

Abstract
Every human is unique. We differ in our genomes, environment, behavior, disease history, and past and current medical treatment—a complex catalog of differences that often leads to variations in the way each of us responds to a particular therapy. We argue here that true personalization of drug therapies will rely on “virtual patient” models based on a detailed characterization of the individual patient by molecular, imaging, and sensor techniques. The models will be based, wherever possible, on the molecular mechanisms of disease processes and drug action but can also expand to hybrid models including statistics/machine learning/artificial intelligence-based elements trained on available data to address therapeutic areas or therapies for which insufficient information on mechanisms is available. Depending on the disease, its mechanisms, and the therapy, virtual patient models can be implemented at a fairly high level of abstraction, with molecular models representing cells, cell types, or organs relevant to the clinical question, interacting not only with each other but also the environment. In the future, “virtual patient/in-silico self” models may not only become a central element of our health care system, reducing otherwise unavoidable mistakes and unnecessary costs, but also act as “guardian angels” accompanying us through life to protect us against dangers and to help us to deal intelligently with our own health and wellness.
Keywords: cancer, drug response, mechanistic model, next-generation sequencing, patient model, virtual clinical trial
Abstract
Cada ser humano es único, Nos diferenciamos en nuestros genomas, ambiente, conductas, historia de enfermedades y, tratamiento médico actual y pasado; un complejo catálogo de diferencias que a menudo conducen a variaciones en la forma cómo cada uno responde a una terapia específica. En este artículo se argumenta que la verdadera personalización de las terapias farmacológicas se basará en modelos de “pacientes virtuales” de acuerdo a una detallada caracterización del paciente individual mediante técnicas moleculares, de imágenes y de sensores, Los modelos se basarán, siempre que sea posible, en mecanismos moleculares de los procesos patológicos y la acción de fármacos, pero también se pueden expandir a modelos híbridos que incluyan elementos basados en la inteligencia artificial, el aprendizaje automático (o de máquinas) y las estadísticas a partir de los datos disponibles para abordar áreas terapéuticas o terapias para las cuales no se dispone de suficiente información sobre esos mecanismos.
Dependiendo de la enfermedad, sus mecanismos y la terapia, los modelos de paciente virtual se pueden implementar en un nivel bastante alto de abstracción, con modelos moleculares que representen células, tipos celulares u órganos relevantes para la pregunta clínica, que puedan interactuar no solo entre sí, sino que también con el ambiente. A futuro, los modelos de “paciente virtual/realizado in-silico (en computadora)” pueden no solo llegar a ser un elemento central de nuestro sistema de atención de salud, reduciendo de alguna manera los errores inevitables y los costos innecesarios, sino también actuar como “ángeles guardianes” que nos acompañen a través de la vida para protegernos contra los peligros y ayudar a manejarnos inteligentemente con nuestra propia salud y bienestar.
Abstract
Chaque être humain est unique. Nous différons dans nos génomes, notre environnement, notre comportement, nos antécédents médicaux et dans notre traitement médical, passé et actuel et ce catalogue complexe de différences mène souvent à des variations sur la façon de chacun de répondre à un traitement particulier. Dans cet article, nous expliquons que pour réellement personnaliser les traitements médicamenteu. Il faudra s'appuyer sur des modèles de « patients virtuels », basés sur une caractérisation détaillée de chaque patient par des techniques moléculaires, d'imagerie et de capteurs. À chaque fois que cela sera possible, les modèles se fonderont sur les mécanismes moléculaires des processus pathologiques et de l'action du médicament mais ils pourront aussi s'étendre à des modèles hybrides y compris des éléments basés sur l'intelligence artificielle, les appareils d'apprentissage et les statistiques. Ces éléments sont formés sur des données disponibles pour traiter des domaines thérapeutiques ou des traitements pour lesquels les informations sur les mécanismes sont lacunaires. Selon la maladie, ses mécanismes et le traitement, les modèles de patient virtuel peuvent être concrétisés avec un niveau assez élevé d'abstraction, les modèles moléculaires représentant les cellules, les types cellulaires, ou les organes se rapportant au problème clinique, interagissant non seulement entre eux mais aussi avec l'environnement. À l'avenir, les modèles « patient virtuel/in silico » seront non seulement au centre de notre système de santé en diminuant les erreurs inévitables en situation réelle et les coûts superflus, mais aussi des « anges gardiens »nous accompagnant au cours de notre vie pour nous protéger des dangers et nous aider à gérer intelligemment notre santé et notre bien-être.
Introduction
Personalized medicine aims to treat every patient optimally on the basis of each individual's disease and biological characteristics.1-4 Truly personalized therapy is standard in many areas of medicine, eg, surgery, where the straightforward interpretation of a wealth of data for the individual patient guides therapy choice. In contrast, drug therapies act on the complex molecular networks inside cells, as well as on the interactions between cells and organs. Historically, doctors have lacked both information on the detailed biological network in a patient and the capability to correctly predict the response of this complicated system to the complex action of a drug, essentially ruling out true personalization of drug therapy with current techniques.
To improve the chance that a specific therapy will help a patient, we have used stratification approaches based on statistical correlations between specific biomarkers5 or signatures and a positive or negative outcome of therapy, albeit with serious limitations. Despite intensive research, few biomarkers have shown real clinical benefit,6,7 often with relatively modest sensitivity/specificity values. In this regard, we have to distinguish between “causal” and “network” biomarkers. Causal biomarkers directly recognize a unique molecular state that is directly addressed by a selective drug; cancer-specific examples include the presence of a fusion protein against which a specific drug is available, in the case of crizotinib (Xalkori),8,9 and the use of ERBB2 (formerly HER2 or HER2/neu) amplification status as a biomarker fortrastuzumab, a drug targeting human epidermal growth factor receptor 2 (IIER2).10 Such directly targetable mechanisms are not very common, leaving many patients without this option. Network biomarkers are not directly linked to a drug target, and uncertainties are often much greater, further reducing the correlation between the biomarker result and drug response. Even when we have a biomarker, serious problems exist, including uncertainty in the prediction due to the enormous complexity and heterogeneity of diseases—eg, the usually unknown heterogeneity of the tumor and the rapid recurrence of tumors in resistant form even after (temporary) “miracle” cures.11-13 True personalization of drug therapy will therefore require much more than current attempts at stratification.
In similarly complex situations with dangerous and/ or expensive consequences, we characterize the situation in sufficient detail to make predictions possible and essentially rebuild the situation as exactly as necessary in mechanistic models of reality in the computer, allowing us to make unavoidable mistakes safely, quickly, and inexpensively on the computer rather than in reality. Such mechanistic models require three basic components to make accurate predictions of the future development of complex systems (with or without disturbances): (i) information about the detailed rules governing the processes to be modeled; (ii) the necessary computing power; and (iii) a detailed characterization of the situation at the start of the modeling.
From decades of research, there is a growing knowledge base on the processes relevant for a number of diseases (some more than others), with cancer being a particularly good example.14,15 Computing power has continued to increase by a factor of roughly a thousand every 10 years, providing rapidly increasing computing resources.16 However, every model is only as good as the data available to initialize it1—data that up to a short time ago simply have not been available on individual patients and their diseases. However, this has changed, primarily due to developments in DNA-sequencing technologies. Whereas the first human genome sequence took more than 10 years and billions of dollars to complete, we can now determine multiple genomes per machine run at a cost close to $1000 per genome.18 DNA sequencing can be used to decipher the patient genome, transcriptome, epigenome, and metagenome and thus directly determine much of the information we need to characterize the relevant biological networks in any disease.19-23 Sequencing techniques can also be applied to analyze other types of biological information (eg, levels of proteins, protein modifications, protein complexes, status of the immune system) by appropriate experimental strategies.24-27
This type of information about a disease in an individual patient allows us, for the first time, to develop personalized computer models of many different diseases. For example, in oncology, we generate computer models of the individual tumor and patient on the basis of a comprehensive omics analysis of both, allowing us to predict effects and side effects of (mechanistic) drugs. However, the approach is not restricted to cancer as long as the same three requirements above are fulfilled, albeit with different challenges; eg, information on molecular mechanisms of disease and drug therapy are, in general, available in cancer but often missing in neuropsychiatry diseases. Moreover, a sufficiently detailed molecular characterization of the disease in an individual patient is usually possible in cancer (eg, via biopsies) but may be more difficult in brain diseases due to sampling issues. Nevertheless, there is very encouraging progress in these disease areas, including from a number of large research projects that are characterizing basic mechanisms (eg, the Human Brain Project), as well as the development of innovative solutions for sampling issues.28-30 Thus, the virtual patient model outlined here, exemplified using cancer because of the advanced knowledge in the field on potential disease mechanisms, holds promise as a model for neuropsychiatry and other complex diseases, as well as for prevention and wellness applications.
The way forward: what information do we need?
In many diseases, there is an enormous complexity of processes we are trying to affect by drugs, which in turn have complex mechanisms of action.31 Oncology is an obvious first model for this approach as, unlike for other diseases, a large amount of information on the biological networks and molecular processes acting in tumors (and other tissues) is available, helping us to understand the disturbed processes in tumors. Also, we often have access to the diseased tissue—or, in the form of a liquid biopsy, to tumor-derived material32-35—and can therefore analyze its biology by high-throughput (omics) techniques. Such analyses often identify somatic changes that have dramatic effects, which either lead to already well-characterized (and published) consequences or induce functional changes, which can often be deduced by predicting changes in protein structure and the expected functional consequences.24,25,36,37
We have made huge leaps forward in the diagnosis and treatment of cancer, but with the significant molecular heterogeneity apparent between individual patients, their tumors, and even between cells within a tumor, the enormity of the task of “curing cancer” is becoming apparent. The possibility to generate individual models of cancer patients has, in particular, been driven by the development of next-generation sequencing (NGS) techniques. NGS can be used to analyze the genome/exome of both tumor and patient, providing information on sequence variants arising by a range of mechanisms, eg, mutation, deletions/insertions, or rearrangements. Sequencing or chip-based analyses are typically used to characterize genome methylation patterns, whereas RNA sequencing is the best method to analyze the abundance and processing of the different types of RNA molecules (coding, noncoding, microRNA).23,36,37 Similarly, a number of approaches can be used to analyze proteins and protein modification states (eg, mass-spectrometry techniques, reverse-phase protein arrays, proximity extension/ligation assays), as well as metabolite levels, providing key information on the results of posttranscriptional regulation in the biological networks.25,26,38-42
Spatially resolved and single-cell analysis techniques
Nevertheless, even in tumors, bulk omics analyses only provide part of the information we would need. Tumors can differ from the normal cell by hundreds to thousands of potentially relevant changes, in combinations that are unique to the tumor and often even to tumor cells. Combined with differences in the underlying germ-line genome, as well as in the epigenome and transcriptome of the different somatic cell types from which the tumor has originated, we have an irreducible complexity that can only be ignored if we accept correspondingly lower success rates in selecting an optimal therapy for the individual patient. Moreover, many tumors are heterogeneous, with differences affecting the predicted therapy response, rendering smaller or larger fractions of tumor cells inherently resistant to a therapy.11,12 If only a fraction of the tumor cells stop growing, resistant cell populations will continue to multiply, counteracting the effect of the treatment.13
In an ideal situation, all analyses mentioned above would be carried out either in a spatially resolved fashion or on the single-cell level by disaggregating the tumor. Protocols for sequencing and imaging-based spatially resolved transcriptome sequencing have been developed.43-46; moreover, genome and transcriptome analyses and protein detection by proximity extension assays have been successfully carried out at the single-cell level.46-50
Immune therapies, immune status
With recognition of the key role the immune system plays in many disease processes, the field of immunotherapeutics has recently emerged.51 We are still in the discovery phase in identifying potential biomarkers predictive of response to these drugs beyond obvious candidates, such as the number and expression levels of mutations altering peptide sequences potentially displayed by human leukocyte antigen (HLA),as well as the possible effect of programmed death-ligand 1 (PDL1) expression on the response to anti-PDLl treatment.52,53 Characterization of an individual's immune system would also be important in attempts to truly personalize therapies in many diseases, including cancer53 and neuropsychiatric disorders such as depression and schizophrenia; neuropsychiatry disorders are a particular focus given the increasing interest in the role of the immune system in these disorders.
As a step in this direction, we have developed a new approach to identify the combination of antibody (heavy and light chain) or T-cell-receptor α and β chains in a blood sample.27 This method is based on the amplification of RNA from microsomes in an emulsion polymerase chain reaction system, under conditions in which the primary amplification products (heavy and light chains for B cells, α and β chains for T cells) join during the amplification step, providing a single, sequenceable DNA molecule containing the relevant information from both chains. Deep sequencing (sequencing of the transcriptome at sufficient depth to provide information on both the sequence and frequency of the messenger RNAs [mRNAs] present) of this material will not only provide information on the sequence of the mRNAs coding for relevant antibodies/T-cell receptors but also on the combination of chains present in the analyzed cells.
Haplotype sequencing
We have access to molecular data on tens of thousands of human genomes (healthy and diseased), but most of these are generated as “mixed diploid” sequences. Because of the diploid nature of the human genome, if we really want to understand how genetic variation is linked to gene and genome function, phenotype, and disease, then determining each of the haplotype sequences of a genome is essential. Anticipating the importance of analyzing “phase-sensitive” variation (ie, reflecting the inherently diploid biology of genes and genomes) for personal genomics and individualized medicine, investigators have carried out a recent analysis of multiple haplotype-resolved genomes, revealing the extent of sequence variation that exists between the two alleles of human individuals, with many genes harboring sequence variants that are apparent at the amino acid level, increasing the repertoire of proteins that are expressed.54
Model-driven personalized therapy in other disease areas
After cancer, it is expected that many other diseases (and prevention of diseases) will be addressed. For neuropsychiatry disorders and neurodegenerative diseases, challenges to establishing individual patient models remain. One major bottleneck is a lack of information on disease mechanisms, almost completely missing for disorders like schizophrenia, and partially missing for many neurodegenerative diseases, eg, Huntington and Alzheimer disease. Even for Huntington disease (the first neurodegenerative disease for which the primary cause was identified, approximately 20 years ago55), we still do not understand the translation of the primary cause into the disease phenotype. A much deeper understanding of the molecular basis of such diseases is required before we can apply the same technologies. To this end, large-scale initiatives—eg, the Human Brain Project, the Blue Brain Project, The Human Connectome Project, and the Virtual Brain—have been initiated, and efforts to generate omics data, an integral part of disease characterization, are gaining traction, albeit not yet on the scale we see for cancer. Until more mechanistic information is available, we should be able to use hybrid models combining a mechanistic core with statistics/machine learning components to compensate for the current lack of mechanistic knowledge.
A more serious problem could be the difficulty of accessing diseased tissues, eg, the brain, to generate molecular data. If data from other, more easily accessible tissues such as blood are not sufficient, alternative strategies are required. Owing to rapid progress in single-cell sequencing, molecular data from hard-to-sample areas could be generated from the few cells clinging to tools used to introduce electrodes into affected areas, eg, in brain-stimulation therapies.28 In parallel, surrogate tissues could be developed from individual patients by reprogramming induced pluripotent stem (iPS) cells; eg, skin cells could be developed from neuronal cells for further molecular analysis.29,30 Such data would be complemented by imaging and increasingly powerful sensor data, the latter providing large amounts of information, possibly on a continuous basis.
Despite these hurdles, the knowledge base is substantially increasing, thus expanding the possible uses of modeling to develop a mechanistic understanding—the first crucial step in the personalization of therapy in these diseases.
Data, data, data
Although in oncology much of the mechanistic information we consider in virtual patient models will be molecular (simply because drugs act as molecular entities), there is also other mechanistic information (eg, on blood flow and blood pressure) of high relevance to modeling patients in other disease areas (eg, heart disease). Comprehensive clinical data—including information on factors that may influence disease trajectory and response to drugs, such as lifestyle factors, comorbidities, and the microbiome—all act to complement the existing molecular-level data to construct a more comprehensive picture of an individual's disease. Imaging data, an ever more relevant data source in neuroscience, will become increasingly incorporated into virtual patient models. Neuroimaging and sensor techniques are providing a route, not only toward measuring and monitoring important functions within the body and brain, including behavioral responses, but also for assessing functional changes resulting from disease processes, injury, environmental stimuli, or response to treatment, for example.56,57 In oncology, imaging technologies are generating data that can be easily associated with molecular data.58; for instance, metabolic activity data from specific tumors generated using positron emission tomography scans can be straightforwardly linked to molecular data generated both on bulk tumor and single cells, eg, spatially resolved transcript-tome analyses from biopsies from the same tumor (see above). The merging of data from different sources will help to improve the mechanistic basis of the computational model and to optimize the predictions made.
The “virtual patient/in-silico self” model
The best (and probably only) way to integrate the very large data sets, necessary to be generated for every single patient as a prerequisite for truly personalized therapy and prevention, will involve the generation and use of sufficiently detailed computational models of the disease process in the individual patient. These computational models can then be “virtually” treated with all drugs or drug combinations. For this, we have developed PyBiosS (http://pybios.molgen.mpg.de),59,60 a modeling engine written in Python, an object-oriented modeling language, with objects corresponding to the components of the biological networks we are trying to model. Using PyBios3, we have developed a large and comprehensive model of cell signaling transduction and associated processes—ModCellTM.60-63
This system simplifies the construction of the very large models we need to represent the complex processes in the patient by allowing the assembly of larger models from submodels representing individual pathways or sections of pathways. Models initially represent the biological networks of normal cells and have two major types of components: (i) the basic structure of the model, eg, protein species and their biochemical complexes, phosphorylation, activation/inhibition, transcriptional induction; and (ii) the appropriate kinetic laws and (if available) parameter values derived from, eg, pathway databases and/or the scientific literature.
Model individualization
Models are individualized using omics (and other) data generated through a comprehensive, highly qualitative, and deep characterization of a patient's samples (for cancer patients, in particular the tumor). For the characterization of cell signal transduction pathways, measurements on multiple layers is helpful: whole-exome sequencing provides insight into loss- or gain-of-function mutations; the proteome offers a comprehensive picture of protein expression, whereas the phosphorproteome indicates the activity state of the signaling pathways; and transcriptome data helps to estimate synthesis and decay rates of mRNAs and proteins and will enable evaluation of turnover rates for different molecular species. Moreover, transcriptional signatures can be linked to modules of genes that are located downstream of a specific signaling pathway or indicate crosstalk effects between different upstream pathways.
However, the full range of data types mentioned above are not always available for every patient. For cancer patients, we typically use genomic data (eg, exome or whole-genome sequence data) from both tumor and germ-line samples, complemented by transcriptome data (ideally from RNA sequencing [RNA-Seq]experiments), allowing the expression of any identified mutations and other alterations to be cross-checked. The integration of other data types from the same patient and samples (eg, epigenome, proteome, and metabolome) further defines the individual disease at the molecular level and provides further data to initialize the models.
The molecular alterations identified within the tumor (by comparison, eg, with the patient's genome) are included in the model, resulting in objects with altered/ inactivated function; eg, if an activating ras mutation is identified in the tumor genome and the mutation is expressed in the RNA sequencing data set (and we have no protein data indicating otherwise), we assume that a mutant ras protein is present in the tumor, represented in the model by a corresponding object with appropriate changes in its function. Drugs are also objects that are added to the model on the basis of their known dissociation constants and target protein(s), resulting in, eg, inactive target-drug complexes.
To identify the optimal treatment for a specific patient, a variety of computational indicators are used; for instance, in cancer patients, the computed levels of MYC or phosphorylation status of tumor protein 53 (p53), cleavage of poly(ADP-ribose) polymerase 1 (PARP1), and GTP-loading status of Ras-related C3 botulinum toxin substrate 1 (RAC1)and cell division control protein 42 homolog (CDC42)serve as proxies for corresponding phenotypic effects, such as cell proliferation, senescence, migration, and apoptosis induction.
Beyond mechanistic information: hybrid models of the virtual patient
Although oncology provides a wealth of mechanistic information to incorporate into models, this is not sufficient for many applications. Although we can mechanistically predict the concentrations of protein and protein modification states involved in apoptosis, we do not have accurate mechanistic models describing the phenotypic response of cells to a specific molecular configuration. This is even worse in many other disease areas. For many neuropsychiatric diseases, we are still far from having molecular models describing the disease process and drug action. Many nondrug-based treatments, eg, electroconvulsive therapy and psychotherapy, do not act through well-defined molecular mechanisms; the drug therapies used in parallel act through known molecular mechanisms. To incorporate such nonmechanistic components into our mechanistic models, we can define “pseudo-objects” as part of the object-oriented modeling system, encapsulating, eg, neuronal or Bayesian networks trained to translate nonmechanistic components into mechanistic consequences. As our knowledge increases, these can be replaced by more robust mechanistic model components.
Model optimization and validation
To be able to represent relevant changes observed in the individual tumors and the different targets for many drugs, models have to be quite detailed. This information may not be directly perceivable or may be hard to obtain experimentally (eg, drug-binding affinities, dephosphorylation, and degradation).
As models grow in complexity, more parameters have to be specified; eg, the large-scale network represented by ModCellTM, covering over 45 signaling pathways and hundreds of genes and their interactions, leads to tens of thousands of parameters.60-63 For this, we are increasingly relying on reverse engineering/parameter optimization strategies, minimizing the difference between prediction and experimental results, eg, drug testing on experimental models (patient-derived organoids, xenografts, and iPS cells3,29,30), and as far as possible, patient treatment results. Results from such experimental models have a crucial role to play in refining the accuracy of and validating model predictions, generating invaluable data on disease mechanisms and response to drugs. This process is still at an early stage, but as time goes on will deliver increasingly accurate predictions in a self-learning model.
To optimize the choice of parameter, statistical techniques are deployed, eg, memetic algorithms based on local search chains (MA-SW-Chain),64 to minimize the difference between predicted and experimental data, as well as derived predictions, improving the choice of parameter space and thus the accuracy of the model. To minimize the effect of overtraining, observed in many situations in which we have many free parameters but comparatively few observations, the available data are divided into training sets, used to identify optimized parameter vectors, and test sets, providing an unbiased estimate of the performance of these vectors on an independent data set. In tandem, uncertainty and sensitivity analyses can be conducted to assess how reliable model predictions are. Uncertainty analysis exploits statistical techniques, such as Bayesian and frequentist estimation methods, to assess the effects of lack of knowledge or potential errors in the model.65 Sensitivity analysis allows the identification of key data and assumptions that have most influence on model outputs.66
Evaluation of this strategy has revealed the potential of the mechanistic modeling approach, based on comprehensive molecular characterization of patient and tumor, to identify patient-specific responses to microRNA-based treatments for colon cancer61 and is generating promising results within clinical studies (eg, TREAT20[Tumor REsearch And Treatment - 20 patient pilot] and its follow-up project, TREAT20plus].
Treating the virtual tumor with a drug or drug combination
Within the model, a drug is represented as an object that binds to one or more (usually many) target objects with specific dissociation constants, forming a drugtarget object with specific changes. This change can be simple, eg, a protein kinase inhibitor binding to specific protein kinases forming inactive complexes, but can also be more complex, as in the case of a drug blocking activation of its target protein. Drug combinations, probably to become increasingly important in clinical practice,67 will result in the formation of the appropriate complexes for both drugs. Specific schedules of administering drugs in a combination therapy can similarly be represented in the modeling of the tumor response.
Role of the liver: pharmacogenomics and the tumor model
Individual patients react differently to drugs, due to differences in the metabolism or activation of the drug, determined by their cytochrome C genes68 and other variants in their genome. To represent this, we can introduce a “liver” model into the overall system, which will metabolize the drug appropriately, mimicking the multifactorial processes determining drug bioavailability and degradation. Drug and drug metabolite concentrations generated over time then interact with the “tumor” model.
Drugs also affect normal tissues, not just tumors
To estimate possible (side) effects of drugs or drug combinations on normal tissues of the body, we can model the different normal cell types of the patient by combining information on relevant variants, deletions, and copy number variants, etc, identified in the patient genome with information on tissue- or cell-specific gene expression patterns (eg, from the Illumina Body map). As with the tumor, these tissue/cell models can then be (virtually) exposed to the drug and drug metabolites in the virtual circulation to identify potentially unacceptable side effects of specific therapies.
Interactions of tumor cells with each other, soma cells, and the immune system
As knowledge accumulates on the molecular mechanisms underlying interactions between tumor cells, their surrounding soma cells, and infiltrating cells of the immune system, “virtual” tumors will increase in complexity, represented by individual cellular models interacting through the exchange of signals, etc. Also, different regions of tumors are exposed to different oxygen and nutrient levels, depending on proximity to blood vessels, and will therefore differ in their biology. As we learn more about interactions between the immune system and tumors, this knowledge can be represented in future models, based on the growing information base and the increasing use of immunomodulation in oncology.51,52
Synthesis A: a truly personalized medicine
We are confident that a truly personalized drug therapy in oncology (outlined in (Figure 1). based on a deep omics analysis of tumor and patient will rapidly become the standard for cancer patients and will serve as a model for applying similar techniques in many other therapeutic areas. This will not only allow the identification of unexpected drugs (not normally used for cancer treatment), but also the identification of patient-specific combination therapies. New, model-based tools can allow the doctor treating the patient to visualize and interactively explore the effects of drugs/drug combinations at the molecular level within individual patients (Figure 2). The modeling approach provides information on the underlying biological pathways relevant for many diseases, with potential benefits for identifying specific disease endophenotypes and associated biomarkers, as well as comorbidities that have a genetic rather than an environmental/lifestyle basis. An integrated approach is required, comprising information on multiple levels, from clinical data and lifestyle factors to imaging techniques that provide a more global view of interactions and help to refine the model's underlying knowledge base.
Figure 1. Path to prediction. Tumor biopsies and control (usually blood) samples are taken from individual patients for deep molecular analysis. Complex omics data are generated (eg, genome/exome, transcriptome, possibly proteome) and analyzed comprehensively for tumor-specific alterations, eg, mutations and gene fusions, loss of heterozygosity, ploidy, and 3D protein modeling of protein mutations, etc. Data generated are mined for clinically relevant features, such as diagnostic or prognostic markers. The patient-specific data are mapped to cancer-relevant pathways within a large-scale mechanistic computational model of cell signaling transduction and associated processes (eg, ModCellTM60-63). Individualized models are used to predict in silico the response of individual tumors to drug(s), singly or in combination, to identify the optimal therapeutic strategy for a specific patient. LOH, loss of heterozygosity; NGS, next-generation sequencing.
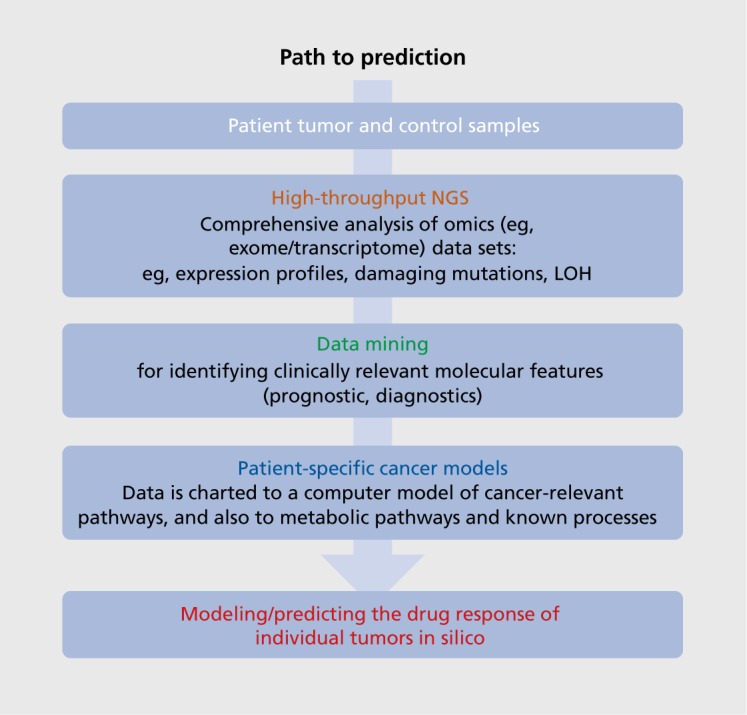
Figure 2. (Opposite and overleaf) Visualizing drug action. Illustration of the effect of afatinib on receptor tyrosine signaling (eg, EGFR) and major downstream pathways, such as the MAP kinase cascade and PI3K/AKT signaling. (A) Diagram of the model network, illustrating its complexity. Colors of nodes represent fold change in concentration of model species (see scale) due to inhibition by afatinib as predicted by the in silico model. (B) Afatinib's major target is the EGF/EGFR signaling pathway. Visualization of one branch of this pathway shows the strong inhibitory effect of afatinib on downstream network components (shown in green). ABL1, Abelson murine leukemia viral oncogene homolog 1; ADAM10, A Disintegrin and metalloproteinase domain-containing protein 10; AKT, protein kinase B; EGFR, epidermal growth factor receptor; ERBB2, formerly HER2 or HER2/neu; MAP, mitogen-activated protein; PI3K, phosphoinositide 3-kinase; PLCG1, phospholipase C γ 1 SHC1, SHC (Src homology 2 domain containing) transforming protein 1; STAT, signal transducer and activator of transcription.

However, a molecular model of even a single cell can be daunting in its complexity. It is therefore hard to imagine that we could, in the foreseeable future, represent the patient completely with an estimated 1014 interacting cells. We will have to compromise and model the patient as interacting molecular models representing the essential components that probably affect treatment success. In cancer, these would represent the different cell populations of the tumor (representing tumor heterogeneity, stroma, invading immune cells, blood vessels, etc); the liver as one single idealized cell carrying out drug metabolism and activation, depending on the cytochrome C alleles and other variants in the patient genome; and the most important cell types of the body (eg, neuronal cells, heart cells), to identify possible side effects of drugs in normal tissues. Ideally, the patient's immune system would also be represented, essential for autoimmune or infectious diseases, but also of probable relevance for predicting response of tumors to immunotherapeutics. As our knowledge and technical capabilities increase, such virtual patient models will increase in complexity. In the future, it could become routine practice to generate “preclinical” body maps from every patient (from iPS cells) for molecular analysis. Data of this type could become enormously important for further improvement of the individual patient models, but it would also provide access to “surrogate” brain samples for molecular analysis.
The virtual patient/in-silico self models could also find applications in addition to personalized therapy and prevention. If sufficiently predictive, they could become an important component of our health care system and also our lifestyle, ultimately developing into a “guardian angel,” accompanying every individual from before birth into old age, updated both from medical and molecular data generated at intervals at, eg, medical visits, but also more or less continuously through increasingly powerful sensor systems. Such in-silico self models have the potential to not only help doctors identify the optimal therapy and develop truly personalized prevention, but also to allow us to interact more intelligently with our bodies and possibly even our minds (see www.healthcarecompacteurope.eu and www.futurehealtheurope.eu for further information regarding potential impact on health care systems).
Synthesis B: virtual clinical trials, virtualization of drug development
The same technology can also be used to virtualize large parts of the drug development process, allowing virtual clinical trials as soon as molecular data on the binding specificity of a drug candidate are available (through the use of docking programs, possibly even on candidates before synthesis). These virtual clinical trials can increase the effectiveness of real clinical trials and improve the likelihood that drugs that have already failed during late stage clinical development due to lack of efficacy in a nonstratified clinical trial are approved rapidly and (relatively) cheaply in patients selected as responders to the drug.69 Again, virtualization based on detailed mechanistic models is likely to happen first in cancer and other diseases where we have detailed knowledge of molecular and cellular disease mechanisms; nevertheless, a similar approach to diseases other than cancer, such as neuropsychiatry disorders, will hopefully follow relatively soon given the enormous impact these diseases have on individuals, their families, and on society as a whole.
Acknowledgments
I want to thank a large number of people and institutions that support this effort and have helped, through discussions, to form the paper. In particular, I would like to thank my colleagues at Alacris Theranostics GmbH and the Max Planck Institute for Molecular Genetics, especially Bodo Lange and Marie-Laure Yaspo, as well as Nora Benhabiles at the French Alternative Energies and Atomic Energy Commission (CEA) and Georges Dagher at the French National Institute of Health and Medical Research (INSERM). I would also like to thank Lesley Ogilvie for her extensive and critical help in completing the manuscript, and Margret Hoehe for many helpful comments and suggestions. Hans Lehrach receives funding from TREAT20plus (031 A 512C) through the German Federal Ministry for Research (BMBF).
Conflict of interest/disclosure statement: Hans Lehrach is founder and scientific advisor of Alacris Theranostics GmbH, a company that aims to develop “virtual patient” models for use in therapy choice and drug development.
REFERENCES
- 1.Brand A. Public health genomics - public health goes personalized? Eur J Public Health. 2011;21(1):2–3. doi: 10.1093/eurpub/ckq197. [DOI] [PubMed] [Google Scholar]
- 2.Hood L., Friend SH. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat Rev Clin Oncol. 2011;8(3):184–187. doi: 10.1038/nrclinonc.2010.227. [DOI] [PubMed] [Google Scholar]
- 3.Henderson D., Ogilvie LA., Hoyle N., Keilholz U., Lange B., Lehrach H. Personalized medicine approaches for colon cancer driven by genomics and systems biology: OncoTrack. Biotechnol J. 2014;9(9):1104–1114. doi: 10.1002/biot.201400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pangalos MN., Schechter LE., Hurko O. Drug development for DNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov. 2007;6(7):521–532. doi: 10.1038/nrd2094. [DOI] [PubMed] [Google Scholar]
- 5.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69(3):89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 6.Taube SE., Clark GM., Dancey JE., McShane LM., Sigman CC., Gutrnan SI. A perspective on challenges and issues in biomarker development and drug and biomarker codevelopment. J Natl Cancer Inst. 2009;101(21):1453–463. doi: 10.1093/jnci/djp334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poste G. Bring on the biomarkers. Nature. 2011;469(7329):156–157. doi: 10.1038/469156a. [DOI] [PubMed] [Google Scholar]
- 8.Forde PM., Rudin CM. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. 2012;13(8):1195–1201. doi: 10.1517/14656566.2012.688029. [DOI] [PubMed] [Google Scholar]
- 9.Roberts PJ. Clinical use of crizotinib for the treatment of non-small cell lung cancer. Biologics. 2013;7:91–101. doi: 10.2147/BTT.S29026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hicks DG., Kulkarni S. Trastuzumab as adjuvant therapy for early breast cancer: the importance of accurate human epidermal growth factor receptor 2 testing. Arch Pathol Lab Med. 2008;132(6):1008–1015. doi: 10.5858/2008-132-1008-TAATFE. [DOI] [PubMed] [Google Scholar]
- 11.Lengauer C., Kinzler KW., Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396(6712):643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 12.Meacham CE., Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burrell RA., Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014;8(6):1095–1111. doi: 10.1016/j.molonc.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanahan D., Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 15.Hanahan D., Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Prometeus GmbH: TOP500. Available at: http://www.top500.org/statistics/perfdevel/. Published biannually. Accessed April 2016 [Google Scholar]
- 17.Haby J. Forecast model data input. Available at: www.theweatherprediction.com/habyhints/60/. Accessed April 2016 [Google Scholar]
- 18.Illumina Website. Available at: www.illumina.com. Accessed April 2016 [Google Scholar]
- 19.Lehrach H. DNA sequencing methods in human genetics and disease research. F1000Prime Rep. 2013;5:34. doi: 10.12703/P5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGinn S., Bauer D., Brefort T., et al New technologies for DNA analysis - a review of the READNA Project. N Biotechnol. 2015;33(3):311–330. doi: 10.1016/j.nbt.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Weischenfeldt J., Simon R., Feuerbach L., et al Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in earlyonset prostate cancer. Cancer Cell. 2013;23(2):159–170. doi: 10.1016/j.ccr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Jones DT., Jäger N., Kool M., et al Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hovestadt V., Jones DT., Picelli S., et al Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014;510(7506):537–541. doi: 10.1038/nature13268. [DOI] [PubMed] [Google Scholar]
- 24.Markiv A., Rambaruth ND., Dwek MV. Beyond the genome and proteome: targeting protein modifications in cancer. Curr Opin Pharmacol. 2012;12(4):408–413. doi: 10.1016/j.coph.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Spurrier B., Ramalingam S., Nishizuka S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat Protoc. 2008;3(11):1796–1808. doi: 10.1038/nprot.2008.179. [DOI] [PubMed] [Google Scholar]
- 26.Fredriksson S., Gullberg M., Jarvius J., et al Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 27.Warnatz HJ., Glökler J., Lehrach H. Method for linking and characterising linked nucleic acids in a composition. European Patent Application EP 2626433 A1. August 14 2013. Available at: https://www.google.com/ patents/EP2626433A1 cl=en&dq=Hans+Lehrach&hl=de&sa=X&ved=0ahll KEwjCg5_Yz9nOAhWKCywKHX9YDIwQ6AEISjAF. Accessed April 2016 [Google Scholar]
- 28.Zaccaria A., Bouamrani A., Chabardes S., et al Deep brain stimulationassociated brain tissue imprints: a new in vivo approach to biological research in human Parkinson's disease. Mol Neurodegener. 2016;11:12. doi: 10.1186/s13024-016-0077-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinton DA., Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295–305. doi: 10.1038/nature10761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corti S., Faravelli I., Cardano M., Conti L. Human pluripotent stem cells as tools for neurodegenerative and neurodevelopmental disease modeling and drug discovery. Expert Opin Drug Discov. 2015;10(6):615–629. doi: 10.1517/17460441.2015.1037737. [DOI] [PubMed] [Google Scholar]
- 31.Imming P., Sinning C., Meyer A. Opinion: drugs, their targets and the nature and number of drug targets. Nat Rev Drug Discov. 2006;5(10):821–834. doi: 10.1038/nrd2132. [DOI] [PubMed] [Google Scholar]
- 32.Bidard FC., Peeters DJ., Fehm T., et al Clinical validity of circulating tumour cells in patients with metastatic breast cancer: a pooled analysis of individual patient data. Lancet Oncol. 2014;15(4):406–414. doi: 10.1016/S1470-2045(14)70069-5. [DOI] [PubMed] [Google Scholar]
- 33.Pantel K., Alix-Panabieres C. Circulating tumour cells in cancer patients: challenges and perspectives. Trends Mol Med. 2010;16(9):398–406. doi: 10.1016/j.molmed.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Crowley E., Di Nicolantonio F., Loupakis F., Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol. 2013;10(8):472–484. doi: 10.1038/nrclinonc.2013.110. [DOI] [PubMed] [Google Scholar]
- 35.Alix-Panabieres, Pantel K. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013;7(21):6384–6388. doi: 10.1158/0008-5472.CAN-13-2030. [DOI] [PubMed] [Google Scholar]
- 36.Sultan M., Schulz MH., Richard H., et al A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321(5891):956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 37.Lin CY., Erkek S., Tong Y., et al Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature. 2016;530(7588):57–62. doi: 10.1038/nature16546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wegner KD., Linden S., Jin Z., et al Nanobodies and nanocrystals: highly sensitive quantum dot-based homogeneous FRET-imrnunoassay for serum-based EGFR detection. Small. 2014;10(4):734–740. doi: 10.1002/smll.201302383. [DOI] [PubMed] [Google Scholar]
- 39.GeiBler D., Stufler S., Löhmannsröben HG., Hildebrandt N. Six-color time-resolved Forster resonance energy transfer for ultrasensitive multiplexed biosensing. J Am Chem Soc. 2013;135(3):1102–1109. doi: 10.1021/ja310317n. [DOI] [PubMed] [Google Scholar]
- 40.GeiBler D., Charbonniere LJ., Ziessel RF., et al Quantum dot biosensors for ultra-sensitive multiplexed diagnostics. Angew Chem Int Ed Engl. 2010;49(8):1396–1401. doi: 10.1002/anie.200906399. [DOI] [PubMed] [Google Scholar]
- 41.Morgner F., GeiBler D., Stufler S., et al A quantum-dot-based molecular ruler for multiplexed optical analysis. Angew Chem Int Ed Engl. 2010;49(41):7570–7574. doi: 10.1002/anie.201002943. [DOI] [PubMed] [Google Scholar]
- 42.Soderberg O., Gullberg M., Jarvius M., et al Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 43.Ke R., Mignardi M., Pacureanu A., et al In situ sequencing for RNA analysis in preserved tissue and cells. Nat Methods. 2013;10(9):857–860. doi: 10.1038/nmeth.2563. [DOI] [PubMed] [Google Scholar]
- 44.Lee J., Daugharthy E., Scheiman J., et al Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343(6177):1360–1363. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee J., Daugharthy ER., Scheiman J., et al Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. NatProtoc. 2015;10(3):442–458. doi: 10.1038/nprot.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crosetto N., Bienko M., van Oudenaarden A. Spatially resolved transcriptomics and beyond. Nat Rev Genet. 2015;16(1):57–66. doi: 10.1038/nrg3832. [DOI] [PubMed] [Google Scholar]
- 47.Mali P., Aach J., Lee J., et al Barcoding cells using cell-surface programmable DNA-binding domains. Nat Methods. 2013;10(5):403–406. doi: 10.1038/nmeth.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weibrecht I., Lundin E., Kiflemariam S., et al In situ detection of individual mRNA molecules and protein complexes or post-translational modifications using padlock probes combined with the in situ proximity ligation assay. Nat Protoc. 2013;8(2):355–372. doi: 10.1038/nprot.2013.006. [DOI] [PubMed] [Google Scholar]
- 49.Soderberg O., Gullberg M., Jarvius M., et al Direct observation of individual endogenous protein complexes in situ by proximity ligation. Wat. Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 50.Weibrecht I., Gavrilovic M., Lindbom L., Landegren U., Wählby C., Söderberg O. Visualising individual sequence-specific protein-DNA interactions in situ. N Biotechnol. 2012;29(5):589–598. doi: 10.1016/j.nbt.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Mahoney KM., Rennert PD., Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Wat. Rev Drug Discov. 2015;14(8):561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 52.Schreiber RD., Old LJ., Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 53.Patel SP., Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 54.Hoehe MR., Church GM., Lehrach H., et al Multiple haplotype-resolved genomes reveal population patterns of gene and protein diplotypes. Nat. Commun. 2014;5:5569. doi: 10.1038/ncomms6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.MacDonald ME., Scott HS., Whaley WL., et al Huntington disease-linked locus D4S111 exposed as the alpha-L-iduronidase gene. Somat Cell Mol Genet. 1991;17(4):421–425. doi: 10.1007/BF01233067. [DOI] [PubMed] [Google Scholar]
- 56.Politis M. Neuroimaging in Parkinson disease: from research setting to clinical practice. Nat Rev Neurol. 2014;10(12):708–722. doi: 10.1038/nrneurol.2014.205. [DOI] [PubMed] [Google Scholar]
- 57.Ecker M., Murphy D. Neuroimaging in autism—from basic science to translational research. Nat Rev Neurol. 2014;10(2):82–91. doi: 10.1038/nrneurol.2013.276. [DOI] [PubMed] [Google Scholar]
- 58.Chowdhury R., Ganeshan B., Irshad S., et al The use of molecular imaging combined with genomic techniques to understand the heterogeneity in cancer metastasis. Br J Radiol. 2014;87(1038):20140065. doi: 10.1259/bjr.20140065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wierling C., Herwig R., Lehrach H. Resources, standards and tools for systems biology. Brief Funct Genomic Proteomic. 2007;6(3):240–251. doi: 10.1093/bfgp/elm027. [DOI] [PubMed] [Google Scholar]
- 60.Wierling C., Kuhn A., Hache H., et al Prediction in the face of uncertainty: a Monte Carlo-based approach for systems biology of cancer treatment. MutatRes. 2012;746(2):163–170. doi: 10.1016/j.mrgentox.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Röhr C., Kerick M., Fischer A., et al High-throughput miRNA and mRNA sequencing of paired colorectal normal, tumor and metastasis tissues and bioinformatic modeling of miRNA-1 therapeutic applications. PloS One. 2013;8(7):e67461. doi: 10.1371/journal.pone.0067461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wierling C., Kessler T., Ogilvie LA., Lange BM., Yaspo ML., Lehrach H. Network and systems biology: essential steps in virtualising drug discovery and development. Drug Discov Today Technol. 2015;15:33–40. doi: 10.1016/j.ddtec.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 63.Ogilvie LA., Wierling C., Kessler T., Lehrach H., Lange BM. Predictive modeling of drug treatment in the area of personalized medicine. Cancer Inform. 2015;14(suppl 4):95–103. doi: 10.4137/CIN.S19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Molina D., Lozano M., Garcia-Martinez C., Herrera F. Memetic algorithms for continuous optimisation based on local search chains. Evol Comput. 2010;18(1):27–63. doi: 10.1162/evco.2010.18.1.18102. [DOI] [PubMed] [Google Scholar]
- 65.Helton JC., Davis FJ. Latin hypercube sampling and the propagation of uncertainty in analyses of complex systems. Reliability Eng Syst Safety. 2003;81(1):23–69. [Google Scholar]
- 66.Rodriguez-Fernandez M., Banga JR. SensSB: a software toolbox for the development and sensitivity analysis of systems biology models. Bioinformatics. 2010;26(13):1675–1676. doi: 10.1093/bioinformatics/btq242. [DOI] [PubMed] [Google Scholar]
- 67.Al-Lazikani B., Banerji U., Workman P. Combinatorial drug therapy for cancer in the post-genomic era. Nat Biotechnol. 2012;30(7):679–692. doi: 10.1038/nbt.2284. [DOI] [PubMed] [Google Scholar]
- 68.Ma Q., Lu AY. Pharmacogenetics, pharmacogenomics, and individualized medicine. Pharmacol Rev. 2011;63(2):437–459. doi: 10.1124/pr.110.003533. [DOI] [PubMed] [Google Scholar]
- 69.Lehrach H. Virtual clinical trials, an essential step in increasing the effectiveness of the drug development process. Public Health Genomics. 2015;18(6):366–371. doi: 10.1159/000441553. [DOI] [PubMed] [Google Scholar]