

Abstract
Objective
Cardiac natriuretic peptides (NPs) bind to two receptors (NPRA‐mediator of signaling; NPRC‐clearance receptor) whose ratio, NPRR (NPRA/NPRC), determines the NP bioactivity. This study investigated the relationship of NP receptor gene expression in adipose tissue and muscle with obesity and glucose intolerance. Prospectively, the study also assessed whether changes in NP receptor expression and thermogenic gene markers accompanied improvements of insulin sensitivity.
Methods
A cross‐sectional study of subjects with a wide range of BMI and glucose tolerance (n = 50) was conducted, as well as a randomized 12‐week trial of subjects with type 2 diabetes mellitus (T2DM) treated with pioglitazone (n = 9) or placebo (n = 10).
Results
NPRR mRNA was significantly lower in adipose tissue of subjects with obesity when compared with lean subjects (P ≤ 0.001). NPRR decreased with progression from normal glucose tolerance to T2DM (P < 0.01) independently of obesity. Treatment of subjects with T2DM with pioglitazone increased NPRR in adipose tissue (P ≤ 0.01) in conjunction with improvements in insulin sensitivity and increases of the thermogenic markers PPARγ coactivator‐1α and uncoupling protein 1 (P ≤ 0.01).
Conclusions
Decreased adipose tissue NPRR was associated with obesity, glucose intolerance, and insulin resistance. This relationship was not observed for skeletal muscle NPRR. Pharmacological improvement of insulin sensitivity in subjects with T2DM was tied to improvement in NPRR and increased expression of genes involved in thermogenic processes.
Keywords: natriuretic peptide receptors, adipose tissue, obesity, type 2 diabetes mellitus
Introduction
The cardiac peptides atrial natriuretic peptide (ANP) and B‐type natriuretic peptide (BNP) are best known for their role as key hormones in cardiorenal homeostasis 1, 2. ANP and BNP bind and activate a membrane receptor, NP receptor A (NPRA), which has intrinsic guanylyl cyclase activity 3, 4. The cGMP generated activates protein kinase G (PKG) resulting in phosphorylation of its targets. Along with their proteolysis by endopeptidase, one of the physiological pathways by which NPs are removed from circulation is via receptor‐mediated internalization by the NP receptor C (NPRC), the so‐called clearance receptor 5. The relative level of NPRA to NPRC is thought to be a major determinant of NP system activity 6, 7.
Almost two decades ago, NP receptors were unexpectedly found to be expressed in the adipose tissue of both rats 8 and humans 9, suggesting that there may be a metabolic role for NPs. ANP and BNP were reported to induce a strong lipolytic effect in cultured human adipocytes with potency similar to catecholamine activation of the β‐adrenergic receptors (β‐AR; ref. 10). Metabolic and cardiorenal disorders, often coupled with hypertension, have been associated with dysregulation of the NP system at the level of the circulating peptides 11 and the signaling pathways mediating their biological effects 12. For example, in large‐scale studies of subjects from the Framingham Heart Study, plasma levels of NPs were lower in subjects with obesity 13 and those with metabolic risk factors 14.
Recently, we showed that NPs can activate a brown fat gene expression pattern to increase energy expenditure 15. In human adipocytes and animal models, similar to the process driven by β‐AR agonists, NPs promote transcriptional regulation of genes involved in mitochondrial biogenesis and uncoupled respiration, such as PPARγ coactivator‐1α (PGC‐1α) and uncoupling protein 1 (UCP‐1; ref. 15). NPs have also been reported to enhance fat oxidation in human skeletal muscle myotubes 16. Consistent with these cell culture studies, mice overexpressing BNP or PKG had increased PGC‐1α and PPARγ, mitochondrial biogenesis, and fat oxidation in skeletal muscle and were resistant to diet‐induced obesity and glucose intolerance 12.
To translate these findings into humans, we compared the expression of NPRA and NPRC in adipose tissue and skeletal muscle from subjects with normal weight and subjects with obesity who exhibited a range of glucose tolerance and insulin resistance. We also prospectively investigated whether an intervention that improves insulin sensitivity changed the expression of NP receptors, the NPRA/NPRC ratio (NPRR), and the expression of thermogenic marker genes.
Methods
Design of clinical studies
Two distinct populations were investigated: 1) a cross‐sectional study of subjects with a wide range of BMI and glucose tolerance, and 2) a cohort of subjects with type 2 diabetes mellitus (T2DM) treated with pioglitazone or placebo (ClinicalTrials.gov #NCT00656864). All aspects of the studies were approved by the Institutional Review Boards of Florida Hospital (study 1) and the University of Vermont (study 2). All study participants provided written informed consent prior to participation.
Study 1: Cross‐sectional study
We initially performed a cross‐sectional study of 50 subjects (32 females and 18 males) with a wide range of BMI (18.3‐60.3 kg/m2) who were weight stable (< 3 kg change in the 8 weeks prior to study). Subjects were divided according to BMI into two groups: 24 subjects with normal weight (Lean; BMI ≤ 25 kg/m2) and 26 subjects with obesity (Obese; BMI ≥ 30 kg/m2; Table 1). Subjects were further categorized according to their glucose tolerance status (based on historical diagnosis, glucose tolerance test, and HbA1C) into three groups: normal glucose tolerance (NGT), impaired glucose metabolism (IGM), and type 2 diabetes mellitus (T2DM). Percutaneous needle biopsies of subcutaneous abdominal adipose tissue (SAAT) and the vastus lateralis muscle were performed to obtain tissue samples for analysis. Biopsies of SAAT were performed on the right or left lower quadrant using a Mercedes Liposuction needle (Miami Fat Supply, Sunrise, FL, USA). Skeletal muscle biopsies were performed on the right leg using the Bergstrom technique (Millennium Surgical Corp., Narbeth, PA, USA). Detailed phenotyping of the participants included the following: anthropometric measures (weight, height, and waist circumference), fasting glucose and lipid profile, body composition measured by dual‐energy X‐ray absorptiometry (DXA) using a GE Lunar iDXA whole‐body scanner (Lunar iDEXA, GE, Madison, WI, USA), and resting metabolic rate and respiratory quotient measured using a MAX‐II metabolic cart with a canopy attachment (AEI Technologies, Pittsburgh PA, USA). On separate days, a standard 75 g oral glucose tolerance test (OGTT) and an intravenous glucose tolerance test (IVGTT) were performed.
Table 1.
Clinical parameters and measures of insulin sensitivity—Study 1: Cross‐sectional study
| Entire group, | Lean, | Obese, | |
|---|---|---|---|
| n = 50 | n = 24 | n = 26 | |
| NGT/IGM/T2DM (n) | 31/10/9 | 18/5/1 | 13/5/8 |
| Age (years) | 43.8 ± 1.9 | 40.6 ± 3.3 | 46.7 ± 2.2a |
| BMI (kg/m2) | 30.4 ± 1.3 | 22.2 ± 0.4 | 37.7 ± 1.2c |
| Waist circumference (cm) | 96.3 ± 3.2 | 77.1 ± 1.2 | 114.8 ± 3.2c |
| Lean mass (kg) | 49.2 ± 1.8 | 43.7 ± 1.9 | 53.3 ± 2.7b |
| Fat mass (kg) | 33.1 ± 2.6 | 18.0 ± 0.8 | 47.6 ± 2.9c |
| Total body fat (%) | 38.5 ± 1.5 | 29.6 ± 1.4 | 47.3 ± 1.1c |
| Triglycerides (mmol/l) | 1.3 ± 0.1 | 1.1 ± 0.1 | 1.5 ± 0.1b |
| HDL cholesterol (mmol/l) | 1.5 ± 0.1 | 1.8 ± 0.1 | 1.2 ± 0.1c |
| LDL cholesterol (mmol/l) | 2.8 ± 0.1 | 2.8 ± 0.2 | 2.7 ± 0.2 |
| HbA1C (%) | 5.7 ± 0.1 | 5.5 ± 0.1 | 5.9 ± 0.1a |
| Fasting glucose (mmol/l) | 5.3 ± 0.1 | 4.9 ± 0.1 | 5.7 ± 0.2b |
| Fasting insulin (mIU/l) | 5.64 ± 1.14 | 1.35 ± 0.1 | 9.44 ± 1.89c |
| HOMA‐IR | 1.47 ± 0.34 | 0.29 ± 0.02 | 2.5 ± 0.57c |
| QUICKI | 0.42 ± 0.01 | 0.49 ± 0.01 | 0.36 ± 0.01c |
| REE FFM (kcal/24h/kg) | 29.9 ± 1.2 | 30.2 ± 1.5 | 29.7 ± 1.9 |
| Fasting RQ | 0.879 ± 0.007 | 0.882 ± 0.01 | 0.876 ± 0.01 |
Study 1: Cross‐sectional study of 50 subjects (32 females and 18 males) stratified into two groups: subjects with normal weight (Lean; BMI ≤ 25 kg/m2) and subjects with obesity (Obese; BMI ≥ 30 kg/m2).
Data are presented as means ± SEM. Significant difference between Lean group and Obese group at the level of a P < 0.05; b P < 0.01; and c P < 0.001.
IGM, impaired glucose metabolism; NGT, normal glucose tolerance; HOMA‐IR, homeostasis model assessment of the insulin resistance index; QUICKI, quantitative insulin sensitivity check index; REE FFM, resting energy expenditure adjusted for fat free mass; RQ, respiratory quotient; T2DM, type 2 diabetes mellitus.
Study 2: Interventional study
Nineteen subjects (11 females and 8 males, BMI = 34.8 ± 8.4) with well‐controlled T2DM (HbA1C < 7%) on diet and exercise (n = 7) or a stable dose of metformin (n = 12, MET) were recruited for a randomized, double‐blind, and placebo‐controlled study. Subjects underwent an OGTT and SAAT biopsy at baseline and after 12 weeks of treatment with placebo (n = 10) or pioglitazone (45 mg/day; n = 9) using methods described for study 1. The primary endpoint was changes in NP receptor expression (as a ratio of NPRA and NPRC, i.e., NPRR) in SAAT of subjects with T2DM treated with pioglitazone when compared with controls. With the final sample size of nine subjects in the active treatment group and ten subjects in the control group, the study had >90% power to detect 40% difference in NPRR using a two‐sided test.
Blood analyses
Fasting blood samples were collected after an overnight fast in tubes containing EDTA, and plasma was frozen at −80°C until analyses. HbA1C was measured on a COBAS INTEGRA 800 (Roche Diagnostics, Mannheim, Germany) automated analyzer, and serum insulin was measured using the MSD Human Insulin Assay (Meso Scale Discovery, Rockville, MD, USA). Blood glucose concentrations were measured using YSI 2300 STAT Plus Glucose and l‐Lactate Analyzer (YSI Incorporated, Yellow Springs, OH, USA). Plasma N‐terminal fragment of proBNP (NT‐proBNP) was measured by the MSD Human NT‐proBNP Assay (Meso Scale Discovery, Rockville, MD, USA). BNP was measured in plasma samples using the Alere Triage BNP Test on an Alere Triage®MeterPro (Alere, Waltham, MA, USA). Homeostasis model assessment of the insulin resistance index (HOMA‐IR) was calculated as follows: [(fasting insulin in mU/l) × (fasting glucose in mmol/l)/22.5] 17. Quantitative insulin sensitivity check index (QUICKI) was calculated using the inverse of the sum of the logarithms of the fasting insulin and fasting glucose 18. Data from the IVGTT were used to calculate the insulin sensitivity index (Si) using the Minimal Model method of Bergman (MINMOD‐Millennium,© R. Bergman; ref. 19).
Gene expression analysis
Total RNA from adipose tissue was isolated using an RNeasy Lipid Tissue Mini kit and from skeletal muscle tissue using RNeasy Fibrous Tissue kit (Qiagen, Hilden, Germany). cDNA was synthesized with a high‐capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA, USA), and mRNA levels were measured by quantitative real‐time PCR using a ViiA™ 7 Real‐Time PCR System (Applied Biosystems) with TaqMan® Gene Expression Assays (Applied Biosystems; Supporting Information Table SR1). GAPDH, cyclophilin B, and 18S were used as endogenous controls for adipose tissue; and GAPDH and RPLPO (large ribosomal protein) were used for muscle gene expression.
Western blotting
Samples of SAAT (100 mg) from four lean subjects, four subjects with obesity, and six subjects with T2DM from the cross‐sectional study (study 1) were used to measure NPRA and NPRC proteins. Adipose tissue was homogenized and sonicated in buffer containing 25 mM HEPES (pH 7.4), 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 5 mM glycerophosphate, 0.9% Triton X‐100, 0.1% IGEPAL CA630, 5 mM sodium pyrophosphate, and 10% glycerol, with complete protease inhibitor cocktail (Roche Diagnostics). A total of 50 µg protein was resolved in 10% Tris‐glycine gels, transferred to nitrocellulose membranes (Bio‐Rad, Hercules, CA, USA), and incubated overnight at 4°C with primary antibodies against NPRA (#31333; Novus Biologicals, Littleton, CO, USA), NPRC (#31365; Novus Biologicals), or β‐actin (#75186; Abcam, Cambridge, MA, USA). Secondary antisera conjugated with either alkaline phosphatase or HRP were used for detection. NP receptor quantification normalized to β‐actin was performed using ImageJ software.
Statistical analysis
Data are presented as means ± SEM. Data were analyzed with Statistical Analysis System (SAS) version 9.3 (SAS Institute, Cary, NC, USA) and presented using GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA, USA). The normality of the distribution of each variable was evaluated using the Shapiro‐Wilk test. Correlation analyses were performed by calculating Pearson's correlation coefficients, and a step‐down Bonferroni procedure was used to control the familywise error rate. Differences between groups were analyzed by t‐test. One‐way ANCOVA with post hoc group comparisons using the Tukey‐Kramer adjustment were used to determine specific differences across groups. Repeated‐measures analyses with post hoc step‐down Sidak adjustment for multiple comparisons were used to examine main effects and their interactions. Power was calculated using The POWER Procedure with SAS (SAS Institute) for paired means. Significant differences were set at P < 0.05.
Results
Study 1: Cross‐sectional study
The main clinical characteristics of the subjects are summarized in Table 1. As expected, subjects with obesity exhibited features of IGM (higher levels of fasting plasma glucose and insulin) and dyslipidemia (higher triglycerides and lower HDL cholesterol) and were more insulin resistant than subjects with normal weight. NPRA gene expression in SAAT was significantly lower in subjects with obesity when compared with subjects with normal weight, with the reverse relationship for NPRC levels (P ≤ 0.0001 and P = 0.0004, respectively; Figure 1A). There were no differences in the expression levels of either receptor between males and females or after adjusting for age. In addition to the BMI, the mRNA levels of both NP receptors correlated with other measures of adiposity such as body fat distribution (waist circumference) and body composition (fat mass from DXA), as well as measures of lipid metabolism (triglycerides and HDL cholesterol) and glucose metabolism (HbA1C and HOMA‐IR; Table 2). In relation to glucose tolerance, there was a decrease of NPRA and an increase of NPRC transcripts in adipose tissue of subjects with T2DM when compared with subjects with NGT (Figure 1B). There were no differences in muscle NPRA and NPRC mRNA levels between the BMI groups (NPRA: 0.81 ± 0.08 vs. 0.75 ± 0.06; NPRC: 0.46 ± 0.12 vs. 0.56 ± 0.22 for LEAN vs. OBESE) or glucose tolerance groups (NPRA: 0.74 ± 0.05, 0.84 ± 0.15, and 0.81 ± 0.08; NPRC: 0.63 ± 0.25, 0.38 ± 0.12, and 0.37 ± 0.09 for NGT, IGM, and T2DM, respectively). To verify the mRNA expression data from adipose tissue, we measured NPRA and NPRC protein levels by Western blot analysis from lysates of SAAT from the LEAN and OBESE cohorts with NGT versus individuals with T2DM (Figure 1C). As shown in Figure 1D, we observed a significant reduction in NPRA levels in the OBESE and T2DM cohort relative to the LEAN cohort, whereas NPRC was increased in the OBESE cohort.
Figure 1.
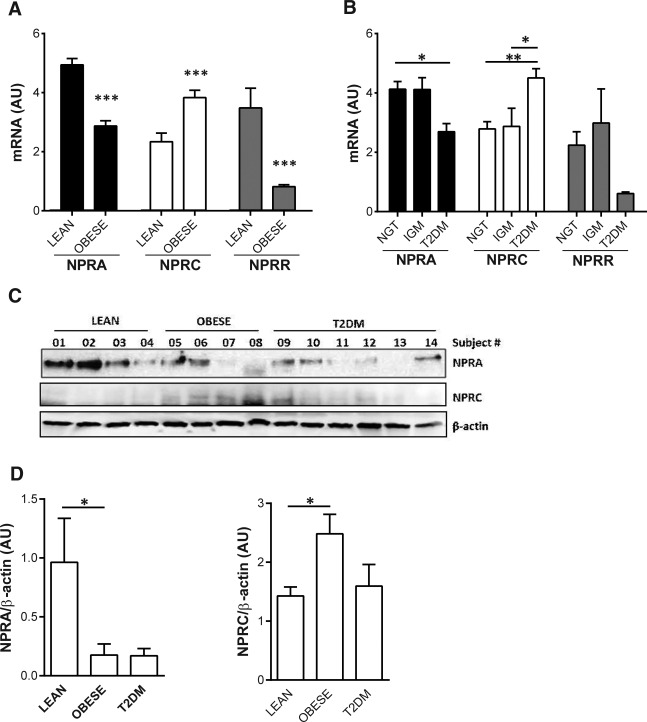
Gene expression levels (mRNA) of NPRA and NPRC and the ratio of NPRA and NPRC (NPRR) in subcutaneous adipose tissue between (A) BMI groups (LEAN and OBESE) and (B) glucose tolerance groups (NGT, IGM, and T2DM). (C) Lysates of human subcutaneous adipose from four LEAN subjects, four OBESE subjects with NGT, and six subjects with T2DM were subjected to Western blotting as described in Methods. Samples were processed to assess membrane receptors NPRA and NPRC. β‐actin is measured as the control for total protein applied to the gel. (D) Bar graphs show quantification of NPRA or NPRC normalized to β‐actin. *Significant difference between groups at the level of *P < 0.05; **P < 0.01; and ***P < 0.001. AU, arbitrary units.
Table 2.
Correlations of NPRA and NPRC mRNA levels in adipose tissue and the relative ratio of NPRA to NPRC (NPRR) with clinical parameters
| NPRA | NPRC | NPRR | NPRR P‐values | NPRR P‐values after BC | |
|---|---|---|---|---|---|
| BMI (kg/m2) | −0.736b | 0.321a | −0.637b | <0.0001 | <0.0001 |
| Waist circumference (cm) | −0.752b | 0.295a | −0.637b | <0.0001 | <0.0001 |
| Fat mass (kg) | −0.710b | 0.327a | −0.664b | <0.0001 | <0.0001 |
| Lean mass (kg) | −0.351a | −0.122 | −0.107 | 0.463 | 0.463 |
| Triglycerides (mmol/l) | −0.399a | 0.309a | −0.426a | 0.002 | 0.006 |
| HDL cholesterol (mmol/l) | 0.657b | −0.316a | 0.543b | <0.0001 | 0.0004 |
| HbA1C (%) | −0.352a | 0.383a | −0.487a | 0.0003 | 0.002 |
| Fasting glucose (mmol/l) | −0.354a | 0.364a | −0.454a | 0.001 | 0.004 |
| Fasting insulin (mIU/l) | −0.548b | 0.258 | −0.495a | 0.0003 | 0.002 |
| HOMA‐IR | −0.519b | 0.26 | −0.479a | 0.0005 | 0.0025 |
| Systolic BP (mmHg) | −0.181 | 0.341a | −0.286a | 0.044 | 0.089 |
Data are presented as Pearson's correlation coefficients: a P < 0.05 and b P < 0.0001. NPRR data were log transformed for the statistical analysis. After adjustment for multiple comparisons with step‐down Bonferroni correction (BC), the correlations for NPRC were not significant but did not change significantly for NPRR (viz., NPRR P‐values vs. NPRR P‐values after BC).
The relative ratio of NPRA to NPRC mRNA levels (NPRR), which determines the strength of NP signaling 6, was significantly lower in T2DM when compared with NGT and IGM (T2DM vs. NGT, P = 0.008; T2DM vs. IGM, P = 0.003). Irrespective of BMI, levels of NPRA and NPRC in adipose tissue and their relative ratio were highly correlated with the insulin Si assessed by the IVGTT (Figure 2A‐C). Within this cohort, the subject with the highest Si (= 15.8) appears to be an outlier but may represent a small percentage of subjects in the overall population. Excluding this subject did not change the results (NPRA: r = 0.640, P < 0.001; NPRC: r = −0.383, P = 0.012; NPRR: r = 0.584, P < 0.001; and PGC‐1α: r = 0.613, P < 0.001).
Figure 2.
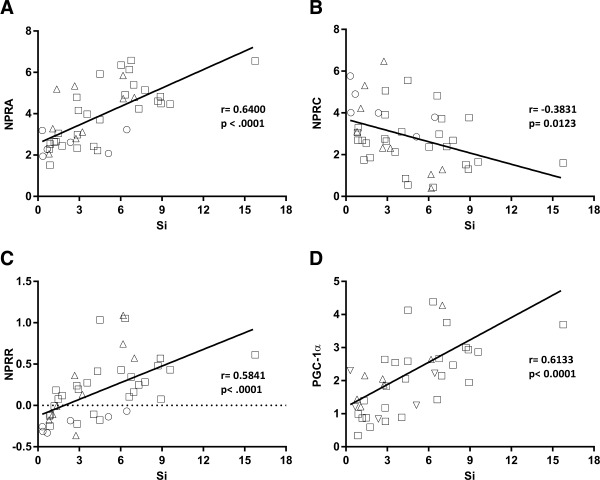
Graphical correlations between adipose tissue mRNA levels of (A) NPRA, (B) NPRC, (C) NPRR, and (D) PGC‐1α with insulin Si assessed by IVGTT. r represents Pearson's (parametric) correlation coefficient. NPRR data are log transformed. Squares (□) represent subjects with NGT; triangles (Δ) represent subjects with IGM; and circles (○) represent subjects with T2DM.
PGC‐1α is a transcriptional coregulator that is a master driver of mitochondrial biogenesis and enriched in tissues with high oxidative metabolism 20. In response to NP signaling, its expression is increased in adipocytes 15. We found that independent of age and gender, PGC‐1α mRNA was lower in the adipose tissue of subjects with obesity versus normal‐weight individuals (P < 0.001). This ratio was also strongly correlated with insulin sensitivity (Figure 2D) and remained significant after the adjustment for measures of adiposity (total body fat mass: r = 0.376, P = 0.024; waist circumference: r = 0.335, P = 0.046). PGC‐1α transcripts in adipose tissue showed a significant positive correlation with NPRA mRNA (r = 0.697, P < 0.001) and a negative correlation with NPRC mRNA (r = −0.468, P = 0.0015). These results are consistent with the notion that improved NP signaling is accompanied by enhanced respiratory ability in adipose tissue.
There were no significant differences in plasma levels of BNP or NT‐proBNP between our cohort of LEAN and OBESE (Figure 3A) or between glucose tolerance groups. This is most likely due to the small sample size of the current study when compared with prior studies 11, 13, 14. However, even with this small number of subjects plasma, BNP levels were negatively correlated with fasting glucose (r = −0.456, P = 0.013) and HbA1C (r = −0.559, P = 0.0016) and positively correlated with resting energy expenditure (REE FFM; r = 0.411, P = 0.037).
Figure 3.
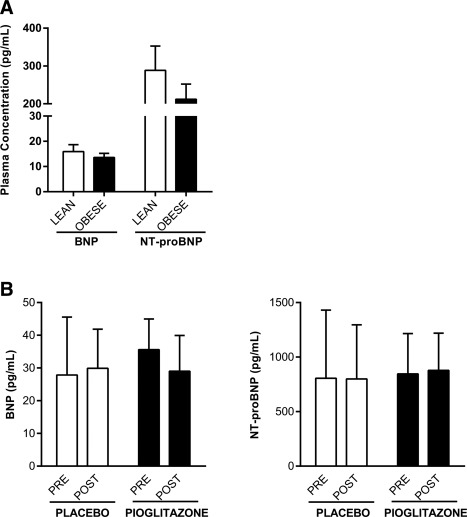
Circulating variables of the NP system. Plasma concentrations of BNP and NT‐proBNP were measured in fasting plasma samples and compared between (A) BMI groups (LEAN and OBESE) in the cross‐sectional study and (B) at baseline (PRE) and after 12 weeks of treatment (POST) with PLACEBO or PIOGLITAZONE in the interventional study.
Study 2: Interventional study
The main clinical characteristics of the subjects at baseline and after treatment with placebo or the PPARγ agonist pioglitazone are summarized in Table 3. As expected, treatment with pioglitazone improved fasting insulin, fasting glucose, HOMA‐IR, and QUICKI, whereas no significant changes were observed in subjects on placebo. Pioglitazone‐treated subjects gained more weight than those on placebo (P = 0.007), as is well established for this therapy 21. Treatment with pioglitazone was associated with significantly lowered levels of NPRC mRNA in adipose tissue (P = 0.046), along with a tendency toward increased NPRA gene expression (P = 0.068; Figure 4A and 4B). Together with a favorable shift in NPRR (P < 0.01; Figure 4C), the pioglitazone‐treated group showed a significantly higher level of PGC‐1α mRNA in adipose tissue (P = 0.002; Figure 4D) and increased UCP‐1 gene expression (P = 0.0015; Figure 4E), whereas no such changes were found in the placebo group. These effects were independent of gender, metformin, total fat mass, or changes in BMI. Plasma levels of BNP and NT‐proBNP did not change after pioglitazone treatment (Figure 3B).
Table 3.
Clinical parameters and measures of insulin sensitivity—Study 2: Interventional study
| Placebo (n = 10) | Pioglitazone (n = 9) | |||
|---|---|---|---|---|
| Pre‐treatment | Post‐treatment | Pre‐treatment | Post‐treatment | |
| BMI (kg/m2) | 33.5 ± 1.9 | 33.3 ± 1.8 | 36.4 ± 3.5 | 37.5 ± 3.7b,I |
| Waist circumference (cm) | 111.6 ± 4.2 | 111.8 ± 4.0 | 110.7 ± 4.6 | 112.1 ± 4.4 |
| Lean mass (kg) | 58.1 ± 4.7 | 57.9 ± 4.6 | 56.4 ± 2.9 | 56.8 ± 2.9 |
| Fat mass (kg) | 40.7 ± 5.9 | 40.2 ± 5.8 | 36.4 ± 4.1 | 37.9 ± 4.2 |
| Total body fat (%) | 39.9 ± 3.5 | 39.6 ± 3.3 | 39.8 ± 3.1 | 40.6 ± 3.2 |
| Triglycerides (mmol/l) | 4.7 ± 1.0 | 3.8 ± 0.4 | 3.9 ± 0.5 | 2.9 ± 0.4 |
| HDL cholesterol (mmol/l) | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.3 ± 0.1 |
| LDL cholesterol (mmol/l) | 2.4 ± 0.1 | 2.6 ± 0.1 | 2.5 ± 0.3 | 2.4 ± 0.2 |
| HbA1C (%) | 6.4 ± 0.2 | 6.6 ± 0.2 | 6.4 ± 0.1 | 6.2 ± 0.1 |
| Fasting glucose (mmol/l) | 6.5 ± 0.4 | 6.7 ± 0.5 | 6.6 ± 0.5 | 5.6 ± 0.2a,II |
| Fasting insulin (mIU/l) | 15.0 ± 3.9 | 13.6 ± 2.3 | 13.3 ± 6.0 | 7.2 ± 2.4a,III |
| HOMA‐IR | 4.0 ± 0.9 | 3.9 ± 0.7 | 3.7 ± 1.4 | 1.8 ± 0.6a,IV |
| QUICKI | 0.33 ± 0.02 | 0.32 ± 0.01 | 0.34 ± 0.01 | 0.39 ± 0.03a,V |
Study 2: Interventional study of 19 subjects with T2DM (11 females and 8 males) at baseline (Pre‐treatment) and after 12 weeks of treatment (Post‐treatment) with Placebo (n = 10) or Pioglitazone (n = 9).
Fasting insulin and HOMA‐IR data were log transformed for the statistical analysis. Post hoc group comparisons with step‐down Sidak adjustment were used to control familywise error rates (adjustment for multiple comparisons) in both Placebo (Pre‐ vs. Post‐ comparison) and Pioglitazone (Pre‐ vs. Post‐ comparison) groups. Data are presented as means ± SEM. a,bSignificant difference between Pre‐treatment versus Post‐treatment at the level of a P < 0.05 and b P < 0.01, and the actual P‐values are represented by roman numerals: I P = 0.007; II P = 0.034; III P = 0.035; IV P = 0.013; and V P = 0.017.
Figure 4.
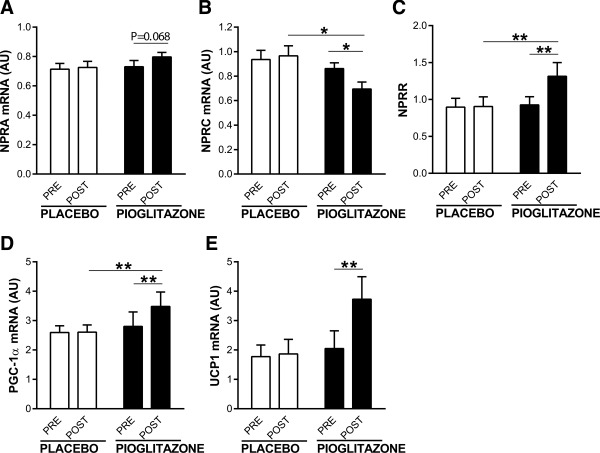
Effect of PIOGLITAZONE versus PLACEBO on mRNA levels of (A) NPRA, (B) NPRC, (C) NPRR, (D) PGC‐1α, and (E) UCP‐1. *Significant difference between groups at the level of *P < 0.05 and **P < 0.01. AU, arbitrary units. UCP‐1 data were not normally distributed and were log transformed for the statistical test.
Discussion
More than a decade ago, ANP was shown to stimulate lipolysis in human adipocytes in a manner similar to the catecholamines 10. Other studies reported increased lipid mobilization and postprandial fat oxidation when NPs were infused acutely into subjects 22, 23. Recently, and again similar to the catecholamines, we showed that NPs affect energy balance by stimulating the amount and activity of brown adipocytes 6. We illustrated the importance of the relative amounts of NPRA to NPRC in metabolism and obesity in mice lacking one or the other of these receptors. Npra−/− mice have increased adiposity, whereas Nprc−/− mice have reduced fat mass and express higher levels of brown adipocyte markers 15. Altogether, these findings suggested that the ratio of NPRA to NPRC is a key component in the ability of NPs to regulate metabolic activity and energy expenditure 15. These intriguing findings prompted us to examine NP receptor expression as well as downstream targets in adipose tissue and skeletal muscle in humans with a range of obesity and glucose intolerance.
Our results show that in the SAAT of subjects with obesity, the ratio of NPRA to NPRC was shifted toward reduced NPRA and increased NPRC expression when compared with that in the lean group. Interestingly, we also found that when subjects were categorized by their degree of glucose tolerance, there was a decline in adipose tissue NPRA and an increase in NPRC levels from NGT to T2DM. Consistent with this, our findings also show that NP receptor in adipose tissue was strongly associated with insulin sensitivity. Notably, 12 weeks of treatment with pioglitazone significantly increased the NPRA/NPRC ratio in parallel with improvements in insulin sensitivity in patients with T2DM. Thiazolidinediones such as pioglitazone bind and activate PPARγ in adipocytes initiating a series of transcriptional changes that influence insulin sensitivity, adiponectin secretion, and promote fat deposition 21.
In parallel with the differences in the NPRA/NPRC ratio in both the cross‐sectional and intervention study, we found differences in the expression of the important transcriptional coregulator PGC‐1α in adipose tissue. PGC‐1α was lower in adipose tissue of subjects with obesity when compared with subjects with normal weight, similar to findings in adipose tissue of subjects with morbid obesity 24; however, no difference in PGC‐1α gene expression levels was found in muscle in the two groups. In the subjects with T2DM treated with pioglitazone, we found increases in PGC‐1α, as well as increased expression of UCP‐1. Although we cannot directly ascribe these changes in PGC‐1α and UCP‐1 to changes in NP signaling, they are consistent with our previous data in human adipocytes and in mouse models 15 and fit well with the fact that reduced levels of PGC‐1α are related to markers of insulin resistance and cardiovascular risk 25.
Earlier studies with mouse adipocytes reported that insulin dose‐dependently and reciprocally regulated the expression of NP receptors by reducing NPRA expression while increasing NPRC 7, and it was suggested that this was part of the mechanism by which insulin may efficiently promote lipogenesis in adipocytes and reduce the lipolytic action of NPs. In humans, insulin levels have been correlated with NPRC expression in the subcutaneous fat of subjects with obesity 26. In addition, NPRC expression was found to be higher in adipose tissue of hypertensive subjects with obesity when compared with lean controls 27. In our study, we observed a more pronounced effect of pioglitazone treatment on NPRC levels. However, at this point, it would be premature to make any conclusions about the cause and effect between insulin sensitivity and NPRC levels.
The increase in levels of NPRC in adipose tissue in the obese state has led to speculation that adipose tissue might be a site of NP clearance and contribute to observed reductions in circulating levels of NPs in plasma in obesity and impaired NP responses 13, 14, 28. Although we did not observe significant differences in either BNP or NT‐proBNP in plasma of subjects within the different BMI groups or among the glucose tolerance groups, this is most likely due to the small sample size of our cohorts, which is a limitation of our study. For example, in the study by Wang et al. 13, they had ∼1,000 subjects in each of the lean versus overweight versus obese cohort, from which they detected a significant difference in BNP levels (18.4 vs. 15.9 vs. 14.8 pg/mL, respectively). Similarly, in another large‐scale study of ∼1,700 randomly selected subjects from a general population, a relative deficiency of circulating NPs with a counter‐regulation by higher aldosterone levels was recognized and associated with cardiorenal and metabolic disease and increased mortality 11. Although levels of BNP in our study were not different, the more important functional consequence in obesity may be the reduced NPRA/NPRC ratios in adipose tissue. As shown in the case of mice lacking NPRC 29, there need not be significant changes in circulating levels to affect activity. Total levels of ANP and BNP between wild‐type and Nprc−/− mice were not different; instead, the circulating half‐life of ANP in Nprc−/− mice was significantly longer 29. Nevertheless, these mice have remarkable increases in brown adipocytes in their white fat depots 15, resist diet‐induced obesity, and retain improved insulin sensitivity (unpublished observations).
We investigated the subcutaneous fat depot, but not other adipose depots such as intra‐abdominal. Adipose tissue depots differ in their morphology, physiology as well as activity and may have distinct responses to various stimuli 30 and can differ between species. For example, in mice, the subcutaneous white adipose depot is more highly susceptible to “browning” processes in response to conditions such as cold, β‐adrenergic agonists or other hormonal stimuli than a commonly studied visceral depot in mice such as epididymal 31, 32, 33. Despite this limitation, we found that human subcutaneous adipose tissue exhibits marked changes in components of the NP pathway with respect to obesity and T2DM, specifically, changes in the expression levels of the NP receptors and downstream genes that are regulated by their activation.
In conclusion, our study supports the hypothesis that the cardiac NP system is dysregulated in obesity, insulin resistance, and T2DM. Although we do not know whether the reciprocal regulation of NPRA and NPRC levels in human subcutaneous adipose tissue in subjects with normal weight and individuals with obesity is a cause or a consequence of their insulin sensitivity and overall metabolic status, studies in animal models suggest that activation of the NP system in adipocytes can directly impact energy metabolism and insulin sensitivity 15. Thus, the NP receptors and the ability to convey the NP signal to target cells via NPRA raise the question as to whether they represent a novel pathway for interventions targeting metabolic disorders, including obesity and insulin resistance, in addition to cardiorenal conditions.
Supporting information
Supporting Information
Acknowledgments
The authors thank the study volunteers for their participation and the TRI‐MD clinical research staff for their contributions. The authors thank Eric Weatherford for discussions regarding the study and results.
Funding agencies: This work was supported by institutional funds from Florida Hospital Translational Research Institute for Metabolism and Diabetes; institutional funds from Sanford Burnham Prebys Medical Discovery Institute; and an investigator initiated grant from Takeda Pharmaceuticals USA, Inc.
Disclosure: R.E.P. has received research funding from Takeda Pharmaceuticals USA, Inc. S.C. has received research funding from Novo Nordisk and AstraZeneca. The other authors declared no conflict of interest.
Author contributions: Z.K. performed experiments, researched and analyzed the data, and wrote the manuscript. W.G.T. assisted in the conduct of the clinical study 2 and data analysis. H.X. performed the statistical data analysis. W.W. and D.L. performed experiments. S.C. contributed to study design and discussions and reviewed and edited the manuscript. R.E.P. designed and conducted the clinical studies, reviewed/edited the manuscript, and contributed to discussion. R.E.P. and S.C. are the guarantors of this work, have full access to the data in the study, and take responsibility for the integrity of the data and the accuracy of the data analysis.
Clinical trial registration: ClinicalTrials.gov identifier NCT00656864
References
- 1. Pandey KN. Guanylyl cyclase/atrial natriuretic peptide receptor‐A: role in the pathophysiology of cardiovascular regulation. Can J Physiol Pharmacol 2011;89:557‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Potter LR, Abbey‐Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate‐dependent signaling functions. Endocr Rev 2006;27:47‐72. [DOI] [PubMed] [Google Scholar]
- 3. Potter LR, Hunter T. Guanylyl cyclase‐linked natriuretic peptide receptors: structure and regulation. J Biol Chem 2001;276:6057‐6060. [DOI] [PubMed] [Google Scholar]
- 4. Lafontan M, Moro C, Berlan M, Crampes F, Sengenes C, Galitzky J. Control of lipolysis by natriuretic peptides and cyclic GMP. Trends Endocrinol Metab 2008;19:130‐137. [DOI] [PubMed] [Google Scholar]
- 5. Potter LR. Natriuretic peptide metabolism, clearance and degradation. FEBS J 2011;278:1808‐1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collins S. A heart‐adipose tissue connection in the regulation of energy metabolism. Nat Rev Endocrinol 2014;10:157‐163. [DOI] [PubMed] [Google Scholar]
- 7. Nakatsuji H, Maeda N, Hibuse T, et al. Reciprocal regulation of natriuretic peptide receptors by insulin in adipose cells. Biochem Biophys Res Commun 2010;392:100‐105. [DOI] [PubMed] [Google Scholar]
- 8. Sarzani R, Paci VM, Dessi‐Fulgheri P, Espinosa E, Rappelli A. Comparative analysis of atrial natriuretic peptide receptor expression in rat tissues. J Hypertens Suppl 1993;11:S214‐S215. [PubMed] [Google Scholar]
- 9. Sarzani R, Dessi‐Fulgheri P, Paci VM, Espinosa E, Rappelli A. Expression of natriuretic peptide receptors in human adipose and other tissues. J Endocrinol Invest 1996;19:581‐585. [DOI] [PubMed] [Google Scholar]
- 10. Sengenes C, Berlan M, De Glisezinski I, Lafontan M, Galitzky J. Natriuretic peptides: a new lipolytic pathway in human adipocytes. FASEB J 2000;14:1345‐1351. [PubMed] [Google Scholar]
- 11. Buglioni A, Cannone V, Cataliotti A, et al. Circulating aldosterone and natriuretic peptides in the general community: relationship to cardiorenal and metabolic disease. Hypertension 2015;65:45‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miyashita K, Itoh H, Tsujimoto H, et al. Natriuretic peptides/cGMP/cGMP‐dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009;58:2880‐2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang TJ, Larson MG, Levy D, et al. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004;109:594‐600. [DOI] [PubMed] [Google Scholar]
- 14. Wang TJ, Larson MG, Keyes MJ, Levy D, Benjamin EJ, Vasan RS. Association of plasma natriuretic peptide levels with metabolic risk factors in ambulatory individuals. Circulation 2007;115:1345‐1353. [DOI] [PubMed] [Google Scholar]
- 15. Bordicchia M, Liu D, Amri EZ, et al. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J Clin Invest 2012;122:1022‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Engeli S, Birkenfeld AL, Badin PM, et al. Natriuretic peptides enhance the oxidative capacity of human skeletal muscle. J Clin Invest 2012;122:4675‐4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412‐419. [DOI] [PubMed] [Google Scholar]
- 18. Katz A, Nambi SS, Mather K, et al. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 2000;85:2402‐2410. [DOI] [PubMed] [Google Scholar]
- 19. Pacini G, Bergman RN. MINMOD: a computer program to calculate insulin sensitivity and pancreatic responsivity from the frequently sampled intravenous glucose tolerance test. Comput Methods Programs Biomed 1986;23:113‐122. [DOI] [PubMed] [Google Scholar]
- 20. Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC‐1 family of transcription coactivators. Cell Metab 2005;1:361‐370. [DOI] [PubMed] [Google Scholar]
- 21. Fonseca V. Effect of thiazolidinediones on body weight in patients with diabetes mellitus. Am J Med 2003;115 (Suppl 8A):42S‐48S. [DOI] [PubMed] [Google Scholar]
- 22. Birkenfeld AL, Boschmann M, Moro C, et al. Lipid mobilization with physiological atrial natriuretic peptide concentrations in humans. J Clin Endocrinol Metab 2005;90:3622‐3628. [DOI] [PubMed] [Google Scholar]
- 23. Birkenfeld AL, Budziarek P, Boschmann M, et al. Atrial natriuretic peptide induces postprandial lipid oxidation in humans. Diabetes 2008;57:3199‐3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Semple RK, Crowley VC, Sewter CP, et al. Expression of the thermogenic nuclear hormone receptor coactivator PGC‐1α is reduced in the adipose tissue of morbidly obese subjects. Int J Obes Relat Metab Disord 2004;28:176‐179. [DOI] [PubMed] [Google Scholar]
- 25. Ruschke K, Fishbein L, Dietrich A, et al. Gene expression of PPARγ and PGC‐1α in human omental and subcutaneous adipose tissues is related to insulin resistance markers and mediates beneficial effects of physical training. Eur J Endocrinol 2010;162:515‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pivovarova O, Gogebakan O, Kloting N, et al. Insulin up‐regulates natriuretic peptide clearance receptor expression in the subcutaneous fat depot in obese subjects: a missing link between CVD risk and obesity? J Clin Endocrinol Metab 2012;97:E731‐E739. [DOI] [PubMed] [Google Scholar]
- 27. Dessi‐Fulgheri P, Sarzani R, Rappelli A. The natriuretic peptide system in obesity‐related hypertension: new pathophysiological aspects. J Nephrol 1998;11:296‐299. [PubMed] [Google Scholar]
- 28. Sarzani R, Salvi F, Dessi‐Fulgheri P, Rappelli A. Renin‐angiotensin system, natriuretic peptides, obesity, metabolic syndrome, and hypertension: an integrated view in humans. J Hypertens 2008;26:831‐843. [DOI] [PubMed] [Google Scholar]
- 29. Matsukawa N, Grzesik WJ, Takahashi N, et al. The natriuretic peptide clearance receptor locally modulates the physiological effects of the natriuretic peptide system. Proc Natl Acad Sci USA 1999;96:7403‐7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cinti S. The adipose organ at a glance. Dis Model Mech 2012;5:588‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cohen P, Levy JD, Zhang Y, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 2014;156:304‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barbatelli G, Murano I, Madsen L, et al. The emergence of cold‐induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am J Physiol Endocrinol Metab 2010;298:E1244‐E1253. [DOI] [PubMed] [Google Scholar]
- 33. Fisher FM, Kleiner S, Douris N, et al. FGF21 regulates PGC‐1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 2012;26:271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information