

Graphical abstract
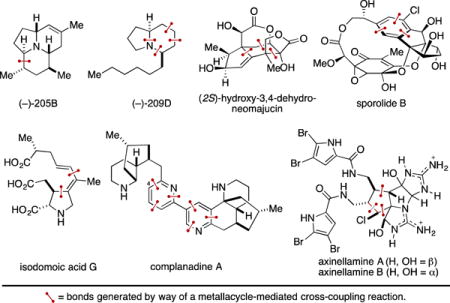
Keywords: Metallacycle-mediated cross-coupling, Convergent C–C bond formation, [2+2+1], [2+2+2], Catalyzed Alder-ene, Natural product synthesis
1. Introduction
Reaction processes that forge carbon–carbon bonds in a highly selective fashion sculpt the landscape of organic chemistry and play a profound role in natural product synthesis. In turn, it is well appreciated that total synthesis defines a stringent test for reaction methods and serves as a measure of their power to affect chemo- and stereoselective transformations in complex and diverse settings.1 As eloquently noted by Professor K. C. Nicolaou, “ever since the first laboratory construction of a carbon–carbon bond by Kolbe in his historic synthesis of acetic acid in 1845, carbon–carbon bond-forming reactions have played an enormously decisive and important role in shaping chemical synthesis.”2 In fact, new C–C bond-forming processes that offer unique retrosynthetic relationships can have a profound impact on the future of chemical synthesis, as well as neighboring disciplines where organic molecules play an important, if not central, role. While classic examples of powerful C–C bond-forming processes include aldol- and Grignard-type reactions, cycloadditions, sigmatropic rearrangements and other pericyclic processes, cation-olefin and radical cyclizations, modern advances have witnessed the emergence of metal-catalyzed cycloisomerization, cross-coupling (e.g., Pd- and Ni-), and olefin/alkyne metathesis.
Unique among these diverse classes of chemical reactivity are those that can be effectively employed in intermolecular settings, hence providing opportunities for convergent coupling. Convergency is among the most important strategic considerations in complex molecule synthesis, as it provides a means to decrease the longest linear sequence of chemical transformations required to convert simple starting materials into complex structures.3 Because yields of organic reactions are typically less than quantitative, significant contraction in the length of such sequences used to prepare a target can have a profound impact on efficiency. Of additional value, points of convergency in a synthesis pathway offer opportunities to introduce structural variation in a straightforward fashion – a feature of relevance in medicinal pursuits.4 It is instructive to recognize that most classical methods for convergent coupling by C–C bond formation are based on nucleophilic addition to polarized π-bonds (aldol and Grignard-type chemistry, carbonyl olefination, and 1,4-addition) and cycloaddition reactions. Modern advances have introduced distinct strategies for coupling that afford complementary bond constructions to these classic methods.1, 2 Such processes have had a profound impact on organic synthesis, with Pd-catalyzed cross-coupling and olefin metathesis each being recognized with a Nobel Prize.
In this review we place emphasis on an emerging collection of intermolecular C–C bond-forming processes that offer distinct retrosynthetic relationships in comparison to the now well-established strategies discussed above. The collection of reactions of interest here can be broadly characterized as metallacycle-mediated cross-coupling, and our focus is to cover those processes that have been demonstrated to be useful in natural product total synthesis.
At the outset of this discussion we recognize that a great variety of organic reactions are thought to proceed by way of metallacyclic intermediates, perhaps among the most popular of which are metathesis reactions (Figure 1A). These processes have had a transformative impact on organic synthesis, with their power often being wielded in intramolecular ring-forming chemistry (reactions that, by definition, are not convergent – rather, they are unimolecular). The development of versions of metathesis chemistry that allow for effective and stereoselective cross-coupling, and the application of such techniques to achieve convergent coupling in natural product synthesis, is certainly of great current interest, but such chemistry has been reviewed elsewhere.1
Figure 1.
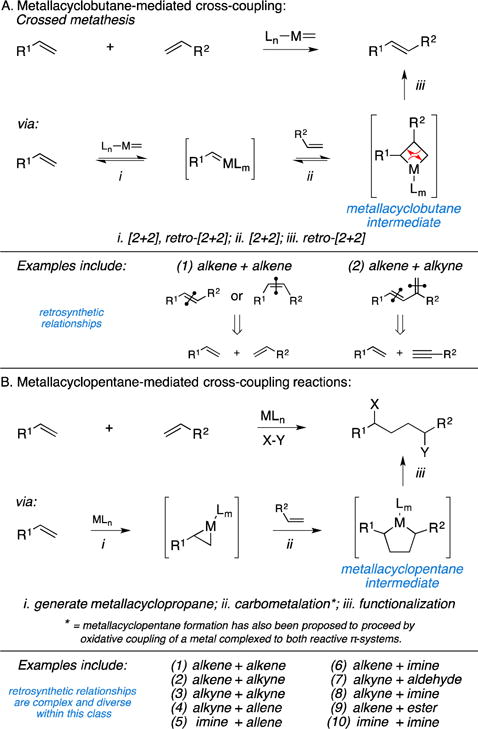
Introduction to metallacycle-mediated cross-coupling.
Here, we focus on a related and diverse collection of metallacycle-mediated coupling reactions that are thought to proceed through distinct metallacyclic intermediates – metallacyclopentanes (and analogs thereof) rather than metallacyclobutanes (Figure 1B). A large variety of coupling partners have been demonstrated to be compatible with this class of reactivity and, in some cases, many different stereoselective methods are available for each. In many instances the final organometallic intermediates generated in the course of these cross-coupling reactions can be functionalized in a number of different ways to establish a variety of diverse retrosynthetic relationships that derive from a common class of chemical reactivity.
From a historical perspective, this class of coupling chemistry was described nearly 70 years ago with the trimerization of acetylene as an approach to the formation of benzene (the Reppe reaction).5 Nearly thirty years after this discovery, the first demonstrations of powerful metallacycle-mediated cross-coupling chemistry began to appear in the context of natural product synthesis – Vollhardt’s use of Co-mediated [2+2+2] chemistry for the synthesis of estrone in 1980.6 While the latter part of the 20th century was marked by spectacular contributions at the interface of organometallic and synthetic organic chemistry, the last two decades have witnessed a substantial increase in contributions that feature the development of metallacycle-mediated cross-coupling chemistry and application of these processes in natural product synthesis.
We have organized this report in a manner that is primarily focused on the nature of the coupling partners employed in the key metallacycle-mediated cross-coupling step (i.e., alkyne–alkyne, alkyne–alkene, alkene–imine, etc.). Attention will be paid to chronology within each subset of coupling processes discussed, but the overall presentation will follow a sequence that we believe is appropriate from a pedagogical standpoint. In some cases we offer a brief discussion of the proposed mechanism and origin of selectivity associated with the metallacycle-mediated coupling process of relevance.
Overall, we hope that this Report serves to unify a class of eclectic, yet powerful, C–C bond-forming reactions as belonging to a broader, single class of chemical reactivity: “metallacycle-mediated cross-coupling.” Our focus on discussing applications in natural product synthesis is meant to demonstrate the great power that these reactions have in complex and diverse molecular settings, while providing a collection of examples that serve to educate the reader as to where such reactions have been employed in the context of larger molecular challenges.
2. General Considerations
In planning to use any metallacycle-mediated cross-coupling reaction in natural product synthesis, one must be familiar with general considerations associated with this class of reactivity. The first of these is simple reactivity. While many examples of metallacycle-mediated C–C bond formation have appeared in the context of intramolecular chemistry, achieving success in intermolecular settings is substantially more challenging (Figure 2A). In fact, there are many intramolecular bond-forming processes in this class that are extremely challenging to accomplish in an intermolecular fashion (i.e., alkene–alkene and alkyne–alkene). Once confidence is gained for being able to achieve desired levels of reactivity, one must pay attention to regioselection (Figure 2B). With unsymmetrically substituted π-systems, as one would anticipate encountering in most natural product synthesis settings, the most useful reactions in this class must proceed with site-selectivity (i.e., which carbon of each unsymmetrical π-system gains a C–C bond). Next, depending on the nature of the coupling partners employed, one may need to consider diastereoselectivity (Figure 2C) and enantioselectivity. Finally, with a plan to employ metallacycle-mediated cross-coupling reactions in complex settings, one must also consider chemoselectivity and functional group compatibility. As will be evident in the various sections of this review, great advances have been made that address these considerations, revealing coupling reactions that are quite useful in highly complex settings.
Figure 2.
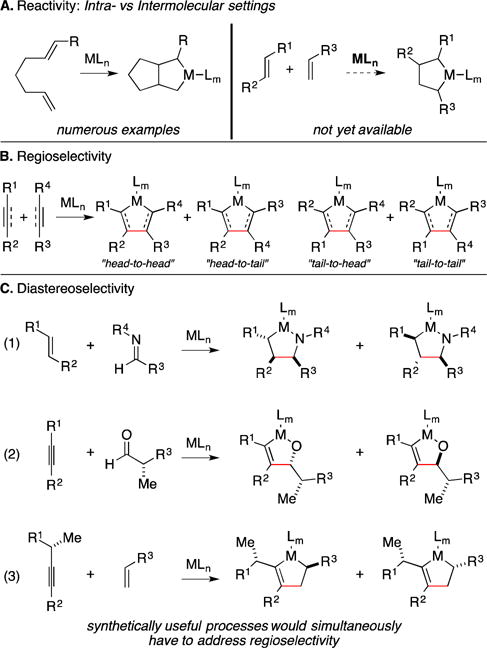
General considerations – reactivity and selectivity.
3. Alkyne–Alkyne Cross-Coupling
Introduction
Alkynes have played a prominent role in the development of metallacycle-mediated cross-coupling chemistry. The earliest reports embraced homo-trimerization5 and homo-dimerization7 en route to benzene and metallacyclopentadienes of Group IV metals. Subsequent achievements by Professors Buchwald, Negishi and Takahashi in the late 1980’s and mid-1990’s led to the realization of methods to cross-couple alkynes through processes that delivered Cp2Zr-based metallacyclopentadienes.8–12
In 1999 a significant departure from Group IV metal–Cp2 complexes appeared and paved a new path for the development of metallacycle-mediated alkyne–alkyne cross-coupling chemistry. From a program aimed at exploring the reactivity of an organometallic intermediate generated from the combination of Ti(Oi-Pr)4 and 2RMgX (conditions previously explored for the Kulinkovich reaction13, 14), Professor Sato described a simple process for the regioselective coupling of internal alkynes with terminal alkynes (Figure 3A).15 Regioselectivity in this process was controlled using a sterically or electronically biased internal alkyne as the first coupling partner (TMS-substituted or highly polarized, respectively) and a terminal alkyne as the second coupling partner. As such, highly selective versions of this process required forming 1,3-dienes that possess these groups at the 1-position.
Figure 3.
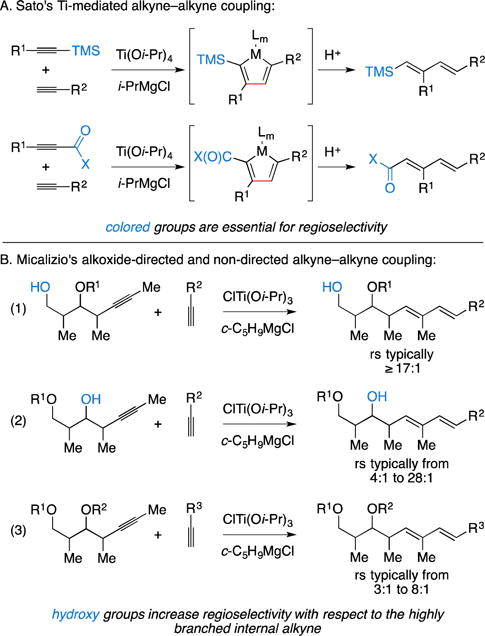
Ti-alkoxide-mediated alkyne–alkyne coupling.
In efforts aimed at establishing metallacycle-mediated cross-coupling chemistry that could address 1,3-diene motifs that are encountered in a wide range of polyketide-derived targets, the Micalizio laboratory investigated the coupling reactions summarized in Figure 3B.16 Their studies ultimately led to the conclusion that a distal hydroxyl group imparts a substantial bias to control regioselectivity of the process, independent of the relative stereochemistry of the internal alkyne coupling partner (eq. 1). These studies also revealed a moderate directing effect of homopropargylic alcohols (eq. 2) and described the inherent regioselectivity of coupling reactions of internal alkyne substrates that lack a free hydroxyl directing group (eq. 3). In these latter two classes of coupling reactions (eqs. 2 and 3), relative stereochemistry of the internal alkyne coupling partner was found to also impact regioselectivity of the process.16, 17 Despite the variable nature of regioselectivity observed, this type of coupling process has been employed in a variety of natural product settings, vide infra.
A. Total Synthesis of Callystatin A
In 2008, the first use of a metallacycle-mediated alkyne–alkyne cross-coupling reaction in natural product synthesis was published.18 This demonstration appeared in efforts targeting the total synthesis of callystatin A (Figure 4A), a rare and highly cytotoxic member of the leptomycin class of natural products.19 The Micalizio laboratory reported a highly convergent synthesis that proceeded by way of a coupling reaction that forged the C13–C14 bond and established the accompanying (E,E)-1,3-diene motif. Their study was complemented by concise 7- and 6-step syntheses of coupling partners 1 and 2 and highlighted the use of a Ti-mediated alkyne–alkyne coupling to generate the functionalized late-stage intermediate 3 (76% yield, rr = 5:1; Figure 4B). The success of this coupling reaction is notable, as it demonstrated chemoselectivity for engaging the terminal alkyne of polyunsaturated substrate 2, while also showing compatibility with a dienylic ether and allylic acetal.
Figure 4.
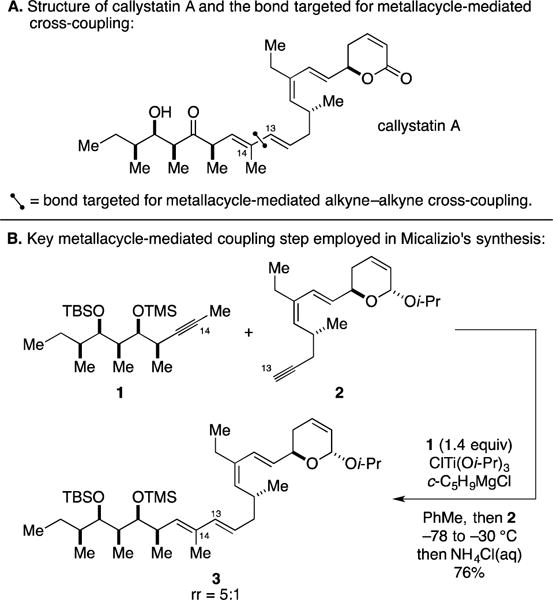
Total synthesis of callystatin A.
Finally, it is worth pointing out that this bond construction delivers a stereodefined 1,3-diene in an efficient, convergent manner without requiring stereodefined olefinic coupling partners – motifs that are typically required for Pd-catalyzed cross-coupling chemistry.
B. Formal Total Synthesis of Dictyostatin
Dictyostatin is a rare macrolide from marine origin (most recently isolated from the Caribbean sponge Corallistidae sp.) that exhibits potent anticancer properties (Figure 5A).20 Related to the marine natural product discodermolide, this agent retains activity against multi-drug resistant cell lines. With the withdrawal of discodermolide from clinical trials in 2005, significant interest has been directed toward dictyostatin, particularly since the natural supply is vanishingly scarce. These considerations and the potential to develop new applications of metallacycle-mediated cross-coupling chemistry led the Micalizio group to pursue its synthesis.21
Figure 5.
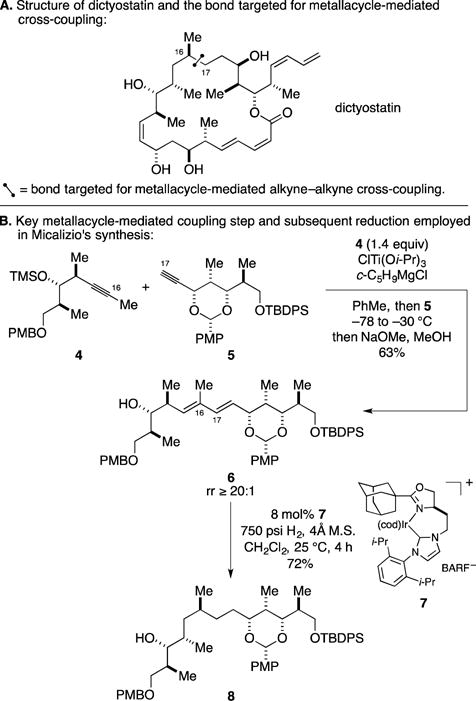
Formal total synthesis of dictyostatin.
In contrast to their use of alkyne–alkyne coupling to address the central 1,3-diene of callystatin A, previously discussed, here they opted to explore the use of this type of cross-coupling chemistry to establish the stereodefined northern hemisphere of the target – a structure that lacks a 1,3-diene. As illustrated in Figure 5A, they set their sights on the C16–C17 bond and relied on subsequent stereoselective hydrogenation to generate the requisite stereochemistry at C16. As summarized in Figure 5B, Ti-mediated coupling of the internal alkyne 4 with terminal alkyne 5 proceeded well and, after selective desilylation, delivered the 1,3-diene-containing product 6 in 63% yield (rr ≥ 20:1). Subsequent use of a stereoselective hydrogenation then delivered fragment 8. Seven additional steps were employed to advance this species to an intermediate that Paterson converted to dictyostatin through nine subsequent chemical transformations.22
C. Total Synthesis of Virginiamycin M2
The virginiamycins are a class of antibiotics produced by Streptomyces that have been used as dietary supplements to accelerate the growth of animals and to prevent and treat bacterial infections. Natural and synthetic derivatives of the virginiamycins have been found to possess potent antibiotic activity against methicillin-, erythromycin-, and vancomycin-resistant S. aureus.23 In 2010, Panek reported a total synthesis of virginiamycin M2 employing a metallacycle-mediated cross-coupling as a key step (Figure 6A).24, 25 With a bold plan in mind, Panek and coworkers targeted the establishment of the C8–C9 bond by Ti-mediated union of the propargylic alcohol 9 with terminal alkyne 10 (Figure 6B). Notably, both coupling partners possessed unprotected hydroxyl- groups, and the terminal alkyne component also contained an α,β-unsaturated secondary amide. Demonstrating the functional group compatibility of this class of metallacycle-mediated cross-coupling, they reported a Ti-mediated reaction process that delivered triene 11 in 58% yield, and with outstanding levels of regioselectivity.
Figure 6.
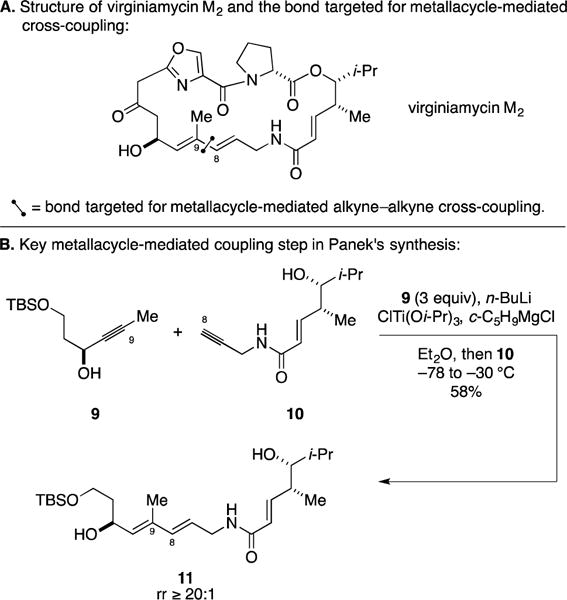
Total synthesis of virginiamycin M2.
D. Total Synthesis of the Proposed Structure of Heronamide C
Heronamide C is a member of a large class polyene macrocyclic lactam-containing natural products produced from Streptomyces sp. that has been reported to have a pronounced and unique effect on the morphology of cultured cells.26 Pursuing their interest in the chemistry and biology of polyene macrolactams, the Kanoh laboratory at Tohoku University targeted the total synthesis of the proposed structure of heronamide C (Figure 7A).27 In 2014, the authors reported the Ti-mediated coupling of propargylic ether 12 with conjugated enyne 13 – a process that delivered the stereodefined conjugated triene 14 in 64% yield and that proceeded with good levels of regioselectivity (rr = 12:1). Notably, their success with this coupling reaction highlighted the ability to avoid elimination chemistry possible with both the intermediate Ti-complex of 12 (see Figure 7C), and the metallacyclopentadiene formed upon alkyne–alkyne coupling.28
Figure 7.
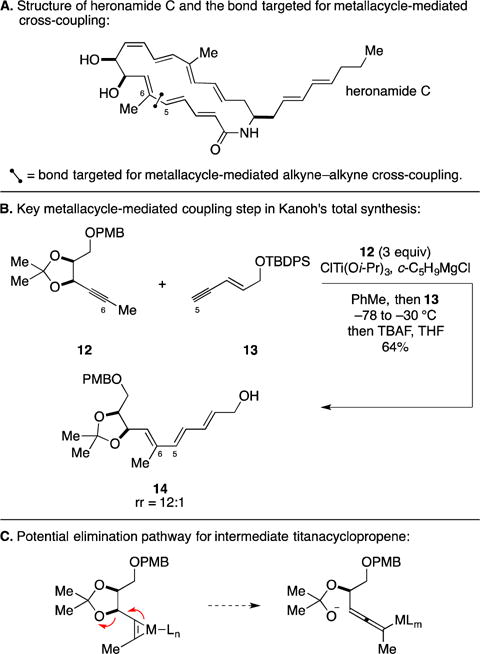
Total synthesis of the proposed structure of heronamide C.
E. Synthesis and Discovery of Valerjesomycin
While not a natural product, Micalizio’s function-oriented synthesis efforts in the area of benzoquinone ansamycins embraced the application of a metallacycle-mediated alkyne–alkyne coupling as a key convergent step. This chemistry fueled an SAR study that ultimately led to the discovery of valerjesomycin (Figure 8A) as a benzoquinone ansamycin-inspired agent with exquisite paralog selectivity for Hsp90α and Hsp90β over Grp94.29 As illustrated in Figure 8B, these investigations targeted the construction of the C7–C8 bond by metallacycle-mediated cross-coupling between 15 and 16. After hydrolysis of the ester, this sequence delivered seco-acid 17 in 52% yield as an 8:1 mixture of regioisomers. Their success in this process demonstrated the ability to generate a Ti–alkyne complex on a substrate that contains a free aniline (15) and revealed chemoselectivity for reaction of the Ti-alkyne complex of 15 with the terminal alkyne of 16 in preference to the α,β-unsaturated ethyl ester.
Figure 8.
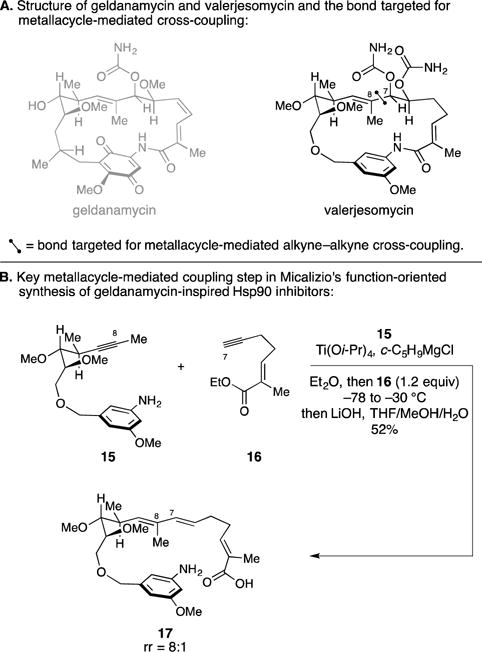
Function-oriented synthesis – discovery of valerjesomycin as a paralog selective natural product-inspired Hsp90 inhibitor.
4. Alkene–Alkyne Cross-Coupling
Introduction
In early investigations of the potential utility of metallacycle-mediated bond-formation in organic synthesis, intramolecular reactions took center stage. It was here where the first examples of alkene–alkyne coupling processes emerged in the context of forming carbo- and heterocyclic systems.30–33 While adopted by the synthetic organic chemistry community in short order, these processes remained limited to intramolecular settings – for the generation of 5- and 6-membered rings (Figure 9A). The challenge of accomplishing alkene–alkyne cross-coupling (intermolecular) is related to the low reactivity of substituted alkenes in the intermolecular carbometalation step and the established instability of the metallacyclopentene intermediates that result from cross-coupling.34, 35 As illustrated in Figure 9B, a central feature of Negishi’s alkyne–alkyne coupling process took advantage of this instability. Treatment of an alkyne with the organometallic species generated from addition of 2 equiv. of EtMgBr to Cp2ZrCl2 generates a zirconacyclopentene intermediate. This species serves as an effective source of a Zr-alkyne complex (with loss of ethylene) that can participate in coupling with a variety of different π-systems (e.g., alkynes, nitriles, aldehydes).31
Figure 9.
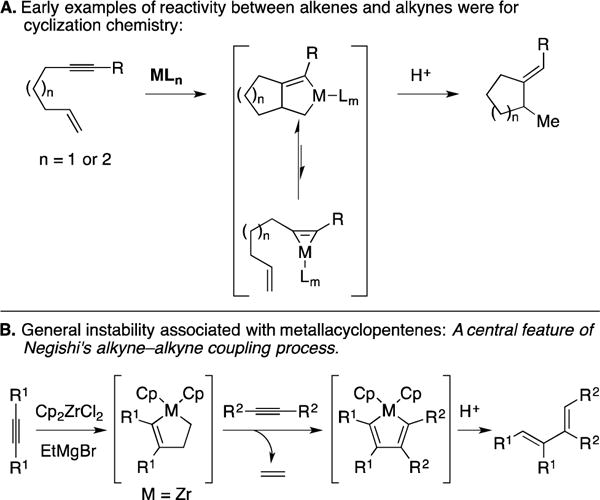
Early investigations of methods that proceed by way of metallacyclopentenes.
Despite the instability of metallacyclopentene intermediates, methods have become available for the regio- and stereoselective metallacycle-mediated cross-coupling of alkenes with alkynes. An early example, that has had a significant impact in natural product synthesis, is Trost’s Ru-catalyzed Alder-ene reaction (Figure 10).36, 37 Here, metallacycle-formation is followed by trapping of the unstable organometallic intermediate by regio- and stereoselective β-hydride elimination and reductive elimination – a sequence that results in a 1,4-diene-containing product and regenerates the active catalyst.
Figure 10.
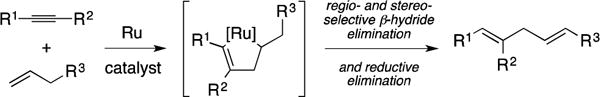
Ru-catalyzed Alder-ene reaction.
Recently, Group IV metal-mediated coupling chemistry has emerged as an alternative approach to alkene–alkyne coupling that affords complementary bond-constructions to the Ru-catalyzed Alder-ene reaction. The first of these was offered by Professor Takahashi and proceeded through a pathway that trapped the metallacyclopentene intermediate by elimination (Figure 11A).38–40 While effective, this approach afforded products as mixtures of alkene stereoisomers – an undesirable trait that has likely resulted in a lack of adoption of this reaction in natural product synthesis. In a departure from the use of Cp2M-based species, and in the context of studies aiming to render the metallacycle-mediated cross-coupling alkoxide-directed, Micalizio and Cha described highly stereoselective alkyne–allylic alcohol coupling processes that established (E)-disubstituted or (Z)-trisubstituted alkenes (Figure 11B).41, 42 Here, reversible docking of the alkene substrate to the preformed metallacyclopropene by Ti–O bond formation is proposed to result in an opportunity for intramolecular carbometalation through a boat-like transition state. Minimization of allylic strain (1,3- or 1,2-) was then rationalized as the means by which one can attain high levels of stereocontrol for the formation of 1,4-dienes containing either an (E)-disubstituted or (Z)-trisubstituted alkene.
Figure 11.
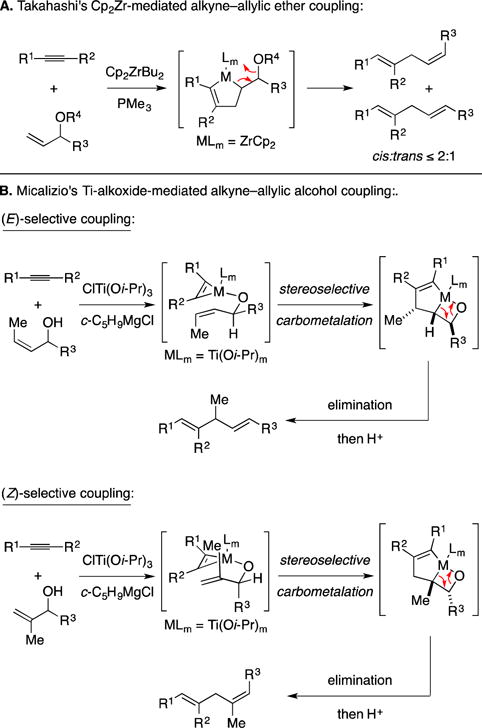
Group IV metal-mediated alkyne–allylic alcohol coupling.
The Ru-catalyzed Alder-ene reaction and alkoxide-directed Ti-mediated alkyne–allylic alcohol coupling have been used in a variety of natural product syntheses.
A. Total Synthesis of Acetogenins
The first examples that revealed the great power of Ru-catalyzed Alder-ene reactions in total synthesis were focused on the assembly of members of the acetogenin family of natural products. The version of this reaction that was central to these campaigns involved the coupling of alkynoates to terminal alkenes. As illustrated in Figure 12A, the relevant metallacycle-mediated alkyne–alkene coupling reaction is thought to proceed by regioselective formation of a ruthenium-based metallacyclopentene that, after β-hydride elimination and reductive elimination, unveils a species that promptly cyclizes to generate a γ-lactone.43 Regioselectivity with respect to the alkyne reaction partner has been proposed to derive from preferential coordination of the alcohol to the metal center.
Figure 12.
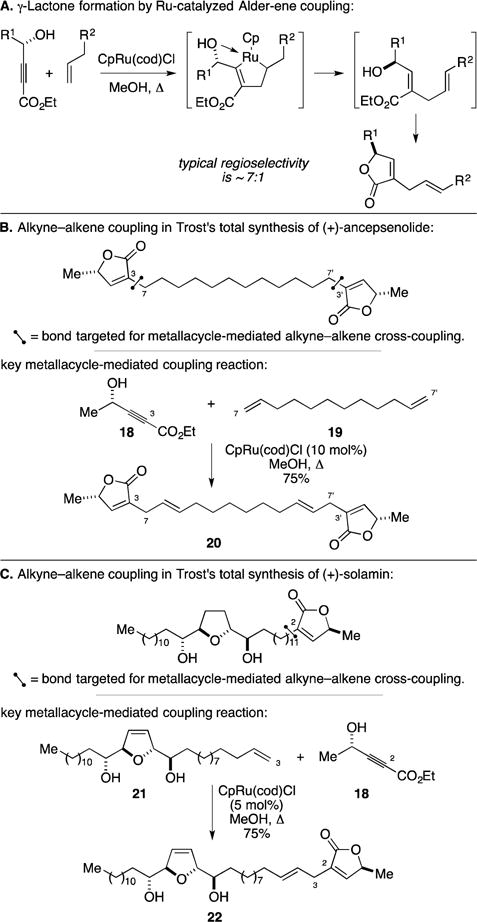
Ru-catalyzed Alder-ene reaction for the synthesis of acetogenins.
Two examples where this variant of the Alder-ene reaction has been particularly effective are in the total syntheses of acetogenins (+)-ancepsenolide and (+)-solamin, highlighted in Figure 12B and Figure 12C, respectively. In studies directed toward the total synthesis of ancepsenolide, Trost demonstrated the ability to employ a Ru-catalyzed Alder-ene reaction in a two-directional fashion (Figure 12B).44 Union of 18 with 19 delivered tetraene 20 in 75% yield, and this species was converted to the natural product by chemoselective reduction of the electron rich disubstituted alkenes. In a similar manner, solamin was prepared by a related Alder-ene reaction of 18 with 21 (Figure 12C).45 Here, the key metallacycle-mediated coupling process was demonstrated to be chemoselective, engaging only the terminal alkene of 21 in the C–C bond-forming event. Overall, these examples highlight the utility of Ru-catalyzed Alder-ene reaction for the installation of γ-lactones in complex settings.46
B. Total Synthesis of the Proposed Structure of Trocheliophorolide B
In 2012, an interesting application of the Ru-catalyzed Alder-ene reaction appeared in the context of Trost’s studies directed toward synthesis of the proposed structure of trocheliophorolide B (Figure 13A).47 As illustrated in Figure 13B, a γ-lactone-forming process was executed between alkyne 18 and allyl alcohol (23). Here, the Alder-ene process delivered an enol after β-hydride elimination – a product that then isomerized to an aldehyde. This version of metallacycle-mediated alkyne–alkene coupling led to the synthesis of the proposed structure of trocheliophorolide B in just three addition steps.
Figure 13.
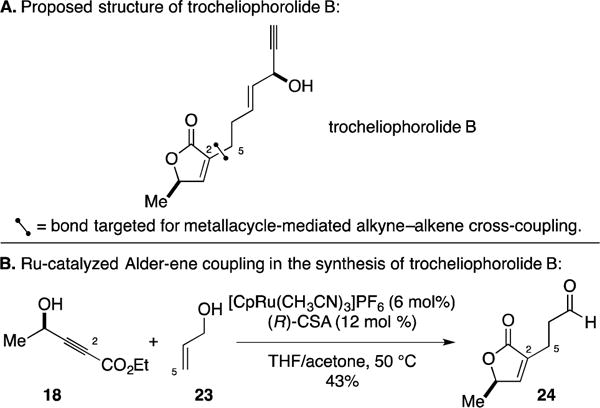
Synthesis of the proposed structure of trocheliophorolide B.
C. Total Synthesis of Deschlorocallipeltoside A
Callipeltoside A is a cytotoxic marine-derived polyketide that inhibits in vitro proliferation of NSCLC-N6 human bronchopulmonary non-small-cell lung carcinoma and P388 cells (Figure 14A).48 In a program aimed at defining a synthetic means to access this natural product and synthetic analogs with potential medicinal value, Trost described the application of Ru-catalyzed alkene–alkyne coupling to establish the C11–C12 bond in a sequence that first led to the production of deschlorocallipeltoside A.49 This alkyne–alkene coupling is notably different than that previously employed by Trost for γ-lactone synthesis in that the alkyne was not significantly polarized. As illustrated in Figure 14B, coupling of alkyne 25 with the protected homoallylic alcohol 26 generated the 1,4-diene product 27 in 85% yield. Regioselectivity in this case was proposed to be influenced by the ability of the propargylic methyl ether to co-ordinate to ruthenium in the intermediate metallacyclopentene. Notably, Trost’s efforts directed toward the synthesis of deschlorocallipeltoside A paved the way for his group’s success in achieving a total synthesis of callipeltoside A – an achievement that resulted in the correct assignment of the absolute and relative stereochemistry of this potent marine-derived toxin.50, 51
Figure 14.
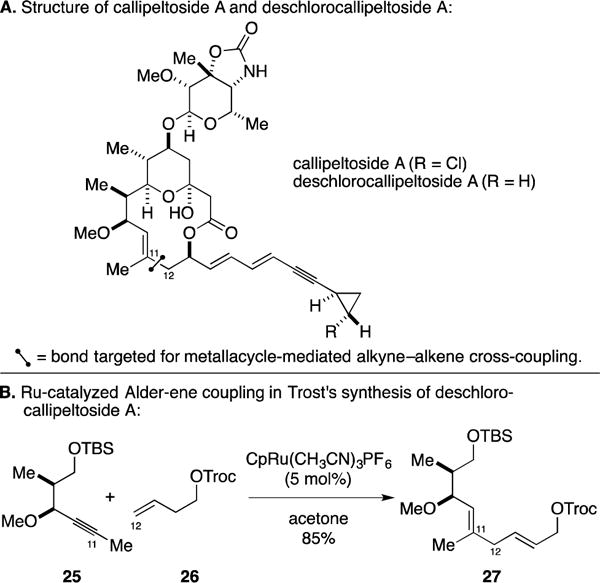
Synthesis of deschlorocallipeltoside A.
D. Formal Total Synthesis of Alternaric Acid
Alternaric acid is a polyunsaturated and stereochemically rich natural product that was first isolated in 1949 from Alternaria solani (Figure 15A).52 Aside from its interesting structure, this compound was found to possess highly selective activity for inhibiting germination of certain fungal strains as well as demonstrating activity as a selective phytotoxic agent.53 Trost reported a total synthesis of this agent via application of his Ru-catalyzed alkyne–alkene coupling method, here through regioselective coupling of a terminal alkene (28) with a terminal alkyne (29; Figure 15B).54 This coupling process proceeded in 65% yield and delivered the 1,4-diene product 30 with 13:1 regioselectivity.
Figure 15.
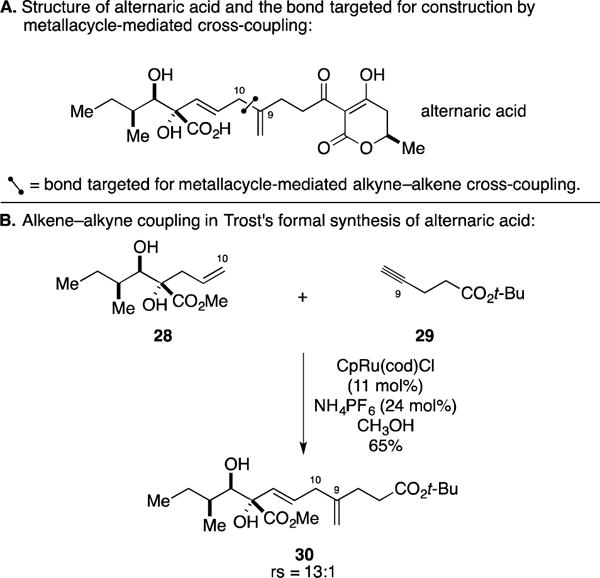
Synthesis of alternaric acid.
E. Formal Total Synthesis of Mycalamide A
Mycalamide A is a marine sponge-derived natural product that possesses potent antiviral and antitumor properties as a result of its ability to arrest protein synthesis (Figure 16A).55 Trost and coworkers accomplished a formal total synthesis of this agent that focused on preparation of the pederic acid unit via construction of the C4–C5 bond.56 As illustrated in Figure 16B, Ru-catalyzed alkyne–alkene coupling between 31 and 32 proceeded in a regioselective fashion, with C–C bond formation occurring distal to the TMS group of the alkyne. The Alder-ene process delivered, as anticipated, the (E)-α,β-unsaturated ester-containing diene product 33.
Figure 16.
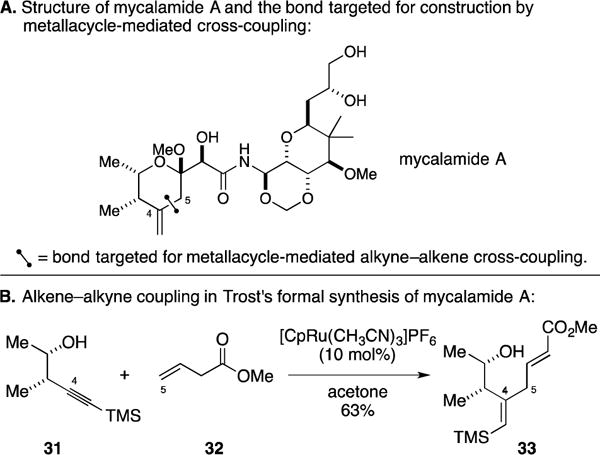
Formal total synthesis of mycalamide A.
F. Total Synthesis of Amphidinolides A and P
The amphidinolides are a family of structurally diverse cytotoxic natural products from symbiotic dinoflagellates of the genus Amphidinium.57 Members of this class have served to inspire a variety of campaigns in total synthesis, two of which marked the exploration of Ru-catalyzed alkyne–alkene coupling. In Trost’s synthesis of amphidinolide A (Figure 17A), an effort that led to the structure elucidation of the natural product, two impressive applications of metallacycle-mediated C–C bond formation were explored: (1) alkyne–alkene cross-coupling to forge the C6–C7 bond, and (2) macrocyclization by a Ru-mediated Alder-ene reaction.58–60 The metallacycle-mediated cross-coupling that was successful in these campaigns is illustrated in Figure 17B. In this remarkable process, the polyunsaturated alkyne 34 was effectively coupled to the 1,4-diene 35. While the product 36 was isolated in only 23% yield, this example supports the great chemoselectivity associated with Ru-catalyzed coupling processes and demonstrates that polymerization of 34 can be avoided (it contains both functionality required for Alder-ene chemistry – the reactive alkyne and the terminal alkene).
Figure 17.
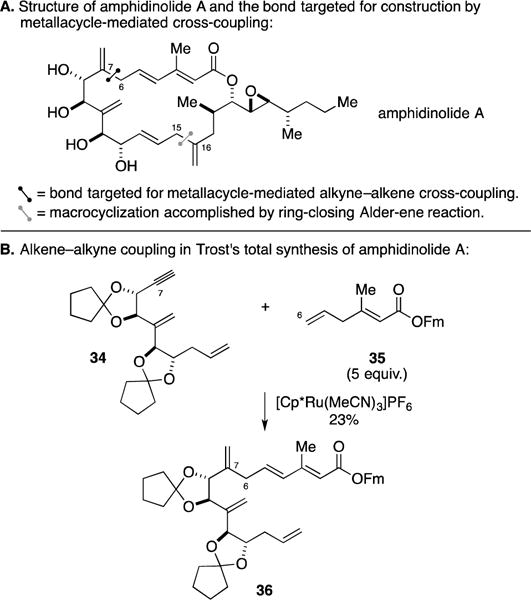
Total synthesis of amphidinolide A.
Amphidinolide P also proved a worthy target for the Trost group (Figure 18).61, 62 Here, they targeted alkene–alkyne coupling technology for the establishment of the C10–C11 bond. As illustrated in Figure 18B, success was seen in the union of β-lactone containing enyne 37 with terminal alkyne 38 – a process that delivered 39 in 75% yield with high levels of selectivity. The observed regioselectivity with respect to the alkyne is likely attributed to non-bonded steric interactions between the substituent on the terminal alkyne and the ligands on ruthenium during formation of the metallacyclopentene intermediate.
Figure 18.
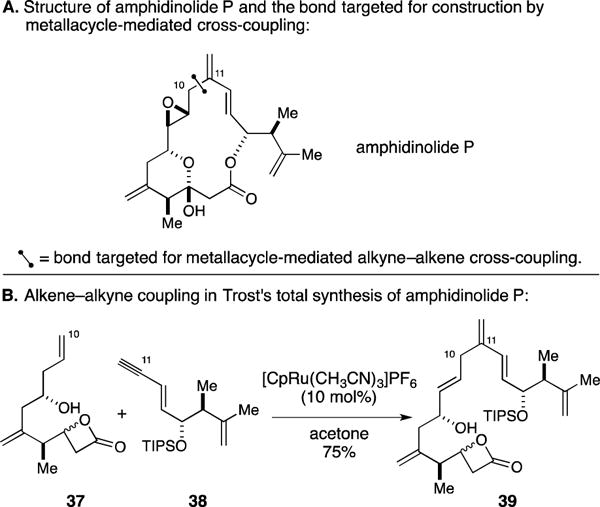
Total synthesis of amphidinolide P.
G. Total Synthesis of (−)-Dactylolide
Dactylolide is a natural product that has attracted a significant amount of attention as a target for total synthesis, not only because of its unique structure – it selectively inhibits the growth of a subset of human tumor cell lines (Figure 19A).63 Lee’s synthesis of (−)-dactylolide focused on the use of a variety of metal-catalyzed transformations, not the least of which included two Ru-catalyzed alkene–alkyne coupling reactions to forge the C5–C6 and the C13–C14 bonds.64 The former of these was accomplished through a pathway that likely does not involve the intermediacy of a metallacyclopentene. The latter, however, embraces the Ru-catalyzed Alder-ene process. As illustrated in Figure 19B, coupling of the protected homoallylic alcohol 40 with the homopropargylic alcohol 41 generated the 1,4-diene-containing product 42 – a compound that was suitably functionalized for a Pd-catalyzed etherification reaction en route to the stereodefined 2,6-cis-disubsituted pyran 43.
Figure 19.
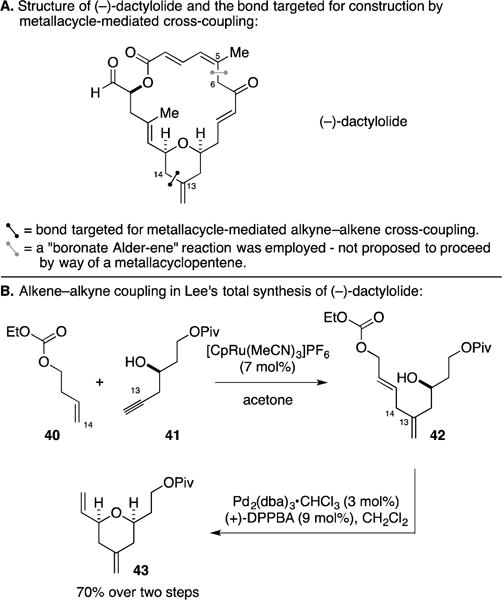
Total synthesis of (−)-dactylolide.
H. Total Synthesis of Bryostatin 16
The bryostatins are a collection of rare, marine-derived macrocyclic lactones that have attracted substantial interest from members of the scientific community. They were originally isolated from the marine bryozoan Bugula neritina and were found to exhibit exceptional biological activity, most notably their anticancer activity in vivo.65 Perhaps not surprisingly, some of the world’s leaders in organic chemistry have spent substantial energy devising strategies to prepare members of this class.
In Trost’s impressive contribution, he set his sights on bryostatin 16 (Figure 20A).66, 67 Here, a highly convergent metallacycle-mediated alkyne–alkene coupling process was explored for construction of the C12–C13 bond. As illustrated in Figure 20B, union of the β,γ-unsaturated ketone 44 with the TMS-alkyne 45 proceeded to deliver the metastable intermediate 46, which was suitably functionalized to undergo stereoselective intramolecular 1,4-addition. Overall, the coupling process led to the generation of a substantial substructure of the natural product target (47) in 34% yield.
Figure 20.
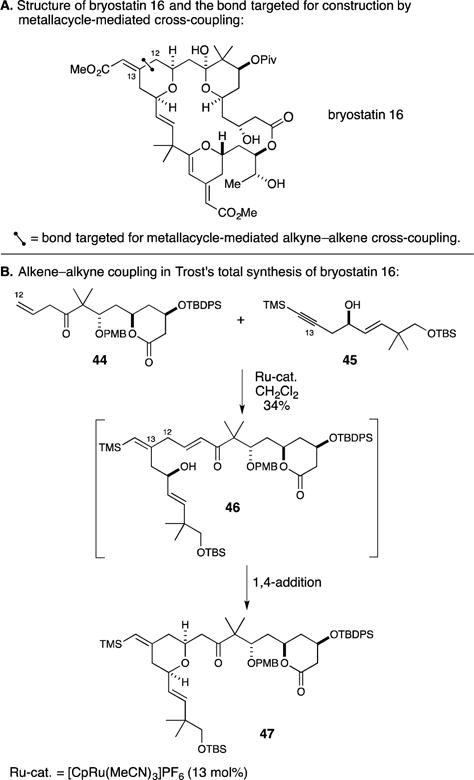
Total synthesis of bryostatin 16.
I. Total Synthesis of Lehualide B
The lehualides are a family of pyrone-containing marine-derived natural products that were originally described as having moderate and selective cytotoxity in vitro (IGROV-ET and K562 cell lines).68 The skipped polyunsaturated nature of lehualide B (Figure 21A) attracted the attention of the Micalizio laboratory, posing what appeared to be a suitable target for application of a recently developed alkoxide-directed titanium-mediated allylic alcohol–alkyne coupling reaction.69 Their synthesis strategy explored the use of such a metallacycle-mediated coupling to establish the C14–C15 bond in concert with setting the stereochemistry of the two trisubstituted olefins of the local 1,4-diene motif. As illustrated in Figure 21B, the polyunsaturated pyrone-containing allylic alcohol 48 was coupled to the TMS-alkyne 49 to deliver the stereodefined product 50 in 61% yield, with ≥20:1 regioselectivity and 7:1 selectivity in favor of the desired Z-trisubstituted alkene. These efforts resulted in the first total synthesis of lehualide B and the discovery that this agent has impressive and highly selective activity against multiple myeloma cell lines (EC50 = 2.9 nM and 2.1 nM in U266 and RPMI 8226 cells, respectively). While not depicted in Figure 21, the Micalizio group executed a function-oriented synthesis study targeting synthetic analogues of lehaulide B – a program that explored a number of variants of the allylic alcohol–alkyne coupling process and produced numerous natural product analogs with potent and selective cytotoxicity toward multiple myeloma cell lines.70
Figure 21.
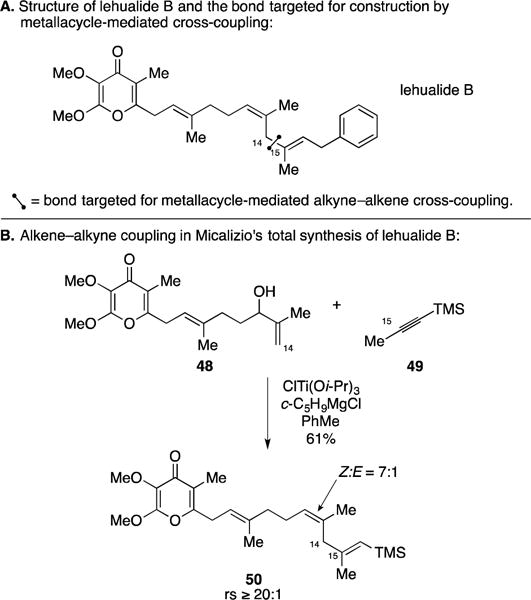
Total synthesis of lehualide B.
J. Total Synthesis of (−)-Vittatalactone
In efforts to explore the use of Ti-mediated allylic alcohol–alkyne coupling chemistry for the convergent synthesis of deoxypropionates, the Micalizio laboratory pursued the synthesis of (−)-vittatalactone (Figure 22A) – a deoxypropionate natural product that was isolated in a search for sex pheromones specific to the cucumber beetle Acylymma vittatum (an insect that causes major damage to the cucurbit crops in North America).71 Their efforts focused on establishment of the C6–C7 bond and featured the metallacycle-mediated cross-coupling depicted in Figure 22B. Union of alkyne 51 and allylic alcohol 52, followed by desilylation, resulted in the formation of 1,4-diene 53 in 63% yield. The central metallacycle-mediated coupling process proceeded with exquisite regio- and stereocontrol, delivering a single 1,4-diene product. Conversion of diene 53 to the deoxypropionate motif central to the vittatalactone structure (54) was then affected by simple hydroxyl-directed hydrogenation.
Figure 22.
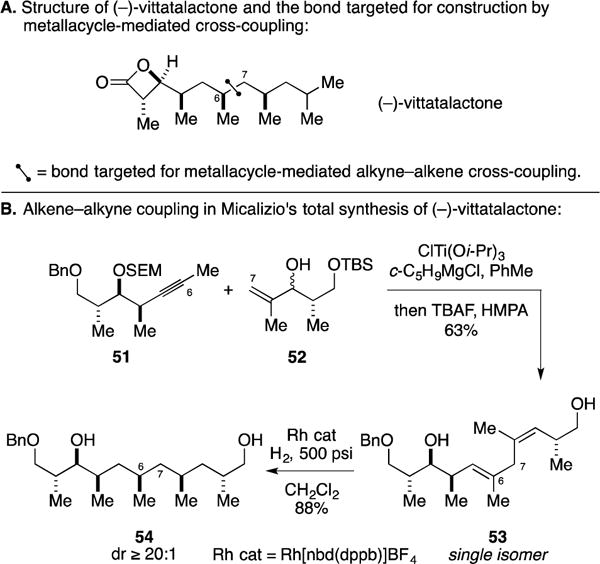
Total synthesis of (−)-vittatalactone.
K. Total Synthesis of Phorbasin C
The phorbasins are a class of structurally unique diterpenes that have been isolated from a southern Australian marine sponge (Phorbas sp.).72–76 Members of this class have been reported to possess a range of antibiotic and cytotoxic properties, with phorbasin C attracting the attention of the Micalizio laboratory (Figure 23A). While the structure of the target was not clear from the reported isolation, the Micalizio laboratory began a synthesis campaign that targeted the development of a regio- and stereoselective allylic alcohol–alkyne coupling process for establishment of the C6–C7 bond.77 As illustrated in Figure 23B, this pursuit led to the successful metallacycle-mediated coupling of cyclohexene 56 to TMS-alkyne 49. Here, alkoxide-directed Ti-mediated coupling proceeded with exquisite regio- and stereochemical control, delivering the 1,4-diene-containing product 57 as a single isomer in 47% yield.
Figure 23.
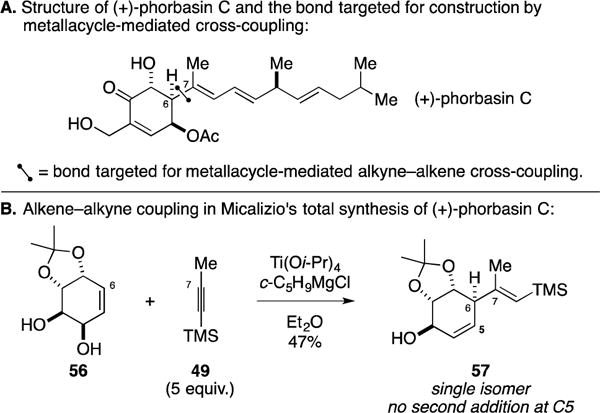
Total synthesis of (+)-phorbasin C.
While demonstrating the ability to achieve stereoselectivity in the formation of the C6 stereocenter, their success with this metallacycle-mediated coupling reaction also revealed the significant influence of non-bonded steric interactions in affecting the rate of C–C bond formation. No product was seen where a second equivalent of the Ti–alkyne complex engaged the allylic alcohol of 57 in a second C–C bond-forming process. Given the requirement for hydroxyl direction in this class of coupling reactions (low levels of reactivity are encountered with substrates that lack a directing group), this second coupling process would require C–C bond-formation from the same face of the alkene as the C6 substituent. Apparently, the steric impediment imposed by this group was sufficient to dissuade a second coupling process.
5. Alkyne–Aldehyde Cross-Coupling
Introduction
Unlike metallacycle-mediated coupling chemistry between alkenes and alkynes, where substantial barriers to achieving suitable reactivity are further complicated by challenges associated with controlling regioselectivity with respect to both reactive π-systems, reductive coupling processes between alkynes and aldehydes are relatively straightforward. In this subset of metallacycle-mediated cross-coupling reactions, reactivity is not a substantial issue, leaving one only to deal with addressing regioselectivity with only one π-component (the alkyne; Figure 24), and stereoselectivity associated with the allylic alcohol product. While a variety of methods are available to accomplish alkyne–aldehyde reductive cross-coupling in a highly selective fashion (regio- and stereo-),78–100 a relatively small subset of these reaction processes have been successfully employed in natural product synthesis. The following examples are illustrative.
Figure 24.
Introduction to metallacycle-mediated alkyne–aldehyde cross-coupling.
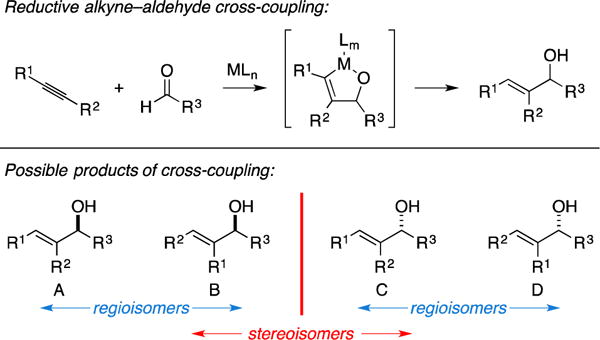
A. Total Synthesis of (−)-Terpestacin
Terpestacin, a structurally complex sesterterpene natural product that selectively inhibits angiogenesis, has risen as an attractive target for total synthesis (Figure 25A).101–107 In 2003, Jamison reported an enantioselective synthesis of this natural product that featured two different metallacycle-mediated coupling reactions: (1) an early example of metallacycle-mediated alkyne–aldehyde coupling in natural product synthesis, and (2) an intermolecular Pauson–Khand annulation (a special type of metallacycle-mediated alkyne–alkene coupling that will be discussed later in this Report).108, 109 As illustrated in Figure 25B, Ni-catalyzed coupling of alkyne 58 to aldehyde 59 delivered the desired product 60 in 41% isolated yield, yet proceeded with only moderate levels of selectivity (rs = 2.6:1, ds = 2:1).
Figure 25.
Total synthesis of (−)-terpestacin.
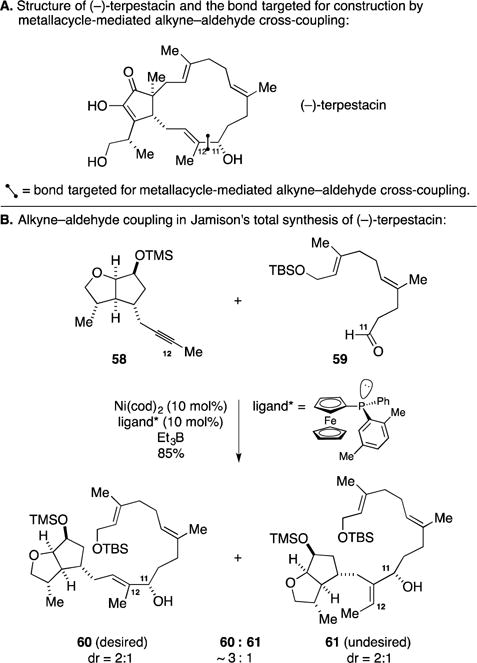
B. Total Synthesis of Macbecin I
The benzoquinone ansamycins are stereochemically complex macrocyclic lactams that have surfaced as attractive targets for synthesis due not only to their structures but also because of their properties as Hsp90 inhibitors. Shortly after studying the factors that influence regioselectivity in Ti-mediated alkyne–aldehyde coupling reactions, Micalizio reported a step-economical synthesis of one of the most active members of this class, macbecin I (Figure 26A).110 The targeted metallacycle-mediate alkyne–aldehyde coupling aimed to forge the C7–C8 bond and realize the highly convergent process depicted in Figure 26B. Preformation of a Ti-alkyne complex of 62 was followed by addition of the highly unstable polyunsaturated aldehyde 63. After protonation of the intermediate oxametallacyclopentene, a mixture of regio- and stereoisomeric products was advanced through a saponification and macrolactamization process to afford the macrocyclic lactam 64 in 44% yield. While the authors could not assess the product mixture formed from the first step in this sequence (the alkyne–aldehyde coupling), they did report a related coupling reaction with a more stable aldehyde where they observed 7:1 regioselectivity and 3:1 diastereoselectivity. Overall, the successful application of metallacycle-mediated alkyne–aldehyde cross-coupling in the context of this synthesis of macbecin I shed light on the functional group compatibility and chemoselectivity of the process, revealing the ability to (1) form a Ti-alkyne complex in the presence of an unprotected aniline, and (2) selectivity in addition to an aldehyde in the presence of a dienylic ester.
Figure 26.
Total synthesis of macbecin I.
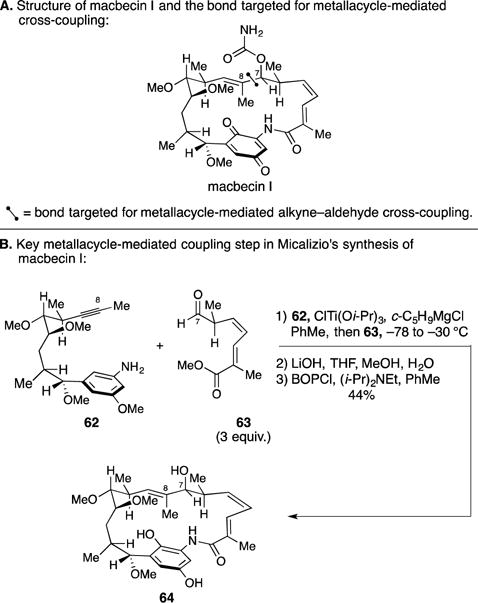
C. Total Synthesis of D-Erythro-Sphingosine
In studies that originated from their interest in expanding the scope and utility of regio- and stereoselective Ni-catalyzed alkyne–aldehyde coupling reactions, Montgomery and coworkers demonstrated the use of this process in the total synthesis of D-erythro-sphingosine (Figure 27A).111 Here, the authors employed their N-heterocyclic carbene-modified Ni-catalyzed process to couple Garner aldehyde 65 to TMS-alkyne 66 (Figure 27B). Overall, they achieved C3–C4 bond-formation with exquisite levels of regio- and stereocontrol en route to 67. Only two additional steps were required to complete the total synthesis.
Figure 27.
Total synthesis of D-erythro-sphingosine.
D. Total Synthesis of Bryostatin 7
As previously discussed in the section of this Report covering Trost’s synthesis of bryostatin 16, the bryostatins have attracted a substantial amount of attention from many members of the scientific community due to their profound biological properties, interesting structures, and low natural abundance. Bryostatin 7 captured the attention of the Krische group and served as a target that provided an opportunity for them to explore the application of a number of catalytic transformations that have been developed in their laboratory (Figure 28A).112 Notably, their synthesis strategy targeted generation of the C20–C21 bond by a metallacycle-mediated alkyne–aldehyde cross-coupling reaction. As illustrated in Figure 28B, they employed a Rh-catalyzed metallacycle-mediated cross-coupling reaction between the highly electron deficient aldehyde 68 and enyne 69, and realized a process that forged the C20–C21 bond and established the C20 stereocenter (dr = 7:1), producing allylic alcohol 62 in 77% yield.
Figure 28.
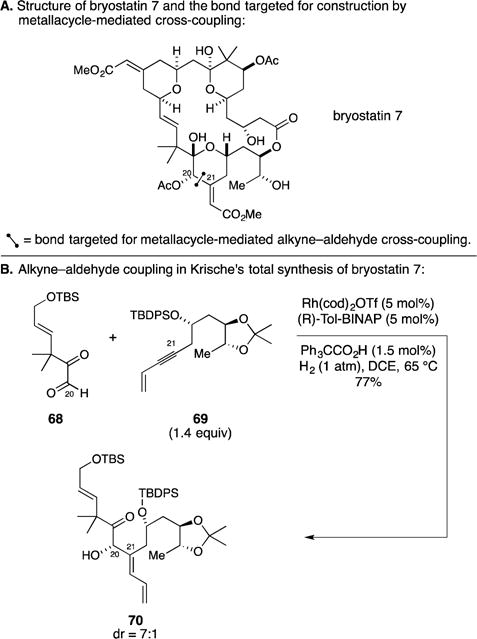
Total synthesis of bryostatin 7.
E. Total Synthesis of Trienomycin A
Ansatrienins are a subset of “ansamycins” – polyketide-derived natural products that are known to possess an important portfolio of antibiotic properties. Trienomycins are members of the ansatrienin class that have antineoplastic activity.113–115 Trienomycins A and F were selected by Krische and coworkers as targets for synthesis, where the C3–C4 bond was selected for establishment by application of their unique Ru-catalyzed acetylene–aldehyde coupling reaction (Figure 29A).116 As depicted in Figure 29B, metallacycle-mediated coupling of aldehyde 72 with acetylene resulted in stereoselective synthesis of the dienylic alcohol 73 in 83% yield (85% ee, Z:E ≥ 20:1). This process is thought to proceed through a mechanism quite distinct from other metallacycle-mediated “alkyne”–aldehyde cross-coupling reactions. Ru-catalyzed dimerization of acetylene is reasoned to first result in the formation of metallacyclopentadiene that then engages the aldehyde in C–C bond-formation.117 While the process was quite effective at selectively generating the C4–C5 (Z)-disubstituted alkene, it would have to later be isomerized to the requisite (E)-disubstituted olefin of the natural product. Nevertheless, this reaction proved to be part of an effective strategy to generate trienomycin A and F, as well as the core of cytotrienin A (not depicted).118
Figure 29.
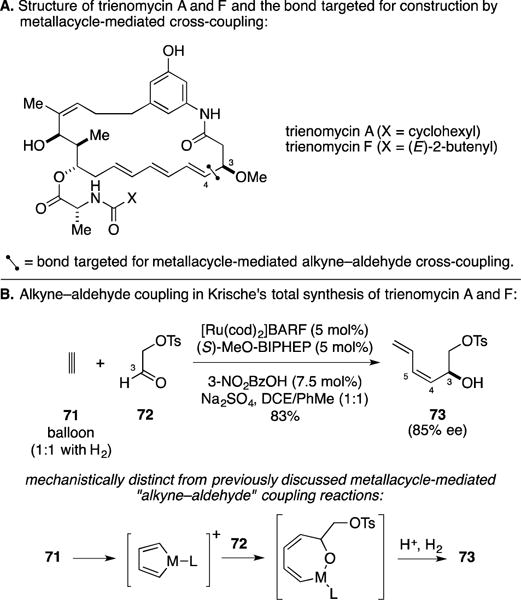
Total synthesis of trienomycin A.
6. Alkyne–Epoxide Cross-Coupling
Introduction
While most metallacycle-mediated cross-coupling chemistry engages the π-system of reaction partners in the course of generating a metallacyclopentane-motif, Ni-catalysis has proven to be an effective means of accomplishing coupling reactions between alkynes and epoxides (Figure 30). While being distinct from other reactions discussed in this report, as they are thought to proceed by way of oxametallocyclohexenes,119 this class of metallacycle-mediated cross-coupling has proven to be useful in natural product synthesis. General issues regarding selectivity are wholly focused on regioselectivity with respect to both coupling partners (i.e., A – D). Currently, products of this reaction derive from C–C bond-formation at the unsubstituted carbon of the epoxide, with regioisomers being possible from C–C bond formation at either carbon of the alkyne (leading to homoallylic alcohols A and/or B).
Figure 30.
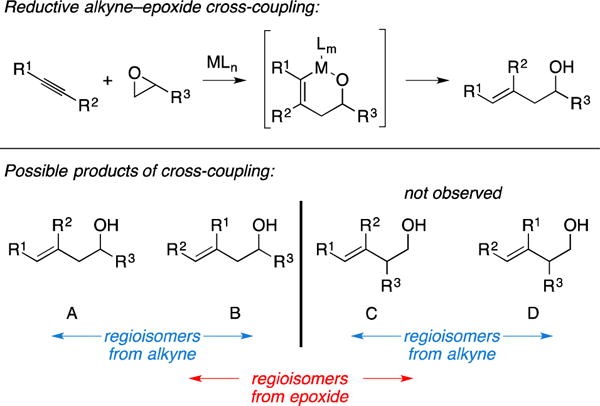
Introduction to metallacycle-mediated alkyne–epoxide cross-coupling.
A. Total syntheses of Amphidinolide T1 and T4
The amphidinolides are a family of marine-derived natural products that have been shown to have a diverse array of potent cytotoxic properties.120, 121 Amphidinolide T natural products surfaced as interests to the Jamison group, perhaps due to their expectation that recently developed Ni-catalyzed transformations may be particularly effective at addressing a subset of the structural motifs associated with them.122, 123 In particular, Jamison approached the total syntheses of amphidinolides T1 and T4 with the planned use of two unique Ni-catalyzed C–C bond-forming processes: (1) an alkyne–epoxide cross-coupling, and (2) macrocyclization by alkyne–aldehyde coupling (Figure 31A). As illustrated in Figure 31B, the metallacycle-mediated cross-coupling reactions that were effectively wielded to accomplish the total syntheses of these two amphidinolides embrace the use of conjugated alkynes in C–C bond-formation – such alkynes are known to participate in Ni-catalyzed coupling reactions with a high degree of regioselectivity.119 Interestingly, in one of these coupling reactions the authors describe the chemoselective functionalization of a diyne. When diyne 77 was coupled to epoxide 75, the homoallylic alcohol 78 was obtained in 40–44% yield. While the 1H NMR spectrum of the crude material indicated some polymerization of the diyne (the limiting reagent), the authors reported that no evidence was found for engaging the TMS-alkyne in the Ni-catalyzed coupling process.
Figure 31.
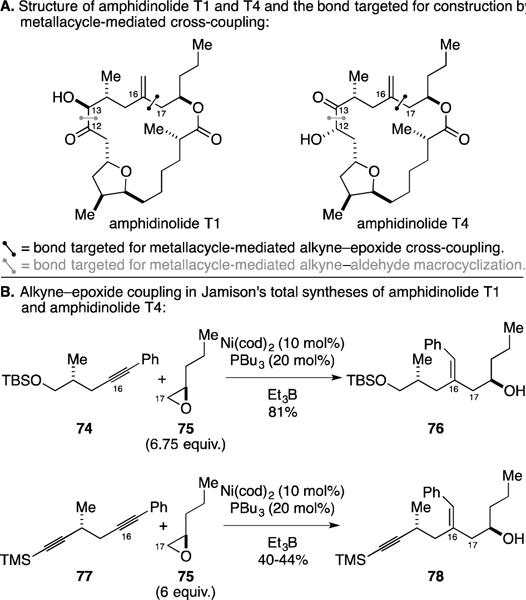
Total synthesis of amphidinolide T1 and T4.
7. Alkene–Imine Cross-Coupling
Introduction
Metallacycle-mediated alkene–imine cross-coupling is a relatively complex process, as issues concerning regioselectivity (with respect to the alkene) and facial selectivity (with respect to both coupling partners) are coupled to the problem of engaging a coupling partner that typically is not highly reactive in metallacycle-mediated coupling chemistry (substituted alkenes) (Figure 32). To date, simple diastereoselection has been observed to highly favor the anti-stereoisomers A and B over the syn-isomers C and D.
Figure 32.
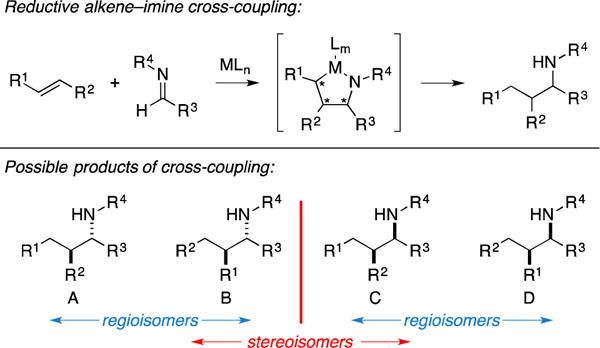
Introduction to metallacycle-mediated alkene–imine cross-coupling.
As previously discussed in the context of alkoxide-directed alkene–alkyne coupling (Figure 11B), this class of metallacycle-mediated cross-coupling has been particularly effective with allylic alcohols.124–127 In such cases, alkoxide-directed metallacycle-mediated cross-coupling is followed by elimination to afford a homoallylic amine (Figure 33). While allylic metals are frequently employed to gain access to homoallylic amines, this class of coupling reaction provides a complementary pathway for the synthesis of such products that does not require the intermediacy of an allylic organometallic reagent and that can deliver products that possess alkene substitution and stereochemistry not easily attainable with allylmetal addition chemistry.128, 129
Figure 33.
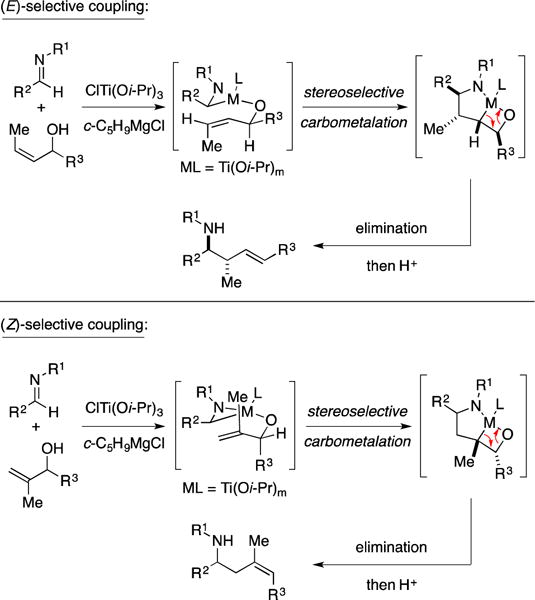
Alkoxide-directed metallacycle-mediated cross-coupling.
A. Total Synthesis of Alkaloid (−)-205B
Alkaloid (−)-205B is a representative member of a large class of alkaloids isolated by Professor Daly in his decades long investigation of natural products from neotropical frogs (Figure 34A).130 It is unique among the natural product class in its ring structure and substitution and, while little is known about the biological activity of this agent, the unnatural antipode (prepared by Toyooka)131 was found to inhibit the α7 nicotinic acetylcholine receptor in a selective fashion.132 In a program that began with the development of metallacycle-mediated alkene–imine coupling chemistry,124, 125, 133 the Micalizio laboratory embraced (−)-205B as a target for synthesis.134 As illustrated in Figure 34B, their efforts focused on chemoselective union of the chiral allylic alcohol 80 with a TMS-imine generated in situ from aldehyde 79 (by treatment with LiHMDS at low temperature). Notably, this variation of alkene–imine coupling proved to be enantiospecific and stereoselective, furnishing the homoallylic primary amine 81 in 57% yield as a single diastereomer, single olefin isomer, and with complete retention of absolute stereochemical information.
Figure 34.
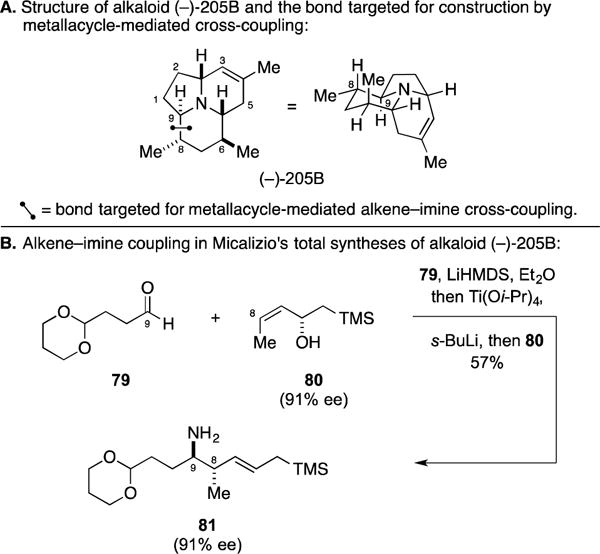
Total synthesis of alkaloid (−)-205B.
B. Total Synthesis of 7-exomethylene Indolizidine 223D
This target (Figure 35A) is a natural product analog and has been included to highlight an interesting chemoselectivity associated with metallacycle-mediated alkene–imine coupling. As illustrated in Figure 35B, the Micalizio laboratory coupled the allylic alcohol 82 with imine 83 as a means to generate homoallylic amine 84. Here, bond-formation was dictated by the allylic alcohol rather than the allylic silane (i.e., no product of aza-Sakurai allylation was observed).135 This chemoselective metallacycle-mediated cross-coupling set the stage for an acid-mediate cyclization cascade that, in one step, resulted in the formation of the target with ≥ 20:1 diastereoselectivity.
Figure 35.
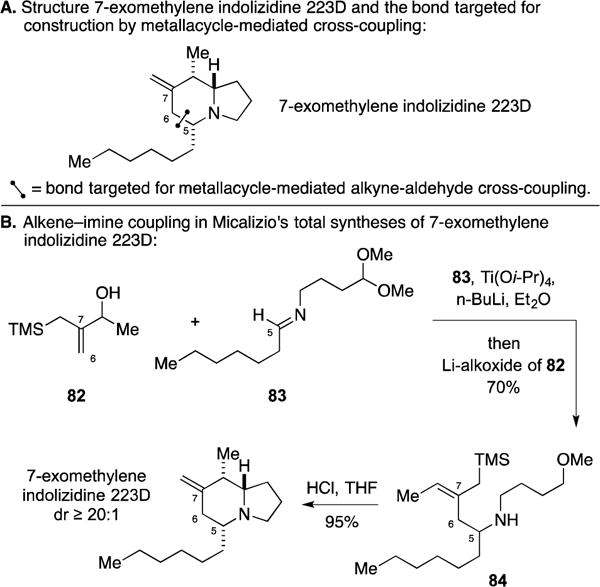
Total synthesis of 7-exomethylene indolizidine 223D.
8. Metallacycle-mediated Cross-Coupling by [2+2+1]
Introduction
Among the most widely employed metallacycle-mediated C–C bond-forming reactions in natural product synthesis is the Pauson-Khand reaction – a process that forges cyclopentenones by metallacycle-mediated alkene–alkyne coupling followed by carbonylation of the intermediate metallacyclopentene (Figure 36). To date, the vast majority of these applications have been conducted in intra- rather than intermolecular settings.136 That said, there have been notable examples of the use of intermolecular Pauson-Khand reactions in natural product synthesis. These processes are, as illustrated in the bottom portion of Figure 36, quite complex. Not only does one need to confront the challenge of engaging substituted alkenes in the key metallacyclopentene-forming step, there is the ever-present challenge of simultaneously controlling regioselectivity in the functionalization of both coupling partners, as well as addressing π-facial selectivity. In selecting examples that highlight cross-coupling-based Pauson-Khand reactions in natural product synthesis, attention has been paid to highlight those that use coupling partners more complex than ethylene.137–139
Figure 36.
Intermolecular Pauson-Khand reaction – an annulation that proceeds by alkene–alkyne coupling.
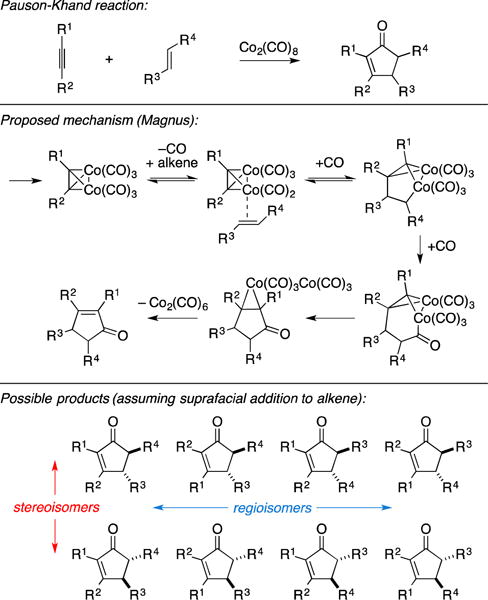
A. Formal Total Synthesis of Brefeldin A
An early example of the use of intermolecular Pauson-Khand annulation in the context of natural product synthesis appeared in Greene’s formal total synthesis of brefeldin A (Figure 37A).140 Here, the authors pursued application of their recently developed diastereoselective version of the intermolecular Pauson-Khand reaction to establish the cyclopentane core of the natural product. As depicted in Figure 37B, union of chiral TMS-alkyne 85 with excess norbornadiene 86 resulted in the formation of cyclopentenone 87 in 82% yield (dr = 24:1).
Figure 37.
Formal total synthesis of brefeldin A.
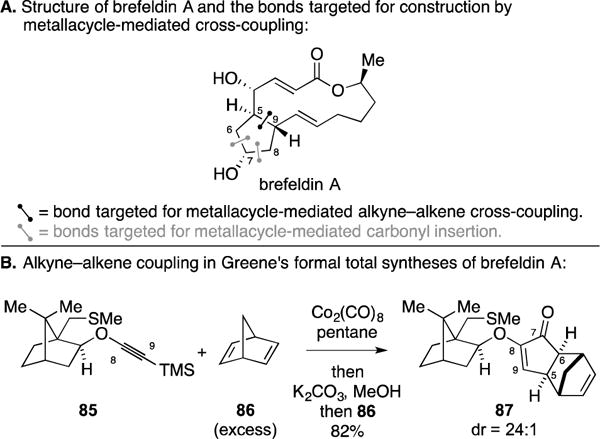
B. Total Synthesis of Asteriscanolide
Asteriscanolide is a cyclooctane-containing sesquiterpene lactone that served as a target of interest to the Krafft laboratory (Figure 38A).141, 142 To address the cyclopentane motif of the natural product, they explored the use of an intermolecular Pauson–Khand reaction between propene (88) and alkynoate 89 (Figure 38B). The authors reported the generation of cyclopentenone 90 in 92% yield. Regioselectivity for the annulation process was rationalized by electronic and steric arguments, respectively.143, 144 The authors noted that the Pauson–Khand reaction was not successful with isobutene or allylic thioalkyl derivatives. As such, the authors found a different pathway for installing the desired quaternary center of the targeted 5-membered ring (by enolization of 90 and subsequent methylation).
Figure 38.
Total synthesis of asteriscanolide.
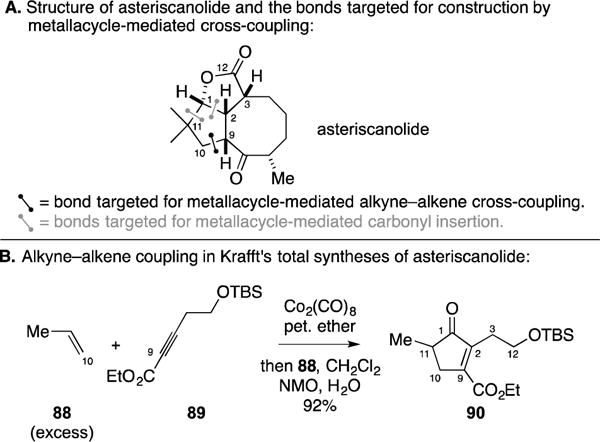
C. Total Synthesis of (−)-Terpestacin
While previously discussed in the context of metallacycle-mediated alkyne–aldehyde cross-coupling, Jamison’s synthesis of (−)-terpestacin (Figure 39A)108, 109 also featured an intermolecular Pauson–Khand reaction. As illustrated in Figure 39B, TMS-acetylene (92) was coupled to stereochemically defined dihydrofuran 91 to deliver the cyclopentenone product 93 as a single isomer in 51% yield.
Figure 39.
Total synthesis of (−)-terpestacin.
D. Total Synthesis of 13,14-Dehydro-12-Oxo-Phytodienoic Acids (deoxy-J1-phytoprostanes)
In efforts to understand whether recently discovered deoxy-J1-phytoprostanes (Figure 40A) are markers of oxidative stress or are biologically active stress metabolites, the Mueller laboratory pursued the total syntheses of members of this class.145 Here, they explored the utility of an asymmetric version of the Pauson–Khand annulation whereby TMS-acetylene was first converted to the chiral Co-complex 94. Subsequent reaction with norbornadiene generated cyclopentenone 95 in 96% yield and with 99% ee.
Figure 40.
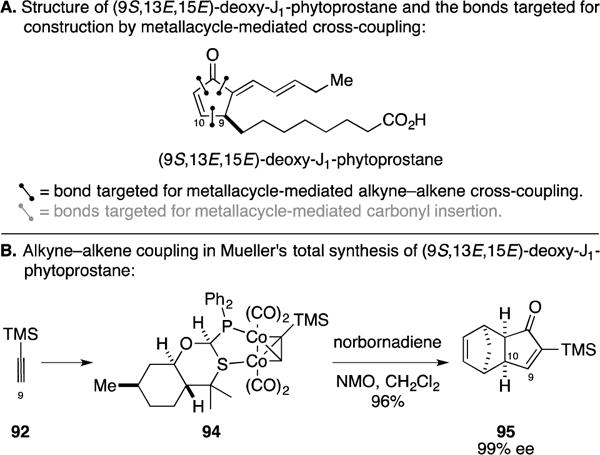
Total synthesis of deoxy-J1-phytoprostanes
E. Total Synthesis of (+/−)-Axinellamines A and B
As part of a program aimed at accomplishing the syntheses of members of the bioactive pyrrole–imidazole marine alkaloid family, the Baran group described an elegant and scalable synthesis of axinellamines A and B (Figure 41A).146 Here, they demonstrated the ability to accomplish a stereo- and regioselective Pauson–Khand-based coupling reaction between the bis-TMS-ether of (E)-butene-1,4-diol (96) and the propargylic Boc-protected amine 97. Their studies led to the discovery of a beneficial role for ethylene glycol in this process and secured a stereoselective entry to cyclopentenone 98 as the first step of their total synthesis campaign.
Figure 41.
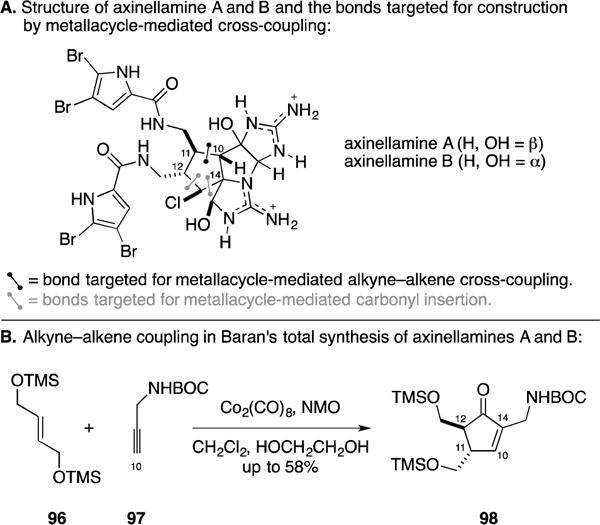
Total synthesis of axinellamines A and B.
9. Metallacycle-mediated Cross-Coupling by [2+2+2]
Introduction
The use of metallacycle chemistry for the union of three unsaturated systems has been known since the middle of the 20th century with Reppe’s pioneering report on the Ni-catalyzed trimerization of acetylene for the synthesis of benzene (Figure 42A).5 While metallacycle-mediated cross-coupling chemistry has taken many different paths that diverge from Reppe’s reaction as a means to accomplish a great variety of coupling processes (many have been presented earlier in this Report), there remains great interest in the development of [2+2+2] chemistry for regio- and stereoselective cross-coupling. With the advent of new metal-promoted or -catalyzed transformations of this type, a wide range of π-systems have been found to participate in related processes, and methods have since been described for the synthesis of a range of carbo- and heterocyclic systems. The proposed mechanisms for these processes are typically based on one of two pathways that diverge from an intermediate metallacyclopentadiene (or related structure) by either: (1) insertion (carbometalation) and reductive elimination, or (2) [4+2] followed by cheletropic extrusion (Figure 42B). Given that our focus is on the application of various metallacycle-mediated cross-coupling reactions in natural product synthesis, we will not discuss mechanistic intricacies associated with each application, unless deemed appropriate. Also, we note that the application of intermolecular versions of [2+2+2] annulation chemistry in natural product synthesis typically takes the form of bimolecular, rather than trimolecular, coupling (Figure 42C).
Figure 42.
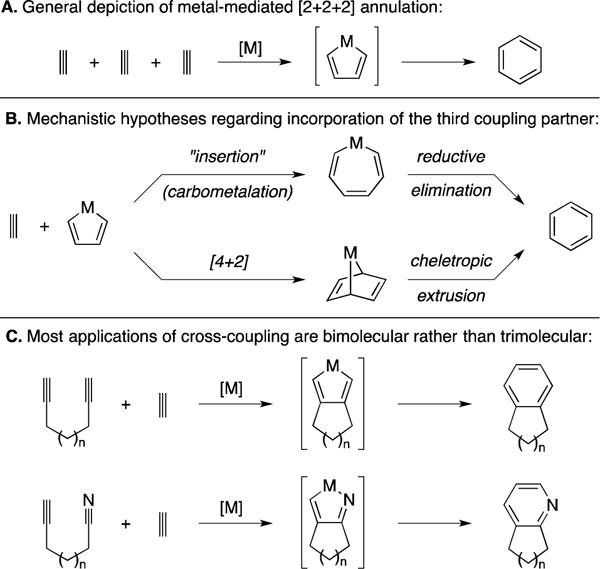
Introduction to metallacycle-mediated cross-coupling by [2+2+2].
This section will be organized by the nature of the π-systems used in the annulation process, beginning with alkyne + alkyne + alkyne and finishing with annulation chemistry that employs an alkene as one of the coupling partners. We note that [2+2+2] chemistry has been the focus of numerous reviews and, as such, we do not aim to be comprehensive in this presentation of the application of [2+2+2] reactions to natural product synthesis. Rather, we share some historic and recent examples that provide a glimpse into the power that such chemistry has for cross-coupling.147, 148
9.1. [Alkyne + Alkyne + Alkyne]
Any discussion of the role of [2+2+2] chemistry in natural product synthesis should begin with the impressive contributions of Professor Vollhardt.149 His research group pioneered the development of Co-mediated [2+2+2] annulation chemistry and has described a number of total syntheses where this reactivity plays a central role. Two syntheses will be presented to highlight particular features of the annulation: (1) the total synthesis of estrone, and (2) the synthesis of the protoberberine nucleus.
A. Total Synthesis of Estrone
Vollhardt’s synthesis of estrone is widely considered to be a classic in natural product synthesis, where a metallacycle-mediated coupling reaction played a profound role in accessing the steroid nucleus (Figure 43A).6, 150, 151 Co-catalyzed coupling of bis-trimethylsilyl acetylene (99) with the D-ring-containing diyne 100 was observed to proceed with chemoselectivity, engaging the diyne in [2+2+2] and generating benzocyclobutene 101 (Figure 43B). Under the reaction conditions, benzocyclobutene 101 was found to undergo electrocyclic ring opening to generate a substituted ortho-xylylene (102) that subsequently participated in a stereoselective intramolecular Diels-Alder reaction en route to 103, the final product generated in this process and isolated in 71% yield. While demonstrating an early example of [2+2+2] chemistry in natural product synthesis, this clever application avoided any concerns with regioselectivity due to the symmetry of 99. Subsequent steps in Vollhardt’s synthesis were used to differentiate the two trimethylsilyl groups located on the steroidal A-ring.
Figure 43.
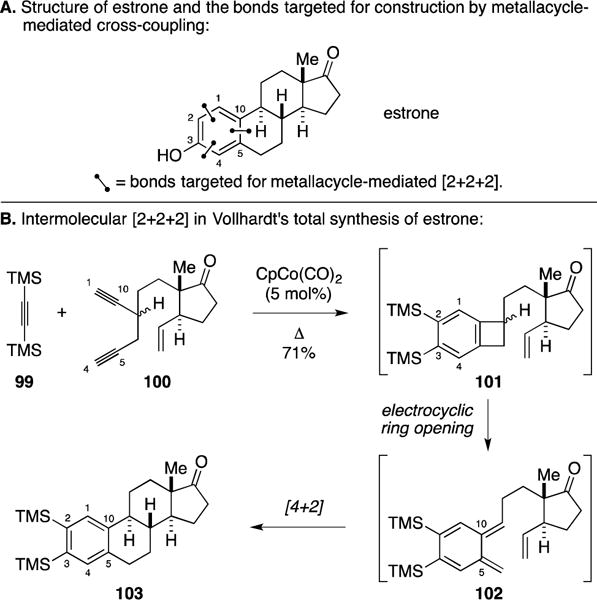
Total synthesis of estrone.
B. Synthesis of the Protoberberine Nucleus
Shortly following their triumph in applying Co-mediated [2+2+2] annulation for the synthesis of estrone, Vollhardt and coworkers described a variant of [2+2+2] chemistry in the context of a synthesis of the protoberberine nucleus (Figure 44A).152 As illustrated in Figure 44B, annulation between the TMS-alkyne-containing diyne 104 and alkyne 105 promoted by one equivalent of CpCo(CO)2 produced the tetracycle 106 in 58% yield as a single regioisomer. Here, the TMS-group of alkyne 105 was reasoned to play a critical role in the regiochemical course of the [2+2+2] reaction.
Figure 44.
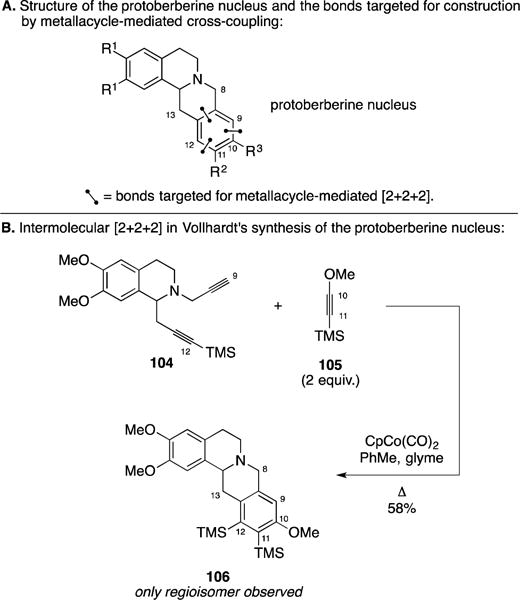
Synthesis of the protoberberine nucleus.
C. Total Synthesis of Sporolide B
In efforts stimulated by the unprecedented molecular structures of the sporolides, natural products isolated from the marine actinomycete Salinospora tropica,153 Professor Nicolaou and coworkers pursued the application of a highly convergent metallacycle-mediated coupling process to gain access to a late stage intermediate in their total synthesis of sporolide B (Figure 45A).154, 155 As illustrated in Figure 45B, Ru-catalyzed coupling of the terminal alkyne 107 with the chlorinated diyne 108 proceeded to deliver 109 in 87% yield as a single regioisomer – the product derived from selective reaction of alkyne 107 at the less hindered C–Ru bond of the metallacyclopentadiene (generated from 108) where alkyne 107 is oriented in such a way to minimize unfavorable steric interaction with ligands at the Ru-metal center.
Figure 45.
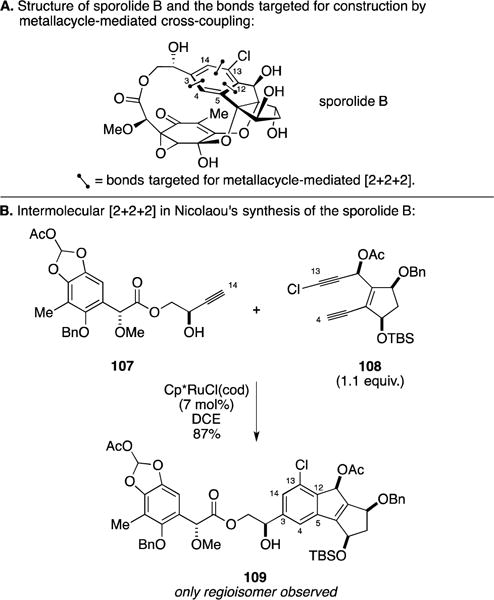
Total synthesis of sporolide B.
D. Total Synthesis of Alcyopterosin A
In efforts focused on exploring the utility of their recently described metalative Reppe reaction, Professor Sato’s research group described the total synthesis of alcyopterosin A (Figure 46A).156 As illustrated, their synthesis employed a Ti-mediated coupling of diyne 111 to alkynoate 110 – a process that generated the bicyclic product 112 in 73% yield (Figure 46B). Here, the mechanistic course of the annulation was reasoned to be distinct from the paths first introduced in this section of the Report (Figure 42B). As illustrated in Figure 46C, initial reaction between the Ti–alkyne complex of 110 with 111 is proposed to generate a functionalized metallacyclopentadiene where regioselection mirrors that which had been described earlier for alkyne–alkyne coupling reactions (Figure 3A). Moving on, the metallacycle engages the pendant alkyne in an intramolecular [4+2] cycloaddition. It has been proposed that the resulting bridged bicyclic metallacyclopentene then rearranges by elimination and isomerization to deliver a benzyltitanium species, protonation of which then delivers the observed product of [2+2+2] and sets the stage for completion of a total synthesis of alcyopterosin A.
Figure 46.
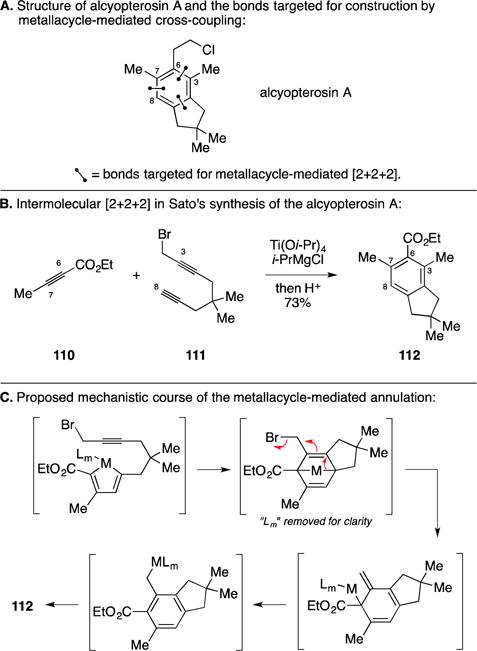
Total synthesis of alcyopterosin A.
E. Total Synthesis of Hyellazole
In 2002 Witulski and coworkers described an approach to substituted carbazoles based on crossed [2+2+2] annulation chemistry and, in the course of these studies, described a convergent synthesis of hyellazole (Figure 47A).157 As depicted in Figure 47B, their key step was the Rh-catalyzed coupling between diyne 113 and alkyne 114 en route to 115. The reaction proceeded with good efficiency (89%) and outstanding regioselectivity (30:1), while demonstrating the utility of a metallacycle-mediated [2+2+2] annulation with heteroatom-substituted alkynes for the synthesis of carbazoles.
Figure 47.
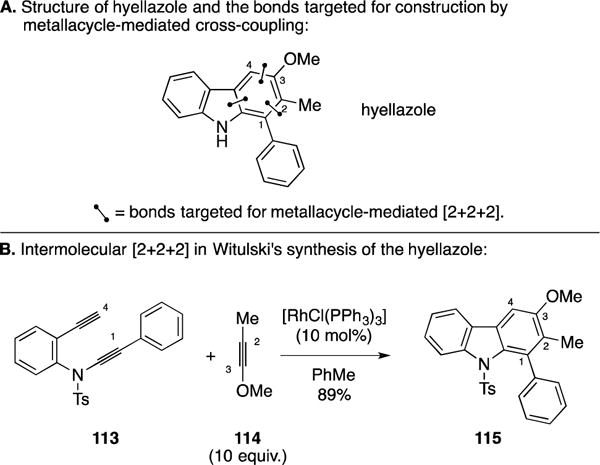
Total synthesis of hyellazole.
9.2. [Alkyne + Alkyne + Nitrile]
This version of the [2+2+2] annulation process has found numerous applications in natural product synthesis. The first of these is discussed below and is followed by more recent examples.
A. Total Synthesis of Vitamin B6
Shortly after their report that described the synthesis of steroids by Co-mediated [2+2+2] annulation between an alkyne and a diyne, the Vollhardt group accomplished a total synthesis of vitamin B6 that demonstrated the potential of a heteroatom analogue of their Co-mediated process to forge densely functionalized pyridines (Figure 48A).158 In a very clever strategy, Vollhardt designed the synthesis to proceed through union of the symmetrical diyne 116 with acetonitrile (117). Due to the symmetry of 116, there is no opportunity to deliver a regioisomer in the [2+2+2]. Fortunately, after annulation, the pyridine product was found to undergo selective mono-destannylation on purification to deliver 118 in 45% yield. Notably, this basic strategy for pyridine synthesis, that employs a symmetric diyne and a nitrile, has been adopted for the synthesis of other alkaloid targets.159
Figure 48.
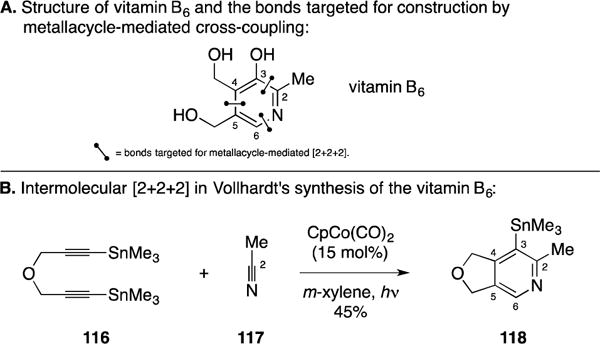
Total synthesis of vitamin B6.
B. Total Synthesis of (+/−)-Lysergene
Moving forward from their vitamin B6 synthesis, the Vollhardt laboratory designed an alkyne + alkyne + nitrile metallacycle-mediated cross-coupling reaction for the synthesis of lysergene (Figure 49A).160 The key annulation reaction is highlighted in Figure 49B and features the coupling of an alkynyl nitrile (119) with TMS-alkyne 120. In comparison to their earlier work targeting vitamin B6, this effort was substantially more complex owing to the unsymmetric nature of the TMS-alkyne coupling partner (120). Fortunately, they were able to employ a Co-promoted [2+2+2] annulation to produce the tetracyclic products 121 and 122 in a combined 49% yield. The desired regioisomer was found to readily desilylate en route to producing 121 in a 38% isolated yield.
Figure 49.
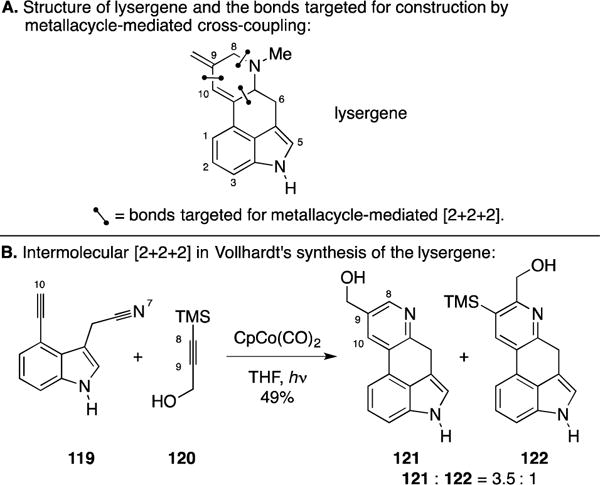
Total synthesis of lysergene.
C. Synthesis of Lavendamycin Methyl Ester
Lavendamycin is an antitumor antibiotic isolated from Streptomyces lavendulae161 that was shown to have activity against several cancer cell lines as well as topoisomerase 1 (Figure 50A).162 In efforts targeting the synthesis of lavendamycin, Nissen and Detert described a variant of an alkyne/alkyne/nitrile [2+2+2] annulation that employed an alkynyl sulfonamide.163 As illustrated in Figure 50B, a Ru-catalyzed [2+2+2] annulation between diyne 123 and methyl cyanoformate (124) resulted in a highly efficient and regioselective [2+2+2] process leading to production of 127 in 92% yield. Mechanistically distinct from examples described in this section, the origin of regioselection was proposed to derive from the [2+2] addition product of methyl cyanoformate (124) with an intermediate metallacycle. Preferential addition occurs to the Ru–C-α bond to form the least hindered cycloadduct. Interestingly, Cp*RuCl(cod) was uniquely effective in accomplishing this regioselective annulation reaction (the authors have moved forward with similar annulation reactions in natural product synthesis).164
Figure 50.
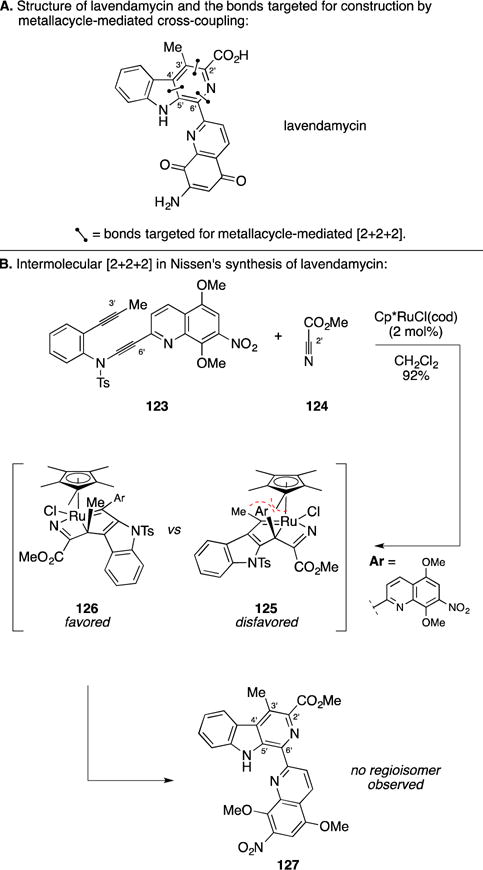
Total synthesis of lavendamycin methyl ester.
D. Total Synthesis of (+)-Complanadine A
Complanadine A is a fascinating alkaloid that possesses an interesting structure and displays unique and potentially valuable medicinal properties, in particular the ability to induce the secretion of neurotrophic factors from 1321N1 cells and result in the differentiation of PC-12 cells (Figure 51A).165 Recently, the Siegel laboratory described a highly convergent assembly of complanadine A by application of a sequence of alkyne/alkyne/nitrile-based [2+2+2] annulation reactions, defining perhaps one of the most elegant applications of this type of metallacycle-mediated cross-coupling reaction in natural product synthesis.166 As illustrated in Figure 51B, CpCo(CO)2-mediated union of 128 with 129 proceeded in 82% yield and delivered the desired pyridine with 25:1 regioselectivity. Moving on, they addressed the synthesis of the second pyridine of complanadine A with another application of an alkyne/alkyne/nitrile [2+2+2] annulation that united the two polycyclic hemispheres of the target. In this latter case (i.e., union of alkyne 131 with alkynyl nitrile 132), modified reaction conditions were required to favor the formation of the desired regioisomer.
Figure 51.
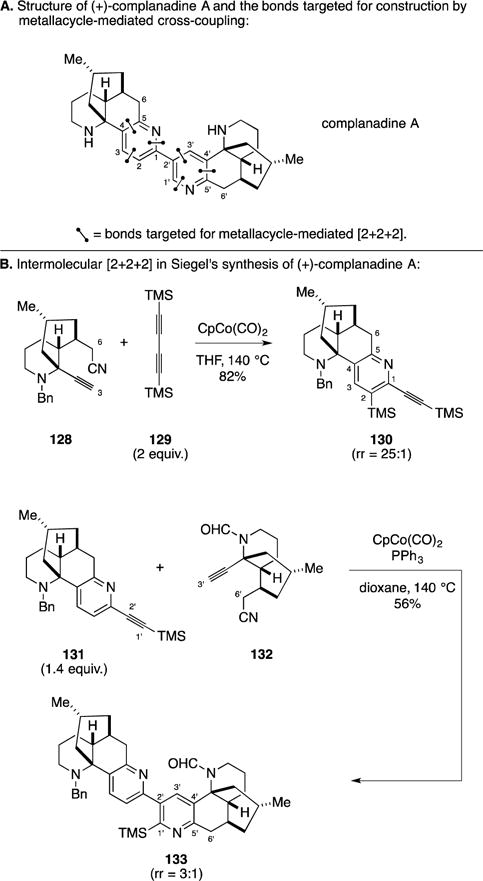
Total synthesis of (+)-complanadine A.
Overall, this [2+2+2] annulation strategy has since been utilized to prepare lycodine (and analogs thereof) with the aim of evaluating its effects on neuronal growth alongside its pseudosymmetric dimer, complanadine A.167, 168
9.3. [Alkyne + Alkyne + Isocyanate]
Among the collection of contributions that blossomed fromed the Vollhardt group, it was found that the Co-mediated [2+2+2] annulation chemistry can be conducted with isocyanates as one of the participating π-systems.169 Specifically, Vollhardt described the intermolecular coupling of 5-isocyanatoalkynes with alkynes en route to the formation of 5-indolizinones (Figure 52). The reaction was found to proceed with good chemo- and regioselectivity when TMS-alkynes are used – the silyl group emerges α to the amide linkage.
Figure 52.
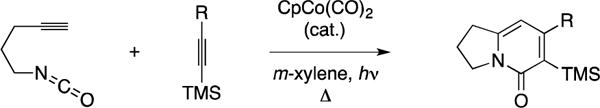
Co-catalyzed [2+2+2] of isocyanatoalkynes with TMS-alkynes.
A. Formal Total Synthesis of Camptothecin
Camptothecin is a pentacyclic alkaloid that possesses potent cytotoxic activity against a variety of tumor cell lines, and synthetic derivatives of this agent have been demonstrated to be useful in the clinic (Figure 53A). In efforts that were sprung from their development of an isocyanate variant of their Co-catalyzed [2+2+2] annulation reaction, the Vollhardt laboratory demonstrated the utility of this reaction process in the formal total synthesis of camptothecin.169 As illustrated in Figure 53B, Co-catalyzed annulation between isocyanate 134 and TMS-alkyne 135 resulted in the formation of the indolizinone 136 – a species that was subsequently converted into an intermediate that had previously been employed in a total synthesis of camptothecin.
Figure 53.
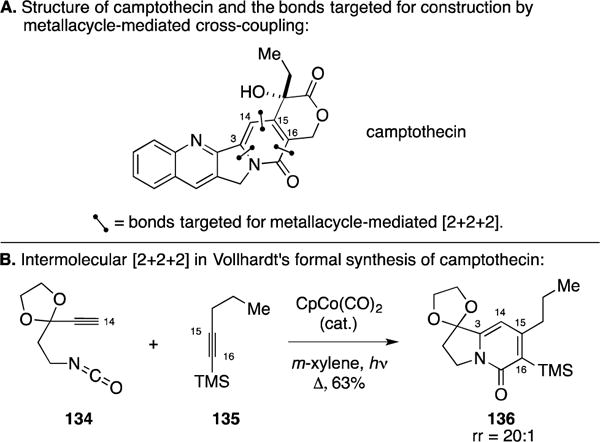
Formal total synthesis of camptothecin.
9.4. [Alkyne + Alkene + Isocyanate]
In a surprising turn that diverges from the [2+2+2] reaction of isocyanatoalkynes, Professor Rovis described an interesting version of this type of annulation with alkenyl isocyanates. As illustrated in Figure 54, Rh-catalyzed [2+2+2] between a terminal alkyne and an alkenyl isocyanate delivers an annulation product (139) that has different connectivity then what was previously described by Vollhardt in his Co-catalyzed annulation reaction. Rovis has proposed a mechanism whereby initial metallacycle-mediated alkyne–isonitrile coupling is followed by CO migration, alkene insertion and reductive elimination.
Figure 54.
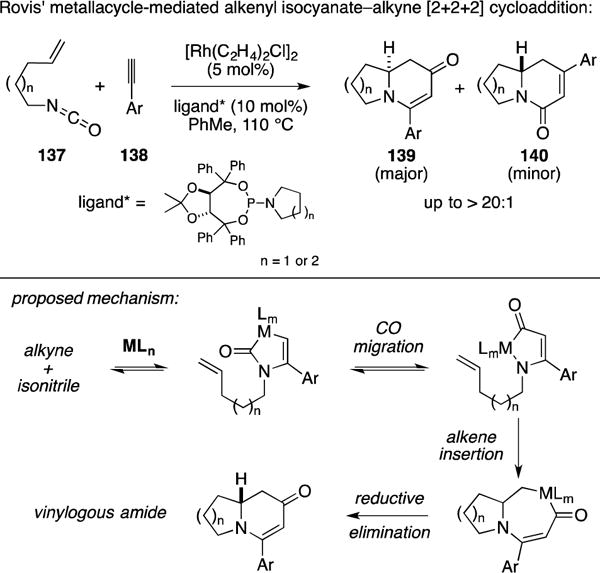
Rh-catalyzed alkenyl isocyanate – alkyne [2+2+2].
A. Total Synthesis of (+)-Lasubine II
During the course of their development of the enantioselective alkenyl isocyanate – alkyne coupling reaction, the Rovis laboratory investigated application of this technology to the total synthesis of (+)-lasubine II (Figure 55A).170 Due to the CO-migration associated with this enantioselective [2+2+2] annulation, the bonds targeted for formation with this process are a bit different than typically seen in [2+2+2] annulation methods. Here, the four C–C bonds highlighted in Figure 55A were those that would derive from the annulation process. As illustrated in Figure 55B, union of alkenyl isocyanate 141 with alkyne 142 established the entire carbon skeleton of the natural product, producing 143 in 62% yield and 98% ee. This intermediate was converted to (+)-lasubine II in just three additional steps.
Figure 55.
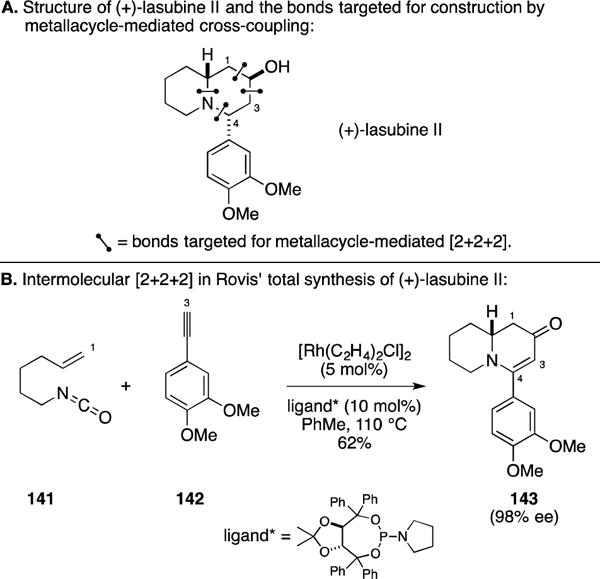
Total synthesis of (+)-lasubine II.
B. Total Synthesis Alkaloid (−)-209D
In studies springing from their initial discovery of Rh-cat. [2+2+2] annulation between terminal alkynes and alkenyl isocyanates, where the vinylogous amide product was favored when aryl-alkynes were employed as coupling partners, the Rovis laboratory targeted the development of a variant of this annulation method that would be effective with alkyl-substituted terminal alkynes.171 In the course of these studies, they demonstrated the utility of this method in the context of a concise, asymmetric total synthesis of alkaloid (−)-209D (Figure 56A). As illustrated in Figure 56B, union of 144 with terminal alkyne 145, under the action of a chiral Rh-catalyst derived from a distinct phosphoramidite ligand than previously explored in the context of the lasubine II synthesis (Figure 55B), resulted in the formation of the indolizidine 146 in 66% yield and 91% ee (the unsaturated lactam product corresponding to 140 in Figure 54 was seen as a minor byproduct in this reaction, with the vinylogous lactam being favored in a ratio of 6:1).
Figure 56.
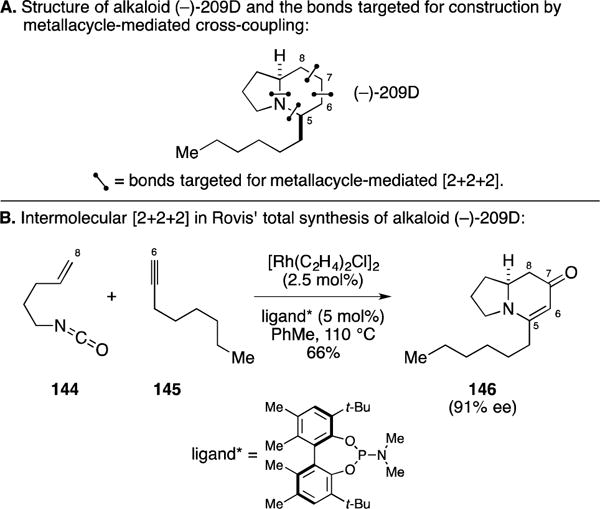
Total synthesis of alkaloid (−)-209D.
9.5. [Alkyne + Alkyne + Alkene]
The final collection of methods to be discussed in this section of the Report are based on metallacycle-mediated intermolecular [2+2+2] annulation methods that engage two alkynes and one alkene during the course of generating a cyclohexadiene motif (Figure 57). In all cases where such chemistry has been employed in the context of natural product synthesis, these annulation procedures take the form of cross-coupling a suitably functionalized enyne with an alkyne.
Figure 57.
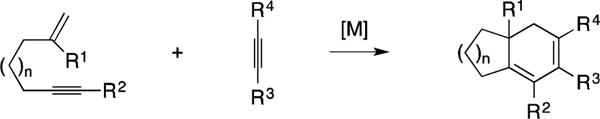
Metallacycle-mediated cross-coupling between enynes and an alkyne by [2+2+2] annulation.
A. Formal Total Synthesis of γ-Lycorane
An early example of the use of metallacycle-mediated cross-coupling between an enyne and an alkyne appeared in Vollhardt’s formal total synthesis of the alkaloid γ-lycorane (Figure 58A).172 Here, their efforts were focused on the use of an enamine double bond in the [2+2+2] annulation, realizing the union of 147 with bis-TMS-acetylene 99 – a process that produced 148 in 65% yield. Related to other elegant synthesis strategies devised by Professor Vollhardt, there was no issue with regioselectivity in this annulation process due to the symmetry of 99.
Figure 58.
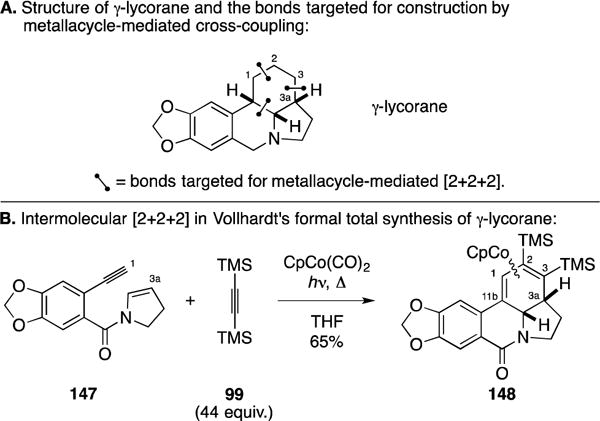
Total synthesis of γ-lycorane.
B. Formal Total Synthesis of Strychnine
Since R. B. Woodward’s pioneering synthesis of strychnine,173, 174 this alkaloid has stood as a classic target in natural product synthesis (Figure 59A).175–185 Moving forward with the development of methods for Co-mediated [2+2+2] annulation between enynes and alkynes, Vollhardt’s group demonstrated the ability to employ an indole double bond in the annulation process and highlighted the application of this coupling chemistry in the context of a formal total synthesis of strychnine.186, 187 As illustrated in Figure 59B, union of 149 with acetylene (71) delivered the tetracyclic product 150 in 46% yield. As has previously been demonstrated in numerous Vollhardt-designed synthesis strategies employing [2+2+2] annulation chemistry, the key annulation employed in the strychnine campaign avoids any issues with regioselectivity due to the symmetry of the alkyne coupling partner (acetylene).
Figure 59.
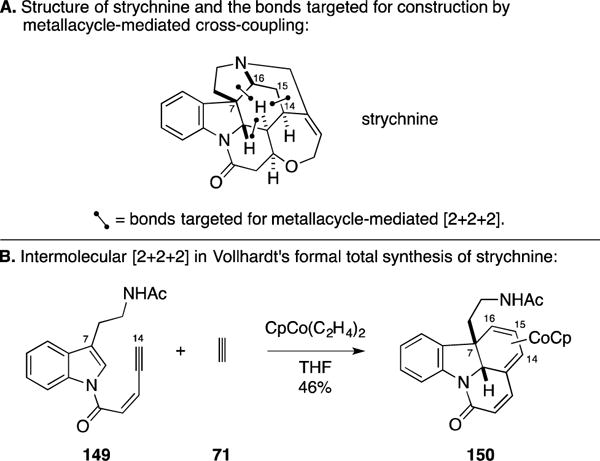
Formal total synthesis of strychnine.
C. Total Synthesis of (2S)-Hydroxy-3,4-Dehydroneomajucin
In efforts focused on the exploration of chemoselectivity in their hydroxyl-directed alkyne–alkene and alkyne–alkyne coupling reactions, the Micalizio laboratory discovered a highly regio- and stereoselective [2+2+2] annulation en route to angularly substituted hydrindanes.188–191 This reaction played a central role in their recently described synthesis of (2S)-hydroxy-3,4-dehydroneomajucin – a molecule that belongs to a larger class of sesquiterpenes that have been shown to possess potent and unique activity as neurotrophic agents (Figure 60A).192 Here, union of enyne 151 with the unsymmetrical alkyne 152 proceeded to deliver the angularly substituted hydrindane 153 in 73% yield (rs ≥ 20:1; ds ≥ 20:1). This process is unique with respect to other intermolecular enyne + alkyne [2+2+2] reactions employed in natural product synthesis in that the alkyne component is unsymmetrical and the annulation proceeds with exquisite regioselectivity, while also establishing an angular quaternary center (C9). In addition to moving this material forward to complete the first total synthesis of (2S)-hydroxy-3,4-dehydroneomajucin, the Micalizio laboratory also demonstrated the ability to advance this intermediate to (1R,10S)-2-oxo-3,4-dehydroneomajucin and (−)-jiadifenin.
Figure 60.
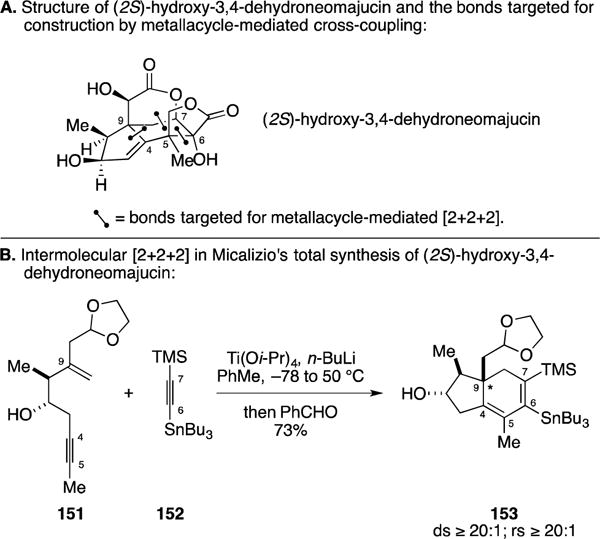
Total synthesis of (2S)-hydroxy-3,4-dehydroneomajucin.
10. Other Intermolecular C–C Bond-Forming Reactions that Derive from the Chemistry of Metallacyclopentanes
Introduction
At the start of this Report, we set the stage for a discussion that was centered on an inclusive definition of metallacycle-mediated cross-coupling. This perspective united crossed metathesis chemistry with the diverse collection of coupling reactions that are thought to proceed through metallacyclopentane-like intermediates, while also including an example of a process that is thought to proceed by way of an oxametallacyclohexene intermediate (Jamison’s alkyne–epoxide coupling). While most of the syntheses described earlier clearly highlight intermolecular C–C bond formation during the metallacycle-forming step, the latter section on [2+2+2] annulation chemistry presents applications where the sequence of events is less clear. Perhaps in a number of cases (not including the process depicted in Figure 60), the metallacyclopentane-forming step occurs intramolecularly, with the intermolecular process taking place after formation of the metallacyclopentane intermediate. We recognize the ambiguity with these processes, and decided to include them due to the overall intermolecular nature of the metallacycle-mediated transformations. Because of this ambiguity, we are compelled to share several examples of natural product syntheses that employ a metallacycle-mediated C–C bond-forming event, but where the intermolecular processes are proposed to occur after an initial metallacycle-mediated cyclization. This collection of reactions is viewed as somewhat off-topic, but we have selected to highlight a few modern examples that speak to a different role of metallacycle chemistry in intermolecular C–C bond-formation.
A. Total Syntheses of Kainoids
Professor John Montgomery has been a leader in the development of Ni-catalyzed metallacycle-mediated C–C bond-forming reactions and the use of these processes in natural product synthesis. One of his early contributions was the discovery of a Ni-catalyzed cyclization that is terminated by a cross-coupling event with another organometallic species. In this way, his annulation chemistry provides a means to forge two C–C bonds, one from the metallacycle-forming intramolecular reaction and one from the capture of this species by cross-coupling with a different organometallic species. A beautiful collection of syntheses highlight the broad generality of this type of chemistry while also establishing a highly stereoselective entry to kainoid natural products (Figure 61A).
Figure 61.
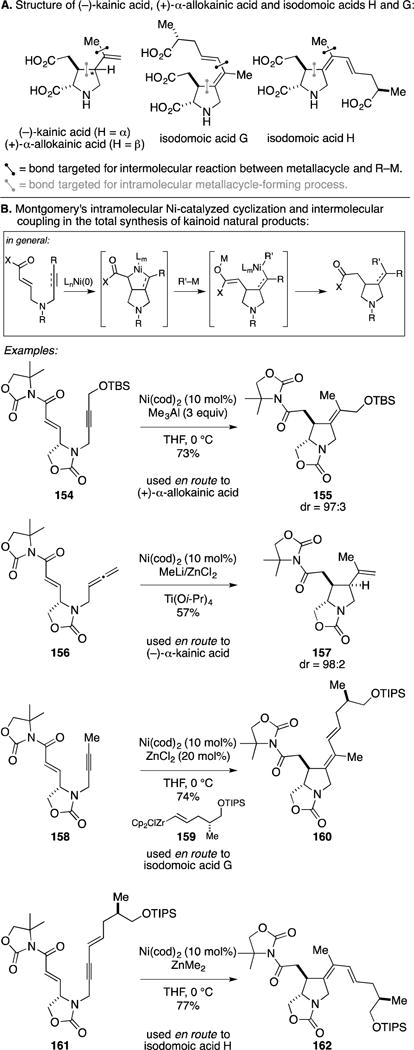
Total synthesis of kainoids by sequential Ni-catalyzed cyclization and intermolecular coupling.
A general depiction of the reaction process that has been employed in these syntheses is presented in the top section of Figure 61B, and the specific examples reported in the context of total syntheses follow [for (−)-kainic acid, (+)-α-allokainic acid, and isodomoic acids G and H].193–196 Overall, this collection of examples highlights the strategic utility of Montgomery’s Ni-catalyzed transformations to address challenging and complex problems in stereoselective synthesis. While not selected for discussion here, Montgomery has also employed related strategies for the synthesis of (+)-testudinariol A (by allene–aldehyde cyclization and coupling with Me2Zn) and (+/−)-deformyl-isogeissoschizine (by terminal alkyne–α,β-unsaturated imide cyclization and coupling with Me2Zn).197, 198
B. Total Synthesis of Carbacyclin
Professor Sato took the early lead in developing metallacycle-mediated coupling chemistry promoted by reagents prepared from the combination of Ti(Oi-Pr)4 and two equivalents of a Grignard reagent (reaction conditions originally used for the Kulinkovich reaction).199, 200 Among the many contributions that Professor Sato made to this area of reaction methodology were collections of intramolecular metallacycle-forming processes that took advantage of the resulting titanacyclopentane-type intermediate in subsequent bond-forming processes. One of these was highlighted in the total synthesis of (+/−)-carbacyclin (Figure 62A).201–203 As depicted in Figure 62B, Ti-promoted intramolecular diene cyclization was followed by isomerization to a Ti-enolate and intermolecular aldol reaction. The reaction process was concluded with intramolecular addition of the resulting organometallic species to the pendant ester (intramolecular nucleophilic acyl substitution – “INAS”). This elegant cascade establishes both rings of the carbacyclin structure and the functionalized side chain in a single step.
Figure 62.
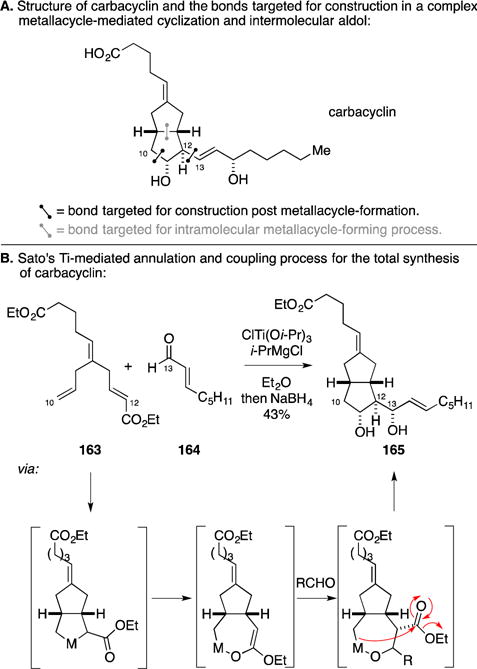
Total synthesis of (+/−)-carbacyclin.
C. Total Synthesis of 9-Hydroxysargaquinone
Professor Whitby has established a collection of strategies for C–C bond formation that stem from the reactivity of zirconacyclopentane intermediates.204, 205 As illustrated in Figure 63, a lithiated allylic chloride is employed in initial nucleophilic addition to Zr and the resulting ate-complex undergoes ring expansion by a 1,2-shift to generate a zirconacyclohexane. This species, also an allylzirconium, can be used in subsequent bond forming reactions – i.e. addition to an aldehyde.
Figure 63.
Whitby’s application of allylic carbenoid species to convert zirconacyclopentanes to ring-expanded allylzirconiums.
In 2005, Whitby described a zirconium-mediated synthesis of 9-hydroxysargaquinone that proceeded through a zirconacycle-mediated coupling process that generated three C–C bonds of the target structure (Figure 64A).206 While we have excluded summarizing natural product syntheses that proceed by simple metallacycle-mediated addition of Grignard reagents or organolithium species to an alkene or alkyne, we have included the synthesis of 9-hydroxysargaquinone described by Whitby because of the multicomponent nature of the process. As illustrated in Figure 64B, coupling of tributylstannyl propyne (166) to geranial (167) proceeded to deliver the functionalized tetraene 168 possessing a vinyltin species suitable for subsequent functionalization. Notably, this process demonstrated the ability to selectively expand the zirconacyclopentene intermediate by migration of the sp3, rather than sp2, carbon.
Figure 64.
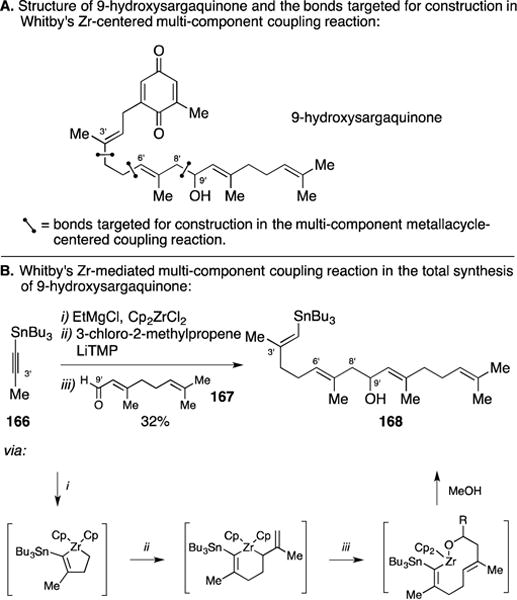
Synthesis of 9-hydroxysargaquinone.
D. Total Synthesis of (+/−)-Acetoxyodontoschismenol
In pursuits focused on the total synthesis of acetoxyodontoschismenol, Whitby pursued a different variant of this multi-component coupling reaction (Figure 65A).207 Here, intramolecular diene cyclization was used as the initial step to generate the reactive zirconacyclopentane species – an intermediate whose carbenoid-based ring expansion would not be as straightforward as the example previously described in the context of 9-hydroxysargaquinone. As illustrated in Figure 65B, the hybridization of both carbons bound to zirconium is sp3, setting up a question regarding selectivity of the subsequent carbenoid insertion step. Fortunately, Whitby observed product derived from migration of the neopentyl sp3 carbon, followed by allylation upon addition of the α,β-unsaturated aldehyde and, finally, iodination of the remaining sp3 C–Zr bond. Overall, the metallacycle-mediated coupling process delivered the product in 65% yield, whereby the quaternary center resident in the cyclopentane was established with 4:1 selectivity.
Figure 65.
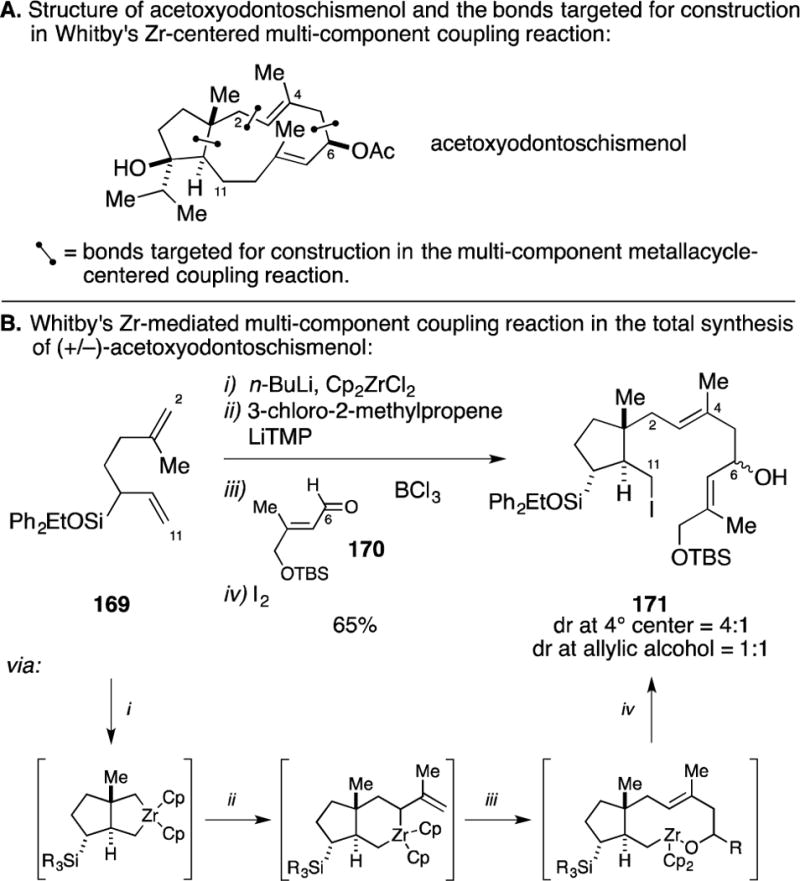
Synthesis of (+/−)-acetoxydontoschismenol.
E. Total Synthesis of Fluvirucin B1
Admittedly not quite fitting seamlessly in this section of the Report, we conclude with a discussion that highlights the application of Professor Hoveyda’s Zr-catalzyed carbomagnesiation chemistry in natural product synthesis. The general process, first described in 1983 for addition of ethylmagnesium bromide to simple alkenes,208 features the use of Cp2ZrCl2 in a catalytic process for net regioselective hydroxyalkylation (Figure 66).209, 210
Figure 66.
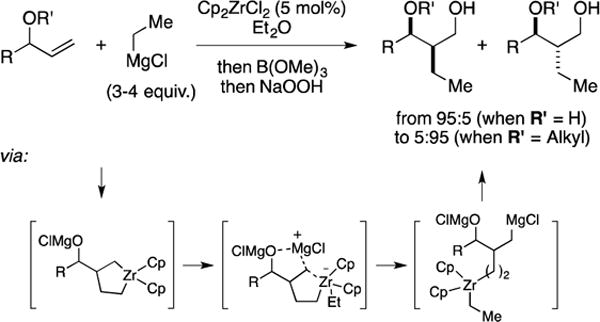
Hoveyda’s Zr-catalyzed regio- and stereoselective carbomagnesiation.
In 1995, the Hoveyda group described a route to fluvirucin B1 that employed two variations of their Zr-catalyzed carbomagnesiation process (Figure 67A).211, 212 As illustrated in Figure 67B, one of these employed the diastereoselective ethylmagnesiation of an allylic alcohol (172) and was terminated by reaction with a tosylated aziridine. The other demonstrated the utility of their catalytic asymmetric ethylmagnesiation. Here enantioselective addition to 3-dihydrofuran (174) resulted in an organometallic intermediate that underwent ring-opening elimination to deliver homoallylic alcohol 175 with outstanding levels of enantioselectivity.
Figure 67.
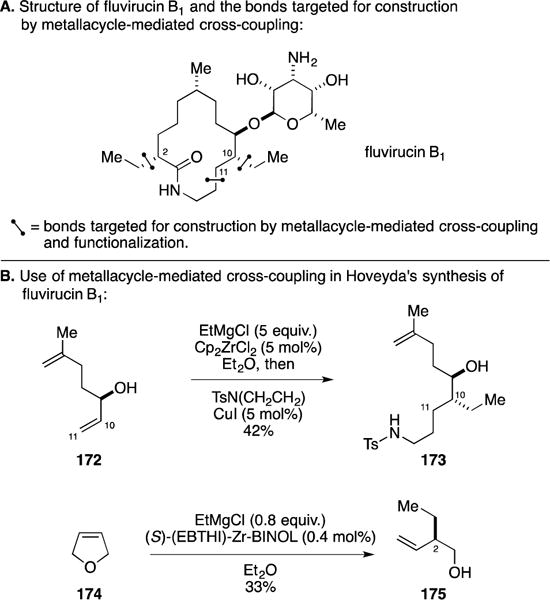
Synthesis of fluvirucin B1.
11. Intermolecular Kulinkovich Reactions in Natural Product Synthesis
The use of the Kulinkovich reaction in natural product synthesis was reviewed in 2012 and, as such, we will only highlight some recent examples that employ intermolecular Kulinkovich reactions in the context of natural product synthesis.13
A. Total Synthesis of (+)-Spirolaxine Methyl Ether
In 2007, Professor Phillips described the application of the intermolecular Kulinkovich reaction for subunit coupling in the total synthesis of (+)-spirolaxine methyl ether (Figure 68A).213 As illustrated in Figure 68B, these efforts highlighted the union of terminal alkene 176 with the chiral lactone 177 en route to the hydroxycyclopropane product 178. Exposure of this product to the combination of Fe(NO3)3 and Bu3SnH resulted in ring opening to generate the corresponding ketone, and subsequent desilylation resulted in a triol that spontaneously cyclized to the spiroketal-containing product 179.
Figure 68.
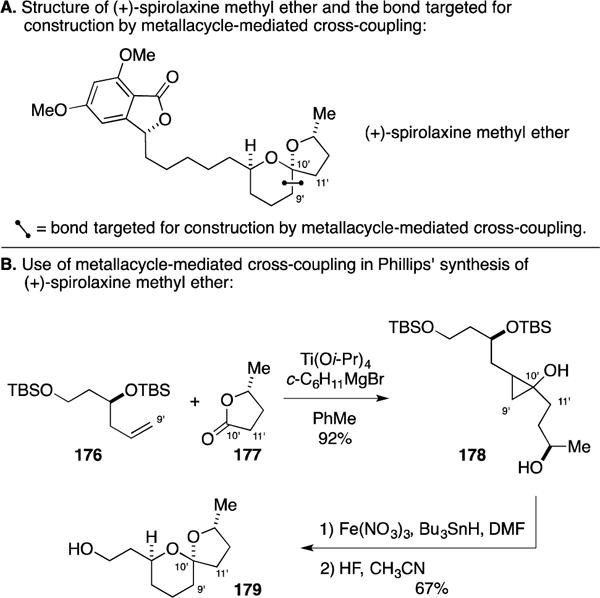
Synthesis of (+)-spirolaxine methyl ether.
B. Total Synthesis of Demethyl (C-11) Cezomycin
Recently, a related approach to subunit coupling was described in Wu’s total synthesis of the anti-fungal antibiotic demethyl (C-11) cezomycin (Figure 69A).214 Here, intermolecular Kulinkovich reaction between ester 180 and terminal alkene 181 was employed to generate hydroxycylopropane product 182 that, in subsequent steps, was cleaved by the action of Fe(NO3)3 and advanced to the central spiroketal motif of the target (Figure 69B).
Figure 69.
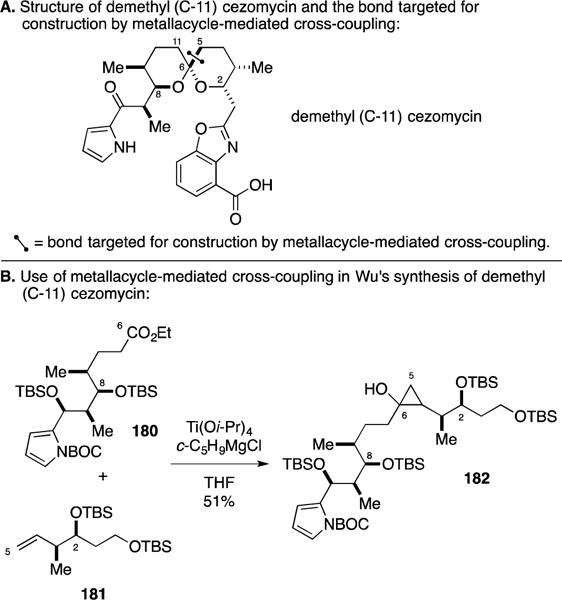
Synthesis of dimethyl (C-11) cezomycin.
12. Summary and Outlook
Metallacycle-mediated cross-coupling is broadly defined here as an intermolecular C–C bond-forming process that proceeds by way of a metallacyclic intermediate. Since the middle of the 20th century, such chemistry has grown far beyond simple alkyne trimerization, and has established a large collection of highly selective synthetic methods that can be employed to realize unique retrosynthetic relationships of relevance to the total synthesis of terpenes, alkaloids and polyketides. Coupling partners can now include a variety of participating π-systems, and intermolecular C–C bond formation can occur during the formation of a metallacyclic intermediate, or after – where the metallacycle-forming step is first embraced in an intramolecular fashion. It is becoming increasingly clear that the transformations accessible with this style of chemistry are not only complex, offering the opportunity to forge numerous C–C bonds and stereochemical relationships in a single reaction process, they are diverse and can be wielded to address a large variety of structural challenges in synthetic organic chemistry.
Until recently, many reactions that we now consider “metallacycle-mediated cross-coupling” were characterized independently, with focus given to the nature of the metal employed, and/or whether a reaction component can be used in a substoichiometric fashion. We believe that unifying what can appear as a collection of disparate chemical transformations as belonging to a larger class of reactivity will have a beneficial pedagogical impact and define a robust foundation on which to build a bright future for this area of organometallic chemistry. With a focus on addressing problems in chemical synthesis, we believe that metallacycle-mediated cross-coupling will continue to define fertile ground for discovery, with advances enabling ever more efficient syntheses of exquisitely complex targets.
While there is much cause for enthusiasm regarding the future of metallacycle-mediated cross-coupling, there remain many barriers that we believe should be addressed. Future contributions that would have a profound impact on this area include those that would: (1) provide a means to move away from depending on the use of expensive, rare and toxic metals, (2) avoid the use of “exotic” and costly ligands, (3) realize reaction processes that are user-friendly (i.e. that do not require a rigorously anhydrous and anaerobic environment), and (4) establish a general and selective means of achieving a true “mix-and-match” future for the coupling of diverse, substituted, and functionalized π-systems – all while being chemo-, regio-, and stereoselective. The last 50 years of chemical research has established a robust foundation in this area and has offered numerous examples that reinforce the great value of metallacycle-mediated cross-coupling in total synthesis. We look forward to seeing this area of chemical reactivity continue to grow in a manner that greatly impacts the future of complex molecule synthesis.
Acknowledgments
The authors acknowledge funding for their program in metallacycle-mediated cross-coupling from the National Institutes of Health – GM080266.
Biographies

Natasha F. O’Rourke received her B.Sc. in Chemistry and Biology from St. Francis Xavier University (NS, Canada) in 2008 under the supervision of Dr. Manuel A. S. Aquino where she worked on the synthesis of diruthenium (II, III) and dirhenium (III, III) tetracarboxylates possessing biologically relevant ligands for potential application as metallotherapeutic agents. In 2014, she received her Ph.D. in Organic Chemistry and Chemical Biology from the University of Victoria (BC, Canada) where she studied under the supervision of Dr. Jeremy E. Wulff. Her graduate studies focused on mechanistic investigation of orthogonal transformations of bis-vinyl ethers and on developing methods to access small molecule disruptors of the PD-1/PD-L1 interaction involved in metastatic cancer. In 2014, she moved to the United States where she joined the group of Prof. Glenn C. Micalizio at Dartmouth College (NH) as a postdoctoral research associate in the Department of Chemistry. Her work has focused on the development of new synthetic methods and strategies for natural product synthesis.

Matthew J. Kier received his B.A. in chemistry from Goucher College in 2006 under Professor George E. Greco where he worked on metal-catalyzed cycloaddition reactions. In 2010 he received an M.S. in chemistry from the University of California, Irvine under the supervision of Professor Christopher D. Vanderwal where he worked on the formal synthesis of porothramycins A and B. Currently, he is a graduate student in the Department of Chemistry at Dartmouth College where his research in the laboratory of Professor Glenn C. Micalizio is focused on developing new metallacycle-centered chemistry for applications in natural product total synthesis.

Professor Micalizio trained as a Fellow of the Helen Hay Whitney Foundation in the laboratories of Professor Stuart L. Schreiber at Harvard University (2001-2003), received his PhD at the University of Michigan (2001) after graduate studies in the laboratories of Professor William R. Roush, and completed undergraduate studies at Ramapo College of NJ (1996). He began his independent academic career as an Assistant Professor in the Department of Chemistry at Yale University (2003), was later recruited to join the Department of Chemistry at the Scripps Research Institute and, most recently Dartmouth College where he is currently the New Hampshire Professor of Chemistry. His research interests are focused on the development and application of new methods and synthesis strategies in organic chemistry and the application of these toward the discovery of molecules with interesting biological properties.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]
- 2.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4442–4489. doi: 10.1002/anie.200500368. [DOI] [PubMed] [Google Scholar]
- 3.Velluz L, Valls J, Mathieu J. Angew Chem Int Ed. 1967;6:778–789. [Google Scholar]
- 4.Beeler AB, Schaus SE, Porco JA. Curr Opin Chem Biol. 2005;9:277–284. doi: 10.1016/j.cbpa.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Reppe W, Schweckendick WJ. Justus Liebigs Ann Chem. 1948;560:104–116. [Google Scholar]
- 6.Funk RL, Vollhardt KPC. J Am Chem Soc. 1980;102:5253–5261. [Google Scholar]
- 7.Vol’pin ME, Dubovitskii VA, Nogina OV, Kursanov DN. Dokl Akad Nauk Sssr. 1963;151:1100–1103. [Google Scholar]
- 8.Buchwald SL, Lum RT, Dewan JC. J Am Chem Soc. 1986;108:7441–7442. [Google Scholar]
- 9.Buchwald SL, Watson BT. J Am Chem Soc. 1987;109:2544–2546. [Google Scholar]
- 10.Buchwald SL, Nielsen RB. J Am Chem Soc. 1989;111:2870–2874. [Google Scholar]
- 11.Takahashi T, Kageyama M, Denisov V, Hara R, Negishi E. Tetrahedron Lett. 1993;34:687–690. [Google Scholar]
- 12.Xi Z, Hara R, Takahashi T. J Org Chem. 1995;60:4444–4448. [Google Scholar]
- 13.Haym I, Brimble MA. Org Biomol Chem. 2012;10:7649–7665. doi: 10.1039/c2ob26082d. [DOI] [PubMed] [Google Scholar]
- 14.Kulinkovich OG, Sviridov SV, Vasilevski DA, Pritytskaya TS. J Org Chem USSR (Eng Transl) 1989;25:2027–2028. [Google Scholar]
- 15.Hamada T, Suzuki D, Urabe H, Sato F. J Am Chem Soc. 1999;121:7342–7344. [Google Scholar]
- 16.Perez LJ, Shimp HL, Micalizio GC. J Org Chem. 2009;74:7211–7219. doi: 10.1021/jo901451c. [DOI] [PubMed] [Google Scholar]
- 17.Shimp HL, Micalizio GC. Org Lett. 2005;7:5111–5114. doi: 10.1021/ol052241n. [DOI] [PubMed] [Google Scholar]
- 18.Reichard HA, Rieger J, Micalizio GC. Angew Chem Int Ed. 2008;47:7837–7840. doi: 10.1002/anie.200803031. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi M, Higuchi K, Murakami N, Tajima H, Aoki S. Tetrahedron Lett. 1997;38:2859–2862. [Google Scholar]
- 20.Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE. Biochem Pharmacol. 2003;66:75–82. doi: 10.1016/s0006-2952(03)00192-8. [DOI] [PubMed] [Google Scholar]
- 21.Shimp HL, Micalizio GC. Tetrahedron. 2009;65:5908–5915. [Google Scholar]
- 22.Paterson I, Britton R, Delgado O, Meyer A, Poullennec KG. Angew Chem Int Ed. 2004;43:4629–4633. doi: 10.1002/anie.200460589. [DOI] [PubMed] [Google Scholar]
- 23.Mukhtar TA, Wright GD. Chem Rev. 2005;105:529–542. doi: 10.1021/cr030110z. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Panek JS. Angew Chem Int Ed. 2010;49:6165–6168. doi: 10.1002/anie.201002220. [DOI] [PubMed] [Google Scholar]
- 25.Wu J, Panek JS. J Org Chem. 2011;76:9900–9918. doi: 10.1021/jo202119p. [DOI] [PubMed] [Google Scholar]
- 26.Raju R, Piggott AM, Conte MM, Capon RJ. Org Biomol Chem. 2010;8:4682–4689. doi: 10.1039/c0ob00267d. [DOI] [PubMed] [Google Scholar]
- 27.Sakanishi K, Itoh S, Sugiyama R, Nishimura S, Kakeya H, Iwabuchi Y, Kanoh N. Eur J Org Chem. 2014:1376–1380. [Google Scholar]
- 28.Nakagawa T, Kasatkin A, Sato F. Tetrahedron Lett. 1995;36:3207–3210. [Google Scholar]
- 29.Jeso V, Iqbal S, Hernandez P, Cameron MD, Park H, LoGrasso PV, Micalizio GC. Angew Chem Int Ed. 2013;52:4800–4804. doi: 10.1002/anie.201301323. [DOI] [PubMed] [Google Scholar]
- 30.RajanBabu TV, Nugent WA, Taber DF, Fagan PJ. J Am Chem Soc. 1988;110:7128–7135. [Google Scholar]
- 31.Negishi E, Cederbaum FE, Takahashi T. Tetrahedron Lett. 1986;27:2829–2832. [Google Scholar]
- 32.Negishi E, Holmes SJ, Tour JM, Miller JA. J Am Chem Soc. 1985;107:2568–2569. [Google Scholar]
- 33.Negishi E, Swanson DR, Cederbaum FE, Takahashi T. Tetrahedron Lett. 1987;28:917–920. [Google Scholar]
- 34.Reichard HA, Micalizio GC. Chem Sci. 2011;2:573–589. doi: 10.1039/C0SC00394H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Micalizio GC. Early Transition Metal-Mediated Reductive Coupling Reactions. In: Molander GA, Knochel P, editors. Comprehensive Organic Synthesis II. 2nd. Vol. 5. Oxford: Elsevier; 2014. pp. 1660–1737. [Google Scholar]
- 36.Trost BM, Indolese A. J Am Chem Soc. 1993;115:4361–4362. [Google Scholar]
- 37.Trost BM, Indolese AF, Muller TJJ, Treptow B. J Am Chem Soc. 1995;117:615–623. [Google Scholar]
- 38.Suzuki N, Kondakov DY, Takahashi T. J Am Chem Soc. 1993;115:8485–8489. [Google Scholar]
- 39.Takahashi T, Suzuki N, Kageyama M, Kondakov DY, Hara R. Tetrahedron Lett. 1993;34:4811–4814. [Google Scholar]
- 40.Suzuki N, Kondakov DY, Kageyama M, Kotora M, Hara R, Takahashi T. Tetrahedron. 1995;51:4519–4540. [Google Scholar]
- 41.Kolundzic F, Micalizio GC. J Am Chem Soc. 2007;129:15112–15113. doi: 10.1021/ja075678u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lysenko IL, Kim K, Lee HG, Cha JK. J Am Chem Soc. 2008;130:15997–16002. doi: 10.1021/ja806440m. [DOI] [PubMed] [Google Scholar]
- 43.Trost BM, Müller TJJ. J Am Chem Soc. 1994;116:4985–4986. [Google Scholar]
- 44.Trost BM, Müller TJJ, Martinez J. J Am Chem Soc. 1995;117:1888–1899. [Google Scholar]
- 45.Trost BM, Shi ZQ. J Am Chem Soc. 1994;116:7459–7460. [Google Scholar]
- 46.Trost BM, Calkins TL, Bochet CG. Angew Chem Int Ed. 1997;36:2632–2635. [Google Scholar]
- 47.Trost BM, Quintard A. Org Lett. 2012;14:4698–4700. doi: 10.1021/ol302074h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zampella A, DAuria MV, Minale L, Debitus C, Roussakis C. J Am Chem Soc. 1996;118:11085–11088. [Google Scholar]
- 49.Trost BM, Gunzner JL. J Am Chem Soc. 2001;123:9449–9450. doi: 10.1021/ja011424b. [DOI] [PubMed] [Google Scholar]
- 50.Trost BM, Dirat O, Gunzner JL. Angew Chem Int Ed. 2002;41:841–842. doi: 10.1002/1521-3773(20020301)41:5<841::aid-anie841>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 51.Trost BM, Gunzner JL, Dirat O, Rhee YH. J Am Chem Soc. 2002;124:10396–10415. doi: 10.1021/ja0205232. [DOI] [PubMed] [Google Scholar]
- 52.Brian PW, Curtis PJ, Hemming HG, Unwin CH, Wright JM. Nature. 1949;164:534–534. [Google Scholar]
- 53.Brian PW, Curtis PJ, Hemming HG, Jefferys EG, Unwin CH, Wright JM. J Gen Microbiol. 1951;5:619–632. doi: 10.1099/00221287-5-4-619. [DOI] [PubMed] [Google Scholar]
- 54.Trost BM, Probst GD, Schoop A. J Am Chem Soc. 1998;120:9228–9236. [Google Scholar]
- 55.Burres NS, Clement JJ. Cancer Res. 1989;49:2935–2940. [PubMed] [Google Scholar]
- 56.Trost BM, Yang H, Probst GD. J Am Chem Soc. 2004;126:48–49. doi: 10.1021/ja038787r. [DOI] [PubMed] [Google Scholar]
- 57.Ishibashi M, Kobayashi J. Heterocycles. 1997;44:543–572. [Google Scholar]
- 58.Trost BM, Harrington EA. J Am Chem Soc. 2004;126:5028–5029. doi: 10.1021/ja049292k. [DOI] [PubMed] [Google Scholar]
- 59.Trost BM, Harrington EA, Chisholm JD, Wrobleski ST. J Am Chem Soc. 2005;127:13598–13610. doi: 10.1021/ja053365y. [DOI] [PubMed] [Google Scholar]
- 60.Trost BM, Wrobleski ST, Chisholm JD, Harrington EA, Jung M. J Am Chem Soc. 2005;127:13589–13597. doi: 10.1021/ja0533646. [DOI] [PubMed] [Google Scholar]
- 61.Trost BM, Papillon JPN. J Am Chem Soc. 2004;126:13618–13619. doi: 10.1021/ja045449x. [DOI] [PubMed] [Google Scholar]
- 62.Trost BM, Papillon JPN, Nussbaumer T. J Am Chem Soc. 2005;127:17921–17937. doi: 10.1021/ja055967n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cutignano A, Bruno I, Bifulco G, Casapullo A, Debitus C, Gomez-Paloma L, Riccio R. Eur J Org Chem. 2001:775–778. doi: 10.1021/np010053+. [DOI] [PubMed] [Google Scholar]
- 64.Yun SY, Hansen EC, Volchkov I, Cho EJ, Lo WY, Lee D. Angew Chem Int Ed. 2010;49:4261–4263. doi: 10.1002/anie.201001681. [DOI] [PubMed] [Google Scholar]
- 65.Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat Prod Rep. 2002;19:413–453. doi: 10.1039/b009211h. [DOI] [PubMed] [Google Scholar]
- 66.Trost BM, Dong GB. Nature. 2008;456:485–488. doi: 10.1038/nature07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trost BM, Dong GB. J Am Chem Soc. 2010;132:16403–16416. doi: 10.1021/ja105129p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sata N, Abinsay H, Yoshida WY, Horgen FD, Sitachitta N, Kelly M, Scheuer PJ. J Nat Prod. 2005;68:1400–1403. doi: 10.1021/np0500528. [DOI] [PubMed] [Google Scholar]
- 69.Jeso V, Micalizio GC. J Am Chem Soc. 2010;132:11422–11424. doi: 10.1021/ja104782u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jeso V, Yang C, Cameron MD, Cleveland JL, Micalizio GC. ACS Chem Biol. 2013;8:1241–1252. doi: 10.1021/cb300582s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diez PS, Micalizio GC. Angew Chem Int Ed. 2012;51:5152–5156. doi: 10.1002/anie.201200035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vuong D, Capon RJ. J Nat Prod. 2000;63:1684–1685. doi: 10.1021/np000239t. [DOI] [PubMed] [Google Scholar]
- 73.McNally M, Capon RJ. J Nat Prod. 2001;64:645–647. doi: 10.1021/np0005614. [DOI] [PubMed] [Google Scholar]
- 74.Zhang H, Capon RJ. Org Lett. 2008;10:1959–1962. doi: 10.1021/ol8004744. [DOI] [PubMed] [Google Scholar]
- 75.Zhang H, Major JM, Lewis RJ, Capon RJ. Org Biomol Chem. 2008;6:3811–3815. doi: 10.1039/b808866g. [DOI] [PubMed] [Google Scholar]
- 76.Lee HS, Park SY, Sim CJ, Rho JR. Chem Pharm Bull. 2008;56:1198–1200. doi: 10.1248/cpb.56.1198. [DOI] [PubMed] [Google Scholar]
- 77.Macklin TK, Micalizio GC. J Am Chem Soc. 2009;131:1392–1393. doi: 10.1021/ja809491b. [DOI] [PubMed] [Google Scholar]
- 78.Bahadoor AB, Flyer A, Micalizio GC. J Am Chem Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]
- 79.Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065–9066. [Google Scholar]
- 80.Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 81.Montgomery J. Angew Chem Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]
- 82.Sa-ei K, Montgomery J. Org Lett. 2006;8:4441–4443. doi: 10.1021/ol061579u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568–9569. doi: 10.1021/ja072992f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montgomery J, Sormunen GJ. Top Curr Chem. 2007;279:1–23. [Google Scholar]
- 85.Malik HA, Sormunen GJ, Montgomery J. J Am Chem Soc. 2010;132:6304–6305. doi: 10.1021/ja102262v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang W-S, Chan J, Jamison TF. Org Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]
- 87.Miller KM, Huang W-S, Jamison TF. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 88.Miller KM, Jamison TF. J Am Chem Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 89.Luanphaisarnnont T, Nbubaku CO, Jamison TF. Org Lett. 2005;7:2937–2940. doi: 10.1021/ol050881k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598–7610. [Google Scholar]
- 91.Huddleston RR, Jang H-Y, Krische MJ. J Am Chem Soc. 2003;125:11488–11489. doi: 10.1021/ja030415v. [DOI] [PubMed] [Google Scholar]
- 92.Jang H-Y, Huddleston RR, Krische MJ. J Am Chem Soc. 2004;126:4664–4668. doi: 10.1021/ja0316566. [DOI] [PubMed] [Google Scholar]
- 93.Jang H-Y, Krische MJ. Acc Chem Res. 2004;37:653–661. doi: 10.1021/ar020108e. [DOI] [PubMed] [Google Scholar]
- 94.Cho C-W, Krische MJ. Org Lett. 2006;8:891–894. doi: 10.1021/ol052976s. [DOI] [PubMed] [Google Scholar]
- 95.Cho C-W, Krische MJ. Org Lett. 2006;8:3873–3876. doi: 10.1021/ol061485k. [DOI] [PubMed] [Google Scholar]
- 96.Kong J-R, Ngai M-Y, Krische MJ. J Am Chem Soc. 2006;128:718–719. doi: 10.1021/ja056474l. [DOI] [PubMed] [Google Scholar]
- 97.Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. [Google Scholar]
- 98.Takayanagi Y, Yamashita K, Yoshida Y, Sato F. Chem Comm. 1996:1725–1728. [Google Scholar]
- 99.Yamashita K, Sato F. Tetrahedron Lett. 1996;37:7275–7278. [Google Scholar]
- 100.Yamashita K, Urabe H, Sato F. Tetrahedron Lett. 1997;38:4619–4622. [Google Scholar]
- 101.Jin YH, Qiu FYG. Org Biomol Chem. 2012;10:5452–5455. doi: 10.1039/c2ob25940k. [DOI] [PubMed] [Google Scholar]
- 102.Trost BM, Dong GB, Vance JA. Chem Eur J. 2010;16:6265–6277. doi: 10.1002/chem.200903356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berger GO, Tius MA. J Org Chem. 2007;72:6473–6480. doi: 10.1021/jo070923d. [DOI] [PubMed] [Google Scholar]
- 104.Trost BM, Dong GB, Vance JA. J Am Chem Soc. 2007;129:4540–4541. doi: 10.1021/ja070571s. [DOI] [PubMed] [Google Scholar]
- 105.Berger GO, Tius MA. Org Lett. 2005;7:5011–5013. doi: 10.1021/ol052015d. [DOI] [PubMed] [Google Scholar]
- 106.Tatsuta K, Masuda N. J Antibiot. 1998;51:602–606. doi: 10.7164/antibiotics.51.602. [DOI] [PubMed] [Google Scholar]
- 107.Jung HJ, Lee HB, Kim CJ, Rho JR, Shin JH, Kwon HJ. J Antibiot. 2003;56:492–496. doi: 10.7164/antibiotics.56.492. [DOI] [PubMed] [Google Scholar]
- 108.Chan J, Jamison TF. J Am Chem Soc. 2003;125:11514–11515. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]
- 109.Chan J, Jamison TF. J Am Chem Soc. 2004;126:10682–10691. doi: 10.1021/ja0470968. [DOI] [PubMed] [Google Scholar]
- 110.Belardi JK, Micalizio GC. Angew Chem Int Ed. 2008;47:4005–4008. doi: 10.1002/anie.200800400. [DOI] [PubMed] [Google Scholar]
- 111.Sa-ei K, Montgomery J. Tetrahedron. 2009;65:6707–6711. doi: 10.1016/j.tet.2009.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lu Y, Woo SK, Krische MJ. J Am Chem Soc. 2011;133:13876–13879. doi: 10.1021/ja205673e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Umezawa I, Funayama S, Okada K, Iwasaki K, Satoh J, Masuda K, Komiyama K. J Antibiot. 1985;38:699–705. doi: 10.7164/antibiotics.38.699. [DOI] [PubMed] [Google Scholar]
- 114.Komiyama K, Hirokawa Y, Yamaguchi H, Funayama S, Masuda K, Anraku Y, Umezawa I, Omura S. J Antibiot. 1987;40:1768–1772. doi: 10.7164/antibiotics.40.1768. [DOI] [PubMed] [Google Scholar]
- 115.Funayama S, Anraku Y, Mita A, Yang ZB, Shibata K, Komiyama K, Umezawa I, Omura S. J Antibiot. 1988;41:1223–1230. doi: 10.7164/antibiotics.41.1223. [DOI] [PubMed] [Google Scholar]
- 116.Del Valle DJ, Krische MJ. J Am Chem Soc. 2013;135:10986–10989. doi: 10.1021/ja4061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kong JR, Krische MJ. J Am Chem Soc. 2006;128:16040–16041. doi: 10.1021/ja0664786. [DOI] [PubMed] [Google Scholar]
- 118.Rössle M, Del Valle DJ, Krische MJ. Org Lett. 2011;13:1482–1485. doi: 10.1021/ol200160p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Molinaro C, Jamison TF. J Am Chem Soc. 2003;125:8076–8077. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 120.Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77–93. doi: 10.1039/b310427n. [DOI] [PubMed] [Google Scholar]
- 121.Kobayashi J. J Antibiot. 2008;61:271–284. doi: 10.1038/ja.2008.39. [DOI] [PubMed] [Google Scholar]
- 122.Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2004;126:998–999. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]
- 123.Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297–4307. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Takahashi M, McLaughlin M, Micalizio GC. Angew Chem Int Ed. 2009;48:3648–3652. doi: 10.1002/anie.200900236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Takahashi M, Micalizio GC. J Am Chem Soc. 2007;129:7514–7516. doi: 10.1021/ja071974v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen MZ, McLaughlin M, Takahashi M, Tarselli MA, Yang DX, Umemura S, Micalizio GC. J Org Chem. 2010;75:8048–8059. doi: 10.1021/jo101535d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lysenko IL, Lee HG, Cha JK. Org Lett. 2009;11:3132–3134. doi: 10.1021/ol901006c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Denmark SE, Almstead NG. Allylation of Carbonyls: Methodology and Stereochemistry. In: Otera J, editor. Modern Carbonyl Chemistry. Wiley-VCH; Weinheim: 2000. pp. 299–401. [Google Scholar]
- 129.Chemler SR, Roush WR. Recent Applications of the Allylation Reaction to the Synthesis of Natural Products. In: Otera J, editor. Modern Carbonyl Chemistry. Wiley-VCH; Weinheim: 2000. pp. 403–490. [Google Scholar]
- 130.Daly JW, Spande TF, Garraffo HM. J Nat Prod. 2005;68:1556–1575. doi: 10.1021/np0580560. [DOI] [PubMed] [Google Scholar]
- 131.Toyooka N, Fukutome A, Shinoda H, Nemoto H. Angew Chem Int Ed. 2003;42:3808–3810. doi: 10.1002/anie.200351906. [DOI] [PubMed] [Google Scholar]
- 132.Tsuneki H, You Y, Toyooka N, Kagawa S, Kobayashi S, Sasaoka T, Nemoto H, Kimura I, Dani JA. Mol Pharmacol. 2004;66:1061–1069. doi: 10.1124/mol.104.000729. [DOI] [PubMed] [Google Scholar]
- 133.Tarselli MA, Micalizio GC. Org Lett. 2009;11:4596–4599. doi: 10.1021/ol901870n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang D, Micalizio GC. J Am Chem Soc. 2012;134:15237–15240. doi: 10.1021/ja306362m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yang D, Micalizio GC. J Am Chem Soc. 2009;131:17548–17549. doi: 10.1021/ja908504z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gibson SE, Mainolfi N. Angew Chem Int Ed. 2005;44:3022–3037. doi: 10.1002/anie.200462235. [DOI] [PubMed] [Google Scholar]
- 137.Vázquez-Romero A, Cárdenas L, Blasi E, Verdaguer X, Riera A. Org Lett. 2009;11:3104–3107. doi: 10.1021/ol901213d. [DOI] [PubMed] [Google Scholar]
- 138.Vázquez-Romero A, Verdaguer X, Riera A. Eur J Org Chem. 2013:1716–1725. [Google Scholar]
- 139.Jiang B, Li M, Xing P, Huang Z. Org Lett. 2013;15:871–873. doi: 10.1021/ol400030a. [DOI] [PubMed] [Google Scholar]
- 140.Bernardes V, Kann M, Riera A, Moyano A, Pericás MA, Greene AE. J Org Chem. 1995;60:6670–6671. [Google Scholar]
- 141.Krafft ME, Cheung YY, Abboud KA. J Org Chem. 2001;66:7443–7448. doi: 10.1021/jo010623a. [DOI] [PubMed] [Google Scholar]
- 142.Krafft ME, Cheung YY, Juliano-Capucao CA. Synthesis. 2000:1020–1026. [Google Scholar]
- 143.Krafft ME, Romero RH, Scott IL. J Org Chem. 1992;57:5277–5278. [Google Scholar]
- 144.Krafft ME. Tetrahedron Lett. 1988;29:999–1002. [Google Scholar]
- 145.Iqbal M, Evans P, Lledo A, Verdaguer X, Pericas MA, Riera A, Loeffler C, Sinha AK, Mueller MJ. Chembiochem. 2005;6:276–280. doi: 10.1002/cbic.200400259. [DOI] [PubMed] [Google Scholar]
- 146.Su S, Rodriguez RA, Baran PS. J Am Chem Soc. 2011;133:13922–13925. doi: 10.1021/ja206191g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Dominguez G, Perez-Castells J. [2+2+2] Cycloadditions. In: Molander GA, Knochel P, editors. Comprehensive Organic Synthesis II. Oxford: Elsevier; 2014. pp. 1537–1581. [Google Scholar]
- 148.Dominguez G, Perez-Castells J. Chem Soc Rev. 2011;40:3430–3444. doi: 10.1039/c1cs15029d. [DOI] [PubMed] [Google Scholar]
- 149.Vollhardt KPC. Angew Chem. 1984;96:525–541. [Google Scholar]
- 150.Funk RL, Vollhardt KPC. J Am Chem Soc. 1977;99:5483–5484. doi: 10.1021/ja00458a044. [DOI] [PubMed] [Google Scholar]
- 151.Nicolaou KC, Sorensen EJ. Classics in Total Synthesis. VCH; Weinheim: 1995. p. 798. [Google Scholar]
- 152.Hillard RLI, Parnell CA, Vollhardt KPC. Tetrahedron. 1983;39:905–911. [Google Scholar]
- 153.Buchanan GO, Williams PG, Feling RH, Kauffman CA, Jensen PR, Fenical W. Org Lett. 2005;7:2731–2734. doi: 10.1021/ol050901i. [DOI] [PubMed] [Google Scholar]
- 154.Nicolaou KC, Tang YF, Wang JH. Angew Chem Int Ed. 2009;48:3449–3453. doi: 10.1002/anie.200900264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nicolaou KC, Wang JH, Tang YF, Botta L. J Am Chem Soc. 2010;132:11350–11363. doi: 10.1021/ja1048994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tanaka R, Nakano Y, Suzuki D, Urabe H, Sato F. J Am Chem Soc. 2002;124:9682–9683. doi: 10.1021/ja027008o. [DOI] [PubMed] [Google Scholar]
- 157.Witulski B, Alayrac C. Angew Chem Int Ed. 2002;41:3281–3284. doi: 10.1002/1521-3773(20020902)41:17<3281::AID-ANIE3281>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 158.Parnell CA, Vollhardt KPC. Tetrahedron. 1985;41:5791–5796. [Google Scholar]
- 159.McIver A, Young DD, Deiters A. Chem Commun. 2008:4750–4752. doi: 10.1039/b811068a. [DOI] [PubMed] [Google Scholar]
- 160.Saá C, Crotts DD, Hsu G, Vollhardt KPC. Synlett. 1994:487–489. [Google Scholar]
- 161.Doyle TW, Balitz DM, Grulich RE, Nettleton DE, Gould SJ, Tann CH, Moews AE. Tetrahedron Lett. 1981;22:4595–4598. [Google Scholar]
- 162.Fang YA, Linardic CM, Richardson DA, Cai W, Behforouz M, Abraham RT. Mol Cancer Ther. 2003;2:517–526. [PubMed] [Google Scholar]
- 163.Nissen F, Detert H. Eur J Org Chem. 2011:2845–2853. [Google Scholar]
- 164.Dassonneville B, Witulski B, Detert H. Eur J Org Chem. 2011:2836–2844. [Google Scholar]
- 165.Morita H, Ishiuchi K, Haganuma A, Hoshino T, Obara Y, Nakahata N, Kobayashi J. Tetrahedron. 2005;61:1955–1960. [Google Scholar]
- 166.Yuan C, Chang C-T, Axelrod A, Siegel D. J Am Chem Soc. 2010;132:5924–5925. doi: 10.1021/ja101956x. [DOI] [PubMed] [Google Scholar]
- 167.Yuan CX, Chang CT, Siegel D. J Org Chem. 2013;78:5647–5668. doi: 10.1021/jo400695c. [DOI] [PubMed] [Google Scholar]
- 168.Johnson T, Siegel D. Bioorg Med Chem Lett. 2014;24:3512–3515. doi: 10.1016/j.bmcl.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 169.Earl RA, Vollhardt KPC. J Am Chem Soc. 1983;105:6991–6993. [Google Scholar]
- 170.Yu RT, Rovis T. J Am Chem Soc. 2006;128:12370–12371. doi: 10.1021/ja064868m. [DOI] [PubMed] [Google Scholar]
- 171.Yu RT, Lee EE, Malik G, Rovis T. Angew Chem Int Ed. 2009;48:2379–2382. doi: 10.1002/anie.200805455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Grotjahn DB, Vollhardt KPC. Synthesis. 1993:579–605. [Google Scholar]
- 173.Woodward RB, Cava MP, Hunger A, Ollis WD, Daeniker HU, Schenker K. Tetrahedron. 1963;19:247–&. [Google Scholar]
- 174.Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. J Am Chem Soc. 1954;76:4749–4751. [Google Scholar]
- 175.Cannon JS, Overman LE. Angew Chem Int Ed. 2012;51:4288–4311. doi: 10.1002/anie.201107385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mori M. Heterocycles. 2010;81:259–292. [Google Scholar]
- 177.Beemelmanns C, Reissig HU. Angew Chem Int Ed. 2010;49:8021–8025. doi: 10.1002/anie.201003320. [DOI] [PubMed] [Google Scholar]
- 178.Kaburagi Y, Tokuyama H, Fukuyama T. J Am Chem Soc. 2004;126:10246–10247. doi: 10.1021/ja046407b. [DOI] [PubMed] [Google Scholar]
- 179.Ohshima T, Xu JY, Takita R, Shimizu S, Zhong DF, Shibasaki M. J Am Chem Soc. 2002;124:14546–14547. doi: 10.1021/ja028457r. [DOI] [PubMed] [Google Scholar]
- 180.Bodwell GJ, Li J. Angew Chem Int Ed. 2002;41:3261–3262. doi: 10.1002/1521-3773(20020902)41:17<3261::AID-ANIE3261>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 181.Bonjoch J, Sole D. Chem Rev. 2000;100:3455–3482. doi: 10.1021/cr9902547. [DOI] [PubMed] [Google Scholar]
- 182.Kuehne ME, Xu F. J Org Chem. 1998;63:9427–9433. [Google Scholar]
- 183.Knight SD, Overman LE, Pairaudeau G. J Am Chem Soc. 1995;117:5776–5788. [Google Scholar]
- 184.Magnus P, Giles M, Bonnert R, Johnson G, Mcquire L, Deluca M, Merritt A, Kim CS, Vicker N. J Am Chem Soc. 1993;115:8116–8129. [Google Scholar]
- 185.Magnus P, Giles M, Bonnert R, Kim CS, Mcquire L, Merritt A, Vicker N. J Am Chem Soc. 1992;114:4403–4405. [Google Scholar]
- 186.Eichberg MJ, Dorta RL, Lamottke K, Vollhardt KPC. Org Lett. 2000;2:2479–2481. doi: 10.1021/ol006131m. [DOI] [PubMed] [Google Scholar]
- 187.Eichberg MJ, Dorta RL, Grotjahn DB, Lamottke K, Schmidt M, Vollhardt KPC. J Am Chem Soc. 2001;123:9324–9337. doi: 10.1021/ja016333t. [DOI] [PubMed] [Google Scholar]
- 188.Greszler SN, Reichard HA, Micalizio GC. J Am Chem Soc. 2012;134:2766–2774. doi: 10.1021/ja2105043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Cheng XY, Micalizio GC. Org Lett. 2014;16:5144–5147. doi: 10.1021/ol502496d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kim WS, Aquino C, Mizoguchi H, Micalizio GC. Tetrahedron Lett. 2015;56:3557–3559. doi: 10.1016/j.tetlet.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Jeso V, Aquino C, Cheng XY, Mizoguchi H, Nakashige M, Micalizio GC. J Am Chem Soc. 2014;136:8209–8212. doi: 10.1021/ja504374j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Cheng XY, Micalizio GC. J Am Chem Soc. 2016;138:1150–1153. doi: 10.1021/jacs.5b12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chevliakov MV, Montgomery J. J Am Chem Soc. 1999;121:11139–11143. [Google Scholar]
- 194.Ni Y, Kassab RM, Chevliakov MV, Montgomery J. J Am Chem Soc. 2009;131:17714–17718. doi: 10.1021/ja907931u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ni Y, Amarasinghe KKD, Ksebati B, Montgomery J. Org Lett. 2003;5:3771–3773. doi: 10.1021/ol0355783. [DOI] [PubMed] [Google Scholar]
- 196.ElDouhaibi AS, Kassab RM, Song M, Montgomery J. Chem Eur J. 2011;17:6326–6329. doi: 10.1002/chem.201100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Amarasinghe KKD, Montgomery J. J Am Chem Soc. 2002;124:9366–9367. doi: 10.1021/ja027148y. [DOI] [PubMed] [Google Scholar]
- 198.Fornicola RS, Subburaj K, Montgomery J. Org Lett. 2002;4:615–617. doi: 10.1021/ol017213t. [DOI] [PubMed] [Google Scholar]
- 199.Sato F, Urabe H, Okamoto S. Chem Rev. 2000;100:2835–2886. doi: 10.1021/cr990277l. [DOI] [PubMed] [Google Scholar]
- 200.Sato F, Urabe H, Okamoto S. Synlett. 2000:753–775. [Google Scholar]
- 201.Okamoto S, Subburaj K, Sato F. J Am Chem Soc. 2000;122:11244–11245. [Google Scholar]
- 202.Wolan A, Six Y. Tetrahedron. 2010;66:3097–3133. [Google Scholar]
- 203.Wolan A, Six Y. Tetrahedron. 2010;66:15–61. [Google Scholar]
- 204.Gordon GJ, Luker T, Tuckett MW, Whitby RJ. Tetrahedron. 2000;56:2113–2129. [Google Scholar]
- 205.Dixon S, Fillery SM, Kasatkin A, Norton D, Thomas E, Whitby RJ. Tetrahedron. 2004;60:1401–1416. [Google Scholar]
- 206.Dixon S, Gordon GJ, Whitby RJ. Chem Commun. 2005:4303–4305. doi: 10.1039/b508524a. [DOI] [PubMed] [Google Scholar]
- 207.Baldwin IR, Whitby RJ. Chem Commun. 2003:2786–2787. doi: 10.1039/b309848f. [DOI] [PubMed] [Google Scholar]
- 208.Dzhemilev UM, Vostrikova OS, Sultanov RM. B Acad Sci USSR CH+ 1983;32:193–195. [Google Scholar]
- 209.Hoveyda AH, Xu Z. J Am Chem Soc. 1991;113:5079–5080. [Google Scholar]
- 210.Houri AF, Didiuk MT, Xu Z, Horan NR, Hoveyda AH. J Am Chem Soc. 1993;115:6614–6624. [Google Scholar]
- 211.Xu ZM, Johannes CW, Houri AF, La DS, Cogan DA, Hofilena GE, Hoveyda AH. J Am Chem Soc. 1997;119:10302–10316. [Google Scholar]
- 212.Houri AF, Xu ZM, Cogan DA, Hoveyda AH. J Am Chem Soc. 1995;117:2943–2944. [Google Scholar]
- 213.Keaton KA, Phillips AJ. Org Lett. 2007;9:2717–2719. doi: 10.1021/ol0710111. [DOI] [PubMed] [Google Scholar]
- 214.Li S-G, Wu Y. Chem Asian J. 2013;8:2792–2800. doi: 10.1002/asia.201300623. [DOI] [PubMed] [Google Scholar]