar]
- Moore EM, Riley EP. What Happens When Children with Fetal Alcohol Spectrum Disorders Become Adults? Curr Dev Disord Rep. 2015;2:219–227. doi: 10.1007/s40474-015-0053-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mwansa-Kambafwile J, Cousens S, Hansen T, Lawn JE. Antenatal steroids in preterm labour for the prevention of neonatal deaths due to complications of preterm birth. Int J Epidemiol. 2010;39(Suppl 1):122–33. doi: 10.1093/ije/dyq029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VB, Probyn ME, Campbell F, Yin KV, Samuel CS, Zimanyi MA, Bertram JF, Black MJ, Moritz KM. Low-dose maternal alcohol consumption: effects in the hearts of offspring in early life and adulthood. Physiol Rep. 2014;2 doi: 10.14814/phy2.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary CM, Bower C, Zubrick SR, Geelhoed E, Kurinczuk JJ, Nassar N. A new method of prenatal alcohol classification accounting for dose, pattern and timing of exposure: improving our ability to examine fetal effects from low to moderate alcohol. J Epidemiol Community Health. 2010;64:956–962. doi: 10.1136/jech.2009.091785. [DOI] [PubMed] [Google Scholar]
- Olson HC, Feldman JJ, Streissguth AP, Sampson PD, Bookstein FL. Neuropsychological deficits in adolescents with fetal alcohol syndrome: clinical findings. Alcohol Clin Exp Res. 1998;22:1998–2012. [PubMed] [Google Scholar]
- Ouko LA, Shantikumar K, Knezovich J, Haycock P, Schnugh DJ, Ramsay M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: implications for fetal alcohol spectrum disorders. Alcohol Clin Exp Res. 2009;33:1615–1627. doi: 10.1111/j.1530-0277.2009.00993.x. [DOI] [PubMed] [Google Scholar]
- Painter RC, De Rooij SR, Bossuyt PM, Simmers TA, Osmond C, Barker DJ, Bleker OP, Roseboom TJ. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am J Clin Nutr. 2006;84:322–327. doi: 10.1093/ajcn/84.1.322. quiz 466-7. [DOI] [PubMed] [Google Scholar]
- Parnell SE, Ramadoss J, Delp MD, Ramsey MW, Chen WJ, West JR, Cudd TA. Chronic ethanol increases fetal cerebral blood flow specific to the ethanol-sensitive cerebellum under normoxaemic, hypercapnic and acidaemic conditions: ovine model. Exp Physiol. 2007;92:933–943. doi: 10.1113/expphysiol.2007.038091. [DOI] [PubMed] [Google Scholar]
- Pennington JS, Shuvaeva TI, Pennington SN. Maternal dietary ethanol consumption is associated with hypertriglyceridemia in adult rat offspring. Alcohol Clin Exp Res. 2002;26:848–855. [PubMed] [Google Scholar]
- Probyn ME, Parsonson KR, Gardebjer EM, Ward LC, Wlodek ME, Anderson ST, Moritz KM. Impact of low dose prenatal ethanol exposure on glucose homeostasis in Sprague-Dawley rats aged up to eight months. PLoS One. 2013;8:e59718. doi: 10.1371/journal.pone.0059718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probyn ME, Zanini S, Ward LC, Bertram JF, Moritz KM. A rodent model of low- to moderate-dose ethanol consumption during pregnancy: patterns of ethanol consumption and effects on fetal and offspring growth. Reprod Fertil Dev. 2012;24:859–870. doi: 10.1071/RD11200. [DOI] [PubMed] [Google Scholar]
- Ptashne M. On the use of the word 'epigenetic'. Curr Biol. 2007;17:R233–R236. doi: 10.1016/j.cub.2007.02.030. [DOI] [PubMed] [Google Scholar]
- Ramadoss J, Lunde ER, Chen WJ, West JR, Cudd TA. Temporal vulnerability of fetal cerebellar Purkinje cells to chronic binge alcohol exposure: ovine model. Alcohol Clin Exp Res. 2007a;31:1738–1745. doi: 10.1111/j.1530-0277.2007.00477.x. [DOI] [PubMed] [Google Scholar]
- Ramadoss J, Lunde ER, Pina KB, Chen WJ, Cudd TA. All three trimester binge alcohol exposure causes fetal cerebellar purkinje cell loss in the presence of maternal hypercapnea, acidemia, and normoxemia: ovine model. Alcohol Clin Exp Res. 2007B;31:1252–1258. doi: 10.1111/j.1530-0277.2007.00422.x. [DOI] [PubMed] [Google Scholar]
- Ramadoss J, Magness RR. Vascular effects of maternal alcohol consumption. Am J Physiol Heart Circ Physiol. 2012;303:H414–H421. doi: 10.1152/ajpheart.00127.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Wold LE, Natavio M, Ren BH, Hannigan JH, Brown RA. Influence of prenatal alcohol exposure on myocardial contractile function in adult rat hearts: role of intracellular calcium and apoptosis. Alcohol Alcohol. 2002;37:30–37. doi: 10.1093/alcalc/37.1.30. [DOI] [PubMed] [Google Scholar]
- Riley EP, Infante MA, Warren KR. Fetal alcohol spectrum disorders: an overview. Neuropsychol Rev. 2011;21:73–80. doi: 10.1007/s11065-011-9166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley EP, Mcgee CL. Fetal alcohol spectrum disorders: an overview with emphasis on changes in brain and behavior. Exp Biol Med (Maywood) 2005;230:357–365. doi: 10.1177/15353702-0323006-03. [DOI] [PubMed] [Google Scholar]
- Roseboom TJ, Van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP. Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview. Mol Cell Endocrinol. 2001;185:93–98. doi: 10.1016/s0303-7207(01)00721-3. [DOI] [PubMed] [Google Scholar]
- Sawant OB, Lunde ER, Washburn SE, Chen WJ, Goodlett CR, Cudd TA. Different patterns of regional Purkinje cell loss in the cerebellar vermis as a function of the timing of prenatal ethanol exposure in an ovine model. Neurotoxicol Teratol. 2013;35:7–13. doi: 10.1016/j.ntt.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant OB, Ramadoss J, Hankins GD, Wu G, Washburn SE. Effects of L-glutamine supplementation on maternal and fetal hemodynamics in gestating ewes exposed to alcohol. Amino Acids. 2014;46:1981–1996. doi: 10.1007/s00726-014-1751-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlotz W, Phillips DI. Fetal origins of mental health: evidence and mechanisms. Brain Behav Immun. 2009;23:905–916. doi: 10.1016/j.bbi.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Shen L, Liu Z, Gong J, Zhang L, Wang L, Magdalou J, Chen L, Wang H. Prenatal ethanol exposure programs an increased susceptibility of non-alcoholic fatty liver disease in female adult offspring rats. Toxicol Appl Pharmacol. 2014;274:263–273. doi: 10.1016/j.taap.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Delaney-Black V, Nordstrom B. Fetal alcohol spectrum disorder. JAMA. 2003;290:2996–2999. doi: 10.1001/jama.290.22.2996. [DOI] [PubMed] [Google Scholar]
- Stankiewicz AM, Swiergiel AH, Lisowski P. Epigenetics of stress adaptations in the brain. Brain Res Bull. 2013;98:76–92. doi: 10.1016/j.brainresbull.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Stein CE, Fall CH, Kumaran K, Osmond C, Cox V, Barker DJ. Fetal growth and coronary heart disease in south India. Lancet. 1996;348:1269–1273. doi: 10.1016/s0140-6736(96)04547-3. [DOI] [PubMed] [Google Scholar]
- Stein CE, Kumaran K, Fall CH, Shaheen SO, Osmond C, Barker DJ. Relation of fetal growth to adult lung function in south India. Thorax. 1997;52:895–899. doi: 10.1136/thx.52.10.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streissguth AP, Aase JM, Clarren SK, Randels SP, Ladue RA, Smith DF. Fetal alcohol syndrome in adolescents and adults. JAMA. 1991;265:1961–1967. [PubMed] [Google Scholar]
- Streissguth AP, Barr HM, Kogan J, Bookstein FL. 'Understanding the Occurrence of Secondary Disabilities in Clients with Fetal Alcohol Syndrome (FAS) and Fetal Alcohol Effects (FAE),' Final Report to the Centers for Disease Control and Prevention (CDC) Seattle, Washington: University of Washington Press; 1996. [Google Scholar]
- Tan CH, Denny CH, Cheal NE, Sniezek JE, Kanny D. Alcohol use and binge drinking among women of childbearing age - United States, 2011–2013. MMWR Morb Mortal Wkly Rep. 2015;64:1042–1046. doi: 10.15585/mmwr.mm6437a3. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Wasserman EA, West JR, Goodlett CR. Behavioral deficits induced by bingelike exposure to alcohol in neonatal rats: importance of developmental timing and number of episodes. Dev Psychobiol. 1996;29:433–452. doi: 10.1002/(SICI)1098-2302(199607)29:5<433::AID-DEV3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Tye K, Pollard I, Karlsson L, Scheibner V, Tye G. Caffeine exposure in utero increases the incidence of apnea in adult rats. Reprod Toxicol. 1993;7:449–452. doi: 10.1016/0890-6238(93)90089-p. [DOI] [PubMed] [Google Scholar]
- Ungerer M, Knezovich J, Ramsay M. In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res. 2013;35:37–46. doi: 10.35946/arcr.v35.1.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veazey KJ, Parnell SE, Miranda RC, Golding MC. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics Chromatin. 2015;8:39. doi: 10.1186/s13072-015-0031-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viljoen D, Croxford J, Gossage JP, Kodituwakku PW, May PA. Characteristics of mothers of children with fetal alcohol syndrome in the Western Cape Province of South Africa: a case control study. J Stud Alcohol. 2002;63:6–17. [PubMed] [Google Scholar]
- Wang L, Shen L, Ping J, Zhang L, Liu Z, Wu Y, Liu Y, Huang H, Chen L, Wang H. Intrauterine metabolic programming alteration increased susceptibility to non-alcoholic adult fatty liver disease in prenatal caffeine-exposed rat offspring. Toxicol Lett. 2014;224:311–318. doi: 10.1016/j.toxlet.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Warren KR, Hewitt BG, Thomas JD. Fetal alcohol spectrum disorders: research challenges and opportunities. Alcohol Res Health. 2011;34:4–14. [PMC free article] [PubMed] [Google Scholar]
- Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–388. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- Watkins AJ, Lucas ES, Torrens C, Cleal JK, Green L, Osmond C, Eckert JJ, Gray WP, Hanson MA, Fleming TP. Maternal low-protein diet during mouse pre-implantation development induces vascular dysfunction and altered renin-angiotensin-system homeostasis in the offspring. Br J Nutr. 2010;103:1762–1770. doi: 10.1017/S0007114509993783. [DOI] [PubMed] [Google Scholar]
- Wilcoxon JS, Kuo AG, Disterhoft JF, Redei EE. Behavioral deficits associated with fetal alcohol exposure are reversed by prenatal thyroid hormone treatment: a role for maternal thyroid hormone deficiency in FAE. Mol Psychiatry. 2005;10:961–971. doi: 10.1038/sj.mp.4001694. [DOI] [PubMed] [Google Scholar]
- Wilcoxon JS, Redei EE. Prenatal programming of adult thyroid function by alcohol and thyroid hormones. Am J Physiol Endocrinol Metab. 2004;287:E318–E326. doi: 10.1152/ajpendo.00022.2004. [DOI] [PubMed] [Google Scholar]
- Windham GC, Fenster L, Hopkins B, Swan SH. The association of moderate maternal and paternal alcohol consumption with birthweight and gestational age. Epidemiology. 1995;6:591–597. doi: 10.1097/00001648-199511000-00005. [DOI] [PubMed] [Google Scholar]
- Winsper C, Wolke D, Lereya T. Prospective associations between prenatal adversities and borderline personality disorder at 11–12 years. Psychol Med. 2015;45:1025–1037. doi: 10.1017/S0033291714002128. [DOI] [PubMed] [Google Scholar]
- Wood CE, Keller-Wood M. The critical importance of the fetal hypothalamus-pituitary-adrenal axis. F1000Res. 2016;5 doi: 10.12688/f1000research.7224.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia LP, Shen L, Kou H, Zhang BJ, Zhang L, Wu Y, Li XJ, Xiong J, YU Y, Wang H. Prenatal ethanol exposure enhances the susceptibility to metabolic syndrome in offspring rats by HPA axis-associated neuroendocrine metabolic programming. Toxicol Lett. 2014;226:98–105. doi: 10.1016/j.toxlet.2014.01.023. [DOI] [PubMed] [Google Scholar]
- Yao XH, Gregoire Nyomba BL. Abnormal glucose homeostasis in adult female rat offspring after intrauterine ethanol exposure. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1926–R1933. doi: 10.1152/ajpregu.00822.2006. [DOI] [PubMed] [Google Scholar]
- Zhang CR, Ho MF, Vega MC, Burne TH, Chong S. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin. 2015;8:40. doi: 10.1186/s13072-015-0032-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Sliwowska JH, Weinberg J. Prenatal alcohol exposure and fetal programming: effects on neuroendocrine and immune function. Exp Biol Med (Maywood) 2005;230:376–388. doi: 10.1177/15353702-0323006-05. [DOI] [PubMed] [Google Scholar]