

Abstract
D-dopachrome tautomerase (D-DT) shares amino acid sequence similarity, structural architecture and biological activity with the cytokine MIF. Recent studies show that the two protein homologues also bind to the same cell surface receptor, CD74, to activate the ERK1/2 pathway that ultimately leads to pro-inflammatory and pro-survival gene expression. We recently showed that RTL1000 and DRa1-MOG-35-55, two biological drugs with potent anti-inflammatory properties that treat experimental autoimmune encephalomyelitis (EAE) in mice, bind to the cell surface receptor CD74 with high affinity and compete with MIF for binding to the same regions of CD74. Computational modeling of MIF and RTL1000 binding interactions with CD74 predicted the presence of three CD74 binding regions for each MIF homotrimer. Through a similar approach we have now expanded our work to study the D-DT (MIF2) interaction with CD74 that is mainly defined by three elements scattered throughout the disordered regions of the interacting molecules. The model predicted: a) a hydrophobic cradle between CD74 and D-DT consisting of N-terminal tyrosine residues of three CD74 monomers arranged in a planar alignment interacts with aromatic amino acid residues located in the disordered D-DT C-terminus; b) a triad consisting of the E103 residue on one D-DT monomer in close contact with R179 and S181 on one chain of the CD74 trimer forms an intermolecular salt bridge; and c) amino acid residues on the C-terminus random coil of CD74 chain C form a long interacting area of ~500Å2 with a disordered region of D-DT chain B. These three binding elements were also present in MIF/CD74 binding interactions, with involvement of identical or highly similar amino acid residues in each MIF homotrimer that partner with the exact same residues in CD74. Topologically, however, the location of the three CD74 binding regions of the D-DT homotrimer differs substantially from that of the three MIF binding regions. This key difference in orientation appears to derive from a sequence insertion in D-DT that topologically limits binding to only one CD74 molecule per D-DT homotrimer, in contrast to predicted binding of up to three CD74 molecules per MIF homotrimer. These results have implications for the manner in which D-DT and MIF compete with each other for binding to the CD74 receptor and for the relative potency of DRa1-MOG-35-55 and RTL1000 for competitive inhibition of D-DT and MIF binding and activation through CD74.
Keywords: MIF, D-DT, CD74, predicted binding interactions, inflammatory factors
Graphical Abstract
1. Introduction
Macrophage migration inhibitory factor (MIF) is a 115 amino acid residue protein that is expressed in a variety of tissues and cell types, including leukocytes, corticotrophic pituitary cells, epithelial and endothelial cells, and neurons. MIF is the founding member of a distinct structural superfamily and has a unique biology [1, 2]. It is considered to be a cytokine, a hormone, and a chemokine, and has enzymatic activity [1]. It plays a central role as a pro-inflammatory component of numerous autoimmune diseases and cancer [3]. Upon binding with nM affinity to the CD74/CD44 receptor complex [4, 5], MIF initiates a signaling pathway for sustained activation of ERK1/2 MAPK [5] that ultimately leads to gene transcription and synthesis of pro-inflammatory cytokines and chemokines and increased cell survival and proliferation [6]. In addition to binding to the CD74/CD44 receptor complex, MIF also interacts in a non-cognate manner with CXCR2, CXCR4 and CXCR7 [7–9], triggering trafficking and cell chemotactic responses to the sites of acute and chronic inflammation. The MIF protein has been crystalized and its three-dimensional structure was determined. [10–12] MIF is a trimer of identical subunits in which each monomer contains two antiparallel alpha helices apposed to a 4-stranded beta sheet platform. Additionally, the monomer also has two beta-strands that interact with the beta sheet of adjacent subunits. The beta sheets form a central barrel around a solvent-accessible channel with the alpha helices located in the periphery of the trimer
CD74 is a multifunctional type II transmembrane protein involved in several biological processes including, among others, the folding and subcellular trafficking of MHC Class II glycoproteins. Little is known about the overall structural conformation of CD74, but biochemical data indicate that the first 61 amino acid residues of the extracellular protein of the P31 isoform (CD74 56–117) are highly prone to proteolytic degradation in some unfolded regions (as shown by NMR), indicative of a substantial degree of a flexible random coil conformation [13]. Two discrete regions, the transmembrane domain (residues 32–56) and a membrane-distal trimer entity (residues 118–192), have been demonstrated by NMR and circular dichroism to form a highly ordered trimerization domain (TD) with a high content of alpha-helix conformation [13–16].
A second cognate ligand for CD74 with structural and biochemical similarity to MIF termed D-dopachrome tautomerase (D-DT), was described recently [11, 17]. D-DT and MIF share ~35% identity making them potential homologs [17]. From the structural standpoint the D-DT topology resembles that of MIF and folds to form a homotrimer with extensive contacts between subunits through the lateral beta-sheet strands [11]. D-DT binds as well to CD74 with high affinity (D-DT: KD = 5.42×10−9 M vs. MIF: KD = 1.40×10−9 M) [17] and competes with MIF for binding to the receptor, suggesting that the two homologues might share the same binding site on CD74. Like MIF, D-DT it is capable of triggering the activation of ERK1/2 MAP kinase to similar downstream signaling responses.
Despite the structural and functional knowledge about MIF and D-DT, the molecular parameters governing the interaction of these two cytokines with their shared receptor are not fully understood. Recently, using a protein:protein docking algorithm based on published biochemical and biological studies, a theoretical molecular model of the MIF/CD74 and RTL1000/CD74 complex was described [18]. This study showed that MIF interacts with disordered residues at the N- and the C-termini of the CD74 receptor through residues close to the C-terminus on the sides of the MIF barrel. This model predicted a dodecameric complex in which three binding sites on the MIF trimer each bound to one CD74 trimer [18]. Taking a similar approach we now describe a working model for the structure of the D-DT/CD74 complex. The model predicts that D-DT binds to the CD74 receptor with a different topology than MIF but uses a similar type of interaction and, to some extent similar amino acid residues, emphasizing the role of flexible loops and conserved sequences. The model also identifies residues on both proteins that are susceptible to mutagenesis that could be pivotal in testing respective binding interactions.
2. Materials and Methods
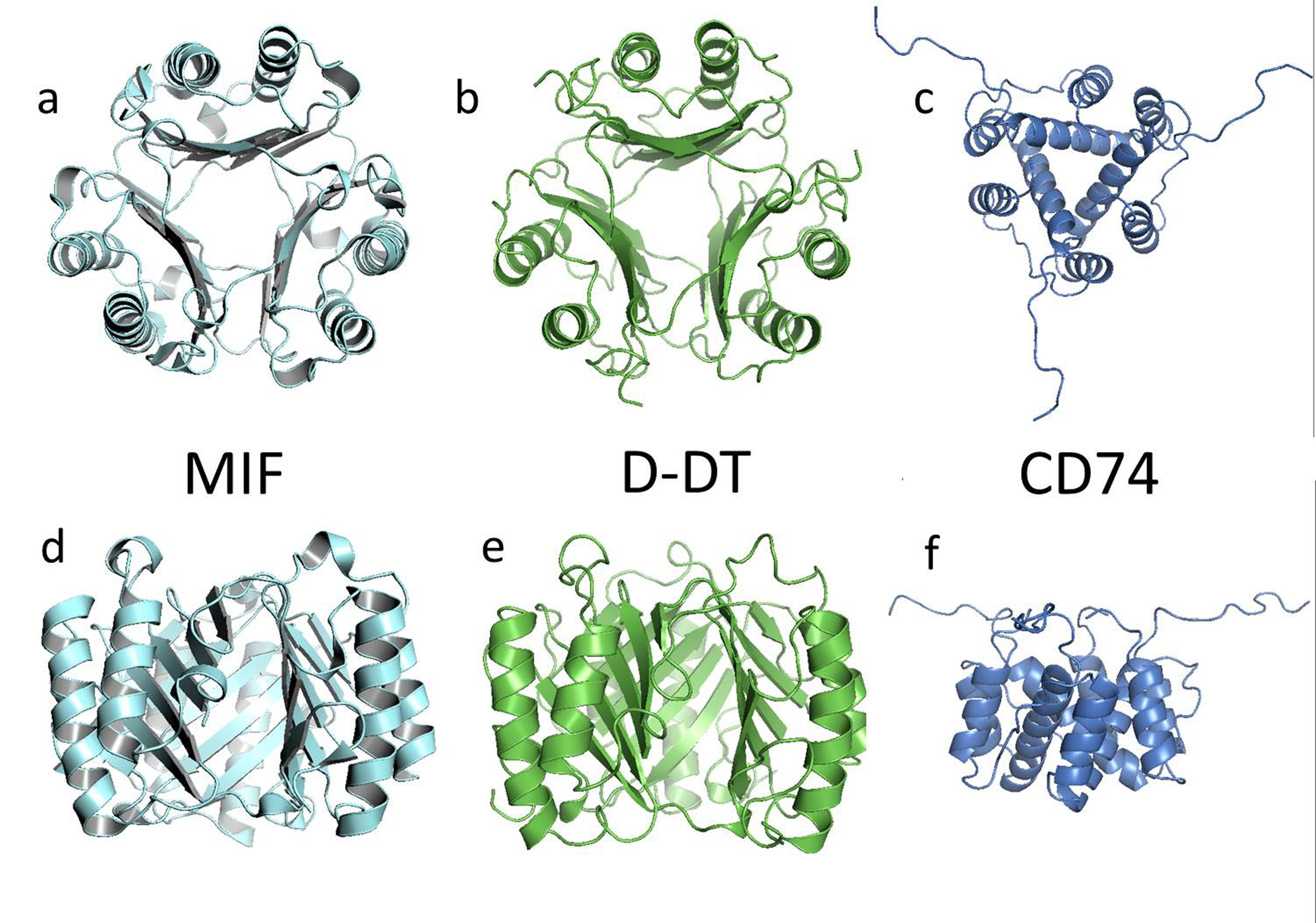
ZDOCK is a rigid-body protein:protein docking program that utilizes the Fast Fourier Transform algorithm to predict potential interactions between two binding partners. In order to grade the quality of the output models, the algorithm uses several variables such as shape complementarity, electrostatics and statistical potential terms [19, 20]. For this purpose we used the X-ray molecular coordinates for D-DT (PDB ID: 1DPT), MIF (PDB ID: 1GD0) and the CD74 partial extracellular domain (PDB ID: 1IIE) deposited in the Protein Data Bank. The structure of the CD74 trimeric domain was determined by NMR and our modeling involved 20 solved individual trimers [16], all of which were used for the docking work in this study. Structural models of MIF, D-DT and CD74 are shown in Figure 1. All structural rendering and molecular visualization was accomplished with PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). Protein sequence alignment was done using the tool Blast [21, 22].
Figure 1.

Cartoon describing the secondary, tertiary and quaternary structures of MIF (1GD0, a and d), D-DT (1DPT, b and e), and the CD74 trimerization domain (1IIE, c and f). Atomic coordinates were retrieved from the Protein Data Bank (PDB) and ribbon-rendered using PyMol. Upper row shows views of the structures from the top. Lower row show a lateral view of the same homotrimers. Molecular image of CD74 is at a smaller scale to allow visualization of the unstructured random coils.
3. Results
3.1 D-DT/CD74 complex docking modeling
The algorithm used in this study, ZDOCK, produced ten slightly different docking models of the D-DT/CD74 interaction, with the first model being the most energetically stable. Based on this model the D-DT homotrimer binds to the CD74-TD close to its N-terminus. A representative rendering of two different views of the optimal model of D-DT/CD74 binding is shown in Figure 2a&b. All ten binding predictions have three features in common: a) a hot spot characterized by a hydrophobic area of contact comprised of the N-terminal Y118 and G119 residues of the CD74 trimer and the C-terminal (mostly hydrophobic) amino acid residues W105, Q106, & 115-TFL-117 of D-DT, protected from the solvent by a seal of hydrophilic residues (Figure 3); b) a hydrophilic interaction by the triad R179 and S181 on CD74 and the D-DT residue E103 (Figure 4); and c) an area of contact in the flexible disordered C-terminal region of both molecules (Figure 5).
Figure 2.

Surface rendered model of the overall topology of binding for D-DT and the CD74 trimerization domain. D-DT binds to CD74 in an angular way. a) CD74 trimer vertical axis contrasts with the D-DT axis tilted about 120°. b) Complex with the D-DT axis maintained in a vertical orientation. The CD74 receptor is in blue and the different D-DT subunits are in light grey, pink and yellow.
Figure 3.

Highlight of the CD74 and D-DT images participating in the binding interface. a) Residues on CD74 are shown in blue (left) and D-DT residues are shown in yellow (right). b) Close up slice view from the top of the hot spot that characterizes the interaction between D-DT and CD74. The bulk of the CD74 structure is shown in grey. Y118 and G119 are colored in blue. Hydrophobic residues contributed from D-DT are colored yellow. c) Side view of a stick-rendered image of the D-DT/CD74 complex at the hot spot interface. Coloring as described for panel b. d) Hydrophilic ring sealing the hydrophobic patch of the interface. Most of the residues are contributed by CD74 protomers, including N120, T122, R179, H180, S181 and E183. D-DT contributes T69 and E71 positioned at the periphery of the interface. This is a typical arrangement found in hot spots. Orange residues are from CD74. CD74 residues Y118 and G119 are in blue and hydrophobic residues from D-DT are in yellow. Molecular images are not at the same scale.
Figure 4.

Potential salt bridges are formed by the interaction of D-DT and CD74. a) E103, R179, and S181 form a triad at the D-DT and CD74 interface, with a salt bridge predicted to form between CD74 R179 and D-DT E103. A second salt bridge forms between a second set of D-DT and CD74 protomers, while S181 and R179 form hydrogen bonds. b) Enlarged view of the salt bridges.
Figure 5.

Depiction of the transient interaction between D-DT and CD74. The CD74 trimer is grey with one binding region on an unstructured C-terminal extension colored in blue. Residues in one of the D-DT protomers involved in the binding is colored in cyan.
Hydrophobic area of contact: hot spot. This is the most extensive of the three interfaces and includes a large hydrophobic patch from each component of the complex, as suggested by the model presented in Figure 3. Each CD74 protomer in the trimer contributes the N-terminal residues Y118 and G119 located in the top center of the trimer (Figure 3a, 3b, 3c & 3d). The Y118-G119 pair from each monomer is arranged in a circular fashion, forming a nest-like area (Figure 3c & 3d) occupying the center of the CD74 binding face. These hydrophobic residues are located in a disordered flexible ribbon that also includes N120 and M121 at the N-terminal region of CD74. When viewed in consecutive models of the CD74 trimer NMR structures, these residues adopt slightly different conformations (Supplementary Figure 1), suggesting that this region of the CD74 trimer is very dynamic with only a few conformers able to establish a productive interaction with D-DT. This nest shelters a group of non-contiguous hydrophobic residues (I64, W105, Q106, 113-VM-114 & 116-FL-117) (Figures 3c, 3d & 3e) located near the C-terminus of one of the D-DT protomers, mostly of hydrophobic nature. Surrounding this hydrophobic contact interface between D-DT and CD74 are several polar amino acid residues contributed mostly by CD74 (120-NMT-123 and R179, S181 & E183) forming a boundary around the hydrophobic area of contact (Figure 3d). There are also polar residues contributed by D-DT (E101 of D-DT chain A and T69 & E71 from D-DT chain C, Figure 3d). In the model, R179 of one CD74 protomer and S181 of a different CD74 protomer are in close contact suggesting that hydrogen bonds might be present. An NMR structure for D-DT is not yet available, but it would be of interest to see how much freedom the C-terminal region of D-DT would display in complex with CD74 and how these different conformers would fit the binding site of CD74.
A second region includes polar and charged amino acid residues from each subunit of the interacting chains: CD74 179-RHS-181 on chain C and E103 on D-DT chain A (Figure 4a & 4b). The model presented here shows that the distance between the charged guanidinium group of CD74 R179 and the side chain carboxy-groups of the D-DT E101 are approximately 1.7–2.8 Å apart, suggesting that they might form a salt bridge. It is important to note that S181 on CD74 also participates in this polar triad through the OH− group from the serine side chain that shares a hydrogen bond with one of the NH3+ side chains of the R179 guanidinium group (Figure 4b). These charged residues appear to be partially exposed to the solvent in the non-complexed trimers but they are buried in the structure suggesting that the formation of a salt bridge would take a desolvation free energy of high magnitude that would be energetically unfavorable. The insertion of a salt bridge in a semi-hydrophobic environment as proposed by this model might ease this requirement. An additional possible interaction between charged residues in this complex involves R179 of CD74 chain B and E71 on D-DT chain C (Figure 4a & 4b). In the model these residues appear to be closer than 4Å, which is the average distance between charged residues in salt bridges. Given the exposure to the hydrophilic environment and the highly negative ΔG of solvation of these residues, it appears unlikely for them to participate in a salt bridge (Figure 4b).
The architecture of the third interaction is presented in Figure 5. The amino acid residues 185-KPTDA-189 located near the C-terminus on CD74 (chain C in this model) make close contact with amino acid residues 63-SIGVVGT-69 positioned in an unstructured loop connecting beta-strand number 4 and alpha helix 4 of the D-DT chain B, thus forming a contact area in which the MIF protomer wraps around the CD74 disordered segment. The physical contacts between polar amino acid residues on CD74 to nitrogen atoms in the main chain of D-DT (CD74 D188 carboxy group to one of the N atoms in the main chain) are not well defined and might be transient as has been documented for other proteins. Residues 101-PLE-103 located between β-strand 5 and the short alpha helix 6 also appear to contribute to the interaction, suggesting more of a role in stability of the complex than in the binding energy.
3.2 Model of the MIF/CD74 complex
In our previous work on the MIF/CD74 structure [18] we reported that the docking analysis predicted two regions in the CD74-TD trimer involved in complex formation: the N-terminal region involving 118-YGNMT-122 and a C-terminal sequence that includes 179-RHSLE-183. A closer inspection of this interaction shows that residues Y118-G119 in the N-terminal region of CD74-TD provide most of the area of contact, thus forming the hydrophobic nest described above for the D-DT/CD74 complex formation (Figure 6a). During binding MIF engages CD74 in a lateral manner exactly at the interface of two protomers (Figure 6a & 6b). Residues that shape this hydrophobic interface with CD74 include F49, P91 on MIF chain A and Y36, I64, W108, N109, F113 & A114 on MIF chain B) in the lateral area of the cylinder (Figure 6c). This interface resembles the hydrophobic nest shaped by the D-DT/CD74 main interface (Figure 3d). MIF and D-DT being homologs, it is not surprising that both proteins utilize the same (or highly conserved) amino acid residues to bind to the same receptor. In addition to this nest there is a hydrophilic ring of amino acid residues (N120, T122, R179, S181 & E183) surrounding the hydrophobic cradle formed by the contribution of each CD74 protomer pictured in Figure 6c. This hydrophilic ring is kept in place by several hydrogen bonds established between R179 and S181. The amino acid residues E54, S74, S76, K177, S90 & D92 from one MIF protomer and T112 from a second MIF protomer also are involved in this hydrophilic structure. The interaction between the peripheral CD74 residues and those of MIF form a seal that might protect the hydrophobic hot spot from contact with the solvent, thereby increasing the energy of binding. CD74 R179 from protomer A forms a hydrogen bond with T178 in CD74 protomer C. Interestingly, this residue also is making contact with the Y36 OH− group of MIF protomer C while the electronic cloud of the Y36 aromatic ring is kept inside the CD74 tyrosine nest of the binding interface. CD74 R179 from protomer B is making contact with the T178 of CD74 protomer A and finally CD74 R179 of protomer C is establishing hydrogen bonds with T178 on protomer B of CD74 (see Figure 6c).
Figure 6.

Overall topology of the MIF/CD74 complex as predicted by ZDOCK. a) Amino acid residues involved in the hot spot contributed by CD74 (left panel, residues Y118 and G119 in blue and surrounding hydrophilic residues in orange) and the corresponding MIF binding site for CD74 colored in yellow (right panel). Models of the whole molecules are not at the same scale. b) Overall topology of the MIF (yellow) and CD74 (blue) complex. c) Amino acid residues Y118 and G119 involved in the hot spot contributed by CD74 are in blue. Residues in orange represent the CD74 hydrophilic ring surrounding the hydrophobic core.
4. Discussion
The structures of D-DT and MIF have been described at the atomic level and the coordinates have been deposited in the Protein Data Bank (PDB). At first glance, we can see that at the structural level both proteins fold in a similar way in a very well defined conformation to form a homotrimer with extensive contacts between subunits. Each protomer exhibits two alpha helices on top of a beta-sheet platform. In the homotrimers, the beta strands are located in the center according to a 3-fold rotation axis and form a solvent channel (see Figure 1).
In contrast and because of its molecular complexity, there are separate reports for different portions of the CD74 structure that include the transmembrane domain [15], the trimerization domain [16] including the disordered region 54–115, and the thyroglobulin domain [23]. The coordinates for the CD74 polypeptide as a whole are not available. However, use of heteronuclear nuclear magnetic resonance (hnNMR) [14] allowed the ability to delimit mobile segments of p31, one of the CD74 isoforms in human cells, from the rigid structure of the trimerized domain [16]. Their data from double and triple resonance show that amino acid residues 56–110 and 187–216 are mobile flexible segments with a high degree of conformational enthropy. These data are reinforced by biochemical studies indicating that CD74 is highly susceptible to proteolytic degradation [13] in the intracellular environment. Recently, however, a new truncated serum soluble form of CD74 has been described from patients with autoimmune liver disease [24]. It is important to emphasize that this soluble CD74 survives because of the lack of proteolytic activity in the fluid phase of the bloodstream that might prevent catastrophic consequences in the homeostatic organism. This soluble form of CD74 is able to bind MIF but the kinetic properties of the binding are unknown. Taken collectively, all of this evidence argues in favor of a model of the p31 isoform of CD74 that has a substantial content of a randomly coiled disordered conformation upstream and downstream of the trimerization domain.
In addition to assisting with the folding and trafficking of MHC Class II in the subcellular endosomal compartment, CD74 is also the cognate receptor for MIF and its functional homologue D-DT [4, 17] and requires the association with CD44 at the cell surface to effect downstream MAPK signaling [5]. Upon binding to the CD44/CD74 co-receptor complex, MIF and D-DT initiate the phosphorylation of ERK1/2 [4, 17] that sets in motion a strong pro-inflammatory and proliferative response [25, 26]. Nevertheless, the molecular interaction of these two cytokines with their receptor at the molecular level has not been addressed. Part of the reason for this lack of information is the difficulty to produce substantive amounts of full length and biochemically stable CD74 for crystallographic or NMR studies.
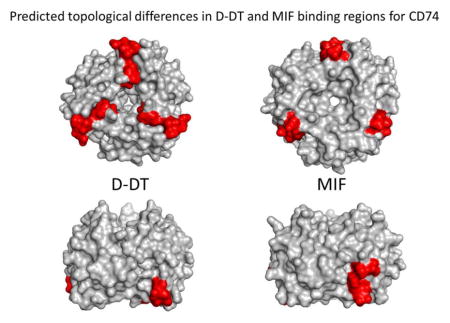
In this work we provide a theoretical rationale for the MIF and D-DT interactions with CD74. Using the ZDOCK docking algorithm we computed the potential sites of binding interface(s) between these two binding pairs. There are no other studies addressing the molecular mechanisms of the D-DT/CD74 interaction. The use of protein:protein docking algorithms is an alternative for exploring and scanning amino acid residue substitution in order to assign those residues for their energetic contribution to the binding. The algorithm predicted 10 different models for the complex CD74/D-DT and all of them shared a common topology of binding at the bottom of the D-DT barrel as shown as shown Figure 7a. D-DT binds to the trimeric CD74 to form a hexameric complex with one CD74 trimer that binds one D-DT trimer. In contrast, one trimer of CD74 binds to each one of the three MIF subunit interfaces, potentially resulting in a dodecameric complex.
Figure 7.

a) Predicted topology of the differing D-DT and MIF binding sites for CD74. Upper panel represents a view of the barrel of the trimers. Lower panel represents a side view of the trimers. b) Proposed alignment between MIF and D-DT. Amino acid residues thought to be inserted or deleted during or after duplication are in blue. c) Orientation of D-DT and MIF binding residues (shown in yellow with the aromatic residues F and W in red) to CD74 hot spot (in blue). d) Aligned ribbon illustration of the C-terminal binding motif for CD74 of MIF and D-DT.
Perhaps the most relevant of the D-DT/CD74 complex interactions is through the hydrophobic interface as described above. As seen in Figures 3a and 6a, the N-terminal Y118 and G119 of CD74 form an aromatic core with hydrophobic properties that nests residues I64, W106, Q107, F117 & L118 located at the very C-terminus segment of one protomer of the D-DT homotrimer. These amino acid residues are homologs to MIF amino acid residues I64, W109, N110, F114 & A115 on MIF. Mutagenesis studies revealed that I64A and W109A partially affected neutrophil-recruiting activity of MIF, lending support to our model that these residues are part of the hydrophobic spot in the interface that might play an important role in MIF activity [27]. In the same study the authors found that Y36A, K66A & N109A (N110 in the alignment shown in Figure 7b) abolished MIF biological activity. In the model proposed here, Y36 is located in the periphery of the hydrophilic seal that surrounds the hot spot and N109 (N110, Fig. 7b) inserts itself in the center space of the CD74 trimer. None of these residues are fully conserved in the D-DT structure (R36, V66 & Q107). Whether or not these residues affect the biological activity of MIF by disrupting the binding to CD74 remains to be seen.
Salt bridges have been defined as interactions that provide stability to tertiary and quaternary structure of proteins [28, 29] but contribute poorly to interface specificity and binding energy. In our model salt bridges are connecting residues of two different trimers suggesting that they might be involved in maintaining complex stability (Figure 4). Likewise, the driving force of the transient interaction described in Figure 5 is mostly through Van der Waals forces, with a few hydrogen bonds (not shown) between side chains of D-DT E103 and CD74 Q184. This region’s energetic contribution to the global energy of binding can be considered transient and in the moderate category when compared to the contribution of hydrophobic interactions. We can speculate that this weak and transient encounter could very well serve as a first point of contact that positions the molecules for more energetically favorable binding modes. This feature might provide for opportunities for different or simultaneous low affinity interactions with the final outcome of a strong global affinity. It is predicted that these weak transient short-lived interactions are characterized by dissociation constants (KD) in the μM range [30].
4.1 Anatomy of MIF and D-DT binding site for CD74: a hot spot?
Using our models as a source, we compared the MIF/CD74 and D-DT/CD74 main interfaces (Figure 7a). According to these models it was clear that this area might play a significant role in the binding of MIF and D-DT to their common receptor. A more detailed inspection of the interfaces showed that the participating side chains in the hydrophobic recognition correspond to the same (or similar) residues on D-DT and MIF: WTFA (and possibly N110) in MIF and WTFL in D-DT (and possibly Q107). Of these four residues, only W is not sequentially adjacent to the other three but spatially is part of the cluster. This mode of binding has been referred to as a hot spot and was initially described for the interaction of the human growth hormone and its receptor [31]. Those authors found that by alanine scanning substitutions on the hormone side, two tryptophan residues accounted for more than 75% of the binding free energy. Later studies extended this concept to a more general paradigm and showed that hot spots are enriched in very specific amino acid side chains like R, D, H, I, W and Y [32]. In the MIF trimer W, I, and Y (three of the more frequently residues found in hot spots) are strategically located inside the hydrophobic core, as predicted by the algorithm and according to the hot spot concept. In the unbound MIF trimer these residues are topologically located on the side of the cylinder at the interface of two subunits in the trimer (Figure 6a). Interestingly, Lolis’ group reported that substitution of I64 or W108 rendered MIF partially biologically inactive [27]. In our predicted model, both amino acid residues are contained in or near the hot spot but the contribution of these two residues to the binding energy of MIF to CD74 remains to be investigated.
In contrast to MIF, the D-DT X-ray structure shows that these residues are located in the bottom of the barrel at the periphery (Figure 7a). The reason for this discrepancy is a potential insertion of an intervening (mostly) hydrophobic segment (108-IGKIGTVM-115, Figure 7b) that pushes the D-DT binding motif downwards and towards the bottom and center of the barrel of the trimer. This difference might explain the ability of D-DT to bind to the CD74 hot spot with a different topology and orientation than MIF (Figure 7c & 7d) and potentially with a different affinity and stoichiometry [17]. There are no biochemical or mutagenesis studies yet available that address these potential D-DT residues involved in the binding. Our model predicts that Y36, I64 and W105 of D-DT (the latter residues structurally equivalent to I64 and W108 in MIF) are part of a hot spot, and may play a key role in the binding free energy of this cytokine to CD74. In the MIF homotrimer this motif is positioned on one side at the interface of two subunits. The chemical nature of the residues involved in the binding of both cytokines to CD74 is similar (see above). MIF as well as D-DT hydrophobic residues are sheltered by the aromatic cradle on CD74 and share basically the same or similar chemical properties. In protein:protein interfaces hot spots contribute up to 75% of the binding energy [31] and correlate with structurally conserved interface residues [33]. Taken into context, we speculate that this hot spot plays a far more important role in the binding of both MIF and D-DT than the salt bridges and that substitutions in CD74 Y118 and/or G119 might have profound effects on the ability of CD74 to bind their ligands. The other two interfaces described here for the D-DT/CD74 pair might contribute to the stability of the complex rather than to the binding free energy. In both cases, the regions contributing to the binding are located at the C-terminal segment on both complexes, a region of great flexibility [10, 12, 14, 34] and we can envision that these residues are free to move about and adopt structures of high conformational enthropy.
Our theoretical model has implications when considering the kinetic parameters governing the D-DT/CD74 or MIF/CD74 complex formation. The models presented in this work predict that a dodecameric structure in which one MIF homotrimer is kept in place by 3 CD74 molecules bound to each of the subunit interfaces would result in a long lasting complex with a slow association and a slow dissociation constant since MIF has three binding sites for CD74. Since the CD74 binding site on MIF is at the subunit interface we might infer that a monomeric MIF would render the cytokine inactive [35]. On the other hand, the interaction between D-DT and CD74 would result in a weaker interaction with a rapid complex formation but also a rapid dissociation rate since, due to stearic limitations, as there is only one predicted binding site for CD74 per every molecule of D-DT. Thus, it is expected that D-DT would not efficiently outcompete MIF to bind the same receptor, in concert with previously reported biochemical data from the Bucala laboratory showing that D-DT has a ~3-fold higher binding rate, but also dissociates much faster than MIF [4, 17]. Because of lack of data, it is challenging to picture how the co-receptors CD44 and CXCR4 might influence the binding properties of MIF or D-DT to CD74 on the cell surface. In this regard, Shi et al. showed that in a transfected COS cell line, MIF bound only to CD74-transfected cells and that CD44 was needed for further signaling [5]. We can speculate that upon MIF binding, CD74 might undergo some structural rearrangement to transmit the “MIF-bound” message through CD44 or CXCR4 [36], two known co-receptors triggering the downstream signaling cascade.
5. Conclusions
In summary, our model provides suggestions for mutagenesis studies to evaluate the role of specific amino acid residues that might be crucial for the binding of CD74 to its ligands, MIF and D-DT.
Supplementary Material
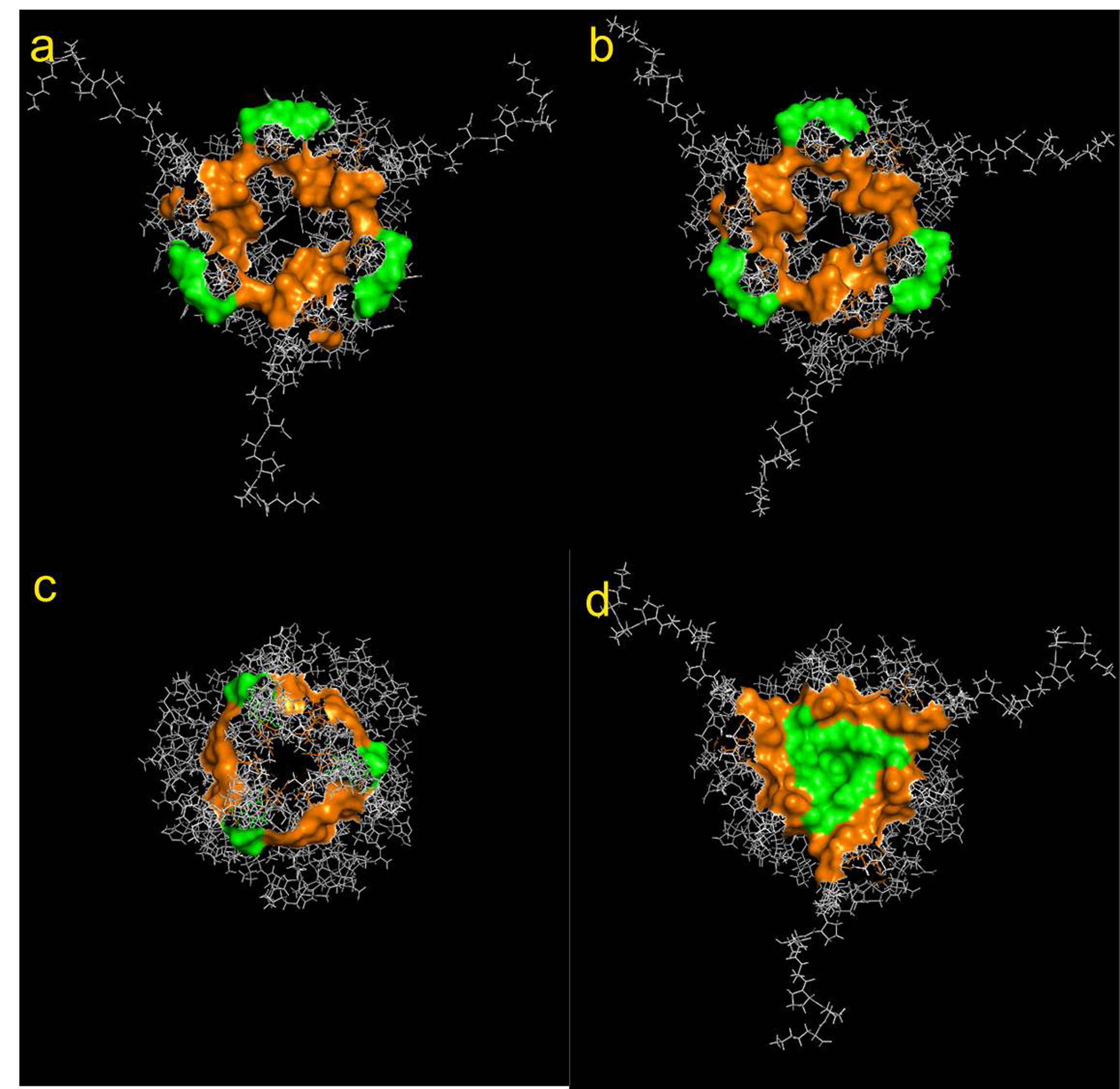
Supplemental Figure 1: Structures of several CD74 conformers as reported in the PDB. View from the MIF and D-DT binding regions. The coordinates deposited in the PDB describe 20 different structures for the CD74 trimerization domain. Residues Y118 and G119 are colored in green. CD74 residues N120, T122, R179, S181 and E183 are colored in orange. Only the structure shown in Panel d) would have a conformation capable of binding to MIF or D-DT.
{kind=link}
{kind=link}
Highlights.
Docking analysis used to study structural orientation of MIF and D-DT binding to CD74
CD74 binding region differed for D-DT vs. MIF due to 8 amino acid insertion in D-DT
Model predicts 3 CD74 binding sites per each MIF and D-DT molecule
D-DT/CD74 and MIF/CD74 binding involves a hot spot, salt bridges and hydrogen bonds
Model is consistent with known binding constants and empirical binding data
Acknowledgments
The authors wish to thank Gail Kent for assistance with manuscript submission.
Funding: This work was supported by the National Institutes of Health [grant AI122574] and National Multiple Sclerosis Society [grant RG-5068-A-6] (AAV); National Institutes of Health [grants AR049610 and AI042310] (LL, RB); National Multiple Sclerosis Society [grant RG5272A1/T] (GB); and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development (AAV). The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Conflict of Interest: Drs. Vandenbark, Benedek, Meza-Romero and OHSU have a significant financial interest in Artielle ImmunoTherapeutics, Inc., a company that may have a commercial interest in the results of this research and technology. This potential conflict of interest has been reviewed and managed by the OHSU and VAPHCS Conflict of Interest in Research Committees. Kelley Jordan and Drs. Leng, Pantouris, Lolis, and Bucala have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bucala R. MIF, MIF alleles, and prospects for therapeutic intervention in autoimmunity. J Clin Immunol. 2013;33(Suppl 1):S72–8. doi: 10.1007/s10875-012-9781-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stamps SL, Fitzgerald MC, Whitman CP. Characterization of the role of the amino-terminal proline in the enzymatic activity catalyzed by macrophage migration inhibitory factor. Biochemistry. 1998;37(28):10195–202. doi: 10.1021/bi9806955. [DOI] [PubMed] [Google Scholar]
- 3.O’Reilly C, Doroudian M, Mawhinney L, Donnelly SC. Targeting MIF in Cancer: Therapeutic Strategies, Current Developments, and Future Opportunities. Med Res Rev. 2016;36(3):440–60. doi: 10.1002/med.21385. [DOI] [PubMed] [Google Scholar]
- 4.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197(11):1467–76. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25(4):595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, Bucala R, Shachar I. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem. 2008;283(5):2784–92. doi: 10.1074/jbc.M703265200. [DOI] [PubMed] [Google Scholar]
- 7.Alampour-Rajabi S, El Bounkari O, Rot A, Muller-Newen G, Bachelerie F, Gawaz M, Weber C, Schober A, Bernhagen J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015;29(11):4497–511. doi: 10.1096/fj.15-273904. [DOI] [PubMed] [Google Scholar]
- 8.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13(5):587–96. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 9.Klasen C, Ohl K, Sternkopf M, Shachar I, Schmitz C, Heussen N, Hobeika E, Levit-Zerdoun E, Tenbrock K, Reth M, Bernhagen J, El Bounkari O. MIF promotes B cell chemotaxis through the receptors CXCR4 and CD74 and ZAP-70 signaling. Journal of immunology. 2014;192(11):5273–84. doi: 10.4049/jimmunol.1302209. [DOI] [PubMed] [Google Scholar]
- 10.Sugimoto H, Taniguchi M, Nakagawa A, Tanaka I, Suzuki M, Nishihira J. Crystallization and preliminary X-ray analysis of human D-dopachrome tautomerase. J Struct Biol. 1997;120(1):105–8. doi: 10.1006/jsbi.1997.3904. [DOI] [PubMed] [Google Scholar]
- 11.Sugimoto H, Taniguchi M, Nakagawa A, Tanaka I, Suzuki M, Nishihira J. Crystal structure of human D-dopachrome tautomerase, a homologue of macrophage migration inhibitory factor, at 1.54 A resolution. Biochemistry. 1999;38(11):3268–79. doi: 10.1021/bi982184o. [DOI] [PubMed] [Google Scholar]
- 12.Sun HW, Bernhagen J, Bucala R, Lolis E. Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc Natl Acad Sci U S A. 1996;93(11):5191–6. doi: 10.1073/pnas.93.11.5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jasanoff A, Park SJ, Wiley DC. Direct observation of disordered regions in the major histocompatibility complex class II-associated invariant chain. Proc Natl Acad Sci U S A. 1995;92(21):9900–4. doi: 10.1073/pnas.92.21.9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jasanoff A, Song S, Dinner AR, Wagner G, Wiley DC. One of two unstructured domains of Ii becomes ordered in complexes with MHC class II molecules. Immunity. 1999;10(6):761–8. doi: 10.1016/s1074-7613(00)80075-8. [DOI] [PubMed] [Google Scholar]
- 15.Kukol A, Torres J, Arkin IT. A structure for the trimeric MHC class II-associated invariant chain transmembrane domain. J Mol Biol. 2002;320(5):1109–17. doi: 10.1016/s0022-2836(02)00563-6. [DOI] [PubMed] [Google Scholar]
- 16.Jasanoff A, Wagner G, Wiley DC. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. EMBO J. 1998;17(23):6812–8. doi: 10.1093/emboj/17.23.6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merk M, Zierow S, Leng L, Das R, Du X, Schulte W, Fan J, Lue H, Chen Y, Xiong H, Chagnon F, Bernhagen J, Lolis E, Mor G, Lesur O, Bucala R. The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF) Proc Natl Acad Sci U S A. 2011;108(34):E577–85. doi: 10.1073/pnas.1102941108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meza-Romero R, Benedek G, Leng L, Bucala R, Vandenbark AA. Predicted structure of MIF/CD74 and RTL1000/CD74 complexes. Metab Brain Dis. 2016;31(2):249–55. doi: 10.1007/s11011-016-9798-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen R, Weng Z. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins. 2002;47(3):281–94. doi: 10.1002/prot.10092. [DOI] [PubMed] [Google Scholar]
- 20.Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–3. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 23.Guncar G, Pungercic G, Klemencic I, Turk V, Turk D. Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J. 1999;18(4):793–803. doi: 10.1093/emboj/18.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, Garcia AB, McCrann C, Chapiro J, Meinhardt A, Mizue Y, Nikolic-Paterson DJ, Bernhagen J, Kaplan MM, Zhao H, Boyer JL, Bucala R. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatology. 2014;59(2):580–91. doi: 10.1002/hep.26664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99(1):345–50. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brock SE, Rendon BE, Xin D, Yaddanapudi K, Mitchell RA. MIF family members cooperatively inhibit p53 expression and activity. PloS one. 2014;9(6):e99795. doi: 10.1371/journal.pone.0099795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pantouris G, Syed MA, Fan C, Rajasekaran D, Cho TY, Rosenberg EM, Jr, Bucala R, Bhandari V, Lolis EJ. An Analysis of MIF Structural Features that Control Functional Activation of CD74. Chem Biol. 2015;22(9):1197–205. doi: 10.1016/j.chembiol.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keskin O, Ma B, Rogale K, Gunasekaran K, Nussinov R. Protein-protein interactions: organization, cooperativity and mapping in a bottom-up Systems Biology approach. Phys Biol. 2005;2(2):S24–35. doi: 10.1088/1478-3975/2/2/S03. [DOI] [PubMed] [Google Scholar]
- 29.Keskin O, Ma B, Nussinov R. Hot regions in protein--protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345(5):1281–94. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 30.Perkins JR, Diboun I, Dessailly BH, Lees JG, Orengo C. Transient protein-protein interactions: structural, functional, and network properties. Structure. 2010;18(10):1233–43. doi: 10.1016/j.str.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–6. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 32.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280(1):1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 33.Li X, Keskin O, Ma B, Nussinov R, Liang J. Protein-protein interactions: hot spots and structurally conserved residues often locate in complemented pockets that pre-organized in the unbound states: implications for docking. J Mol Biol. 2004;344(3):781–95. doi: 10.1016/j.jmb.2004.09.051. [DOI] [PubMed] [Google Scholar]
- 34.Sugimoto H, Suzuki M, Nakagawa A, Tanaka I, Nishihira J. Crystal structure of macrophage migration inhibitory factor from human lymphocyte at 2.1 A resolution. FEBS letters. 1996;389(2):145–8. doi: 10.1016/0014-5793(96)00553-4. [DOI] [PubMed] [Google Scholar]
- 35.Fan C, Rajasekaran D, Syed MA, Leng L, Loria JP, Bhandari V, Bucala R, Lolis EJ. MIF intersubunit disulfide mutant antagonist supports activation of CD74 by endogenous MIF trimer at physiologic concentrations. Proc Natl Acad Sci U S A. 2013;110(27):10994–9. doi: 10.1073/pnas.1221817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583(17):2749–57. doi: 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Structures of several CD74 conformers as reported in the PDB. View from the MIF and D-DT binding regions. The coordinates deposited in the PDB describe 20 different structures for the CD74 trimerization domain. Residues Y118 and G119 are colored in green. CD74 residues N120, T122, R179, S181 and E183 are colored in orange. Only the structure shown in Panel d) would have a conformation capable of binding to MIF or D-DT.