

Abstract
The influence of diet and environment on human health has been known since ages. Plant-derived natural bioactive compounds (phytochemicals) have acquired an important role in human diet as potent antioxidants and cancer chemopreventive agents. In past few decades, the role of epigenetic alterations such as DNA methylation, histone modifications and non-coding RNAs in the regulation of mammalian genome have been comprehensively addressed. Although the effects of dietary phytochemicals on gene expression and signaling pathways have been widely studied in cancer, the impact of these dietary compounds on mammalian epigenome is rapidly emerging. The present review outlines the role of different epigenetic mechanisms in the regulation and maintenance of mammalian genome and focuses on the role of dietary phytochemicals as epigenetic modifiers in cancer. Above all, the review focuses on summarizing the progress made thus far in cancer chemoprevention with dietary phytochemicals, the heightened interest and challenges in the future.
Keywords: cancer chemoprevention, dietary agents, epigenetics, DNA methylation, histone modification, microRNA, plant polyphenols
1.0 Introduction
The term ‘Epigenetics’ was coined by British developmental biologist Conrad Waddington, derived from the combination of word ‘epigenesis’ and ‘genetics’ [1]. Epigenetics is defined as the branch of biology which studies the casual interactions between genes and their products, where ‘Epi’ means above or over and ‘genetics’ reflects the involvement of genes and heredity. Today epigenetics may be defined as the study of chemical changes to the genome that are heritable and modulate gene expression or cellular phenotype through mechanisms involving how DNA is packaged and expressed without any alterations in the gene nucleotide sequence itself [1, 2]. Epigenetic mechanisms have been shown to be essential in regulating normal cellular functions and play important role during developmental stages. In mammals, DNA methylation is associated with several key processes such as genomic imprinting, X-chromosome inactivation and tissue-specific gene expression [3–6]. Histone methylation and acetylation patterns were demonstrated to be closely associated with cognitive functions such as long term memory formation and storage [7–9]. Epigenetic alterations have been implicated in several pathologies such as cancer, metabolic syndrome, Alzheimer’s disease and other neurological disorders [10–13]. Unlike genetic changes in the genome such as mutation, epigenetic modifications are potentially reversible and can be modified by environmental, dietary and lifestyle factors. Epigenetic mechanisms have been implicated in physiological responses to intrinsic and extrinsic environmental stimuli such as nutrition, radiation, exposure to chemicals, toxins and hormones [14, 15]. The epigenetic diet, or the control of epigenetic modifiers through consumption of dietary phytochemicals, is of extreme interest. Several studies suggest that dietary phytochemicals are not only an essential source of nutrients but also important in the elimination of cancer as a life-threatening disease [14, 15]. In this review, we focused on the role of dietary phytochemicals as epigenetic modifiers. We discussed in detail different mechanisms of epigenetic modifications in mammals and present a comprehensive overview of the current state of knowledge on dietary bioactive compounds and their influence on various epigenetic mechanisms highlighting their role as potential anticancer agents.
2.0 Mechanisms of epigenetic modifications
Although a number of epigenetic mechanisms have now been identified, in mammals there are three major epigenetic mechanisms which are known to regulate gene expression. These include DNA methylation, alteration of chromatin structure by post-translational modification of histone or non-histone proteins, and small non-coding microRNAs (miRNAs) that modulate gene expression by either inhibiting translation or causing targeted degradation of specific mRNAs [15, 16].
2.1 DNA methylation
Methylation of cytosine residues within the dinucleotide sequence-CG is one of the most widely studied epigenetic modifications in mammals [17]. Forming an essential component of the cellular epigenetic machinery, DNA methylation in cooperation with histone modification regulates gene expression by modulating DNA packaging and chromatin architecture [18, 19]. DNA methylation is a chemical modification that involves the transfer of a methyl (CH3) moiety from the donor S-adenosyl methionine (SAM) to the 5′ position of cytosine residue that precedes guanine in the CG dinucleotide sequence, forming 5-methyl cytosine and S-adenosyl-L-homocysteine (SAH) [17, 20–22]. The mammalian genome has been reported to harbor 3×107 methylated cytosine residues mostly within CG dinucleotide sequences [21]. Although CG sequences are unevenly distributed throughout the human genome, they are frequently enriched in gene promoters (often referred as CpG islands) and large repetitive sequences such as LINE and ALU retrotransposon elements [23]. In mammals, most CpG islands are methylated on cytosine residues by a group of enzymes known as DNA methyltransferases (DNMTs), whereas CpG islands within gene promoters tend to be protected from methylation [17, 23]. In cancer, increased DNA methylation has been observed in the CpG islands in the promoter region of some genes that function as a ‘switch’ resulting in gene silencing. Although no mutation or deficiency in any DNMTs has been identified as causally linked to tumor development, most likely because of their critical role during embryogenesis. So far, there are three major DNA methyltransferases (DNMT1, DNMT3a and DNMT3b) identified in mammals. DNMT1 enzyme has been demonstrated to have a 5–30 fold more preference for hemi-methylated substrates and therefore popularly designated as maintenance methyltransferase. It preserves the existing methylation patterns in the daughter DNA strands by adding methyl groups to hemi-methylated CpG sequences following replication. DNMT1 has been demonstrated to be involved in de novo methylation activity in embryo lysates and its sequence specificity was shown to be confined to 5′-CpG-3′ dinucleotide sequence with little dependence on sequence context or density [24]. DNMT3a and DNMT3b enzymes are essential for global de novo methylation as they preferentially target unmethylated CpG sequences [25]. Although DNMT3L, the fourth family member, lacks intrinsic DNMT activity by itself, it co-localizes with DNMT3a and DNMT3b to establish genomic imprints in maternal germ line and facilitate methylation of retroposons [26–28]. DNMT2, another enigmatic member of DNMT family, was found to lack any biochemically detectable DNMT activity. Evidences from phenotypic analyses of mice with mutant DNMT genes have provided useful mechanistic insights into the role and establishment of DNA methylation patterns during development [15, 21].
Hypermethylation of CpG islands is usually associated with gene silencing. There are multiple routes through which DNA methylation can suppress transcription. A general mechanism is to exclude binding of proteins that modulate transcription through their DNA binding domains [29]. This mechanism has been demonstrated to be essential for imprinting of Igf2 gene [30]. CpG methylation has also been shown to block the binding of several other transcription factors, however their biological consequences remain unknown [31]. Another mechanism for DNA methylation mediated gene repression involve binding of specialized DNA binding proteins to the methylated CpG stretches, which form repressor complexes with histone deacetylases (HDACs) and cause chromatin compaction [32–34]. In mammals six methyl-CpG-binding proteins have been characterized till date, which include MeCp2, MBD1-4 and Kaiso. Studies demonstrate that all of them (except mammalian MBD3) possess a domain that specifically targets them to methylated CpG regions in vitro and in vivo [33, 35, 36]. Several detailed reviews are available discussing about DNA methylation, their role in cancer and development as biomarkers, which is currently beyond the scope of this review [37–39].
2.2 Histone Modifications
In addition to DNA methylation, post-translational modification of N-terminal histone tails play a significant role in epigenetic regulation of gene expression [40, 41]. A typical nucleosome unit consists of ~146 bp of DNA wrapped around an octamer of histones (H2A, H2B, H3 and H4) represent the fundamental building unit of eukaryotic chromatin. A diverse array of covalent chemical modification of less structured, protruding N-terminal tails of core histones by methylation, acetylation, ubiquitination, phosphorylation, sumoylation and ADP-ribosylation dictate the dynamics of chromatin state [42]. Euchromatin is lightly packed form of chromatin where DNA is accessible for transcription whereas heterochromatin represents tightly packed chromatin state inaccessible to cellular transcriptional machinery. Most of the chemical modifications occur at lysine (K), arginine (R) and serine (S) residues within the histone tails. These distinct histone modifications on one or more histone tails (often referred to as ‘histone code’) which may act sequentially or in combination are recognized by other proteins that signal further downstream events.
A number of enzymes have been implicated in catalyzing (addition or removal) various histone modifications. Examples include histone acetyltransferases (HATs), histone deacetylases (HDACs), histone methyltransferases (HMTs), histone demethylases (HDMs), histone kinases etc. In brief, HATs catalyze the addition of acetyl group on the ε-amino group of lysine residues in the N-terminal tail of histones, which neutralize the positive charge, relax the chromatin and facilitate binding of transcriptional machinery to the DNA [43]. Till date, 25 HATs have been characterized which are divided into four families. Examples include GNAT (hGCN5, PCAF), MYST (MYST, Tip60), p300/CBP (p300/CBP, SRC (SRC-1) and TAFII250 families (TAFII250) [44, 45]. In contrast, HDACs catalyze the removal of acetyl groups from K residues resulting in the compaction of chromatin configuration repressing gene transcription [45]. HDACs are classified into four groups. HDAC-1, -2, -3 and -8 are members of class I HDAC family while HDAC-4, -5, -6, -7, -9 and -10 belong to class II HDAC family. HDAC-11 belongs to class IV HDAC group. Sirtuins which require NAD+ as cofactor for their activity and are structurally unrelated to other HDAC classes, constitute class III HDAC family [46, 47]. HMTs catalyze the addition of methyl groups to K or R residues while HDMs act to remove them [45–47]. Examples of histone lysine methyltransferase include EZH2 (enhancer of zeste homolog 2) and that of histone lysine demethylase include LSD1 (lysine specific demethylase 1) [48, 49]. Depending on the site of lysine methylation (K4, K9, K27 in histone H3) and methylation status (mono, di or tri methylation), histone methylation may have activating or repressive effect on gene expression [41, 49]. H3K4, H3K36 and H3K79 methylation have activating effects on gene transcription whereas methylation of H3K9, H3K27 and H4K20 is generally associated with gene silencing or transcriptional repression [41, 47, 50]. Several excellent reviews are available on each group of histone modifying enzymes, their mechanism of action and various histone modifications for biomarker and assay development, which is beyond the scope of this review [51, 52].
2.3 Non-coding RNAs
Recent evidences indicate that non-coding RNA (ncRNA) transcripts play fundamental role in epigenetic regulation of gene expression and have been implicated in various epigenetic mechanisms such as transposon silencing, X-chromosome inactivation, DNA imprinting, and paramutation [53–55]. In humans, ncRNAs include microRNA (miRNA), small interfering RNA (siRNA), piwi-interacting RNA (piRNA) which accounts for majority of transcripts, representing approximately 98% of all human transcriptional output [56, 57]. Based on the size, ncRNA can be classified into small ncRNA which are generally less than 200 nucleotides in length and long ncRNA (lncRNA) transcripts that are more than 200 nucleotides in length. Small ncRNAs particularly miRNAs regulate key epigenetic mechanisms. MiRNAs are known to regulate various components of cellular epigenetic machinery particularly polycomb complexes and thus affect multiple downstream effects [53, 58–60]. One such example include miR-214 which downregulates EZH2 expression by targeting its 3′-UTR region and accelerates skeletal muscle differentiation and transcription of developmental regulators in embryonic stem cells [61]. There are other miRNAs which have been implicated in the repression of Bmi1, a component of PRC1 complex [62–64]. DNA methylation has also been shown to be modulated by miRNAs. DNMT1 and 3 have been reported to be targeted by the miR-29 family in lung cancer and leukemia cells [65, 66]. In summary, recent evidences suggest that ncRNAs have emerged has key regulators of epigenetic mechanisms and also, that the modulation of these RNA transcripts by the same epigenetic processes may lead to major consequences. Several excellent publications are available on each group of ncRNAs, their mechanism of action affecting gene regulation, which is beyond the scope of this review [67, 68].
3.0 Epigenetic mechanisms regulated by dietary phytochemicals
Unlike genetic modifications, epigenetic states of genes are reversible and can be altered by certain intrinsic and extrinsic factors. This characteristic of epigenetic mechanism may lead to the development of abnormal phenotype as well as regulate physiological response to some environmental stimuli, diet or therapeutic intervention [15]. In past two decades, accumulated evidences show that dietary phytochemicals present in abundance in fruits, vegetables and beverages which were earlier known only for their antioxidant or chemopreventive effects are also potent epigenetic regulators, which could target or revert abnormal epigenetic modifications in various human pathologies including cancer [20, 69–77]. Here we describe some common dietary phytochemicals, their structure and molecular weight, which are potent epigenetic modifiers (Table 1). These dietary phytochemicals have shown potential as DNMT inhibitors, histone modifiers and vary miRNA level altering gene expression and restoring the expression of various tumor suppressor genes which are summarized in Tables 2–4. We also discuss the progress made on some potential dietary phytochemicals in clinical trials (Table 5). Some popular dietary phytochemicals are discussed in detail below.
Table 1.
Structure, IUPAC name and molecular weight of some common dietary phytochemicals.
| Dietary Agent | Structure | IUPAC Name | Molecular Weight (g/mol) |
|---|---|---|---|
|
| |||
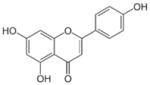
| Apigenin |
|
5,7-dihydroxy-2-(4-hydroxyphenyl)chromen-4-one | 270.23 |
|
| |||
| Curcumin |
|
(1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione | 368.37 |
|
| |||
| Green tea polyphenols | |||
| Epicatechin |
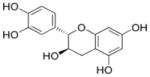
|
(2R,3R)-2-(3,4-dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol | 290.26 |
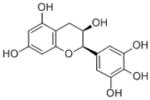
| Epigallocatechin |
|
(2R,3R)-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol | 306.27 |
|
| |||
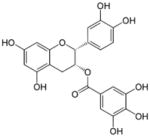
| Epicatechin gallate |
|
[(2R,3R)-2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate | 442.37 |
| Epigallocatechin gallate |
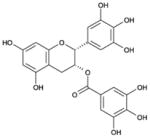
|
[(2R,3R)-5,7-dihydroxy-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-3-yl] 3,4,5-trihydroxybenzoate | 458.37 |
|
| |||
| Soy isoflavones | |||
| Biochanin A |
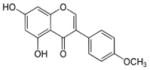
|
5,7-dihydroxy-3-(4-methoxyphenyl)chromen-4-one | 284.26 |
| Daidezein |
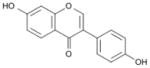
|
7-hydroxy-3-(4-hydroxyphenyl)chromen-4-one | 254.23 |
|
| |||
| Genistein |
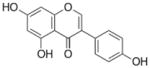
|
5,7-dihydroxy-3-(4-hydroxyphenyl)chromen-4-one | 270.23 |
|
| |||
| Lycopene |
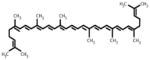
|
(6E,8E,10E,12E,14E,16 E,18E,20E,22E,24E,26E)-2,6,10,14,19,23,27,31-octamethyldotriaconta-2,6,8,10,12,14,16,18,20, 22,24,26,30-tridecaene | 536.87 |
|
| |||
| Quercetin |
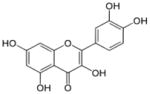
|
2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxychromen-4-one | 302.23 |
|
| |||
| Resveratrol |
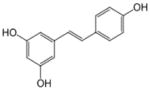
|
5-[(E)-2-(4-hydroxyphenyl)ethenyl]b enzene-1,3-diol | 228.24 |
|
| |||
| Sulforaphane |
|
1-isothiocyanato-4-methylsulfinylbutane | 177.28 |
Table 2.
Dietary phytochemicals altering DNA methylation.
| Dietary Phytochemical | Molecular mechanism(s) | Target genes | Preclinical Model | Disease Type | Dose/Concentration | Ref. |
|---|---|---|---|---|---|---|
|
| ||||||
| Apigenin | DNMT inhibitor | NA | Esophageal cells | NA | 20–50 μM | 85 |
| DNMT inhibitor | Decreases CpG hypermethylation in Nrf2 promoter | Skin cancer (mouse skin) | Cancer | (1.56–50 μM) | 87 | |
|
| ||||||
| Curcumin | DNMT inhibitor | NA | Esophageal Leukemia |
Cancer | 3–50 μM | 85 |
|
| ||||||
| Catechin | DNMT inhibitor | RARβ | Breast | 5–50 μM | 204 | |
| Epicatechin | DNMT inhibitor | Breast | 50 μM | 204, 113 | ||
| Epicatechin-gallate | DNMT inhibitor | Esophageal | 20–50 μM | 204, 113 | ||
| Epigalocatechin-3-gallate | DNMT inhibitor | RARβ, MGMT, MLH1, CDKN2A, RECK,TERT, RXRα,CDX2, GSTP1, W1F1 | Esophageal, Oral, Prostate, Lung, Colon cancer cells | Cancer | 20–100 μM: 0.3–06% | 109, 205, 206, 207, 208 |
|
| ||||||
| Biochanin A | DNMT inhibitor | NA | Esophageal, Prostate | Cancer | 20–100 μM | 85, |
| Daidezein | DNMT inhibitor | NA | Breast | 20–40 μM | 113, 124, 126 | |
| Genistein | DNMT inhibitor |
RARβ, MGMT, CDKN2A, GSTP1, HMGNS, BTG3 RXRα, CDX2, GSTP1, W1F1 |
Esophageal Prostate Tumors (Mice) |
50–300 mg/kg 3.75–100 μM |
85, 124, 126, 127, 128, 129, 131, 135, 209, 210, 211 | |
|
| ||||||
| Lycopene | Unknown | GSTP1, RARβ, HIN-1 | Breast | Cancer | 2 μM | 126 |
| DNMT inhibitor | Activation of GSTP1 promoter | Prostate | 5–40 μM | |||
|
| ||||||
| Quercetin | DNMT inhibitor | CDKN2A | Esophageal Breast Colon |
Cancer | 5–20 μM | 165 |
|
| ||||||
| Resveratrol | DNMT inhibitor | NA | Breast, Lungs | Cancer | 20–40 μM | 212, 213 |
|
| ||||||
| Sulforaphane | DNMT inhibitor | NA | Esophageal, Colon | Cancer | 50 μM | 212 |
NA, information not available
Table 4.
Dietary phytochemicals and changes in miRNA levels.
| Dietary Agent | Disease Type | miRNA | Concentration | Ref. |
|---|---|---|---|---|
|
| ||||
| Apigenin | Lung cancer cells | miR138, miR-125a-5p | 25–100μM | 230 |
| Neuroblastoma cells | miR-138 | 100 μM | 229 | |
|
| ||||
| Curcumin | Bladder cancer cells | miR-203 | 1–20 μM | 231 |
| Lung cancer cells | miR-320, miR-26a, let-7i, miR-130a, mir-16, miR-125b,miR-23a, miR-27b, miR-155, miR-625, miR-576-3p, miR-186, miR-9 | 10–50 μM | 232 | |
| Leukemia | miR-15a, miR-16-1 | 5–20 μM | 233 | |
| Pancreatic cancer cells | miR-103, miR-140, miR-146b, miR-148, miR-15b,miR-181a, miR-181b, miR-181d, miR-195, miR-196a,miR-199a, miR-19a,miR-204, miR-20a, miR-21, miR-22miR-23a,b, miR-24, miR-25, miR-26a, miR-27a, miR-34a,miR-374, miR-510, miR-7, miR-92, miR-93, miR-98 | 10 μmol/L | 234 | |
|
| ||||
| EGCG | Neuroblastoma cells | miR-92, miR-93,miR 106b, miR-34a, miR-99a | 25–100 μM | 228 |
| Hepatocellular carcinoma cells | miR-467b, miR-487b, miR-197, miR-805, miR-374, let-7f, miR-350, miR-24-1, miR-137, miR-335-3p, let-7a, miR-222, miR-26b, miR-30c-1, let-7d, miR-98, miR-30c, miR-30b, miR-32, miR-674, miR-532-5p, let-7g, miR-18a, miR-192, miR-302d, miR-30b, miR-802, let-7e,miR-322, miR-720, miR-146b, miR-340-3p, miR-185, miR-425, miR-10a, miR-126-5p, miR-101a, miR-30e, let-7c, miR-141, miR-33, miR-29a, miR-199b, miR-450a-5p, miR-21, miR-23a, miR-101b, miR-148a, miR-193, miR-23b, miR-107, miR-140, miR-551b, miR-466c-5p, miR-106a, miR-590-3p, miR-875-3p, miR-224, miR-292-5p, miR-678, miR-469, let-7b, miR-463miR-574-3p, miR-201, miR-290-3p, miR-181a, miR-302a, miR-429, miR-133a, miR-190b, miR-710, miR-135b, miR-296-5p, miR-191, miR-188-5p, miR-298, miR-181a-1, miR-466g, miR-26b, miR-466f-3p, miR-29b, miR-1224, miR-291b-5p, miR-324-5p, miR-486, miR-128, miR-450b-3p, miR-135a, miR-294, miR-671-5p, miR-878-3p, miR-801, miR-370, miR-1, miR-494, miR-133b |
100 μM | 235 | |
| Lung carcinoma | let-7c, miR210 | 100 μM | 235 | |
| Pancreatic cancer cells | let-7c | 100 μM | 235 | |
| Colon carcinoma cells | miR-27a, miR-20A, miR-17-5P, miR-21 | 5–30 μM | 236 | |
|
| ||||
| Genistein | Ovarian cancer cells | miR-100, miR-122a, miR-125b, miR-126, miR-135, miR-135b, miR-136, miR-137, miR-141, miR-152,miR-190, miR-196a, miR-196b, miR-204, miR-205, miR-206, miR-217, miR-22, miR-296, miR-30a-3p, miR-30a-5p, miR-331, miR-335, miR-342, miR-362, miR-449b, miR-454, miR-497, miR-500, miR-501, miR-503, miR-515, miR-517c, miR-532, miR-565, miR-578, miR-584, miR-585, miR-590, miR-595, miR-625, miR-647, miR-7, miR-765, miR-766 | 5 μM | 136 |
| Pancreatic cancer cells | miR-200 | 50 μM | 137 | |
|
| ||||
| Indole-3 carbinol Diindolylmethane | Breast cancer cells | miR34a | 25 μM | 137 |
| Prostate cancer cells | miR34a | 10 μM | 238 | |
| miR-21 | 30–60 μM for cells 5mg/kg (mice) |
237 | ||
| Mouse lung tumors | miR-21, mir-31, miR-130a, miR-146b and miR-377 | 100–150 μM | 239 | |
|
| ||||
| Lycopene | Hepa 1–6 cells (hepatic cells) | miR-21 | 0.05% (fed orally to mice) | 240 |
|
| ||||
| Diallyl disulphide | Endothelial progenitor cells | miR-221 | 0–10 μM (10mg/kg/day) | 241 |
| Breast Cancer | miR34a | 25–400 μM | 242 | |
|
| ||||
| Resveratrol | Prostate cancer cells from lymph node | miR-1224-5p, miR-1228, miR-1231, miR-1246, miR-1260, miR-1267, miR-1268, miR-129, miR-1290, miR-1308, miR-1469, miR-149, miR-150, miR-152, miR-15a, miR-17, miR-1825, miR-185, miR-18b, miR-1908, miR-1915, miR-197, miR-1972, miR-1973, miR-1974, miR-1975, miR-1977, miR-1979, miR-20a, miR-20b, miR-24, miR-296-5p, miR-483-5p, miR-513a-5p, miR-548q, miR-572, miR-575, miR-612, miR-638, miR-654-5p, miR-659, miR-671-5p, miR-7, miR-762, miR-764, miR-874, miR-92b, miR-939 | 50 μM | 243 |
| Prostate cancer cells | miR-101, miR-106a, miR-106b, miR-1274b, miR-136, miR-141, miR-145, miR-17, miR-182, miR-1826, miR-200b, miR-200c, miR-20a, miR-20b, miR-21, miR-214, miR-221, miR-222, miR-302c, miR-375, miR-378, miR-720, miR-768-3, miR-93 | 5–100 μM | 244 | |
| Colorectal cancer cells | miR-1, miR-100-1/2, miR-102, miR-103-1, miR-103-2, miR-146a, miR-146b-5p, miR-16-0, miR-17, miR-181a2, miR-194-2, miR-196a1, miR-205, miR-206, miR-21, miR-23a, miR-23b, miR-25, miR-26a, miR-29c, miR-30a-3p, miR-30c-1, miR-30d, miR-30e-5p, miR-323, miR-340, miR-363-5p, miR-424, miR-494, miR-497, miR-560, miR-560, miR-565, miR-565, miR-572, miR-574, miR-594, miR-615, miR-622, miR-629, miR-631, miR-638, miR-639, miR-657, miR-659, miR-663, miR-801, miR-92a-2 | 50 μM | 245 | |
|
| ||||
| Luteolin | Prostate cancer cells | miR-630 | 0–10 μM | 246 |
| Gastric cancer cells | miR-34a | 5–50 μM | 247 | |
|
| ||||
| Phenethyl isothiocyanate | Prostate cancer cells | miR-141 | 0–20 μM | 248 |
Table 5.
Clinical trials conducted with dietary phytochemicals.
| Dietary Agent | Molecular mechanism | Disease conditions | Dose | Endpoints/Outcome | Ref. |
|---|---|---|---|---|---|
|
| |||||
| Curcumin | DNMT inhibitor | Prostate cancer | 0.1 g/day | Curcumin modulates PSA production in combination with other isoflavones | 249 |
| DNMT inhibitor | Colorectal cancer | 0.036–0.18 g/day | Pilot study shows safe administration in patients up to an equivalent dose of 180 mg. It has low bioavailability in humans and undergoes intestinal metabolism. | 250 | |
|
| |||||
| Catechin | DNMT inhibitor | Prostate cancer | NA | May decrease risk for advanced prostate cancer. | 251 |
| Epicatechin | DNMT inhibitor | 25.5 mg | Studies finds low bioavailability and/or bioaccumulation of green tea polyphenols in prostate tissue with statistically insignificant changes in systemic and tissue biomarkers. Green tea polyphenol activity may be indirect or need evaluation with longer intervention durations, repeated dosing, or in patients at earlier stages of the disease. | 252 | |
| Epicatechin-gallate | DNMT inhibitor | 39.8 mg | |||
| Epigalocatechin-3-gallate | DNMT inhibitor | 200 mg | |||
|
| |||||
| Genistein | DNMT inhibitor | Prostate cancer | 30 mg | Genistein intake by early prostate cancer patients modulates several biomarkers and has an inhibitory effect on androgen related biomarker. | 253 |
| Decreases MBD1, MBD4, MeCP2 expression | May induce cell cycle arrest and inhibit proliferation | 254 | |||
|
| |||||
| Daidzein | HDAC inhibitor | Prostate cancer | 0.02 mg | Safety and tolerability of isoflavone intake are proven. The results support the value of isoflavone treatment for prostate cancer risk reduction. | 254 |
|
| |||||
| Indole-3 carbinol | HDAC inhibitor | Prostate cancer | 225 mg | BR-DIM was well tolerated in prostate cancer patients. Showed variable results on PSA levels. | 255 |
|
| |||||
| Lycopene | DNMT inhibitor | Prostate cancer | 20–25 μM | Prostatic lycopene levels were low after supplementation in prostate cancer patients and no change in rate of HGPIN progression to prostate cancer. | 256 |
|
| |||||
| Quercetin | DNMT inhibitor | Renal cancer | 1400 mg/m2 | Plasma levels of quercetin inhibited lymphocyte tyrosine kinase activity, with evidence of antitumor activity. | 257 |
|
| |||||
| Sulforaphane | Decreases DNMT expression | Prostate cancer | 200 μmoles/day | PSA levels were not affected significantly. | 258 |
NA, information not available
3.1 Apigenin
Apigenin is a dietary plant flavonoid, chemically known as 4′,5,7,-trihydroxyflavone, present in common fruits and vegetables such as parsley, onions, oranges, tea, chamomile and wheat sprouts [78]. Previous studies have confirmed that apigenin possess antioxidant, anticancer, anti-inflammatory and anti-mutagenic properties [79–84]. However, the effect of apigenin on epigenetic-related enzymes and their mechanisms was not recognized until recently. Apigenin treatment (20 and 50 μM) was shown to cause a marked decrease in DNMT activity in vitro [85]. Studies from our laboratory demonstrated that apigenin-mediated growth arrest and apoptosis in human prostate cancer PC-3 and 22Rv1 cells was due to inhibition of class I HDACs [86]. In vivo studies using PC-3 xenografts in athymic nude mice further confirmed that oral intake of 20 and 50 μg/day apigenin over an 8-week period reduces tumor burden, HDAC activity and HDAC-1 and HDAC-3 protein levels. HDAC-1 and HDAC-3 mRNA and protein levels were found to be significantly decreased in these cells after 20–40 μM apigenin treatment with consequential increase in global histone H3 and H4 acetylation. A corresponding elevation in p21/waf1 and Bax levels was observed in both in vitro and in vivo studies, which resulted in induction of apoptosis and cell cycle arrest in tumor cells. In a recent study by Paredes-Gonzalez et al. [87], apigenin was shown to reactivate Nrf2 gene which encodes a key transcription factor known to regulate anti-oxidative defense system and skin homeostasis, in mouse skin epidermal JB6 P+ cells via epigenetic mechanisms. Hypermethylation of 15 CpG sites in Nrf2 promoter was demonstrated to be reversed by apigenin treatment in a dose-dependent manner. Apigenin exposure also resulted in decrease expression of DNMT1, DNMT3A, DNMT3B and HDAC 1–8 levels. Furthermore, nuclear localization of Nrf2 was enhanced along with increase expression of Nrf2 and its target gene NQO1 after apigenin treatment.
3.2 Curcumin
Curcumin [1,7-bis (4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione] is the major active component of popular South Asian spice turmeric (Curcuma longa), belonging to Zingiberaceae (ginger) family. The majority of health-benefits associated with turmeric intake has been attributed to curcumin which include anti-microbial, antioxidant, anti-inflammatory, anti-allergic and anti-carcinogenic effects [88–91]. At the molecular level, curcumin has been shown to modulate multiple intracellular pathways associated with proliferation, survival, invasion, apoptosis and inflammation [92]. In the context of epigenetic pathways, several studies have reported curcumin to be a potent modulator of DNMTs, histone modifying enzymes including HDACs and HATs and miRNAs [93]. In silico molecular docking studies of curcumin with DNMT1 revealed its ability to block and/or inhibit the catalytic thiol group of C1226 binding site in the enzyme resulting in decreased DNMT activity [93]. This study was further validated by in vitro experimental studies which showed curcumin to be a potent DNA hypo-methylating agent [94]. Curcumin was reported to be an effective HDAC inhibitor in HeLa nuclear extracts with an IC50 value of 115 μM. Docking studies performed with curcumin binding to HDAC-8 revealed it to be more potent inhibitor of HDAC than known pharmacological inhibitors such as sodium butyrate and valproic acid [95]. Another study reported that curcumin treatment to B-NHL Raji cells reduces HDAC-1, HDAC-3 and HDAC-8 protein expression in a dose-dependent manner and increases H4 acetylation in these cells [96]. Consistence with earlier findings, studies by Chen et al. [97] reported significant reduction in p300/CREB binding protein (CBP), HDAC-1 and HDAC-3 levels after exposure of Raji cells to curcumin. This decrease in HDACs and CBP levels were implicated in curcumin-mediated repression of NF-κB and Notch1, which resulted in decrease proliferation of Raji cells. Studies revealed curcumin as a specific inhibitor of p300/CBP HAT, which emerged as a novel target for cancer treatment [98–100]. Curcumin treatment caused proteasomal degradation of p300 and other closely related CBP proteins with no effect on HATs such as GCN5 and PCAF [100]. In addition, curcumin-mediated inhibition of p300/CBP was found to block HDAC inhibitor MS-275 induced histone hyper-acetylation in prostate cancer PC3-M cells and peripheral blood lymphocytes. Curcumin has been shown to epigenetically up-regulate DLEC1 and reduce CpG methylation in HT29 cells, lowering protein expression of DNMTs and HDACs attenuating clonogenicity of human colorectal adenocarcinoma HT29 cells [101]. In another study, curcumin has been shown to act as dual agent in causing apoptosis and reversal of promoter methylation of p15 gene in ALL leukemia cells [102].
The chemotherapeutic and chemopreventive effects of curcumin has also been closely linked to its ability to modulate miRNAs in cancer cells. A microarray based study of the effect of curcumin (10 μM) on the miRNA profile in pancreatic cancer PxBC-3 cells showed significant changes in the expression of 29 miRNAs (11 upregulated and 18 downregulated) after 72 h treatment [103]. Further studies confirmed that miR-22, which has tumor suppressive function, was upregulated after exposure to curcumin and its downstream target genes SP1 and ESR1 were suppressed in these cells. Ali et al. [104] demonstrated that treatment of pancreatic cancer cells with curcumin and its analogue CDF could induce gemcitabine sensitivity via induction of miR-200 and inactivation of miR-21 expression. These studies underline the importance of curcumin-mediated modulation of miRNAs as an important mechanism for its biological effects. Curcumin has been reported to reduce DNMT3B activation leading to hypomethylation of RARβ resulting in upregulation of its expression [105]. Dendrasomal curcumin has been shown to induce DNA hypomethylation in part via up-regulation of epi-miRs, leading to the re-expression of silenced tumor suppressor genes including MEG3, and suppress oncogenes including HOTAIR, targeting histone modifying complexes in hepatocellular carcinoma HCC cells [106].
3.3 Green tea polyphenols and epigallocatechin-3-gallate (EGCG)
EGCG, the major constituent present in green tea, is widely known for its health-related benefits. Other major constituents present in green tea are epicatechin-3-gallate, epigallocatechin, and epicatechin. These polyphenols have been shown to possess multifaceted effect on various cellular targets at the molecular level including epigenetic pathways or enzymes [107, 108]. Several studies have demonstrated that EGCG is a potent demethylating agent which inhibits enzymes involved in DNA methylation and is an effective histone modifying agent [109–116]. It is well known that CpG island hypermethylation at the promoter region leads to epigenetic repression of several critical tumor suppressor genes during tumorigenesis. Fang et al. [109] reported that EGCG acts as a competitive inhibitor of DNMT (Ki=6.89 μM), which binds to the catalytic pocket and inhibit DNMT activity in a dose-dependent manner. Furthermore, EGCG treatment (5–50 μM for 12–144 h) was found to effectively reactivate methylation-silenced genes viz. p16 (INK4a), retinoic acid receptor beta (RARβ), O(6)-methylguanine methyltransferase (MGMT), and human mutL homologue 1 (hMLH1), in human esophageal cancer KYSE 510 cells. EGCG was also reported to inhibit HDACs and increase permissive or active histone modifications such as histone acetylation at the target gene promoters. Studies from our laboratory by Pandey et al. [110] showed that exposure of prostate cancer cells to green tea polyphenols (GTP) caused re-expression of epigenetically silenced glutathione-S transferase pi (GSTP1) gene which correlated with the promoter demethylation due to DNMT1 inhibition and histone modification at the promoter region. However, GTP treatment did not show any global hypomethylation effect which could result in genomic instability assessed by the methylation status of LINE-1 promoter remained unaffected. GTP treatment decreased mRNA and protein levels of MBD1, MBD4, MeCP2, HDAC 1–3 whereas acetylated histone H3 (LysH9/18) and H4 were found to be elevated. Another study from our group by Thakur et al. [111], demonstrated that GTP treatment caused cell cycle arrest and apoptosis by inducing proteasomal degradation of class I HDACs in human prostate cancer cells. Studies by Li et al. [112] demonstrated that EGCG in combination with class I HDAC inhibitor, trichostatin A (TSA) could synergistically reactivate ERα expression in ERα negative MDA-MB-231 breast cancer cells by modulating histone methylation and acetylation pattern at the gene promoter. In addition, they also reported that treatment with EGCG and/or TSA contributes to transcriptional activation of ERα by causing a decreased binding of transcription repressor complex, Rb/p130-E2F4/5-HDAC1-SUV39H1-DNMT1 to the regulatory region of the gene. Nandkumar et al. [113] demonstrated that EGCG reactivates silenced tumor suppressor genes Cip1/p21 and p16INK4a by reduction in DNA methylation and increase in histone acetylation in human skin cancer cells.
EGCG has been reported to modulate polycomb proteins such as Bmi-1 and EZH2 [114, 115]. EGCG alone or in combination with 3-Deazaneplanocin A (DZNep), an EZH2 inhibitor was shown to decrease polycomb group proteins (PcG) proteins including EZH2, EED, SUZ12, MEL18 and Bmi-1 via a mechanism involving proteasome associated degradation. The reduction in PcG proteins correlated with a decrease in repressive chromatin marks viz. H3K27me3 and H2AK119ub and HDAC-1 whereas accumulation of acetylated H3 levels was found to be elevated in skin cancer SCC-13 and human epidermoid carcinoma A431 cells [114, 115]. In a recent study form our group, Deb et al. [116] reported that EGCG and/or GTP treatment to breast cancer cells induced expression of epigenetically repressed TIMP-3 gene, which is mediated by modulating epigenetic mechanisms involving EZH2 and class I HDACs independent of the promoter DNA methylation. After EGCG or GTP treatment, protein levels of class I HDACs and EZH2 were significantly reduced. Interestingly, transcriptional activation of TIMP-3 was associated with decreased EZH2 localization and H3K27me3 at the promoter with a concomitant elevation in H3K9/18 acetylation levels. Time-dependent exposure to EGCG resulted in reactivation of known tumor-suppressor genes in HeLa cells due to marked changes in the methylation of the promoter regions of these genes [117]. In cystic and adenoid cystic carcinoma SACC83 cells, EGCG significantly enhanced the protein and mRNA expression levels of RECK, and markedly reduced the invasive ability of these cells [118]. Jha et al. [119] demonstrated the dual inhibitory effect of EGCG in oral squamous carcinoma cells. In this study, EGCG has been reported to efficaciously reduce DNA methylation catalyzed by SssI methyltransferase and human DNMT1 and inhibited DNMT activity by increasing the level of SAH in the methylation reaction catalyzed by catechol-O-methyl transferase (COMT). Additionally, EGCG also interacted directly with the catalytic site of human DNMT1. Chen et al. [120] performed a genome wide methylation and mRNA expression screen in oral squamous carcinoma OSCC cells after EGCG treatment that revealed a total of 761 differentially methylated gene loci. Comparison of gene expression profiling revealed 184 transcripts with a significant difference and fold change. Gene ontology analysis of differentially methylated loci and functional annotation of differentially expressed genes indicated that the main pathways involved were metabolic, mitogen activated protein kinase (MAPK), Wnt signaling, and cell cycle pathways suggesting EGCG can affect the methylation status and gene expression.
3.4 Genistein and soy isoflavones
Genistein is a phyto-estrogen belonging to isoflavone group. Isoflavones such as genistein, biochanin A and daidzein occur naturally in plant sources such as soybeans, fava beans, lupine, kudzu, psoralea etc. [121, 122]. Till date, numerous epidemiological and experimental studies have demonstrated the chemopreventive effects of genistein and other isoflavones on various cancer types including breast and prostate cancer [123]. In recent years, the role of genistein and other soy isoflavones as epigenetic modulators regulating gene expression has been widely reported by several studies. Genistein has been shown to be a more potent DNMT inhibitor as compared to biochanin A or daidzein. Fang et al. [124] reported that genistein (2–20 μM) could reactivate methylation-silenced genes such as retinoic acid receptor beta (RARβ), p16INK4a, and MGMT in esophageal squamous carcinoma cells KYSE 510 and prostate cancer LNCaP and PC3 cells. Reactivation of such genes by direct inhibition of DNMT may be one of the factors contributing to the chemopreventive effects of soy isoflavones. In another study, Li et al. [125] demonstrated that genistein treatment in breast MCF10AT benign cells and MCF-7 cancer cells depletes telomerase (hTERT) activity through epigenetic modification which involves genistein-mediated decrease in DNMT1, DNMT3a and DNMT3b levels. Site-specific hypomethylation in hTERT promoter was found to increase the binding of E2F-1 transcription factor. Furthermore, genistein was shown to repress hTERT promoter by chromatin remodeling which involved increase in trimethyl-H3K9 enrichment with a concomitant decrease in dimetyl-H3K4 chromatin marks. Studies by King-Batoon et al. [126] showed that low, non-toxic dose of genistein (3.125 μM, re-supplemented every 48 h for 1 week) could partially demethylate GSTP1, a tumor suppressor gene, in MCF-7, MDA-MB-468 and MCF10A breast cells. Similar in vitro studies on other cancer types provide evidence that genistein is a potent demethylating as well as histone modifying agent, which could reverse the silenced state of critical tumor suppressor genes [127–129]. Genistein, daidzein, daidzein metabolite equol and AglyMax have been reported to induce ERα-mediated histone acetylation via modulation of HAT activity [130]. Majid et al. [131] demonstrated that genistein-mediated epigenetic induction of p21waf1/Cip1 and p16INK4a tumor suppressor genes in human prostate cancer cells involve chromatin modification via upregulation of HATs expression. In another study, Basak et al. [132] demonstrated that androgen receptor (AR) down-regulation in prostate cancer LNCaP cells by genistein was attributed to the inhibition of HDAC6-Hsp90 co-chaperone function, which is required for AR protein stabilization. Genistein and daidzein treatment in prostate cancer cells caused alterations in the methylation profiles of 58 genes [133]. Both these agents caused modifications in the methylation frequencies of MAD1-like 1 (MAD1L1), tumor necrosis factor receptor-associated factor 7 (TRAF7), lysine (K)-specific demethylase 4B (KDM4B), and human telomerase reverse transcriptase (hTERT) genes. Furthermore, genistein treatment in prostate cancer cells increased ER-β levels reducing its promoter methylation that is required in preventing cell proliferation [134]. However, in vivo clinical studies were inconclusive and did not fall in line with the studies performed in cell line models. In a randomized trial study involving thirty four healthy pre-menopausal women to investigate the estrogenic and methylation effect of soy isoflavones (40 mg or 140 mg daily) through one menstrual cycle, Qin et al. [135] reported that considering all subjects or by dose there were no significant changes for any of the cancer related genes (p16INK4a, RASSF1A, RARβ2, ER, and CCND2). In fact, RARβ2 methylation was shown to increase post-treatment at circulating genistein levels greater than 600 ng/ml unlike the effect observed for genistein levels below 600 ng/ml. Similarly, CCND2 methylation was found to increase in subjects with post-treatment genistein levels above 200 ng/ml as opposed to the effects observed below this genistein levels. Changes in the methylation of other genes p16INK4a, RASSF1A and ER did not correlate with post-treatment genistein levels.
Genistein and other soy isoflavones have shown to module miRNAs as well. Parker et al. [136] performed miRNA profiling of genistein treated and untreated ovarian cancer UL-3A and UL-3B cells and found 53 differentially expressed miRNAs. Upregulation of miR-200 and let-7 by isoflavones was shown to down-regulate ZEB1, slug and vimentin and therefore cause reversal of epithelial to mesenchymal transition in gemcitabine resistant pancreatic cancer cells [137]. In human uveal melanoma cells, genistein treatment demonstrated significant growth inhibition by targeting miR-27a and its target ZBTB10 [138]. In colorectal cancer cells, genistein has been shown to reduce miR-95, Akt and SGK1 to inhibit cell growth while in breast cancer cells it increased the levels of miR-23b to attenuate proliferation [139, 140].
3.5 Indole-3-carbinol and diindolylmethane
Indole-3-carbinol (I3C) is a hydrolysis product derived from a glucosinolate catalyzed by the plant enzyme myrosinase, which is further metabolized under the acidic conditions in the stomach to form dimer 3,3′-diindolylmethane (DIM) [141–143]. These phytochemicals are present in cruciferous vegetables particularly Brassica genus which include broccoli, cabbage, cauliflower, brussel sprouts, mustard, radish etc. [144]. Both these phytochemicals have been shown to exert anticancer effects by down-regulating multiple cellular targets and inducing cell cycle arrest and apoptosis in various cancer types [144, 145]. A recent study by Li et al. [146] demonstrated that DIM selectively induce proteasomal degradation of class I HDACs (HDAC-1, -2, -3 and -8) with no effect on class II HDACs, both in human colon cancer cells in vitro and in vivo in tumor xenografts. Furthermore, depletion of class I HDACs was shown to relieve transcription repression of cyclin-dependent kinase inhibitors p21/waf1 and p27/Kip2, which ultimately resulted in G2 arrest and DNA damage triggered apoptosis. In another study, Beaver et al. [147] examined the effects of I3C and DIM on prostate cancer LNCaP and PC-3 cells, which differ in their androgen receptor status. DIM was shown to inhibit HDAC activity in both cell lines; however I3C demonstrated modest inhibition of HDAC activity in androgen-responsive LNCaP cells with no significant effect on androgen-refractory PC-3 cells. A significant decrease in HDAC-2 protein levels was reported in both cell lines after DIM treatment but the levels of HDAC-1, HDAC-3, HDAC-4, HDAC-6, and HDAC-8 remained unaltered. Interestingly, a recent study by Busbee et al. [148] reported that I3C and DIM may decrease activation, proliferation and cytokine production by staphylococcal enterotoxin B-induced T cells in vitro and in vivo by acting as class I HDAC inhibitors.
In a study to examine the effect of DIM on miRNA expression profile between gemcitabine-sensitive and gemcitabine-resistant pancreatic cancer cells, Li et al. [149] reported that DIM treatment caused significant upregulation of members of miR-200 and let-7 families. Furthermore, re-expression of miR-200 in gemcitabine-resistant cells was found to be associated with a corresponding downregulation of mesenchymal markers ZEB1, slug and vimentin and upregulation of epithelial marker E-cadherin, which lead to the reversal of EMT phenotype to epithelial morphology. In another study, DIM treatment was demonstrated to increase the expression of miR-146a which correlated with reduced expression of EGFR, MTA-2, IRAK-1, and NF-κB, ultimately resulting in the inhibition of pancreatic cancer cell invasion [150]. In human breast cancer MCF-7 (estrogen receptor positive) and MDA-MB-468 (p53 mutant) cells, DIM treatment was demonstrated to augment G2/M cell cycle arrest by down-regulating the expression of Cdk-2, Cdk-4 and Cdc25A [151]. In addition DIM treatment was shown to cause upregulation of miR-21 which negatively regulates Cdc25A resulting in decreased breast cancer cell proliferation. Similar studies were reported with I3C which targets the PTEN/AKT signaling pathway and downregulates miR-21. Down-regulation of miR-21 was shown to further reduce resistance of HCC cells to I3C through inhibition of PTEN/AKT pathway in vivo.
3.6 Lycopene
Lycopene is the bright red carotene pigment belonging to tetra terpenoids and a phytochemical which occur naturally in tomatoes, carrot, watermelon, papaya, cherries etc. It is a potent anti-oxidant and has been shown to modulate multiple genes involved in DNA repair, cell cycle control and apoptosis in breast cancer cells [152, 153]. Studies by King-Batoon et al. [126] reported the effects of lycopene on GSTP1 gene in breast cancer cells. Lycopene (2 μM for 1 week) was shown to induce GSTP1 expression and demethylate GSTP1 promoter in MDA-MB-468 cell line but not in MCF-7 breast cancer cells. The expression of other genes such as RARβ2 and HIN1 remained unaltered by lycopene treatment in MCF-7 and MDA-MB-468 breast cancer cells. Although this study has shown that lycopene may be DNA methylating agent but further studies are needed to decipher the lycopene-mediated effects on epigenetic mechanisms. The protective effect of lycopene on the prostate is different between androgen-responsive and androgen-refractory derived prostate cancer cells. Fu et al. [154] demonstrated that lycopene treatment significantly decreased methylation levels of GSTP1 promoter and increased the mRNA and protein levels of GSTP1 in an androgen-refractory PC-3 cells. It also caused decrease in DNMT3A expression in PC-3 cells while no such change were observed in LNCaP cells.
3.7 Organosulfur compounds
Vegetables such as onion, garlic, shallots etc. belonging to allium family possess various water and fat soluble organosulfur compounds (OSCs) which have been widely implicated for numerous health-related benefits including cancer prevention, improved immunity, cardiovascular health and anti–microbial activity [155, 156]. Conversion of alliin to allicin and other alkyl alkane-thiosulfinates by the enzyme alliinase generate OSCs, which further decompose to more stable sulfur compounds such as diallyl sulfide (DAS), diallyl disulfide (DADS) and diallyl trisulfide (DATS). S-allylmercaptocysteine (SAMC) is the active metabolite derived from DADS, both of which are finally converted to allyl mercaptan (AM) and other metabolites [157]. Accumulated evidences suggest that in human cancer cells these OSCs increase histone (H3/H4) acetylation, highlighting HDACs as their potential targets [157–159]. In colon cancer Caco-2 and HT-29 cells, DADS was reported to inhibit cell proliferation and cause G2 cell cycle arrest by inhibiting HDAC activity, inducing histone hyperacetylation as well as p21 (Waf1/cip1) expression [160]. Furthermore, treatment of human colon cancer cells with AM caused accumulation of acetylated histones in cellular chromatin and facilitated binding of transcription factor SP3, followed by recruitment of p53 at the p21/waf1 promoter. Similarly, a study by Nian et al. [161] where several garlic derived OSCs were tested or screened in vitro for their ability to inhibit HDACs. They reported that AM was the most potent HDAC inhibitor. Further experiments which include molecular modeling, structure-activity and enzyme kinetic studies confirmed competitive inhibition of HDAC-8 by AM (Ki=24 μM).
3.8 Phenethyl isothiocyanate (PEITC)
Isothiocyanates are metabolites of glucosinolates which occur naturally in cruciferous vegetables [19]. PEITC is a metabolite of gluconasturtin obtained from water cress, which has shown significant beneficial health-related effects particularly in cancer chemoprevention [162]. Like other natural phytochemicals, PEITC have been demonstrated to regulate epigenetic mechanisms by modulating DNA methylation and histone modifications. Studies by Wang et al. [163] reported dual action of PEITC treatment on DNA methylation as well as histone modifications in epigenetic re-activation of GSTP1 gene in prostate cancer cells. Exposure of prostate cancer LNCaP cells to PEITC inhibited HDAC activity and concurrently demethylate the promoter to restore the GSTP1 levels to that found in normal prostate cells. In another study, exposure of mouse erythroleukemic cells to allyl isothiocyanates was found to increase acetylation of histones, with no significant effect on histone deacetylation [158, 159].
3.9 Quercetin
Quercetin is a dietary flavonoid abundantly present in citrus fruits, onion and buckwheat. Studies have shown that this bio-flavonoid is a potent free radical scavenger or anti-oxidant as well as a chemopreventive agent with pro-apoptotic and growth inhibitory effects on cancer cells [164]. Tan et al. [165] investigated the in vitro effects of quercetin on human colon cancer RKO cells and showed that the hypermethylation status of p16INK4a gene could be completely reversed after quercetin treatment in a dose-dependent manner. In another study, quercetin was shown to downregulate tumor necrosis factor (TNF)-induced interferon-gamma-inducible protein 10 (IP-10) and macrophage inflammatory protein 2 (MIP-2) gene expression in murine intestinal epithelial cells. At the molecular level, quercetin treatment was found to cease recruitment of cAMP response element binding protein (CBP)/p300 and histone H3 phosphorylation/acetylation at the gene promoters as well as inhibition of histone acetyltransferase activity [166]. Studies by Lee et al. [167] demonstrate that in human leukemic HL-60 cells, quercetin treatment induced Fas-L related apoptosis and activation of the ERK (extracellular signal–regulated kinase) and JNK (Jun N-terminus kinase) signaling pathways. Quercetin treatment was found to increase histone H3 acetylation which in turn caused increased expression of Fas-L. Furthermore, quercetin treatment resulted in the activation of HAT and inhibition of HDAC; however HAT activation was found to be only associated with ERK and JNK pathways. Priyadarsini et al. [168] reported a positive correlation between HDAC1 and DNMT1 inhibition by quercetin and its anti-tumor properties such as pro-apoptosis, anti-angiogenesis and anti-invasiveness. In a recent study, Ravichandran et al. [169] proposed a refined pharmacophore model for quercetin binding site of SIRT6 protein. This study was in continuation from a previous study reporting quercetin to be a potent SIRT6 inhibitor and identified a preliminary pharmacophore for quercetin binding (consisting of three hydrogen bond donor and one hydrogen bond acceptor) on the SIRT6 histone deacetylase [170]. Quercetin has been demonstrated to induce histone acetylation (H3K56ac) in cultured cells inhibiting SIRT6 that has a critical role in several types of cancer [171].
Quercetin has shown to module miRNAs in various human cancers as well. Using gastric cancer cells, Du et al. [172] demonstrated that quercetin in the presence of miR-143 caused autophagy inhibition via targeting GABARAPL1. In another study, Sonoki et al. [173] demonstrated that quercetin decreased claudin-2 expression and instability of mRNA in lung adenocarcinoma cells mediated by upregulation of miR-16 expression. Treatment of hepatocellular carcinoma cells with quercetin has been shown to involve miR-34a to enhance apoptosis signal through a p53/miR-34a/SIRT1 signal feedback loop [174]. In another study, modulation of miR-217-KRAS axis in osteosarcoma by quercetin enhanced cisplatin sensitivity, regulating the resistance of cancer to chemotherapeutic drugs [175]. In androgen-refractory prostate cancer cells quercetin treatment caused apoptosis, inhibited invasion and migration capabilities and reduced the expression of numerous prostate tumor associated miRNAs, including miR-21. Quercetin increased the expression of tumor suppressor programmed cell death protein 4, that negatively regulate miR-21 in these cells [176].
3.10 Resveratrol
Resveratrol (trans-3,4′,5-trihydroxystilbene), a plant derived polyphenolic compound present in grapes, wine and root extracts of the weed Polygonum cuspidatum, has been identified as a potent anti-aging, anti-inflammatory and chemopreventive agent affecting various molecular targets [177, 178]. Focusing on epigenetic pathways related cellular targets, SIRT1 and acetyl transferase p300 were reported be the activated by resveratrol [179–181]. Resveratrol was identified as a potent dietary activator of SIRT1, which lowers the Km (Michaelis constant) for both acetylated substrate and NAD+. It was reported to stimulate SIRT1-dependent p53 deacetylation which ultimately contributes to increased cell survival [180, 181]. In a study by Wood et al. [182], resveratrol was shown to activate sirtuins from metazoans - Caenorhabditis elegans and Drosophila melanogaster and delay aging without any effect on fecundity. The anti-tumor effect of resveratrol was reported to be mediated partly by SIRT1 [183]. Studies by Wang et al. [184] using SIRT1 mutant mice demonstrated that disruption of SIRT1 function p53 null background causes tumor formation and resveratrol mediated activation of SIRT1 reduced tumorigenesis. In addition, resveratrol was shown to have a negative effect on Survivin gene expression through histone deacetylation at the gene promoter and display a more profound inhibitory effect on BRCA-1 mutant cells both in vitro and in vivo. Using prostate cancer cells, Kai et al. [185] demonstrated that resveratrol was able to cause downregulation of MTA1 (metastasis associated protein), destabilize the NuRD (nucleosome remodeling deacetylase) complex and thus allowing p53 acetylation. Furthermore, activation of p53 was shown to induce pro-apoptotic pathways causing apoptosis in prostate cancer cells. In triple negative breast cancer cells, combination of resveratrol and pterostilbene administered at close to physiologically relevant doses caused synergistic (CI <1) growth inhibition by downregulating SIRT1. This combination also decreased the expression of γ-H2AX and telomerase expression affecting DNA damage and repair [186]. Resveratrol treatment of human bladder cancer EJ cells was found to lead to remarkable S-phase arrest and apoptotic cell death accompanied by loss of phosphorylation of STAT-3, leading to downregulation of the STAT-3 pathway as well as decreased nuclear translocation of SIRT1 and p53 [187]. Resveratrol has also shown to have a moderate inhibitory effect on DNMTs. Resveratrol treatment led to decreased DNMT1 and DNMT3b expression in vitro. In a rodent model of estrogen dependent mammary carcinoma, resveratrol treatment decreased DNMT3b expression in tumor samples but not in the normal tissues [188]. In another study, the chemopreventive effects of resveratrol were demonstrated in vivo by pre-exposing aromatic hydrocarbon receptor (AhR) agonist-treated pregnant Sprague-Dawley rats with resveratrol, an AhR-antagonist, which led to lesser CpG methylation of the BRCA-1 gene [189].
Studies have demonstrated resveratrol induces apoptosis in a time-dependent and dose-dependent manner through increased expression level of miR-16-1 and miR-15a in leukemia cells [190]. In prostate cancer study with stilbenes has demonstrated miRNA-mediated (miR-17-5p and miR-106a-5p) therapeutic strategy, and proposed circulating miRNAs as predictive biomarkers for clinical development [191].
3.11 Sulforaphane
Sulforaphane (SFN) is an isothiocyanate abundantly present in broccoli, water crass, broccoli sprouts, cabbage and kale. SFN has been shown to modulate various physiological processes and affect various cellular pathways consistent with its chemopreventive effects which include induction of cell cycle arrest, apoptosis and xenobiotic metabolism [192, 193]. Microarray based transcriptome analysis of human colon Caco-2 cells treated with SFN at physiological concentrations showed downregulation of DNMT1 gene as well as other genes known to be associated with carcinogenesis [194]. Studies by Meeran et al. [195] demonstrated that in MCF-7 and MDA-MB-231 breast cancer cells, SFN treatment cause dose-dependent and time-dependent inhibition of hTERT (human telomerase reverse transcriptase) via an epigenetic mechanism involving DNA methylation and histone modifications. SFN treatment was shown to cause downregulation of DNMT1 and DNMT3a, which induced site-specific demethylation at hTERT gene first exon facilitating the binding of CTCF associated with hTERT repression. Furthermore, ChIP analysis of hTERT promoter revealed that active histone chromatin marks such as acetyl-H3, acetyl-H3K9 and acetyl-H4 were increased whereas repressive chromatin marks which include trimethyl-H3K9 and trimethyl-H3K27 were reduced after SFN treatment in a dose-dependent manner. The SFN induced hyper-acetylation was reported to promote the binding of repressor proteins such as MAD1 and CTCF to the hTERT regulatory region. In another study, Myzak et al. [196] reported that SFN metabolites–SFN–cysteine and SFN-N-acetylcysteine were more potent HDAC inhibitors in vitro as compared to SFN or its glutathione conjugate. Furthermore, SFN treatment in HCT116 human colorectal cancer cells increased β-catenin-responsive reporter (TOPflash) activity in a dose dependent manner and inhibited HDAC activity. Consequently, there was an induction in acetylated histone levels bound to p21/waf1 promoter. In human prostate epithelial BPH-1, LNCaP and PC-3 cells, SFN treatment was shown to inhibit HDAC activity, which was accompanied by an increase in acetylated histone levels by 50–100% and a corresponding induction of p21 and Bax expression which lead to downstream events such as cell cycle arrest and apoptosis [197]. In human breast cancer cells, SFN treatment was shown to inhibit HDAC activity but no change in H3 or H4 acetylation was observed [198]. In a recent study, Wong et al. [199] demonstrated the genome wide effects of SFN and DIM on promoter methylation in prostate epithelial as well as cancer cells using methyl-DNA immunoprecipitation followed by genome-wide DNA methylation array. The study reported SFN and DIM treatment induce widespread changes in promoter methylation patterns and reverse the status of several cancer-associated aberrantly methylated genes. SFN enhanced the nuclear translocation of Nrf2 and increased the mRNA and protein levels of the Nrf2 target genes in 12-O-tetradecanoylphorbol-13-acetate (TPA) in mouse skin epidermal JB6 (JB6 P+) cells [200]. Furthermore, SFN reduced the expression of DNMTs and downregulated the expression of HDAC-1, HDAC-2, HDAC-3 and HDAC-4, thereby reactivating Nrf2, a transcription factor for anti-oxidant enzymes. Another recent study by Mileo and Miccadei [201] highlighted the role of SFN and its metabolites sulforaphane-N-acetylcysteine (SFN-NAC) and sulforaphane-cysteine (SFNCys) generated via the mercapturic acid pathway in inducing acetylation of histones and inhibiting HDAC activity in the ileum, colon, and prostate C57BL/6J mice tissues. Another study by Myzak et al. [202] provided first evidence for inhibition of in vivo HDAC activity and suppression of tumorigenesis in APC-minus mice. Further studies demonstrated that daily SFN dose (7.5 μM per animal for 21 days) in the diet significantly reduced the growth of PC-3 cells in male nude mice, which correlated with a decrease in HDAC activity and increased global acetylation levels. Furthermore, in human subjects, single dose of 68 g broccoli sprouts caused a significant decrease in HDAC activity in peripheral blood mononuclear cells within 3–6 h after consumption [203].
4.0 Conclusion, limitation and future direction
The role of dietary phytochemicals as epigenetic modifiers has been well supported by the studies described herein holding great promise in cancer prevention and/or therapy. In addition, this review article provides a comprehensive summary on dietary phytochemicals with potential beneficial effects may in part be attributable to their epigenetic properties, including changes in DNA methylation patterns, histone modifications, and in the expression of miRNAs (Figure 1). The fundamental role of epigenetic mechanisms in the regulation of gene expression and epigenetic aberrations implicated in various pathologies itself underlines the importance of compounds targeting them. It is much more challenging than it seems to develop dietary phytochemicals as effective chemopreventive and/or chemotherapeutic agents despite their widespread recognition and acceptance as evident from a large number of research publications [204–258]. There is still insufficient preclinical and clinical data published on the epigenetic changes induced by some of the dietary polyphenols, which remains a challenge and limitation, of the current data to acquire better insight on the molecular mechanisms responsible for these effects. The next important step would be to determine the most effective doses of these dietary phytochemicals in order to obtain beneficial effects in humans. The doses provided in most of the cell culture studies and in vivo experiments remains pharmacologically irrelevant as such high concentrations of these compounds may not be achieved in humans, and might lack clinical benefit. As such, pre-clinical and clinical studies addressing the relevance and validity of in vitro experimental outcomes as well as analysis of safety profile, dose, bio-availability and routes of administration for these compounds are required for designing better chemopreventive/therapeutic strategies along with appropriate designing of the clinical trial. Based on profound research interest in the emerging area of epigenetics with dietary phytochemicals several directions in future could be pursued:
Figure 1.

Overview of epigenetic mechanisms regulating gene expression. Dietary phytochemicals impart anticancer properties through alteration in DNA methylation patterns, histone modification, and changes in non-coding (micro) RNAs levels. Up and down, arrows and ├ inhibition.
Epigenetic remedy with dietary phytochemicals is an emerging area of research that could be effective and valuable approach in fighting cancer at the level of gene expression. Studies conducted to date have shown significant effects from many dietary compounds that target epigenetic pathways in preventing cancer and additional mechanistic studies are needed, in particular, with untested dietary agents.
The exciting field of epigenetics and nutrigenomics, in particular, addresses how nutrients affect the epigenome by targeting DNA methylation, HDAC activity and miRNAs having chemopreventive potential. The effect of a combination of dietary phytochemicals, their course of action and targets is still at an early stage and could be a promising strategy for epigenetic studies.
The phenotype as a result of complex gene-environment interactions in the current, past and ancestral environment results in lifelong remodeling of the epigenome. As some of the effects of these dietary phytochemicals appear to be cell type or organ specific therefore, understanding the mechanism(s) of these differences is a key in designing a personalized regimen for cancer prevention and/or cure.
Several studies have demonstrated that disruption of epigenetic mechanisms can alter immune function and contribute to various disease phenotypes. Understanding and manipulating the epigenome with dietary phytochemicals with a focus on modulating DNA and histone methylation of key inflammatory genes is an effective approach to target immune-related pathways.
Epigenetic defects may eventually lead to genetic defects which are not reversible, appropriate time of interventions using dietary phytochemicals which might be effective in slowing and/or reversing cancer progression should be pursued as future research.
It is demonstrated that nanomaterials encapsulation technology enhances bioavailability and offers significant promise for chemoprevention. Attempts have been made encapsulating various dietary phytochemicals such as curcumin, resveratrol, EGCG and genistein in nanoparticles to circumvent the issues of poor bioavailability with dietary agents, and may be considered for epigenetic studies.
Studies have highlighted the existence of cancer stem cells in most solid and nonsolid tumors. These cells are considered responsible for tumor relapse and resistance to therapy. It has been shown that dietary phytochemicals can impact cancer stem cell self-renewal related pathways. Additional epigenetic studies are needed to determine the pathways which are altered by this particular pool of cancer stem cells.
In conclusion, our knowledge regarding nutritional and diet-based epigenetics is still limited. In particular, the effects of nutrients or bioactive food components on DNA methylation, miRNA expression histone methylation or chromatin remodeling complexes are largely unknown. In future, with the emergence of novel technologies such as next generation sequencing for the genome-wide assessment of DNA methylation, localization of the histone marks, and development of bioinformatics tools to systematically integrate information can facilitate the analysis of epigenetic studies with dietary phytochemicals.
Table 3.
Dietary phytochemicals and histone modification.
| Dietary Agent | Molecular mechanism(s) | Target genes | In vitro model | In vivo model | Disease Type | Dose/Concentration | Ref. |
|---|---|---|---|---|---|---|---|
|
| |||||||
| Apigenin | HDAC inhibitor | p21 | PC-3 cells | Mouse xenograft | Prostate cancer | 20–50 μM (20–50 μg/day) | 86 |
| HDAC inhibitor | NA | Skin cancer cells (mouse) | 1.56–50 μM | 87 | |||
|
| |||||||
| Curcumin | HAT & HDAC inhibitor | H3/H4 deacetylation GATA4, EOMES, GZMB, PRF1 |
Cervix, HIV, Hepatoma Leukemia, Prostate Brain, Lymphoma |
Plasmodium falciparum; Herpes Virus Mouse: Rat |
Malaria Herpes Epilepsy Diabetes and Heart failure |
0.3–75mg/kg (6.25–135μM) |
96–106 214–222 |
|
| |||||||
| Catechin | HAT inhibitor | NA | Lymphocytes | NA | 100 μM | 223 | |
| Epicatechin | HAT inhibitor | NA | Lymphocytes | NA | 100 μM | 223 | |
| Epicatechin-gallate | HAT inhibitor | NA | Lymphocytes, Colon | NA | 100 μM | 223 | |
|
| |||||||
| Epigalocatechin-3-gallate | HAT inhibitor | H3/H4 acetylation | Lymphocytes, Colon | NA | Cancer | 5–100 μM | 223 |
| HMT inhibitor | H3K27 trimethylation NF-κB, IL-6, BMI-1, EZH2, SUZ12 |
Keratinocytes, Prostate | NA | ||||
|
| |||||||
| Equol | HDAC inhibitor | H2A/H2B/H3/H4 | Drosophila | NA | 12.8 μM | 130 | |
| Genistein | HAT inhibitor | Acetylation H2A/H2B/H3/H4 |
Esophageal; Prostate, Breast, Renal | NA | Cancer | 5–100 μM | 124, 127, 128, 130, 131, |
| HADC inhibitor | Acetylation p21, p16, PTEN, p53 FOXA3, SIRT1, hTERT |
NA | |||||
|
| |||||||
| Daidzein | HDAC inhibitor | Histone acetylation | Esophageal, Prostate | NA | Cancer | 12.8–100 μM | 130, 210 |
|
| |||||||
| Indole-3 carbinol Diindolylmethane | HDAC inhibitor | IFN-γ, TNF-α, IL-6, IL-2, NF-κB, COX-2, iNOS, IL-1β, IL-12 | Prostate cancer | Sprague-Dawley rats | 20–80 μg/rat | 82, 141, 143–150 | |
| Colon Cancer | Nude mice | Cancer | 15–100 μM | ||||
| Spleenocytes (CD3+ cells) | Nude mice | 15–100 μM 100–300 mg/kg 100 μM 40 mg/kg |
|||||
|
| |||||||
| Allicin | unknown | H4 acetylation | Erythroleukemia | NA | 2–200 μM | 157–159 | |
| Allyl mercaptan | SIRT1 inhibitor | H3/H4 acetylation P21/WAF1 |
Erythroleukemia Liver, Colon |
Rat: liver | Cancer | 2–500 μM (100mg/kg) | 157–159, 224 |
| S-allymercapto-Cysteine | HAT inhibitor | H3/H4 acetylation | Erythroleukemia | NA | 20–250 μM | 156, 161 | |
| Diallyl disulphide | HDAC inhibitor | H3/H4 acetylation P21/WAF1 |
Erythroleukemia, Leukemia, Liver, Colon, Prostate, Fibroblast | Rat | Cancer | 20–200 μM (42–200 mg/kg) |
156–159,161 224 |
|
| |||||||
| Quercetin | SIRTI activator | IP-10, MIP-2 | Cervix, Drosophila Small intestine |
Cancer Mouse |
Inflammatory diseases | 100 μM | 164–175, |
| HAT inhibitor | Bowel inflammation | 181,182 | |||||
|
| |||||||
| Resveratrol | SIRTi activator | TNF-α, IL-8, RBP | Cervix, Endothelial Embryonic kidney, Macrophages, Lungs, Liver, Cardiomyocytes | Yeast, Drosophila Mouse Rats |
Colon cancer Lung cancer |
10–200 μM | 177–191 |
|
| |||||||
| Sulforaphane | HDAC inhibitor | H3/H4 acetylation RARβ, HBD-2, p21, Bax |
Prostate, Breast, Colon, Kidney | Mouse, prostate cancer xenografts | Cancer | 15–25 μM (443 μg/kg, 68g) | 193–203, 225–227 |
NA, information not available
Acknowledgments
The original work from author’s laboratory outlined in this review was supported by VA Merit Review 1I01BX002494 and from the United States Public Health Service Grants RO1CA108512, R21CA193080 and R03CA186179 to SG.
Footnotes
CONFLICT OF INTEREST
The authors have no competing interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Henikoff S, Matzke MA. Exploring and explaining epigenetic effects. Trends Genet. 1997;13:293–295. doi: 10.1016/s0168-9525(97)01219-5. [DOI] [PubMed] [Google Scholar]
- 2.Hughes V. Epigenetics: The sins of the father. Nature. 2014;507:22–24. doi: 10.1038/507022a. [DOI] [PubMed] [Google Scholar]
- 3.Payer B, Lee JT. X chromosome dosage compensation: how mammals keep the balance. Annu Rev Genet. 2008;42:733–772. doi: 10.1146/annurev.genet.42.110807.091711. [DOI] [PubMed] [Google Scholar]
- 4.Illingworth R, Kerr A, DeSousa D, Jørgensen H, Ellis P, Stalker J, et al. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008;6:e22. doi: 10.1371/journal.pbio.0060022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- 6.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 7.Jarome TJ, Lubin FD. Histone lysine methylation: critical regulator of memory and behavior. Rev Neurosci. 2013;24:375–387. doi: 10.1515/revneuro-2013-0008. [DOI] [PubMed] [Google Scholar]
- 8.Peixoto L, Abel T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology. 2013;38:62–76. doi: 10.1038/npp.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lubin FD, Gupta S, Parrish RR, Grissom NM, Davis RL. Epigenetic mechanisms: critical contributors to long-term memory formation. Neuroscientist. 2011;17:616–632. doi: 10.1177/1073858411386967. [DOI] [PubMed] [Google Scholar]
- 10.Gos M. Epigenetic mechanisms of gene expression regulation in neurological diseases. Acta Neurobiol Exp (Wars) 2013;73:19–37. doi: 10.55782/ane-2013-1919. [DOI] [PubMed] [Google Scholar]
- 11.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 12.Stilling RM, Fischer A. The role of histone acetylation in age-associated memory impairment and Alzheimer’s disease. Neurobiol Learn Mem. 2011;96:19–26. doi: 10.1016/j.nlm.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Bruce KD, Cagampang FR. Epigenetic priming of the metabolic syndrome. Toxicol Mech Methods. 2011;21:353–361. doi: 10.3109/15376516.2011.559370. [DOI] [PubMed] [Google Scholar]
- 14.Suter MA, Aagaard-Tillery KM. Environmental influences on epigenetic profiles. Semin Reprod Med. 2009;27:380–390. doi: 10.1055/s-0029-1237426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 16.Choudhuri S. From Waddington’s epigenetic landscape to small noncoding RNA: some important milestones in the history of epigenetics research. Toxicol Mech Methods. 2011;21:252–274. doi: 10.3109/15376516.2011.559695. [DOI] [PubMed] [Google Scholar]
- 17.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15:3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dehan P, Kustermans G, Guenin S, Horion J, Boniver J, Delvenne P. DNA methylation and cancer diagnosis: new methods and applications. Expert Rev Mol Diagn. 2009;9:651–657. doi: 10.1586/erm.09.53. [DOI] [PubMed] [Google Scholar]
- 19.Link A, Balaguer F, Goel A. Cancer chemoprevention by dietary polyphenols: promising role for epigenetics. Biochem Pharmacol. 2010;80:1771–1792. doi: 10.1016/j.bcp.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerhauser C. Cancer chemoprevention and nutriepigenetics: state of the art and future challenges. Top Curr Chem. 2013;329:73–132. doi: 10.1007/128_2012_360. [DOI] [PubMed] [Google Scholar]
- 21.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 22.Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;33:9–10. doi: 10.1016/0092-8674(83)90327-6. [DOI] [PubMed] [Google Scholar]
- 23.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 24.Yoder JA, Soman NS, Verdine GL, Bestor TH. DNA (cytosine-5)-methyltransferases in mouse cells and tissues. Studies with a mechanism-based probe. J Mol Biol. 1997;270:385–395. doi: 10.1006/jmbi.1997.1125. [DOI] [PubMed] [Google Scholar]
- 25.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 26.Bourc’his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 27.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 28.Denis H, Ndlovu MN, Fuks F. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep. 2011;12:647–656. doi: 10.1038/embor.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watt F, Molloy PL. Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Genes Dev. 1988;2:1136–1143. doi: 10.1101/gad.2.9.1136. [DOI] [PubMed] [Google Scholar]
- 30.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 31.Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993;3:226–231. doi: 10.1016/0959-437x(93)90027-m. [DOI] [PubMed] [Google Scholar]
- 32.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 33.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 34.Feng Q, Zhang Y. The MeCP1 complex represses transcription through preferential binding, remodeling, and deacetylating methylated nucleosomes. Genes Dev. 2001;15:827–832. doi: 10.1101/gad.876201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roos L, Van Dongen J, Bell CG, Burri A, Deloukas P, Boomsma DI, et al. Integrative DNA methylome analysis of pan-cancer biomarkers in cancer discordant monozygotic twin-pairs. Clin Epigenetics. 2016;8:7. doi: 10.1186/s13148-016-0172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhat S, Kabekkodu SP, Noronha A, Satyamoorthy K. Biological implications and therapeutic significance of DNA methylation regulated genes in cervical cancer. Biochimie. 2016;121:298–311. doi: 10.1016/j.biochi.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 39.Yen CY, Huang HW, Shu CW, Hou MF, Yuan SF, Wang HR, et al. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Lett. 2016;373:185–92. doi: 10.1016/j.canlet.2016.01.036. [DOI] [PubMed] [Google Scholar]
- 40.Luger K, Richmond TJ. The histone tails of the nucleosome. Curr Opin Genet Dev. 1998;8:140–146. doi: 10.1016/s0959-437x(98)80134-2. [DOI] [PubMed] [Google Scholar]
- 41.Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- 42.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 43.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 44.Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 46.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 47.Mottet D, Castronovo V. Histone deacetylases: target enzymes for cancer therapy. Clin Exp Metastasis. 2008;25:183–189. doi: 10.1007/s10585-007-9131-5. [DOI] [PubMed] [Google Scholar]
- 48.Shi Y. Histone lysine demethylases: emerging roles in development, physiology and disease. Nat Rev Genet. 2007;8:829–833. doi: 10.1038/nrg2218. [DOI] [PubMed] [Google Scholar]
- 49.Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol. 2001;13:263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- 50.Upadhyay AK, Cheng X. Dynamics of histone lysine methylation: structures of methyl writers and erasers. Prog Drug Res. 2011;67:107–124. doi: 10.1007/978-3-7643-8989-5_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mir AR, Moinuddin, Islam S. Circulating autoantibodies in cancer patients have high specificity for glycoxidation modified histone H2A. Clin Chim Acta. 2016;453:48–55. doi: 10.1016/j.cca.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 52.Ma F, Zhang CY. Histone modifying enzymes: novel disease biomarkers and assay development. Expert Rev Mol Diagn. 2016;11:1–10. doi: 10.1586/14737159.2016.1135057. [DOI] [PubMed] [Google Scholar]
- 53.Deb G, Singh AK, Gupta S. EZH2: Not EZHY (Easy) to Deal. Mol Cancer Res. 2014;12:639–653. doi: 10.1158/1541-7786.MCR-13-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 55.Suzuki T, Miyata N. Epigenetic control using natural products and synthetic molecules. Curr Med Chem. 2006;13:935–958. doi: 10.2174/092986706776361067. [DOI] [PubMed] [Google Scholar]
- 56.Costa FF. Non-coding RNAs, epigenetics and complexity. Gene. 2008;410:9–17. doi: 10.1016/j.gene.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 57.Peschansky VJ, Wahlestedt C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics. 2014;9:3–12. doi: 10.4161/epi.27473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou H, Hu H, Lai M. Non-coding RNAs and their epigenetic regulatory mechanisms. Biol Cell. 2010;102:645–655. doi: 10.1042/BC20100029. [DOI] [PubMed] [Google Scholar]
- 59.Mattick JS. Non-coding RNAs: the architects of eukaryotic complexity. EMBO Rep. 2001;2:986–991. doi: 10.1093/embo-reports/kve230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szymanski M, Barciszewska MZ, Erdmann VA, Barciszewski J. A new frontier for molecular medicine: noncoding RNAs. Biochim Biophys Acta. 2005;1756:65–75. doi: 10.1016/j.bbcan.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 61.Deb G, Thakur VS, Gupta S. Multifaceted role of EZH2 in breast and prostate tumorigenesis: Epigenetics and beyond. Epigenetics. 2013;8:464–476. doi: 10.4161/epi.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 63.Varambally S, Cao Q, Mani RS, Shankar S, Wang X, Ateeg B, et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695–1699. doi: 10.1126/science.1165395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Juan AH, Kumar RM, Marx JG, Young RA, Sartorelli V. Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell. 2009;36:61–74. doi: 10.1016/j.molcel.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu S, Tetzlaff MT, Cui R, Xu X. miR-200c inhibits melanoma progression and drug resistance through down-regulation of BMI-1. Am J Pathol. 2012;181:1823–1835. doi: 10.1016/j.ajpath.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G, et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res. 2008;68:9125–9130. doi: 10.1158/0008-5472.CAN-08-2629. [DOI] [PubMed] [Google Scholar]
- 67.Liz J, Esteller M. lncRNAs and microRNAs with a role in cancer development. Biochim Biophys Acta. 2016;1859:169–76. doi: 10.1016/j.bbagrm.2015.06.015. [DOI] [PubMed] [Google Scholar]
- 68.Fabris L, Ceder Y, Chinnaiyan AM, Jenster GW, Sorensen KD, Tomlins S, et al. The Potential of MicroRNAs as Prostate Cancer Biomarkers. Eur Urol. 2016 doi: 10.1016/j.eururo.2015.12.054. S0302-2838(16)00012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 70.Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104:15805–15810. doi: 10.1073/pnas.0707628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–6418. doi: 10.1182/blood-2008-07-170589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang J, Plass C, Gerhauser C. Cancer chemoprevention by targeting the epigenome. Curr Drug Targets. 2011;12:1925–1956. doi: 10.2174/138945011798184155. [DOI] [PubMed] [Google Scholar]
- 73.Russo GL, Vastolo V, Ciccarelli M, Albano L, Macchia PE, Ungaro P. Dietary Polyphenols and Chromatin Remodelling. Crit Rev Food Sci Nutr. 2015 Sep 10; doi: 10.1080/10408398.2015.1062353. 0. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 74.Thakur VS, Deb G, Babcook MA, Gupta S. Plant phytochemicals as epigenetic modulators: role in cancer chemoprevention. Aaps j. 2014;16:151–163. doi: 10.1208/s12248-013-9548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hauser AT, Jung M. Targeting epigenetic mechanisms: potential of natural products in cancer chemoprevention. Planta Med. 2008;74:1593–1601. doi: 10.1055/s-2008-1081347. [DOI] [PubMed] [Google Scholar]
- 76.Davis CD, Ross SA. Evidence for dietary regulation of microRNA expression in cancer cells. Nutr Rev. 2008;66:477–482. doi: 10.1111/j.1753-4887.2008.00080.x. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, Kong D, Wang Z, Sarkar FH. Regulation of microRNAs by natural agents: an emerging field in chemoprevention and chemotherapy research. Pharm Res. 2010;27:1027–1041. doi: 10.1007/s11095-010-0105-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel D, Shukla S, Gupta S. Apigenin and cancer chemoprevention: progress, potential and promise (review) Int J Oncol. 2007;30:233–245. [PubMed] [Google Scholar]
- 79.Birt DF, Hendrich S, Wang W. Dietary agents in cancer prevention: flavonoids and isoflavonoids. Pharmacol Ther. 2001;90:157–177. doi: 10.1016/s0163-7258(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 80.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 81.Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr. 2004;79:727–747. doi: 10.1093/ajcn/79.5.727. [DOI] [PubMed] [Google Scholar]
- 82.Birt DF, Walker B, Tibbels MG, Bresnick E. Anti-mutagenesis and anti-promotion by apigenin, robinetin and indole-3-carbinol. Carcinogenesis. 1986;7:959–963. doi: 10.1093/carcin/7.6.959. [DOI] [PubMed] [Google Scholar]
- 83.Wei H, Tye L, Bresnick E, Birt DF. Inhibitory effect of apigenin, a plant flavonoid, on epidermal ornithine decarboxylase and skin tumor promotion in mice. Cancer Res. 1990;50:499–502. [PubMed] [Google Scholar]
- 84.Choi JS, Islam MN, Ali MY, Kim EJ, Kim YM, Jung HA. Effects of C-glycosylation on anti-diabetic, anti-Alzheimer’s disease and anti-inflammatory potential of apigenin. Food Chem Toxicol. 2014;64:27–33. doi: 10.1016/j.fct.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 85.Fang M, Chen D, Yang CS. Dietary polyphenols may affect DNA methylation. J Nutr. 2007;137:223s–228s. doi: 10.1093/jn/137.1.223S. [DOI] [PubMed] [Google Scholar]
- 86.Pandey M, Kaur P, Shukla S, Abbas A, Fu P, Gupta S. Plant flavone apigenin inhibits HDAC and remodels chromatin to induce growth arrest and apoptosis in human prostate cancer cells: in vitro and in vivo study. Mol Carcinog. 2012;51:952–962. doi: 10.1002/mc.20866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paredes-Gonzalez X, Fuentes F, Su ZY, Kong AN. Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P+ cells through epigenetics modifications. AAPS j. 2014;16:727–735. doi: 10.1208/s12248-014-9613-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prasad S, Gupta SC, Tyagi AK, Aggarwal BB. Curcumin, a component of golden spice: from bedside to bench and back. Biotechnol Adv. 2014;32:1053–1064. doi: 10.1016/j.biotechadv.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 89.Bar-Sela G, Epelbaum R, Schaffer M. Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr Med Chem. 2010;17:190–197. doi: 10.2174/092986710790149738. [DOI] [PubMed] [Google Scholar]
- 90.Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41:1955–1968. doi: 10.1016/j.ejca.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 91.Teiten MH, Eifes S, Dicato M, Diederich M. Curcumin-the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins (Basel) 2010;2:128–162. doi: 10.3390/toxins2010128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116:4670–4676. doi: 10.1002/cncr.25414. [DOI] [PubMed] [Google Scholar]
- 93.Medina-Franco JL, Lopez-Vallejo F, Kuck D, Lyko F. Natural products as DNA methyltransferase inhibitors: a computer-aided discovery approach. Mol Divers. 2011;15:293–304. doi: 10.1007/s11030-010-9262-5. [DOI] [PubMed] [Google Scholar]
- 94.Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu S, Yu J, et al. Curcumin is a potent DNA hypomethylation agent. Bioorg Med Chem Lett. 2009;19:706–709. doi: 10.1016/j.bmcl.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 95.Bora-Tatar G, Dayangac-Erden D, Demir AS, Dalkara S, Yelekçi K, Erdem-Yurter H. Molecular modifications on carboxylic acid derivatives as potent histone deacetylase inhibitors: Activity and docking studies. Bioorg Med Chem. 2009;17:5219–5228. doi: 10.1016/j.bmc.2009.05.042. [DOI] [PubMed] [Google Scholar]
- 96.Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin. 2005;26:603–609. doi: 10.1111/j.1745-7254.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 97.Chen Y, Shu W, Chen W, Wu Q, Liu H, Cui G. Curcumin, both histone deacetylase and p300/CBP-specific inhibitor, represses the activity of nuclear factor kappa B and Notch 1 in Raji cells. Basic Clin Pharmacol Toxicol. 2007;101:427–433. doi: 10.1111/j.1742-7843.2007.00142.x. [DOI] [PubMed] [Google Scholar]
- 98.Dekker FJ, Haisma HJ. Histone acetyl transferases as emerging drug targets. Drug Discov Today. 2009;14:942–948. doi: 10.1016/j.drudis.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 99.Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–174. doi: 10.2174/157340606776056133. [DOI] [PubMed] [Google Scholar]
- 100.Balasubramanyam K, Varier RA, Altaf M, Swaminathan V, Siddappa NB, Ranga U, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem. 2004;279:51163–51171. doi: 10.1074/jbc.M409024200. [DOI] [PubMed] [Google Scholar]
- 101.Guo Y, Shu L, Zhang C, Su ZY, Kong AN. Curcumin inhibits anchorage-independent growth of HT29 human colon cancer cells by targeting epigenetic restoration of the tumor suppressor gene DLEC1. Biochem Pharmacol. 2015;9:69–78. doi: 10.1016/j.bcp.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma V, Jha AK, Kumar A, Bhatnagar A, Narayan G, Kaur J. Curcumin-Mediated Reversal of p15 Gene Promoter Methylation: Implication in Anti-Neoplastic Action against Acute Lymphoid Leukaemia Cell Line. Folia Biol (Praha) 2015;61:81–89. doi: 10.14712/fb2015061020081. [DOI] [PubMed] [Google Scholar]
- 103.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7:464–473. doi: 10.1158/1535-7163.MCT-07-2272. [DOI] [PubMed] [Google Scholar]
- 104.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, et al. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–3617. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 105.Jiang A, Wang X, Shan X, Li Y, Wang P, Jiang P, Feng Q. Curcumin Reactivates Silenced Tumor Suppressor Gene RARβ by Reducing DNA Methylation. Phytother Res. 2015;29:1237–45. doi: 10.1002/ptr.5373. [DOI] [PubMed] [Google Scholar]
- 106.Zamani M, Sadeghizadeh M, Behmanesh M, Najafi F. Dendrosomal curcumin increases expression of the long non coding RNA gene MEG3 via up-regulation of epi-miRs in hepatocellular cancer. Phytomedicine. 2015;22:961–7. doi: 10.1016/j.phymed.2015.05.071. [DOI] [PubMed] [Google Scholar]
- 107.Butt MS, Sultan MT. Green tea: nature’s defense against malignancies. Crit Rev Food Sci Nutr. 2009;49:463–473. doi: 10.1080/10408390802145310. [DOI] [PubMed] [Google Scholar]
- 108.Shankar S, Ganapathy S, Srivastava RK. Green tea polyphenols: biology and therapeutic implications in cancer. Front Biosci. 2007;12:4881–4899. doi: 10.2741/2435. [DOI] [PubMed] [Google Scholar]
- 109.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (−)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer Res. 2003;63:7563–7570. [PubMed] [Google Scholar]
- 110.Pandey M, Shukla S, Gupta S. Promoter demethylation and chromatin remodeling by green tea polyphenols leads to re-expression of GSTP1 in human prostate cancer cells. Int J Cancer. 2010;126:2520–2533. doi: 10.1002/ijc.24988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Thakur VS, Gupta K, Gupta S. Green tea polyphenols causes cell cycle arrest and apoptosis in prostate cancer cells by suppressing class I histone deacetylases. Carcinogenesis. 2012;33:377–38. doi: 10.1093/carcin/bgr277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li Y, Yuan YY, Meeran SM, Tollefsbol TO. Synergistic epigenetic reactivation of estrogen receptor-alpha (ERalpha) by combined green tea polyphenol and histone deacetylase inhibitor in ERalpha-negative breast cancer cells. Mol Cancer. 2010;9:274. doi: 10.1186/1476-4598-9-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nandakumar V, Vaid M, Katiyar SK. (−)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011;32:537–544. doi: 10.1093/carcin/bgq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Balasubramanian S, Adhikary G, Eckert RL. The Bmi-1 polycomb protein antagonizes the (−)-epigallocatechin-3-gallate-dependent suppression of skin cancer cell survival. Carcinogenesis. 2010;31:496–503. doi: 10.1093/carcin/bgp314. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 115.Choudhury SR, Balasubramanian S, Chew YC, Han B, Marquez VE, Eckert RL. (−)-Epigallocatechin-3-gallate and DZNep reduce polycomb protein level via a proteasome-dependent mechanism in skin cancer cells. Carcinogenesis. 2011;32:1525–1532. doi: 10.1093/carcin/bgr171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Deb G, Thakur VS, Limaye AM, Gupta S. Epigenetic induction of tissue inhibitor of matrix metalloproteinase-3 by green tea polyphenols in breast cancer cells. Mol Carcinog. 2014;54:485–99. doi: 10.1002/mc.22121. [DOI] [PubMed] [Google Scholar]
- 117.Khan MA, Hussain A, Sundaram MK, Alalami U, Gunasekera D, Ramesh L, et al. (−)-Epigallocatechin-3 gallate reverses the expression of various tumor suppressor genes by inhibiting DNA methyltransferases and histone deacetylases in human cervical cancer cells. Oncol Rep. 2015;33:1976–84. doi: 10.3892/or.2015.3802. [DOI] [PubMed] [Google Scholar]
- 118.Zhou XQ, Xu XN, Li L, Ma JJ, Zhen EM, Han CB. Epigallocatechin-3-gallate inhibits the invasion of salivary adenoid cystic carcinoma cells by reversing the hypermethylation status of the RECK gene. Mol Med Rep. 2015;12:6031–6036. doi: 10.3892/mmr.2015.4213. [DOI] [PubMed] [Google Scholar]
- 119.Jha M, Aggarwal R, Jha AK, Shrivastava A. Natural Compounds: DNA Methyltransferase Inhibitors in Oral Squamous Cell Carcinoma. Appl Biochem Biotechnol. 2015;177:577–94. doi: 10.1007/s12010-015-1768-y. [DOI] [PubMed] [Google Scholar]
- 120.Chen LL, Han WF, Geng Y, Su JS. A genome-wide study of DNA methylation modified by epigallocatechin-3-gallate in the CAL-27 cell line. Mol Med Rep. 2015;12:5886–90. doi: 10.3892/mmr.2015.4118. [DOI] [PubMed] [Google Scholar]
- 121.Coward L, Barnes NC, Setchell KDR, Barnes S. Genistein, daidzein, and their β-glycoside conjugates: antitumor isoflavones in soybean foods from American and Asian diets. J Agric Food Chem. 1993;41:1961–1967. [Google Scholar]
- 122.Kaufman PB, Duke JA, Brielmann H, Boik J, Hoyt JE. A comparative survey of leguminous plants as sources of the isoflavones, genistein and daidzein: implications for human nutrition and health. J Altern Complement Med. 1997;3:7–12. doi: 10.1089/acm.1997.3.7. [DOI] [PubMed] [Google Scholar]
- 123.Banerjee S, Li Y, Wang Z, Sarkar FH. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008;269:226–242. doi: 10.1016/j.canlet.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fang MZ, Chen D, Sun Y, Jin Z, Christman JK, Yang CS. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin Cancer Res. 2005;11:7033–7041. doi: 10.1158/1078-0432.CCR-05-0406. [DOI] [PubMed] [Google Scholar]
- 125.Li Y, Liu L, Andrews LG, Tollefsbol TO. Genistein depletes telomerase activity through cross-talk between genetic and epigenetic mechanisms. Int J Cancer. 2009;125:286–296. doi: 10.1002/ijc.24398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.King-Batoon A, Leszczynska JM, Klein CB. Modulation of gene methylation by genistein or lycopene in breast cancer cells. Environ Mol Mutagen. 2008;49:36–45. doi: 10.1002/em.20363. [DOI] [PubMed] [Google Scholar]
- 127.Majid S, Dar AA, Ahmad AE, Hirata H, Kawakami K, Shahryari V, et al. BTG3 tumor suppressor gene promoter demethylation, histone modification and cell cycle arrest by genistein in renal cancer. Carcinogenesis. 2009;30:662–670. doi: 10.1093/carcin/bgp042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Majid S, Dar AA, Shahryari V, Hirata H, Ahmad A, Saini S, et al. Genistein reverses hypermethylation and induces active histone modifications in tumor suppressor gene B-Cell translocation gene 3 in prostate cancer. Cancer. 2010;116:66–76. doi: 10.1002/cncr.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Day JK, Bauer AM, DesBordes C, Zhuang Y, Kim BE, Newton LG, et al. Genistein alters methylation patterns in mice. J Nutr. 2002;132:2419s–2423s. doi: 10.1093/jn/132.8.2419S. [DOI] [PubMed] [Google Scholar]
- 130.Hong T, Nakagawa T, Pan W, Kim MY, Kraus WL, Ikehara T, et al. Isoflavones stimulate estrogen receptor-mediated core histone acetylation. Biochem Biophys Res Commun. 2004;317:259–264. doi: 10.1016/j.bbrc.2004.03.041. [DOI] [PubMed] [Google Scholar]
- 131.Majid S, Kikuno N, Nelles J, Noonan E, Tanaka Y, Kawamoto K, et al. Genistein induces the p21WAF1/CIP1 and p16INK4a tumor suppressor genes in prostate cancer cells by epigenetic mechanisms involving active chromatin modification. Cancer Res. 2008;68:2736–2744. doi: 10.1158/0008-5472.CAN-07-2290. [DOI] [PubMed] [Google Scholar]
- 132.Basak S, Pookot D, Noonan EJ, Dahiya R. Genistein down-regulates androgen receptor by modulating HDAC6-Hsp90 chaperone function. Mol Cancer Ther. 2008;7:3195–3202. doi: 10.1158/1535-7163.MCT-08-0617. [DOI] [PubMed] [Google Scholar]
- 133.Karsli-Ceppioglu S, Ngollo M, Adjakly M, Dagdemir A, Judes G, Lebert A, et al. Genome-wide DNA methylation modified by soy phytoestrogens: role for epigenetic therapeutics in prostate cancer? OMICS. 2015;19:209–219. doi: 10.1089/omi.2014.0142. [DOI] [PubMed] [Google Scholar]
- 134.Mahmoud AM, Al-Alem U, Ali MM, Bosland MC. Genistein increases estrogen receptor beta expression in prostate cancer via reducing its promoter methylation. J Steroid Biochem Mol Biol. 2015;152:62–75. doi: 10.1016/j.jsbmb.2015.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qin W, Zhu W, Shi H, Hewett JE, Ruhlen RL, MacDonald RS, et al. Soy isoflavones have an antiestrogenic effect and alter mammary promoter hypermethylation in healthy premenopausal women. Nutr Cancer. 2009;61:238–244. doi: 10.1080/01635580802404196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Parker LP, Taylor DD, Kesterson J, Metzinger DS, Gercel-Taylor C. Modulation of microRNA associated with ovarian cancer cells by genistein. Eur J Gynaecol Oncol. 2009;30:616–621. [PubMed] [Google Scholar]
- 137.Li Y, VandenBoom TG, Kong D, Wang Z, Ali S, Philip PA, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–6712. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sun Q, Cong R, Yan H, Gu H, Zeng Y, Liu N, et al. Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression. Oncol Rep. 2009;22:563–567. doi: 10.3892/or_00000472. [DOI] [PubMed] [Google Scholar]
- 139.Qin J, Chen JX, Zhu Z, Teng JA. Genistein inhibits human colorectal cancer growth and suppresses miR-95, Akt and SGK1. Cell Physiol Biochem. 2015;35:2069–77. doi: 10.1159/000374013. [DOI] [PubMed] [Google Scholar]
- 140.Avci CB, Susluer SY, Caglar HO, Balci T, Aygunes D, Dodurga Y, et al. Genistein-induced mir-23b expression inhibits the growth of breast cancer cells. Contemp Oncol (Pozn) 2015;19:32–35. doi: 10.5114/wo.2014.44121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rogan EG. The natural chemopreventive compound indole-3-carbinol: state of the science. In Vivo. 2006;20:221–228. [PubMed] [Google Scholar]
- 142.Minich DM, Bland JS. A review of the clinical efficacy and safety of cruciferous vegetable phytochemicals. Nutr Rev. 2007;65:259–267. doi: 10.1301/nr.2007.jun.259-267. [DOI] [PubMed] [Google Scholar]
- 143.Acharya A, Das I, Singh S, Saha T. Chemopreventive properties of indole-3-carbinol, diindolylmethane and other constituents of cardamom against carcinogenesis. Recent Pat Food Nutr Agric. 2010;2:166–177. doi: 10.2174/2212798411002020166. [DOI] [PubMed] [Google Scholar]
- 144.Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle. 2005;4:1201–1215. doi: 10.4161/cc.4.9.1993. [DOI] [PubMed] [Google Scholar]
- 145.Banerjee S, Kong D, Wang Z, Bao B, Hillman GG, Sarkar FH. Attenuation of multi-targeted proliferation-linked signaling by 3,3′-diindolylmethane (DIM): from bench to clinic. Mutat Res. 2011;728:47–66. doi: 10.1016/j.mrrev.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li Y, Li X, Guo B. Chemopreventive agent 3,3′-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Beaver LM, Yu TW, Sokolowski EI, Williams DE, Dashwood RH, Ho E. 3,3′-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicol Appl Pharmacol. 2012;263:345–351. doi: 10.1016/j.taap.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Busbee PB, Nagarkatti M, Nagarkatti PS. Natural indoles, indole-3-carbinol and 3,3′diindolymethane, inhibit T cell activation by staphylococcal enterotoxin B through epigenetic regulation involving HDAC expression. Toxicol Appl Pharmacol. 2014;274:7–16. doi: 10.1016/j.taap.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li Y, Vandenboom TG, Wang Z, Kong D, Ali S, Philip PA, et al. miR-146a suppresses invasion of pancreatic cancer cells. Cancer Res. 2010;70:1486–1495. doi: 10.1158/0008-5472.CAN-09-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jin Y, Zou X, Feng X. 3,3′-Diindolylmethane negatively regulates Cdc25A and induces a G2/M arrest by modulation of microRNA 21 in human breast cancer cells. Anticancer Drugs. 2010;21:814–822. doi: 10.1097/CAD.0b013e32833e53ea. [DOI] [PubMed] [Google Scholar]
- 151.Wang F, Li S, Wang L, Zong H, Fan Q. DATS suppresses growth of esophageal squamous cell carcinoma by regulation of ERK1/2. Clin Lab. 2015;61:315–22. doi: 10.7754/clin.lab.2014.140806. [DOI] [PubMed] [Google Scholar]
- 152.Chalabi N, Satih S, Delort L, Bignon YJ, Bernard-Gallon DJ. Expression profiling by whole-genome microarray hybridization reveals differential gene expression in breast cancer cell lines after lycopene exposure. Biochim Biophys Acta. 2007;1769:124–130. doi: 10.1016/j.bbaexp.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 153.Chalabi N, Delort L, Le Corre L, Satih S, Bignon YJ, Bernard-Gallon D. Gene signature of breast cancer cell lines treated with lycopene. Pharmacogenomics. 2006;7:663–672. doi: 10.2217/14622416.7.5.663. [DOI] [PubMed] [Google Scholar]
- 154.Fu LJ, Ding YB, Wu LX, Wen CJ, Qu Q, Zhang X, et al. The Effects of Lycopene on the Methylation of the GSTP1 Promoter and Global Methylation in Prostatic Cancer Cell Lines PC3 and LNCaP. Int J Endocrinol. 2014;2014:620165. doi: 10.1155/2014/620165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Iciek M, Kwiecien I, Wlodek L. Biological properties of garlic and garlic-derived organosulfur compounds. Environ Mol Mutagen. 2009;50:247–265. doi: 10.1002/em.20474. [DOI] [PubMed] [Google Scholar]
- 156.Nian H, Delage B, Ho E, Dashwood RH. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009;50:213–221. doi: 10.1002/em.20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lea MA, Randolph VM, Patel M. Increased acetylation of histones induced by diallyl disulfide and structurally related molecules. Int J Oncol. 1999;15:347–352. doi: 10.3892/ijo.15.2.347. [DOI] [PubMed] [Google Scholar]
- 158.Lea MA, Randolph VM, Lee JE, desBordes C. Induction of histone acetylation in mouse erythroleukemia cells by some organosulfur compounds including allyl isothiocyanate. Int J Cancer. 2001;92:784–789. doi: 10.1002/ijc.1277. [DOI] [PubMed] [Google Scholar]
- 159.Lea MA, Rasheed M, Randolph VM, Khan F, Shareef A, desBordes C. Induction of histone acetylation and inhibition of growth of mouse erythroleukemia cells by S-allylmercaptocysteine. Nutr Cancer. 2002;43:90–102. doi: 10.1207/S15327914NC431_11. [DOI] [PubMed] [Google Scholar]
- 160.Druesne N, Pagniez A, Mayeur C, Thomas M, Cherbuy C, Duée PH, et al. Diallyl disulfide (DADS) increases histone acetylation and p21(waf1/cip1) expression in human colon tumor cell lines. Carcinogenesis. 2004;25:1227–1236. doi: 10.1093/carcin/bgh123. [DOI] [PubMed] [Google Scholar]
- 161.Nian H, Delage B, Pinto JT, Dashwood RH. Allyl mercaptan, a garlic-derived organosulfur compound, inhibits histone deacetylase and enhances Sp3 binding on the P21WAF1 promoter. Carcinogenesis. 2008;29:1816–1824. doi: 10.1093/carcin/bgn165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cheung KL, Kong AN. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. Aaps j. 2010;12:87–97. doi: 10.1208/s12248-009-9162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wang LG, Beklemisheva A, Liu XM, Ferrari AC, Feng J, Chiao JW. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol Carcinog. 2007;46:24–31. doi: 10.1002/mc.20258. [DOI] [PubMed] [Google Scholar]
- 164.Gibellini L, Pinti M, Nasi M, Montagna JP, De Biasi S, Roat E, et al. Quercetin and cancer chemoprevention. Evid Based Complement Alternat Med. 2011;2011:591356. doi: 10.1093/ecam/neq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Tan S, Wang C, Lu C, Zhao B, Cui Y, Shi X, et al. Quercetin is able to demethylate the p16INK4a gene promoter. Chemotherapy. 2009;55:6–10. doi: 10.1159/000166383. [DOI] [PubMed] [Google Scholar]
- 166.Ruiz PA, Braune A, Holzlwimmer G, Quintanilla-Fend L, Haller D. Quercetin inhibits TNF-induced NF-kappaB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J Nutr. 2007;137:1208–1215. doi: 10.1093/jn/137.5.1208. [DOI] [PubMed] [Google Scholar]
- 167.Lee WJ, Chen YR, Tseng TH. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol Rep. 2011;25:583–591. doi: 10.3892/or.2010.1097. [DOI] [PubMed] [Google Scholar]
- 168.Priyadarsini RV, Vinothini G, Murugan RS, Manikandan P, Nagini S. The flavonoid quercetin modulates the hallmark capabilities of hamster buccal pouch tumors. Nutr Cancer. 2011;63:218–226. doi: 10.1080/01635581.2011.523503. [DOI] [PubMed] [Google Scholar]
- 169.Ravichandran S, Singh N, Donnelly D, Migliore M, Johnson P, Fishwick C, et al. Pharmacophore model of the quercetin binding site of the SIRT6 protein. J Mol Graph Model. 2014;49:38–46. doi: 10.1016/j.jmgm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Singh N, Ravichandran S, Norton DD, Fugmann SD, Moaddel R. Synthesis and characterization of a SIRT6 open tubular column: predicting deacetylation activity using frontal chromatography. Anal Biochem. 2013;436:78–83. doi: 10.1016/j.ab.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kokkonen P, Rahnasto-Rilla M, Mellini P, Jarho E, Lahtela-Kakkonen M, Kokkola T. Studying SIRT6 regulation using H3K56 based substrate and small molecules. Eur J Pharm Sci. 2014;63:71–76. doi: 10.1016/j.ejps.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 172.Du F, Feng Y, Fang J, Yang M. MicroRNA-143 enhances chemosensitivity of Quercetin through autophagy inhibition via target GABARAPL1 in gastric cancer cells. Biomed Pharmacother. 2015;74:169–77. doi: 10.1016/j.biopha.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 173.Sonoki H, Sato T, Endo S, Matsunaga T, Yamaguchi M, Yamazaki Y, et al. Quercetin Decreases Claudin-2 Expression Mediated by Up-Regulation of microRNA miR-16 in Lung Adenocarcinoma A549 Cells. Nutrients. 2015;7:4578–92. doi: 10.3390/nu7064578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lou G, Liu Y, Wu S, Xue J, Yang F, Fu H, et al. The p53/miR-34a/SIRT1 Positive Feedback Loop in Quercetin Induced Apoptosis. Cell Physiol Biochem. 2015;35:2192–202. doi: 10.1159/000374024. [DOI] [PubMed] [Google Scholar]
- 175.Zhang X, Guo Q, Chen J, Chen Z. Quercetin Enhances Cisplatin Sensitivity of Human Osteosarcoma Cells by modulating microRNA-217-KRAS Axis. Mol Cells. 2015;38:638–42. doi: 10.14348/molcells.2015.0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yang FQ, Liu M, Li W, Che JP, Wang GC, Zheng JH. Combination of quercetin and hyperoside inhibits prostate cancer cell growth and metastasis via regulation of microRNA-21. Mol Med Rep. 2015;11:1085–1092. doi: 10.3892/mmr.2014.2813. [DOI] [PubMed] [Google Scholar]
- 177.Athar M, Back JH, Kopelovich L, Bickers DR, Kim AL. Multiple molecular targets of resveratrol: Anti- carcinogenic mechanisms. Arch Biochem Biophys. 2009;486:95–102. doi: 10.1016/j.abb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Savouret JF, Quesne M. Resveratrol and cancer: a review. Biomed Pharmacother. 2002;56:84–87. doi: 10.1016/s0753-3322(01)00158-5. [DOI] [PubMed] [Google Scholar]
- 179.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell. 2008a;14:312–323. doi: 10.1016/j.ccr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 181.Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res (Phila) 2009;2:409–418. doi: 10.1158/1940-6207.CAPR-08-0160. [DOI] [PubMed] [Google Scholar]
- 182.Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, et al. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature. 2004;430:686–689. doi: 10.1038/nature02789. [DOI] [PubMed] [Google Scholar]
- 183.Boily G, He XH, Pearce B, Jardine K, McBurney MW. SirT1-null mice develop tumors at normal rates but are poorly protected by resveratrol. Oncogene. 2009;28:2882–2893. doi: 10.1038/onc.2009.147. [DOI] [PubMed] [Google Scholar]
- 184.Wang RH, Zheng Y, Kim HS, Xu X, Cao L, Luhasen T, et al. Interplay among BRCA1, SIRT1, and Survivin during BRCA1-associated tumorigenesis. Mol Cell. 2008;32:11–20. doi: 10.1016/j.molcel.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kai L, Samuel SK, Levenson AS. Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer. 2010;126:1538–1548. doi: 10.1002/ijc.24928. [DOI] [PubMed] [Google Scholar]
- 186.Kala R, Shah HN, Martin SL, Tollefsbol TO. Epigenetic-based combinatorial resveratrol and pterostilbene alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including SIRT1-dependent γ-H2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer. 2015;15:672. doi: 10.1186/s12885-015-1693-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu ML, Li H, Yu LJ, Chen XY, Kong QY, Song X, et al. Shortterm resveratrol exposure causes in vitro and in vivo growth inhibition and apoptosis of bladder cancer cells. PLoS One. 2014;9:e89806. doi: 10.1371/journal.pone.0089806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Qin W, Zhang K, Clarke K, Weiland T, Sauter ER. Methylation and miRNA effects of resveratrol on mammary tumors vs. normal tissue. Nutr Cancer. 2014;66:270–277. doi: 10.1080/01635581.2014.868910. [DOI] [PubMed] [Google Scholar]
- 189.Papoutsis AJ, Selmin OI, Borg JL, Romagnolo DF. Gestational exposure to the AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring:Preventive effects of resveratrol. Mol Carcinog. 2015;54:261–269. doi: 10.1002/mc.22095. [DOI] [PubMed] [Google Scholar]
- 190.Azimi A, Hagh MF, Talebi M, Yousefi B, Hossein Pour Feizi AA, Baradaran B, et al. Time - and Concentration - Dependent Effects of Resveratrol on miR 15a and miR16-1Expression and Apoptosis in the CCRF-CEM Acute Lymphoblastic Leukemia Cell Line. Asian Pac J Cancer Prev. 2015;16:6463–8. doi: 10.7314/apjcp.2015.16.15.6463. [DOI] [PubMed] [Google Scholar]
- 191.Dhar S, Kumar A, Rimando AM, Zhang X, Levenson AS. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR-17 family in prostate cancer. Oncotarget. 2015;6:27214–27226. doi: 10.18632/oncotarget.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Clarke JD, Dashwood RH, Ho E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008;269:291–304. doi: 10.1016/j.canlet.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tomczyk J, Olejnik A. Sulforaphane--a possible agent in prevention and therapy of cancer. Postepy Hig Med Dosw. 2010;64:590–603. [PubMed] [Google Scholar]
- 194.Traka M, Gasper AV, Smith JA, Hawkey CJ, Bao Y, Mithen RF. Transcriptome analysis of human colon Caco-2 cells exposed to sulforaphane. J Nutr. 2005;135:1865–1872. doi: 10.1093/jn/135.8.1865. [DOI] [PubMed] [Google Scholar]
- 195.Meeran SM, Patel SN, Tollefsbol TO. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS One. 2010;5:e11457. doi: 10.1371/journal.pone.0011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 197.Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis. 2006;27:811–819. doi: 10.1093/carcin/bgi265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–1021. doi: 10.1158/1535-7163.MCT-06-0494. [DOI] [PubMed] [Google Scholar]
- 199.Wong CP, Hsu A, Buchanan A, Palomera-Sanchez Z, Beaver LM, Andres Houseman E, et al. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS One. 2014;9:e86787. doi: 10.1371/journal.pone.0086787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Su ZY, Zhang C, Lee JH, Shu L, Wu TY, Khor TO, et al. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev Res. 2014;7:319–329. doi: 10.1158/1940-6207.CAPR-13-0313-T. [DOI] [PubMed] [Google Scholar]
- 201.Mileo AM, Miccadei S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxidative Medicine and Cellular Longevity. 2016;2016:6475624. doi: 10.1155/2016/6475624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. Faseb j. 2006;20:506–508. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Myzak MC, Tong P, Dashwood WM, Dashwood RH, Ho E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp Biol Med (Maywood) 2007;232:227–234. [PMC free article] [PubMed] [Google Scholar]
- 204.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol Pharmacol. 2005;68:1018–1030. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 205.Huang HC, Way TD, Lin CL, Lin JK. EGCG stabilizes p27kip1 in E2-stimulated MCF-7 cells through down-regulation of the Skp2 protein. Endocrinology. 2008;149:5972–5983. doi: 10.1210/en.2008-0408. [DOI] [PubMed] [Google Scholar]
- 206.Berletch JB, Liu C, Love WK, Andrews LG, Katiyar SK, Tollefsbol TO. Epigenetic and genetic mechanisms contribute to telomerase inhibition by EGCG. J Cell Biochem. 2008;103:509–519. doi: 10.1002/jcb.21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Navarro-Peran E, Cabezas-Herrera J, Campo LS, Rodriguez-Lopez JN. Effects of folate cycle disruption by the green tea polyphenol epigallocatechin-3-gallate. Int J Biochem Cell Biol. 2007;39:2215–2225. doi: 10.1016/j.biocel.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 208.Kato K, Long NK, Makita H, Toida M, Yamashita T, Hatakeyama D, et al. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. Br J Cancer. 2008;99:647–665. doi: 10.1038/sj.bjc.6604521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tang WY, Newbold R, Mardilovich K, Jefferson W, Cheng RY, Medvedovic M, et al. Persistent hypomethylation in the promoter of nucleosomal binding protein 1 (Nsbp1) correlates with overexpression of Nsbp1 in mouse uteri neonatally exposed to diethylstilbestrol or genistein. Endocrinology. 2008;149:5922–5931. doi: 10.1210/en.2008-0682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Fang MZ, Jin Z, Wang Y, Liao J, Yang GY, Wang LD, et al. Promoter hypermethylation and inactivation of O(6)-methylguanine-DNA methyltransferase in esophageal squamous cell carcinomas and its reactivation in cell lines. Int J Oncol. 2005;26:615–622. [PubMed] [Google Scholar]
- 211.Vandegehuchte MB, Lemière F, Vanhaecke L, Vanden Berghe W, Janssen CR. Direct and transgenerational impact on Daphnia magna of chemicals with a known effect on DNA methylation. Comp Biochem Physiol C Toxicol Pharmacol. 2010;151:278–285. doi: 10.1016/j.cbpc.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 212.Paluszczak J, Krajka-KuŸniak V, Baer-Dubowska W. The effect of dietary polyphenols on the epigenetic regulation of gene expression in MCF7 breast cancer cells. Toxicol Lett. 2010;192:119–125. doi: 10.1016/j.toxlet.2009.10.010. [DOI] [PubMed] [Google Scholar]
- 213.Stefanska B, Rudnicka K, Bednarek A, Fabianowska-Majewska K. Hypomethylation and induction of retinoic acid receptor beta 2 by concurrent action of adenosine analogues and natural compounds in breast cancer cells. Eur J Pharmacol. 2010;638:47–53. doi: 10.1016/j.ejphar.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 214.Singh N, Misra K. Computational screening of molecular targets in Plasmodium for novel nonresistant anti-malarial drugs. Bioinformation. 2009;3:255–262. doi: 10.6026/97320630003255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kang J, Chen J, Shi Y, Jia J, Zhang Y. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol. 2005;69:1205–1213. doi: 10.1016/j.bcp.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 216.Cui L, Miao J, Furuya T, Li X, Su XZ, Cui L. PfGCN5-mediated histone H3 acetylation plays a key role in gene expression in Plasmodium falciparum. Eukaryot Cell. 2007;6:1219–1227. doi: 10.1128/EC.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sng JC, Taniura H, Yoneda Y. Histone modifications in kainate-induced status epilepticus. Eur J Neurosci. 2006;23:1269–1282. doi: 10.1111/j.1460-9568.2006.04641.x. [DOI] [PubMed] [Google Scholar]
- 218.Chiu J, Khan ZA, Farhangkhoee H, Chakrabarti S. Curcumin prevents diabetes-associated abnormalities in the kidneys by inhibiting p300 and nuclear factor-kappaB. Nutrition. 2009;25:964–972. doi: 10.1016/j.nut.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 219.Tikoo K, Meena RL, Kabra DG, Gaikwad AB. Change in post-translational modifications of histone H3, heat-shock protein-27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br J Pharmacol. 2008;153:1225–1231. doi: 10.1038/sj.bjp.0707666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Li HL, Liu C, de Couto G, Ouzounian M, Sun M, Wang AB, et al. Curcumin prevents and reverses murine cardiac hypertrophy. J Clin Invest. 2008;118:879–893. doi: 10.1172/JCI32865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 221.Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–878. doi: 10.1172/JCI33160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Liu HL, Chen Y, Cui GH, Zhou JF. Curcumin, a potent anti-tumor reagent, is a novel histone deacetylase inhibitor regulating B-NHL cell line Raji proliferation. Acta Pharmacol Sin. 2005;26:603–609. doi: 10.1111/j.1745-7254.2005.00081.x. [DOI] [PubMed] [Google Scholar]
- 223.Choi KC, Jung MG, Lee YH, Yoon JC, Kwon SH, Kang HB, et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009;69:583–592. doi: 10.1158/0008-5472.CAN-08-2442. [DOI] [PubMed] [Google Scholar]
- 224.Lea MA, Randolph VM. Induction of histone acetylation in rat liver and hepatoma by organosulfur compounds including diallyl disulfide. Anticancer Res. 2001;21:2841–2845. [PubMed] [Google Scholar]
- 225.Schwab M, Reynders V, Loitsch S, Steinhilber D, Schroder O, Stein J. The dietary histone deacetylase inhibitor sulforaphane induces human beta-defensin-2 in intestinal epithelial cells. Immunology. 2008;125:241–251. doi: 10.1111/j.1365-2567.2008.02834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27:2038–2046. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- 227.Pledgie-Tracy A, Sobolewski MD, Davidson NE. Sulforaphane induces cell type-specific apoptosis in human breast cancer cell lines. Mol Cancer Ther. 2007;6:1013–1021. doi: 10.1158/1535-7163.MCT-06-0494. [DOI] [PubMed] [Google Scholar]
- 228.Chakrabarti M, Khandkar M, Banik NL, Ray SK. Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res. 2012;1454:1–13. doi: 10.1016/j.brainres.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Chakrabarti M, Banik NL, Ray SK. miR-138 overexpression is more powerful than hTERT knockdown to potentiate apigenin for apoptosis in neuroblastoma in vitro and in vivo. Exp Cell Res. 2013;319:1575–1585. doi: 10.1016/j.yexcr.2013.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Jiang L, Huang Q, Chang J, Wang E, Qiu X. MicroRNA HSA-miR-125a-5p induces apoptosis by activating p53 in lung cancer cells. Exp Lung Res. 2011;37:387–398. doi: 10.3109/01902148.2010.492068. [DOI] [PubMed] [Google Scholar]
- 231.Saini S, Arora S, Majid S, Shahryari V, Chen Y, Deng G, et al. Curcumin modulates microRNA-203-mediated regulation of the Src-Akt axis in bladder cancer. Cancer Prev Res (Phila) 2011;4:1698–1709. doi: 10.1158/1940-6207.CAPR-11-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zhang J, Du Y, Wu C, Ren X, Ti X, Shi J, et al. Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186n signaling pathway. Oncol Rep. 2010;24:1217–1223. doi: 10.3892/or_00000975. [DOI] [PubMed] [Google Scholar]
- 233.Gao SM, Yang JJ, Chen CQ, Chen JJ, Ye LP, Wang LY, et al. Pure curcumin decreases the expression of WT1 by upregulation of miR-15a and miR-16-1 in leukemic cells. J Exp Clin Cancer Res. 2012;31:27. doi: 10.1186/1756-9966-31-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sun M, Estrov Z, Ji Y, Coombes KR, Harris DH, Kurzrock R, et al. Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells. Mol Cancer Ther. 2008;7:464–473. doi: 10.1158/1535-7163.MCT-07-2272. [DOI] [PubMed] [Google Scholar]
- 235.Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. J Nutr Biochem. 2010;21:140–146. doi: 10.1016/j.jnutbio.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 236.Gandhy SU, Kim K, Larsen L, Rosengren RJ, Safe S. Curcumin and synthetic analogs induce reactive oxygen species and decreases specificity protein (Sp) transcription factors by targeting microRNAs. BMC Cancer. 2012;12:564. doi: 10.1186/1471-2407-12-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jin Y. 3, 3′-Diindolylmethane inhibits breast cancer cell growth via miR-21-mediated Cdc25A degradation. Mol Cell Biochem. 2011;358:345–54. doi: 10.1007/s11010-011-0985-0. [DOI] [PubMed] [Google Scholar]
- 238.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, et al. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am J Transl Res. 2012;4:14–23. [PMC free article] [PubMed] [Google Scholar]
- 239.Melkamu T, Zhang X, Tan J, Zeng Y, Kassie F. Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol. Carcinogenesis. 2010;31:252–258. doi: 10.1093/carcin/bgp208. [DOI] [PubMed] [Google Scholar]
- 240.Ahn J, Lee H, Jung CH, Ha T. Lycopene inhibits hepatic steatosis via microRNA-21-induced downregulation of fatty acid-binding protein 7 in mice fed a high-fat diet. Mol Nutr Food Res. 2012;56:1665–1674. doi: 10.1002/mnfr.201200182. [DOI] [PubMed] [Google Scholar]
- 241.Chiang EP, Chiu SC, Pai MH, Wang YC, Wang FY, Kuo YH, et al. Organosulfur garlic compounds induce neovasculogenesis in human endothelial progenitor cells through a modulation of MicroRNA 221 and the PI3-K/Akt signaling pathways. J Agric Food Chem. 2013;61:4839–4849. doi: 10.1021/jf304951p. [DOI] [PubMed] [Google Scholar]
- 242.Xiao X, Chen B, Liu X, Liu P, Zheng G, Ye F, et al. Diallyl disulfide suppresses SRC/Ras/ERK signaling-mediated proliferation and metastasis in human breast cancer by up-regulating miR-34a. PLoS One. 2014;9:e112720. doi: 10.1371/journal.pone.0112720. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 243.Dhar S, Hicks C, Levenson AS. Resveratrol and prostate cancer: promising role for microRNAs. Mol Nutr Food Res. 2011;55:1219–1229. doi: 10.1002/mnfr.201100141. [DOI] [PubMed] [Google Scholar]
- 244.Sheth S, Jajoo S, Kaur T, Mukherjea D, Sheehan K, Rybak LP, et al. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PLoS One. 2012;7:e51655. doi: 10.1371/journal.pone.0051655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, Croce CM. Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFbeta signaling pathway in SW480 cells. Biochem Pharmacol. 2010;80:2057–2065. doi: 10.1016/j.bcp.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Sakurai MA, Ozaki Y, Okuzaki D, Naito Y, Sasakura T, Okamoto A, et al. Gefitinib and luteolin cause growth arrest of human prostate cancer PC-3 cells via inhibition of cyclin G-associated kinase and induction of miR-630. PLoS One. 2014;9:e100124. doi: 10.1371/journal.pone.0100124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Wu H, Huang M, Liu Y, Shu Y, Liu P. Luteolin Induces Apoptosis by Up-regulating miR-34a in Human Gastric Cancer Cells. Technol Cancer Res Treat. 2015;14:747–755. doi: 10.7785/tcrt.2012.500434. [DOI] [PubMed] [Google Scholar]
- 248.Xiao J, Gong AY, Eischeid AN, Chen D, Deng C, Young CY, et al. miR-141modulates androgen receptor transcriptional activity in human prostate cancer cells through targeting the small heterodimer partner protein. Prostate. 2012;72:1514–22. doi: 10.1002/pros.22501. [DOI] [PubMed] [Google Scholar]
- 249.Ide H, Tokiwa S, Sakamaki K, Nishio K, Isotani S, Muto S, et al. Combined inhibitory effects of soy isoflavones and curcumin on the production of prostate-specific antigen. Prostate. 2010;70:1127–1133. doi: 10.1002/pros.21147. [DOI] [PubMed] [Google Scholar]
- 250.Sharma RA, McLelland HR, Hill KA, Ireson CR, Euden SA, Manson MM, et al. Pharmacodynamic and pharmacokinetic study of oral Curcuma extract in patients with colorectal cancer. Clin Cancer Res. 2001;7:1894–1900. [PubMed] [Google Scholar]
- 251.Johnson JJ, Bailey HH, Mukhtar H. Green tea polyphenols for prostate cancer chemoprevention: A translational perspective. Phytomedicine. 2010;17:3–13. doi: 10.1016/j.phymed.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Nguyen MM, Ahmann FR, Nagle RB, Hsu CH, Tangrea JA, Parnes HL, et al. Randomized, double-blind, placebo-controlled trial of polyphenon E in prostate cancer patients before prostatectomy: evaluation of potential chemopreventive activities. Cancer Prev Res. 2012;5:290–8. doi: 10.1158/1940-6207.CAPR-11-0306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lazarevic B, Hammarstrom C, Yang J, Ramberg H, Diep LM, Karlsen SJ, et al. The effects of short-term genistein intervention on prostate biomarker expression in patients with localised prostate cancer before radical prostatectomy. Br J Nutr. 2012;108:2138–2147. doi: 10.1017/S0007114512000384. [DOI] [PubMed] [Google Scholar]
- 254.Miyanaga N, Akaza H, Hinotsu S, Fujioka T, Naito S, Namiki M, et al. Prostate cancer chemoprevention study: an investigative randomized control study using purified isoflavones in men with rising prostate-specific antigen. Cancer Sci. 2012;103:125–130. doi: 10.1111/j.1349-7006.2011.02120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Heath EI, Heilbrun LK, Li J, Vaishampayan U, Harper F, Pemberton P, et al. A phase I dose-escalation study of oral BR-DIM in castrate-resistant, non-metastatic prostate cancer. Am J Transl Res. 2010;2:402–411. [PMC free article] [PubMed] [Google Scholar]
- 256.Mariani S, Lionetto L, Cavallari M, Tubaro A, Rasio D, De Nunzio C, et al. Low prostate concentration of lycopene is associated with development of prostate cancer in patients with high-grade prostatic intraepithelial neoplasia. Int J Mol Sci. 2014;15:1433–1440. doi: 10.3390/ijms15011433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Goldberg DM, Yan J, Soleas GJ. Absorption of three wine-related polyphenols in three different matrices by healthy subjects. Clinical Biochemistry. 2003;36:79–87. doi: 10.1016/s0009-9120(02)00397-1. [DOI] [PubMed] [Google Scholar]
- 258.Alumkal JJ, Slottke R, Schwartzman J, Cherala G, Munar M, Graff JN, et al. A phase II study of sulforaphane-rich broccoli sprout extracts in men with recurrent prostate cancer. Investigational New Drugs. 2015;33:480–489. doi: 10.1007/s10637-014-0189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]