

Abstract
This review concerns epigenetic mechanisms and their roles in conferring interindividual differences, especially as related to experientially acquired and genetically driven changes in central nervous system (CNS) function. In addition, the review contains commentary regarding the possible ways in which epigenomic changes may contribute to neuropsychiatric conditions and disorders and ways in which epigenotyping might be cross-correlated with clinical phenotyping in the context of precision medicine. The review begins with a basic description of epigenetic marking in the CNS and how these changes are powerful regulators of gene readout. Means for characterizing the individual epigenotype are briefly described, with a focus on DNA cytosine methylation as a readily measurable, stable epigenetic mark. This background enables a discussion of how “epigenotyping” might be integrated along with genotyping and deep phenotyping as a means of implementing advanced precision medicine. Finally, the commentary addresses two exemplars when considering how epigenotype may correlate with and modulate cognitive and behavioral phenotype: schizophrenia and Rett syndrome. These two disorders provide an interesting compare-and-contrast example regarding possible epigenotypic regulation of behavior: whereas Rett syndrome is clearly established as being caused by disruption of the function of an epigenetic “reader” of the DNA cytosine methylome—methyl-CpG-binding protein 2 (MeCP2)—the case for a role for epigenetic mechanisms in schizophrenia is still quite speculative.
Keywords: autism, DNA methylation, epigenetics, epigenotyping, gene transcription, memory, neuroepigenetics, precision medicine, Rett syndrome, schizophrenia
Abstract
Este comentario describe los mecanismos epigenéticos y sus funciones para adjudicar diferencias interindividuales espetialmente en lo relative a los cambios en la función del sistema nervioso central (SNC) tanto adquiridos por la experiencia, como producidos genéticamente. Además, la revisión especula acerca de las posibles formas en que los cambios epigenómicos pueden contribuir a las condiciones y trastornos neuropsiquiótricos y formas en las cuales el epigenotipado pudiera estar transversalmente correlacionado con el fenotipado clínico en el contexto de la medicina de precisión. La revisión comienza con una descripción básica del marcaje epigenético en el SNC y cómo estos cambios son poderosos reguladores de la lectura génica. Se describen brevemente los medios para la caracterización del epigenotipo individual, enfocándose en la metilación de la citosina del DNA, como un marcador epigenético estable y fácil de medir. Estos antecedentes permiten una discusión acerca de cómo el “epigenotipado” podría estar integrado junto con el genotipado y el fenotipado precisos y completos como un medio para implementar una medicina de precisión avanzada. Por último, el comentario revisa dos ejemplos que consideran cómo el epigenotipo puede correlacionarse con y modular el fenotipo cognitivo y conductual: la esquizofrenia y el síndrome de Rett. Estos dos trastornos proporcionan una interesante comparación y contraste en relación con la posible regulación epigenotípica de la conducta. En el síndrome de Rett se ha establecido que esta regulación está causada por una disrupción de la función de un “lector” epigenético del metiloma de citosina del ADN, el MeCP2; en cambio, para la esquizofrenia el papel de los mecanismos epigenéticos a la fecha todavía es bastante especulativo.
Abstract
Cet article s'intéresse aux mécanismes épigénétiques et à leur implication dans les différences interindividuelles, plus particulièrement lors de modifications de la fonction du système nerveux central (SNC) innées et acquises. On y trouvera également des commentaires sur les différentes façons dont les changements épigénomiques peuvent influer sur les maladies et états neuropsychiatriques et sur les façons dont l'épigénotypage peut être croisé avec le phénotypage clinique en médecine de précision. L'article décrit tout d'abord de façon élémentaire le marquage épigénétique dans le SNC et comment ces modifications sont des régulateurs puissants de la lecture des gènes. Les méthodes de caractérisation d'un épigénotype individuel sont brièvement décrites, notamment la méthylation de l'ADN, qui est un marqueur épigénétique stable et facilement mesurable. Dans ce contexte, il est discuté de la façon dont « l'épigénotypage » pourrait participer avec un génotypage et un phénotypage précis et complet à l'amélioration de la médecine de précision avancée. Enfin, le commentaire aborde la schizophrénie et le syndrome de Rett comme deux exemples de la façon dont l'épigénotypage peut correspondre au et moduler le phénotype comportemental et cognitif. Ces deux troubles représentent un exemple intéressant de comparaison des régulations épigénotypiques possibles du comportement: une mutation d'un « lecteur» épigénétique du méthylome cytosine de l'ADN - gène MeCP2 (methyl-CpG-binding protein 2) - est connue pour entraîner le syndrome de Rett alors que le rôle de mécanismes épigénétiques dans la schizophrénie est encore purement théorique.
Introduction
The existence of pronounced interindividual differences in behavior and cognitive function is abundantly clear, even when observing those individuals around us on a daily basis. Many of these differences in behavior are based on personal experiences, resulting in individual human uniqueness via acquired behavioral change through either conscious learning or unconscious conditioning. The extreme range of interindividual differences in human behavior and cognitive function is further illustrated by considering neuropsychiatric conditions that span a broad range—schizophrenia, affective disorders, intellectual disabilities, autism. This review and commentary considers the broad hypothesis that the individual's epigenetic state, which we will refer to as their “epigenotype,” is a molecular mechanism contributing to interindividual variation in behavior and cognition. Indeed, we propose that the epigenome is a driver of both normal individual variation and neuropsychiatric dysfunction. This is not a new concept; it has been discussed extensively in prior literature, not only in biomedicine but also in that for general readership.1-13 Nevertheless, in this review we will endeavor to provide a succinct overview of epigenetic molecular mechanisms, their role in experience-driven interindividual behavior, and their probable role in neuropsychiatric disorders.
Epigenetics: the interface of genes and experience
The relative contributions of “nature” versus “nurture” to an individual's behavior has been debated for millennia, as exemplified in Western literature by the writings of both Greek and Roman philosophers. Modern terminology and understanding has recast the discussion more along the lines of genes versus environment/experience/learning. However, recent discoveries over the last 2 decades or so, based on studies in a wide range of biological and medical disciplines, have clearly indicated that there is a third component to the broad conceptual organizing scheme regarding “nature” versus “nurture.” That third component is epigenetic molecular mechanisms (Figure 1, see ref 2). Epigenetic mechanisms operate at the interface of genes and environment/experience, as has been illustrated by studies from a diverse set of fields including environmental toxicology, cancer biology, developmental biology, genetics, evolutionary biology, molecular biology, and neuroscience.
Figure 1. The epigenome as an interface between genes and environment/experience, and its role as a driver of individuality. See text for discussion.
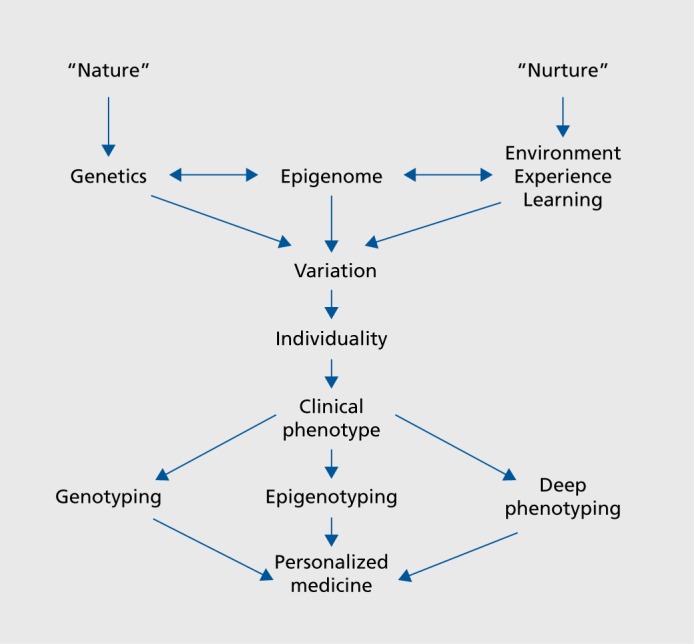
Epigenetic molecular mechanisms confer plasticity on biological systems as broadly defined, as well as on individual cells within an organism. Developmentally, they determine and drive cell fate of individual cells. In an interesting parallel, they also help to generate variance at the organismal level. Thus, it is now becoming clear that at the organismal level epigenetic mechanisms confer variation and uniqueness on single individuals.
Nowhere is this more striking than when considering mammalian central nervous system (CNS) function. Learning and memory clearly utilize epigenetic mechanisms in order for experience to drive lasting behavioral change in an animal.14-18 Epigenetically driven imprinting and modulation of genes influences behavior across generations, as perhaps best illustrated by considering Angelman syndrome and Fragile X mental retardation.11 Environmental influences including neonatal nurturing shape the CNS epigenome, with attendant changes in behavior.7 Drug abuse is closely linked to altered epigenetic marking in the CNS.19 Finally, and most relevant to this commentary, a wide variety of neuropsychiatric disorders are associated with alterations in the CNS epigenome, in both animal models and human studies (see refs 19-62, Table I).
TABLE I. Neuropsychiatric disorders associated with epigenetic mechanisms or epigenetic disruption. This list is not exhaustive, but rather is representative. ATR-X, alpha-thalassemia X-linked mental retardation; PTSD, posttraumatic stress disorder.
| Neurodevelopmental disorders including autism spectrum disorders |
| Rubinstein-Taybi syndrome |
| Angelman syndrome |
| Rett syndrome |
| Pitt-Hopkins syndrome |
| Fragile X mental retardation |
| ATR-X syndrome |
| Coffin-Lowry syndrome |
| Kleefstra syndrome |
| Aging-related and neurodegenerative disorders |
| Cognitive aging and mild cognitive impairment |
| Alzheimer disease |
| Parkinson disease |
| Hereditary sensory-autonomic neuropathy type 1 (HSAN1) |
| Huntington disease |
| Psychosis and affective disorders |
| Schizophrenia |
| Depression |
| Bipolar disorder |
| Stress/hyperactivity |
| PTSD |
| Drug abuse and addiction |
| Cocaine |
| Alcohol |
| Nicotine |
| Amphetamine/methamphetamine |
| Opiates |
Against this backdrop, it is important to have some basic understanding of molecular epigenetic mechanisms that operate in the CNS, especially since these mechanisms are powerful regulators of CNS gene transcription and regulation. CNS epigenetic marking can not only regulate the particular splice variants that are “read out” of the genome within a neuron, but can also completely silence the readout of an entire gene. (or thousands of genes) within a cell. For this reason, in the next section we will review the basic categories of epigenetic molecular marks that have been best established as operating in the CNS.
What is the epigenome?
There are four major categories of epigenetic marks that have been found to operate in the mammalian CNS (thus far, Figure 2):
Figure 2. Schematic representation of epigenetic marks. (A) DNA is condensed within the nucleus through interactions with histones. The DNA-protein complex is referred to as chromatin. (B) The N-terminal tail of a histone has several sites for epigenetic marking that can promote or repress gene transcription. (C) Methylation of DNA in which a methyl group (red diamonds) is transferred to cytosines in genomic regions in and around gene promoters rich in cytosine-guanine nucleotides (CpG islands). (D) A variety of noncoding RNA gene products also confer epigenetic regulation on neural systems.

First, it is important to realize that genes are physically condensed in cells and have a specific three-dimensional structure that is shaped by not only the conformation of the DNA double helix but also the interaction of DNA with histone proteins. The DNA-histone complex is referred to as chromatin, and chromatin structure broadly defined is a powerful regulator of gene readout in the CNS. The nucleosome is the fundamental subunit of chromatin. A nucleosome comprises about two turns of DNA wrapped around a core set of eight histones. Each histone octamer contains two copies each of the histone subunits H2A, H2B, H3, and H4. Chromatin structure can be regulated by processes such as adenosine triphosphate (ATP) - dependent remodeling and by broadly acting, but not well understood, processes of distal interactions between chromatin particles operating across large stretches of the genome.
The amino-terminal tails of individual histones in the histone octamer comprising the core of the chromatin particle are subject to a multitude of posttranslational modifications, including phosphorylation, acetylation, methylation, and ubiquitination. Furthermore, individual subunits within the octamer can be swapped in and out, a process known as histone subunit exchange. All of these modifications can be read out in a combinatorial fashion, either activating or inhibiting transcription.
DNA itself is also subject to direct covalent chemical modification, including having a methyl group added to cytosines within the DNA polymer. Cytosine methylation can occur on either one or both strands of the DNA. DNA methylation is typically suppressive for transcription of the associated gene, but in some instances gene-activating DNA methylation has been observed.
Noncoding RNA transcribed from the genome can also be powerful regulators of gene readout, in most cases through noncoding transcripts interacting directly with messenger RNAs (mRNAs), altering their capacity to be translated into protein products. Types of noncoding RNAs include long noncoding RNAs (IncRNAs), micro RNAs (miRNAs), small noncoding RNAs (snRNAs), silencing RNAs (siRNAs), and RNA interference (RNAi) mechanisms involving double-stranded RNA adducts.
The fact that these mechanisms are in play in the CNS may explain some of the failures in genetic screening in trying to understand genotypes related to neuropsychiatry disorders. There is a major hidden layer of mechanisms, the epigenetic mechanisms, that are operating to control gene function in the CNS, and these mechanisms are not detected by traditional genetic screening processes, genome-wide association study (GWAS) analysis, etc. This is particularly notable to consider for high-incidence and complex neuropsychological disorders such as schizophrenia and autism(s).
For this reason, we (and others) propose that epigenotyping must begin to be implemented as part of large-scale screens to identify “genetic” bases of neuropsychiatry disorders.1,53-62 However, with currently available equipment and infrastructure, many aspects of this approach are technically and technologically daunting. Five limitations are worth highlighting as an introduction to these issues. First is the lack of convenient access to DNA-containing cells other than blood lymphocytes and buccal epithelial cells in patient samples, clearly a limitation when peripheral DNA does not have the same epigenotype as the relevant pathology-associated cells in the CNS. To what extent peripheral DNA will accurately report the CNS epigenotype is an open question at this point. Second, whereas DNA cytosine methylation is highly stable chemically and thus quite amenable to patient sampling, histone and other chromatin modifications are fairly labile and subject to rapid reversal. Third, added to this is the immense heterogeneity of DNA chemical marks, which may each have their own effects on transcription. Two examples to illustrate this point are cytosine methylation versus cytosine hydroxymethylation and the vast number of histone epigenetic marks that an individual cell may accumulate. The high combinatorial variability of histone modifications is particularly relevant in this regard.8,13 Moreover, two or more copies (alleles) of each gene are typically present in a cell, and each allele may not be epigenetically identical. Fourth, at present, there is uncertainty concerning whether any specific DNA methylation change(s) affects gene transcription, and whether they are activating or inactivating depending on genomic context. In other words, no Rosetta Stone algorithm exists concerning how a specific DNA methylation event is going to be read out in terms of gene transcription. Finally, as a practical matter, the expense of epigenotyping is a rate-limiting factor in terms of broad implementation. Methylomic analysis, which requires extensive whole-genome-level DNA sequencing, is the most practical target for epigenotyping at present. Epigenotyping of chromatin-associated proteins will probably be limited to small human population studies and characterization of relatively homogeneous animal models in the near future.
Despite these challenges, technological advances are beginning to make epigenotyping practicable, especially true of DNA cytosine methylation. Anticipated near-future technical and instrumentation advances will make this option increasingly affordable. For these reasons, the exciting prospect exists of applying broad-scale epigenotyping to clinical and basic science studies (see refs 1,53,63 for examples). In the next section, we will address—as a hypothetical—how such an effort might be undertaken.
Integrating epigenetics into personalized medicine
How would our new understanding of the epigenome be integrated into initiatives to realize a personalized medicine approach and implement precision medicine? As illustrated in the bottom portion of Figure 1, individuality encompasses a specific clinical phenotype of a patient or subject. Thus, a comprehensive approach is necessary, involving genotyping, epigenotyping, and deep phenotyping in order to implement advanced precision medicine.
When considering the implementation of this approach for clinical studies, it is highly desirable to organize health care providers involved in assessing and treating the disorder under study into one clinical service in order to provide a single portal of contact/entry for families and subjects to enter the clinical system undertaking the study. Evaluations of all patients should be consistently applied and with high reproducibility, and they should be multidisciplinary, with neurologists, psychiatrists, psychologists, speech and hearing experts, and so forth, on site for evaluating and treating patients in a coordinated fashion. Aspects of this idealized approach may sound like a statement of the obvious, but in many “real-world” cases it is difficult to achieve consistency and multidisciplinarity because of departmental and clinical service boundaries.
The attribute of having a single entry point to the patient care pathway provides an exceptional opportunity for broad-scope clinical characterization and neurobehavioral phenotyping (ie, “deep phenotyping”). Particularly powerful is the opportunity to leverage clinical expertise and infrastructure by combining it with cutting-edge genomic and epigenomic characterization of patients entering the clinical study. Coupling the principles of precision genomic/epigenomic medicine with the broad phenotypic and clinical characterization of study subjects positions the study to clearly define and characterize the neuropsychiatric disorder of interest. Genotypes await discovery for most neuropsychiatric disorders, and in this fashion the genetic and epigenetic mechanisms underlying rare and idiosyncratic disorders can be ascertained in a scope and depth not attainable otherwise. These considerations also set the stage for identifying and exploiting novel therapeutic targets. As well, these discoveries allow development of new genetically engineered vertebrate (rat/mouse/zebrafish) model systems for further in-depth mechanistic, phenotypic, and drug discovery/development endeavors. Needless to say, such a milieu could provide an unparalleled training environment for enabling and shaping the next generation of clinical and scientific leaders in the field of research into neuropsychiatric disorders.
Epigenotyping studies will require technical infrastructure around the molecular genetics components and would probably include collaborations with top-notch genomics facilities. These leveraging technical resources would enable truly cutting-edge and high-throughput next-generation sequencing (NGS) and bioinformatics platforms to be applied to both basic science and clinical research in these endeavors. However, an additional consideration is the need for expert support related to bioinformatics as specifically targeted to the neuropsychiatric disorder under study. Generic bioinformatics resources and platforms are enabling but not sufficient, due to highly specialized types of bioinformatics analysis that are project-specific for neuropsychiatric research.
Patient studies and animal model studies of this sort will enable a new, more detailed understanding of transcriptional and epigenetic disruption in neuropsychiatric disorders and their relationship to human cognitive function, broadly speaking. The scope of the problem of understanding transcriptional and epigenetic dysregulation in neuropsychiatric disorders is immense, and realistically speaking, reduction to simple hypotheses is difficult. For this reason, bioinformatic and computational/mathematical modeling approaches will be required for comprehensive and comprehendible development of theoretical models of transcriptional regulation in neuropsychiatric disorders. Innovative studies combining cutting-edge genomics, epigenomics, and transcriptomics with computational biology will be transformative not only for neuropsychiatric research specifically but also for neuroscience as a discipline.
Also, additional resources will be required in support of developing new approaches to precision behavioral diagnostics and clinical phenotyping of neuropsychiatric disorders in this context.64 Clinical phenotyping involving research to develop and validate objective measures for use in clinical trials, targeting core symptoms of neuropsychiatric disorders, is critical for success. The Research Domain Criteria (RDoC) approach that was initiated by the National Institute of Mental Health (NIMH) has begun to make an impact in these areas, and building on this foundation would enable national-level progress in this domain, especially when coupled with molecular/genetic diagnostics as described above.64-66
There also are additional technological aspects to implementing the approach with regard to the epigenotyping per se. When considering implementation of this approach, and on the basis of currently available criteria, we see that in general there are three categories of molecules related to epigenotyping that can be considered as a complement to now-traditional DNA nucleotide sequence genotyping and whole-exome/transcriptome sequencing: (i) measurements of DNA cytosine methylation; (ii) miRNAs; and (iii) IncRNAs. DNA cytosine methylation can be quantitated genome-wide using bisulfite sequencing approaches or, alternatively, more affordable NGS-based approaches such as methyl binding domain-targeted DNA pull-down plus highthroughput sequencing (MBD-seq). For investigating miRNAs, it is reasonably affordable to use whole-transcriptome small RNA-targeted high-throughput nucleotide sequencing (small RNA-seq) to comprehensively identify and quantitate the small regulatory noncoding RNAs (siRNAs, miRNAs, snRNAs, and Piwi-interacting RNAs [piRNAs]) whose levels are altered in association with the disorder under study. Quantitating these several categories is a nice complement to wholetranscriptome mRNA-targeted high-throughput nucleotide sequencing (mRNA-Seq) to comprehensively identify and quantitate the genes whose transcription are altered (increased or decreased) in response to a neuropsychiatric condition because they potentially provide insight into specific mechanisms that might underlie alterations in gene transcription. Finally, NGS-based sequencing of non-polyA-tailed RNAs to selectively identify IncRNAs can quantitate this important category of epigenetic regulator.
It is important to note that a major limitation to this overall epigenotyping approach is that these studies will not determine in a comprehensive fashion how DNA demethylation and other epigenetic changes at the cellular level gets translated into altered neural and behavioral function. A comprehensive and unbiased genomic and epigenomic approach can identify specific sites of methylation changes, genome-wide and specifically at disease-associated genes. However, these data only allow one to assess correlative changes in DNA methylation and associated transcriptional changes, even though one is using a genome-wide approach. Trying to mechanistically tie these specific changes at single gene exons to complex multicellular, multicomponent processes like neuropsychiatric disorders is quite difficult at this point because of the current limitations in our understanding of the means by which alteration of even a single gene transcript is transduced into functional synaptic changes. This means that the approach is a phenomenological approach to demonstrate that DNA methylation and other epigenetic changes occur and are capable of contributing to the disorder under study. A full understanding of how any change in DNA methylation gets transformed into functional changes in the cell and synapse will require further investigation. In addition, the biological material that is readily accessible in human studies, eg, blood samples and lymphocytes, may not fully or even partially reflect molecular changes in the CNS. Resolution of this issue awaits further detailed study. Finally, there is a plethora of potentially relevant epigenetic changes in a wide variety of cell types in the central nervous system and peripherally.62 This large number of different epigenetic marks will complicate defining specific epigenetic changes that are correlated with, or diagnostic for, specific neuropsychiatric disorders.
However, the proposed approach can yield substantive mechanistic insights allowing the generation of new hypotheses. The studies thereby serve as a foundation for formulating future specific hypotheses concerning how DNA methylation and other epigenetic mechanisms might control persisting changes in synaptic and neural circuit function at the molecular level, and for ultimately understanding how transient or persisting changes in epigenetic marks manifest themselves in altered neural and cognitive function.
Two case studies: schizophrenia and Rett syndrome
In this final section, we will comment on two interesting “case studies” of neuropsychiatric disorders that have been proposed to be (at least partially) mediated by an epigenetic mechanism, namely DNA methylation-related neuroregulation. The two case studies are at opposite ends of the spectrum as related to the quantity and quality of the data available regarding their genetic and epigenetic basis. Schizophrenia is one example; here the genetic basis has been elusive and the epigenetic basis remains quite speculative. The autism spectrum disorder Rett syndrome is the other example; here the genetic basis and epigenetic relevance is clearly and unambiguously defined. In this respect, these two neuropsychiatric disorders serve as a compare-and-contrast example.
Schizophrenia is a case where large-scale human genetic screening has by and large failed to nail down a clear cause based on genetics, genomics, and GWAS studies. Around a decade or more ago there was palpable excitement that a genetic basis for schizophrenia would be quickly revealed in the new genomics era. However, this view has given way to a more realistic understanding that genotyping alone is not sufficient to define schizophrenia or probably even subtypes of schizophrenia. Thus, lately it has been proposed that DNA methylation and other epigenetic marks may be the missing piece of the puzzle and that epigenotyping studies may enable a more complete understanding of genotype/epigenotype/clinical phenotype correlations in schizophrenia.1,52,61,64
This line of thinking regarding an epigenetic hypothesis of psychosis is compellingly motivated by several arguments. As already described, the epigenome, broadly speaking, represents a hitherto unknown molecular interface between experience and the genome. As a potent controller of gene transcription, enabling in many instances the complete silencing of a methylated gene, epigenetic mechanisms represent a very appealing potential mechanism for driving the transcriptional changes documented to occur in schizophrenia. Epigenetic approaches have previously been applied in schizophrenia, scientists having recognized the unique potential of an epigenetic approach in this area.54-56 Many investigators also recognize the critical role of epigenetic mechanisms in modifying genetic expression by environmental events.56,57 A number of human studies have already tried to develop peripheral markers of CNS epigenetic remodeling in psychosis and related disorders.58-61 Thus, given its unknown molecular genetic pathology, schizophrenia is appealing as a candidate neuropsychiatric disorder influenced or driven by epigenetic mechanisms.
This hypothetical scenario with schizophrenia stands in contrast to Rett syndrome, an autism spectrum disorder and neurodevelopmental intellectual disability syndrome for which epigenetic mechanisms are clearly implicated. Rett syndrome has an identified genetic basis, which is mutation of the methyl-CpG binding protein MeCP2, a “reader” of the cytosine methylome. Although the precise mechanisms through which MeCP2 implements the cytosine methylation signal in the genome are not understood, MeCP2 is clearly a mechanistic link between DNA cytosine methylation and neural function in the CNS. Although we will not describe Rett syndrome in detail here because it has been extensively reviewed in the literature,67 Rett syndrome is illustrative of two important take-home messages in the context of this commentary. First, the very existence of the syndrome clearly demonstrates a crucial role for the methylome in regulating behavioral and cognitive function. Second, Rett syndrome is a compelling case illustrating that the epigenome and its read-out can mediate human variation and individuality.
Finally, Rett studies serve as proof of concept that sufficiently deep phenotyping can enable discovery of underlying mechanistic causes of neuropsychiatric disorders. The “syndromic” autism spectrum disorders, by definition, can be delineated from each other and from nonsyndromic autism by sufficiently precise clinical phenotyping. This parsing of, for example, Rett syndrome from Angelman syndrome from Pitt-Hopkins syndrome from Fragile X mental retardation through clinical phenotyping allowed individuating these disorders one from another. This in turn allowed relatively rapid identification of genetic subtypes diagnostic for these disorders, correlated with great precision to specific genetic alterations, within the broad categories of “autism spectrum disorders” or “pervasive developmental delay not otherwise specified.” Similar deep phenotyping at the clinical level of other types of autistic disorders, or indeed even broad categories such as psychotic illnesses or affective disorders, might conceivably enable similar advances.
Summary and closing comments
This review describes the emerging idea that epigenetic mechanisms, especially histone modifications and DNA cytosine methylation, contribute to individual variation in human cognition and CNS function. This idea is fascinating because the same mechanisms are used for triggering and storing long-term “individuality” at the cellular and developmental level, for example, when cells differentiate. We have also emphasized that DNA methylation is a dynamic process in the adult CNS, involved in controlling long-lasting changes in synaptic function and behavior and driving acquired behavioral uniqueness. In addition, these types of findings strongly indicate that we can no longer think of DNA methylation and other epigenetic marks as being static: set up during development as a permanent change. Instead, DNA methylation is subject to active and ongoing regulation in the developing and adult nervous system, controlled by behavioral and environmental experience.
This concept clearly has important implications for the mechanisms potentially contributing to the development and perpetuation of human individuality, as well as individual phenotypes that may present with disease. In this regard then, comprehensive genomic/epigenomic/transcriptomic assessment of the correlates of neuropsychiatric disorders, such as have been delineated in this review, will be necessary for full implementation of precision medicine in the future.
Acknowledgments
The authors thank Kim Strifert, Craig Powell, Eric Nestler, Andrew Kennedy, Jeremy Day, Garrett Kaas, and Kimberly Hawkins for many helpful discussions. They also thank Kimberly Hawkins for help in preparing the figures and manuscript. Research in the authors' laboratories is supported by funds from the NINDS, NIMH, DARPA, and the Pitt-Hopkins Syndrome Foundation. The authors have no conflicts of interest to disclose.
Contributor Information
J. David Sweatt, Department of Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA.
Carol A. Tamminga, Department of Psychiatry, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
REFERENCES
- 1.BrainSeq: A Human Brain Genomics Consortium. BrainSeq: neurogenomics to drive novel target discovery for neuropsychiatric disorders. Neuron. 2015;88(6):1078–1083. doi: 10.1016/j.neuron.2015.10.047. [DOI] [PubMed] [Google Scholar]
- 2.Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry. 2009;65(3):191–197. doi: 10.1016/j.biopsych.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker-Andresen D., Ratnu VS., Bredy TW. Dynamic DNA methylation: a prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013;36(1):3–13. doi: 10.1016/j.tins.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 4.Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;80(3):624–632. doi: 10.1016/j.neuron.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore DS. The Developing Genome: An Introduction to Behavioral Epigenetics. New York, NY: Oxford University Press; 2015 [Google Scholar]
- 6.Francis RC. Epigenetics: How Environment Shapes Our Genes. New York, NY: Norton; 2012 [Google Scholar]
- 7.Meaney MJ., Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7(2):103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood MA., Hawk JD., Abel T. Combinatorial chromatin modifications and memory storage: a code for memory? Learn Mem. 2006;13(3):241–244. doi: 10.1101/lm.278206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levenson JM., Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6(2):108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- 10.Houston I., Peter CJ., Mitchell A., et al Epigenetics in the human brain. Neuropsychopharmacology. 2013;38(1):183–197. doi: 10.1038/npp.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Y., Langley B., Lubin FD., et al Epigenetics in the nervous system. J Neurosci. 2008;8(46):11753–11759. doi: 10.1523/JNEUROSCI.3797-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crick F. Memory and molecular turnover. Nature. 1984;312(5990):101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- 13.Graff J., Mansuy IM. Epigenetic codes in cognition and behaviour. Behav Brain Res. 2008;192(1):70–87. doi: 10.1016/j.bbr.2008.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Barrett RM., Wood MA. Beyond transcription factors: the role of chromatin modifying enzymes in regulating transcription required for memory. Learn Mem. 2008;15(7):460–467. doi: 10.1101/lm.917508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zovkic IB., Paulukaitis BS., Day JJ., Etikala DM., Sweatt JD. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature. 2014;515(7528):582–586. doi: 10.1038/nature13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korzus E., Rosenfeld MG., Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42(6):961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Levenson JM., O'Riordan KJ., Brown KD., et al Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem. 2004;279(39):40545–40559. doi: 10.1074/jbc.M402229200. [DOI] [PubMed] [Google Scholar]
- 18.Miller CA., Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Heller EA., Cates HM., Pena CJ., et al Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–1727. doi: 10.1038/nn.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsankova NM., Berton O., Renthal W., et al Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9(4):519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 21.Kumar A., Choi KH., Renthal W., et al Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron. 2005;48(2):303–314. doi: 10.1016/j.neuron.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 22.Meaney MJ., Szyf M. Environmental programming of stress responses through DNA methylation: life at the interface between a dynamic environment and a fixed genome. Dialogues Clin Neurosci. 2005;7(2):103–123. doi: 10.31887/DCNS.2005.7.2/mmeaney. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weaver IC., Champagne FA., Brown SE., et al Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25(47):11045–11054. doi: 10.1523/JNEUROSCI.3652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alarcon JM., Malleret G., Touzani K., et al Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42(6):947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Abdolmaleky HM., Cheng K., Russo A., et al Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B(1):60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- 26.Akbarian S., Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52(2):293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Borghol N., Suderman M., McArdle W., et al Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol. 2012;41(1):62–74. doi: 10.1093/ije/dyr147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chahrour M., Jung SY., Shaw C., et al MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Y., Dong E., Grayson DR. Analysis of the GAD1 promoter: transacting factors and DNA methylation converge on the 5' untranslated region. Neuropharmacology. 2011;60(7-8):1075–1087. doi: 10.1016/j.neuropharm.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Costa E., Davis JM., Dong E., et al A GABAergic cortical deficit dominates schizophrenia pathophysiology. Crit Rev Neurobiol. 2004;16(1-2):1–23. doi: 10.1615/critrevneurobiol.v16.i12.10. [DOI] [PubMed] [Google Scholar]
- 31.Davies MN., Volta M., Pidsley R., et al Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13(6):R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day JJ., Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70(5):813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eastwood SL., Harrison PJ. Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry. 2003;8(9):769,821–831. doi: 10.1038/sj.mp.4001399. [DOI] [PubMed] [Google Scholar]
- 34.Essex MJ., Thomas Boyce W., Hertzman C., et al Epigenetic vestiges of early developmental adversity: childhood stress exposure and DNA methylation in adolescence. Child Dev. 2011;84(1):58–75. doi: 10.1111/j.1467-8624.2011.01641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fraga MF., Ballestar E., Paz MF., et al Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gluckman PD., Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305(5691):1733–1736. doi: 10.1126/science.1095292. [DOI] [PubMed] [Google Scholar]
- 37.Grayson DR., Jia X., Chen Y., et al Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102(26):9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang HS., Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS One. 2007;2(8):e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang HS., Matevossian A., Whittle C., et al Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci. 2007;27(42):11254–11262. doi: 10.1523/JNEUROSCI.3272-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaminsky ZA., Tang T., Wang SC., et al DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41(2):240–245. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 41.Maze I., Feng J., Wilkinson MB., Sun HS., Shen L., Nestler EJ. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108(7):3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGowan PO., Sasaki A., D'Alessio AC., et al Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meaney MJ., Ferguson-Smith AC. Epigenetic regulation of the neural transcriptome: the meaning of the marks. Nat Neurosci. 2010;13(11):1313–1318. doi: 10.1038/nn1110-1313. [DOI] [PubMed] [Google Scholar]
- 44.Mill J., Dempster E., Caspi A., Williams B., Moffitt T., Craig I. Evidence for monozygotic twin (MZ) discordance in methylation level at two CpG sites in the promoter region of the catechol-O-methyltransferase (COMT) gene. Am J Med Genet B Neuropsychiatr Genet. 2006;141B(4):421–425. doi: 10.1002/ajmg.b.30316. [DOI] [PubMed] [Google Scholar]
- 45.Ng JW., Laura M., Barrett LM., et al The role of longitudinal cohort studies in epigenetic epidemiology: challenges and opportunities. Genome Biol. 2012;13(6):246. doi: 10.1186/gb-2012-13-6-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465(7299):721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 47.Renthal W., Maze I., Krishnan V., et al Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56(3):517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 48.Siegmund KD., Connor CM., Campan M., et al DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2(9):e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tang B., Dean B., Thomas EA. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry. 2011;1:e64. doi: 10.1038/tp.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tyrka AR., Prince LH., Marsit C., Walters OC., Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PloS One. 2012;7(1):e30148. doi: 10.1371/journal.pone.0030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nestler EJ. Epigenetic mechanisms of drug addiction. Neuropharmacology. 2014;76(pt B):259–268. doi: 10.1016/j.neuropharm.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akbarian S. Epigenetics of schizophrenia. Curr Top Behav Neurosciences. 2010;4:611–628. doi: 10.1007/7854_2010_38. [DOI] [PubMed] [Google Scholar]
- 53.Jaffe AE., Gao Y., Deep-Soboslay A., et al Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19(1):40–47. doi: 10.1038/nn.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wockner LF., Noble EP., Lawford BR., et al Genome-wide DNA methylation analysis of human brain tissue from schizophrenia patients. Transl Psychiatry. 2014;4:e339. doi: 10.1038/tp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grayson DR., Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38(1):138–166. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar G., Clark SL., McClay JL., et al Refinement of schizophrenia GWAS loci using methylome-wide association data. Hum Genet. 2015;134(1):7787. doi: 10.1007/s00439-014-1494-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gavin DP., Floreani C. Epigenetics of schizophrenia: an open and shut case. Int Rev Neurobiol. 2014;115:155–201. doi: 10.1016/B978-0-12-801311-3.00005-6. [DOI] [PubMed] [Google Scholar]
- 58.Guidotti A., Auta J., Davis JM., et al Toward the identification of peripheral epigenetic biomarkers of schizophrenia. J Neurogenet. 2014;28(12):41–52. doi: 10.3109/01677063.2014.892485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abdolmaleky HM., Nohesara S., Ghadirivasfi M., et al DNA hypermethylation of serotonin transporter gene promoter in drug naive patients with schizophrenia. Schizophr Res. 2014;152(2-3):373–380. doi: 10.1016/j.schres.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aberg KA., McClay JL., Nerella S., et al Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA. Psychiatry. 2014;71(3):255–264. doi: 10.1001/jamapsychiatry.2013.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Auta J., Smith RC., Dong E., et al DNA-methylation gene network dysregulation in peripheral blood lymphocytes of schizophrenia patients. Schizophr Res. 2013;150(1):312–318. doi: 10.1016/j.schres.2013.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Day JJ., Kennedy AJ., Sweatt JD. DNA methylation and its implications and accessibility for neuropsychiatric therapeutics. Annu Rev Pharmacol Toxicol. 2015;55:591–611. doi: 10.1146/annurev-pharmtox-010814-124527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Franke B., Stein JL., Ripke S., et al Genetic influences on schizophrenia and subcortical brain volumes: large-scale proof of concept. Nat Neurosci. 2016;19(3):420–431. doi: 10.1038/nn.4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clementz BA., Sweeney JA., Hamm JP., et al Identification of distinct psychosis biotypes using brain-based biomarkers. Am J Psychiatry. 2016;173(4):373–384. doi: 10.1176/appi.ajp.2015.14091200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cuthbert BN., Insel TR. Toward new approaches to psychotic disorders: the NIMH Research Domain Criteria project. Schizophr Bull. 2010;36(6):1061–1062. doi: 10.1093/schbul/sbq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Insel T., Cuthbert B., Garvey M., et al Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167(7):748–751. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 67.Lombardi LM., Baker SA., Zoghbi HY. MeCP2 disorders: from the clinic to mice and back. J Clin Invest. 2015;125(8):2914–2923. doi: 10.1172/JCI78167. [DOI] [PMC free article] [PubMed] [Google Scholar]