

Abstract
Elevated levels of circulating asymmetric and symmetric dimethylarginines (ADMA and SDMA) predict and potentially contribute to end organ damage in cardiovascular diseases. Alanine-glyoxylate aminotransferase 2 (AGXT2) regulates systemic levels of ADMA and SDMA, and also of beta-aminoisobutyric acid (BAIB)-a modulator of lipid metabolism. We identified a putative binding site for hepatic nuclear factor 4 α (HNF4α) in AGXT2 promoter sequence. In a luciferase reporter assay we found a 75% decrease in activity of Agxt2 core promoter after disruption of the HNF4α binding site. Direct binding of HNF4α to Agxt2 promoter was confirmed by chromatin immunoprecipitation assay. siRNA-mediated knockdown of Hnf4a led to an almost 50% reduction in Agxt2 mRNA levels in Hepa 1–6 cells. Liver-specific Hnf4a knockout mice exhibited a 90% decrease in liver Agxt2 expression and activity, and elevated plasma levels of ADMA, SDMA and BAIB, compared to wild-type littermates. Thus we identified HNF4α as a major regulator of Agxt2 expression. Considering a strong association between human HNF4A polymorphisms and increased risk of type 2 diabetes our current findings suggest that downregulation of AGXT2 and subsequent impairment in metabolism of dimethylarginines and BAIB caused by HNF4α deficiency might contribute to development of cardiovascular complications in diabetic patients.
Endogenous methylated derivatives of L-arginine, such as asymmetric (ADMA) and symmetric (SDMA) dimethylarginines, have been widely studied as markers and potential mediators of cardiovascular diseases1,2. ADMA has been proposed to directly cause vascular damage via competitive inhibition and uncoupling of nitric oxide synthases (NOS)3. Multiple epidemiological studies reported elevation of plasma ADMA levels in pathological conditions associated with impaired nitric oxide (NO) bioavailability such as atherosclerosis, hypertension and renal failure4. In contrast to ADMA, SDMA cannot directly inhibit NOS, but is nevertheless also independently associated with adverse cardiovascular outcomes5,6. It has been suggested that SDMA contributes to cardiovascular damage by competing with L-arginine for their common transporter7,8 or by affecting lipid metabolism9.
There are two known pathways for enzymatic metabolism of ADMA: hydrolysis to citrulline by dimethylarginine dimethylaminohydrolases 1 and 2 (DDAH1 and DDAH2)10, and conversion to asymmetric dimethylguanidinovaleric acid (ADGV) by alanine-glyoxylate aminotransferase 2 (AGXT2)11. The first pathway is well characterized, while the second one is still poorly understood. Unlike DDAHs, AGXT2 can metabolize not only ADMA, but also SDMA, which leads to formation of the corresponding α-keto-derivative symmetric dimethylguanovaleric acid (SDGV)12. We have demonstrated previously that overexpression of AGXT2 lowers ADMA in vivo and protects endothelial cells from ADMA-induced inhibition of NO production13. Agxt2 knockout mice have elevated plasma levels of both ADMA and SDMA and develop hypertension14. AGXT2 polymorphisms have been linked to elevated systemic SDMA levels in humans15.
In addition to ADMA and SDMA, AGXT2 can also utilize beta-aminoisobutyric acid (BAIB) as a substrate16. AGXT2 polymorphisms lead to increased BAIB levels in urine (hyper-β-aminoisobutyric aciduria), presumably the most common autosomal recessive metabolic trait in humans17 (OMIM 210100). The physiological role of this trait is being currently actively investigated. A recent genome-wide association study revealed a strong association between plasma BAIB levels and serum levels of triglycerides and cholesterol esters17. Subsequent experiments in a zebrafish model suggested that BAIB can directly regulate lipid metabolism17. Indeed, BAIB was shown to reduce body weight and enhance fatty acid oxidation in mice through increased production of leptin by the white adipose tissue18. BAIB has also been proposed to serve as a myokine, which induces browning of white adipose tissue and might be responsible for some of the beneficial effects of exercise19.
However, despite the growing evidence that AGXT2 and its substrates may play an important role in the pathogenesis of cardiovascular and metabolic diseases, the mechanisms of regulation of AGXT2 expression and activity are still unknown. We have identified a highly conserved region in the putative promoter sequences of both human and murine AGXT2. This region contains a predicted binding site for the transcription factor Hepatocyte Nuclear Factor 4 α (HNF4α). Previous results from chromatin immunoprecipitation using an antibody against HNF4α followed by deep sequencing (ChIP-seq) experiments demonstrated HNF4α binding at this locus in the human hepatocyte cell line HepG220,21,22. Moreover, Battle and coauthors demonstrated downregulation of Agxt2 in the liver of Hnf4a conditional knockout mice, which suggest an important role of HNF4a in regulation of AGXT2 expression23. The goal of the current study was to test the hypothesis that HNF4α is a major regulator of AGXT2 expression in vivo.
In this study we demonstrated that HNF4α directly binds to the Agxt2 promoter and serves as a major regulator of Agxt2 expression in vitro and in vivo. It is known from previous studies that severe inborn HNF4α deficiency leads to development of maturity-onset diabetes of the young 1 (MODY1)24, while mild HNF4A polymorphisms are associated with increased risk of type 2 diabetes mellitus and metabolic syndrome25,26. The current findings therefore suggest an intriguing link between type 2 diabetes and AGXT2-mediated impairment of methylarginine, NO and lipid metabolism.
Methods
Plasmids generation
Fragments of the murine Agxt2 gene promoter region were PCR amplified from C57BL/6J wild type mouse genomic DNA, digested with XhoI and HindIII and cloned into the pGL4.10 vector (Promega). Cloning accuracy was validated by sequencing. Plasmids containing mutations in HNF4α binding site were generated using site-directed mutagenesis. Amplicons were then fused using SOE-PCR technique and cloned into pGL4.10 using XhoI and HindIII restriction sites. All PCR primer sequences are listed in the Supplementary Table S1. Endotoxin-free plasmids for cell line transfections were purified using Qiagen Maxi kit (Qiagen).
Cell culture
Hepa 1–6 cells (CLS, Germany) were cultured to 70% confluence in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. NIH3T3 cells (CLS, Germany) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. For transfections cells were seeded at 6000 cells/well in 96-well plate (Greiner Bio-One) and incubated 24 hours.
Animals
Hnf4aF/F;AlbERT2cre mice with tamoxifen-inducible liver-specific Hnf4a deficiency as well as the control Hnf4aF/F mice were previously generated and characterized by Dr. Gonzales’ group27. Animals were fed a diet containing tamoxifen (1 g/kg diet) for four days and returned to regular chow for additional four days. On the eighth day all animals were euthanized and tissues collected for analysis. Mice were housed in a temperature- and light-controlled facility and given food and water ad libitum. All animal studies were performed in accordance with the guidelines and approval of the NCI, National Institutes of Health, Animal Care and Use Committee.
Plasmid transfection and Dual-Luciferase assay
Master mixes of FuGene transfection reagent (Promega) and reporter plasmids in serum-free Opti-MEM medium (Gibco) were added to the cells. Plasmids based on the pGL4.10 vector (Promega) containing parts of murine Agxt2 gene putative promoter region and carrying firefly luciferase as a reporter gene were co-transfected with the pGL4.74 vector (Promega) containing Renilla luciferase gene with HSV TK-promoter as an internal control for transfection efficiency. Cells were assayed with Dual-Luciferase Assay kit (Promega) according to the instructions of the manufacturer twenty four hours after transfection with reporter plasmids. The signal was acquired using a luminometer with auto-injection system Berthold Centro XS3 LB 960. Chemiluminescence intensity variation was then normalized to the Renilla luciferase signal.
siRNA transfection
Lipofectamine RNAiMAX transfection reagent (Invitrogen) and different siRNAs in serum-free Opti-MEM medium (Gibco) diluted 1:1 with water were added to cells. Scrambled oligonucleotides Stealth MediumGC (12935–300, Life Technologies, USA) and Ambion Negative control 1 (AM4611, Life Technologies, USA) were used as negative controls. Cells were incubated for 96 hours without medium change followed by analysis of gene expression.
RNA extraction, cDNA synthesis and qPCR
RNA from siRNA-treated cells was extracted with Cells-to-Ct kit (Ambion), cDNA was subsequently synthesized with the same kit. RNA extraction from murine tissues was performed using RNeasy Plus Universal kit (Qiagen), cDNA was synthesized with High Capacity RNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed using ABI 7300 machine (Applied Biosystems) with PowerSYBR reagent (Applied Biosystems) in 96-tube plates. Standard reaction conditions were used and all genes were assayed on the same plate. The sequences of the primers for qPCR are listed in the Supplementary Table S1.
Immunoblotting
After the 96 hr incubation, cells were washed with PBS, scraped off the plates and centrifuged at 500 g for 10 minutes. Cell pellets were lysed with RIPA buffer containing Roche complete protease inhibitor cocktail (Roche), sonicated and frozen at −80 °C. Protein concentrations in samples were determined using the BCA protein assay (Thermo Scientific, USA). Samples (15 μg of protein/lane) were separated by SDS-PAGE under reducing conditions on 10% polyacrylamide gels and transferred to PVDF membranes (Roti-PVDF, Carl Roth, Germany). Membranes were probed with 1 μg/mL mouse monoclonal anti-HNF4a antibodies clone H1415 (Invitrogen, USA) for 1 h at room temperature followed by incubation with 1 μg/mL HRP-conjugated goat-anti-mouse secondary antibodies (Becton Dickinson) for 1 h at room temperature. To control for sample loading, the membranes were re-probed with 1 μg/ml anti-beta-actin HRP-conjugated mouse monoclonal antibodies (Sigma) for 1 hr at room temperature. Bands were visualized using Lumi-Light Western Blotting Substrate (Roche, Switzerland).
Liver and kidneys isolated from three Cre positive and three Cre negative Hnf4a Flox/Flox mice were homogenized in RIPA buffer containing Halt Protease Inhibitor Cocktail (Thermo Scientific). 50 μg of protein extract was used for SDS-PAGE and Western blot analysis using Biorad TGX precast gels and Trans-Blot Turbo PVDF transfer packs, respectively. Membranes were incubated with antibodies against HNF4A (H1415, Perseus (Tokyo, JP)) then re-probed with β-actin (ab8227, Abcam) as a loading control.
Chromatin immunoprecipitation assay
Chromatin was extracted from 20 million Hepa 1.6 or NIH3T3 cells with ChIP-IT Express Enzymatic kit (Active Motif), according to the manufacturer’s recommendations. Every ChIP reaction was performed with 5 μg of mouse anti-HNF4α antibodies (H1415, Life Technologies). Immunoprecipitated DNA and Input DNA were purified with Chromatin IP DNA Purification Kit (Active Motif). It was subsequently analyzed by qPCR to measure the relative enrichment of the fragments of interest in the total input of ChIP DNA fragments. The negative control ChIP primers were designed to amplify fragments downstream of the Agxt2 predicted transcription start sites. The positive control primers were designed to amplify the region of hepatocyte nuclear factor 1 alpha (Hnf1a) promoter, which has been shown previously to comprise an active HNF4A binding site28. Murine fibroblast cell line NIH 3T3 was used as an additional negative control. ChIP-IT Control qPCR Kit for Mouse (Active Motif) was used as an additional control.
Biochemical measurements
Measurements of ADMA and ADGV in tissue lysates for determination of AGXT2 activity were performed using HPLC-MS-MS as previously described11,29. AGXT2 activity was assessed as a production rate of the AGXT2-specific metabolite of ADMA–asymmetric dymethylguanovaleric acid (ADGV)-per mg of tissue after incubation with isotope-labeled ADMA30. Plasma levels of ADMA, SDMA, ADGV, SDGV and BAIB were determined using the corresponding HPLC-MS-MS methods31,32. Baseline separation was achieved by a HILIC column and identity was confirmed by reference substances and multiple mass transitions. For SDGV, cell-culture-derived isotope labelled [2H6]-SDGV was used as the internal standard, therefore, only relative units (i.e. area ratios) are provided.
Statistics
All quantitative data are presented as mean ± SEM. Normality of distributions was tested using D’Agostino & Pearson omnibus normality test and Shapiro-Wilk normality test. Statistical comparisons between two groups were performed using Student’s t-test and between multiple groups-with Dunnett’s multiple comparison test. Differences were considered statistically significant at p-value < 0.05. All analyses were performed in Prism 6 (GraphPad Software).
Results
HNF4α is a candidate regulator of AGXT2 expression
We performed a conservation analysis of the mammalian sequences upstream of the first exon of AGXT2 gene using the VISTA software33. The only highly conserved region found in mammals within the predicted AGXT2 core promoter region was located −89 bp to −77 bp upstream relative to the murine Agxt2 gene translation start site (according to mm 9 genome annotation, Agxt2 has multiple transcription start sites, therefore all coordinates in this manuscript are reported in relation to the single translation start site). The identified region contained the HNF4α consensus binding sequence34. Furthermore, this region overlapped with the HNF4α footprint from the ChIP-seq analysis using anti-HNF4α antibodies performed in HepG2 cells20,21,22 (Fig. 1a).
Figure 1. Conserved regulatory element in Agxt2 promoter corresponds to HNF4A binding site.

(a) A single conserved element is located in 5′ UTR of human AGXT2 and upstream of mouse Agxt2 gene. UCSC Genome Browser view of human (hg19) AGXT2 and mouse (mm10) Agxt2 loci, with PhyloP conservation tracks shown below. For human locus, ENCODE results identifying HNF4A binding in HepG2 cell lines are shown. For mouse locus, fragment used in luciferase reporter assay (Fig. 2) is indicated by a red bar. Location of primers used for ChIP (Fig. 3b) is indicated with colored vertical bars. (b) mRNA levels of Hnf4a and Agxt2 correlate in mouse tissues according to RNA-seq data. Heat map indicates transcript abundance in fragments per kilobase of exon per million fragments mapped (FPKM)67. (c) Pol2 occupancy at Agxt2 promoter is high in the tissues expressing Hnf4a. UCSC Genome Browser view of mouse Agxt2 locus with corresponding sets of Pol2 ChIP-Seq reads (generated by Bing Ren’s lab)20. Positions of primers, used for ChIP experiments are marked with colored lines, names are in the bottom.
Analysis of the publicly accessible microarray data obtained from 22 C57BL/6 wild type mouse tissues (Gene Expression Omnibus database, dataset GDS314235,36 demonstrated a strong correlation between Hnf4a and Agxt2 expression levels (Pearson’s R = 0.736). Notably, no significant correlation was observed between expression of Hnf4a and the two genes flanking Agxt2 on mouse chromosome 15 (R = 0.02 and −0.14 for upstream Prlr and downstream Dnajc21, respectively), suggesting that HNF4α-dependent regulation is specific to Agxt2. Further, the strongest expression of both Agxt2 and Hnf4a was observed in the liver and the kidney (Fig. 1b). These tissues also showed high RNA polymerase II (Pol II) occupancy at the Agxt2 promoter in the corresponding sets of ChIP-seq reads20, suggesting that HNF4α directly contributes to transcriptional regulation of Agxt2 (Fig. 1c).
Disruption of the predicted HNF4α binding site reduces activity of the Agxt2 promoter
We then tested the functional activity of the predicted HNF4α binding site in the murine Agxt2 promoter using a luciferase reporter assay. The intact construct encoded the predicted core promoter region spanning −211 to +1 of the murine Agxt2 promoter followed by the firefly luciferase reporter. Four mutant constructs (M1–M4) were generated: the M1 and M2 contained previously described functionally significant SNPs incorporated into the HNF4A binding site37,38, while in the M3 and M4 the entire consensus sequence was disrupted34 (Fig. 2a).
Figure 2. HNF4α binding site is necessary for Agxt2 proximal promoter function.

(a) Conserved sequence in the AGXT2 core promoter region (top) matches the known HNF4α binding site consensus sequence (middle)68. Bottom, sequences of disrupted HNF4α binding sites in four mutant constructs (M1–M4), which were tested in the luciferase reporter experiment. Mutated nucleotides are shown in bold red. (b) Disruption of the HNF4α recognition sequence in the Agxt2 core promoter reduced expression of the reporter gene, as demonstrated by the luciferase activity assay in the lysate of the cells expressing the Agxt2 promoter/luciferase reporter constructs. WT-intact fragment containing Agxt2 proximal promoter. M1–M4–mutated constructs, containing the mutations shown in the panel (a). Neg-promoterless luciferase construct (negative control). Average of 9 independent biological replicates, error bars indicate SEM, one-way ANOVA, Dunnett’s multiple comparison test: ***P < 0.0001 for all.
The constructs were expressed in the murine hepatic cell line Hepa 1–6. Firefly luciferase activity measured in the lysates of Hepa 1–6 cells transfected with the mutated constructs was on average 75% lower (maximum 83% in case of the mutated construct 1) than in the lysates of cells transfected with the construct containing the intact promoter (Fig. 2b). The signal of Renilla luciferase used as an internal reference was consistent among the samples.
HNF4α directly binds to Agxt2 promoter
We performed a ChIP assay with anti-HNF4α antibodies to determine whether HNF4α can directly bind to the murine Agxt2 promoter. We chose two cell lines for this experiment: the murine hepatocyte cell line Hepa 1–6, which expresses Hnf4a, and the murine fibroblast cell line NIH 3T3, which does not express Hnf4a (Fig. 3a).
Figure 3. HNF4α directly binds to its consensus site in the Agxt2 promoter.
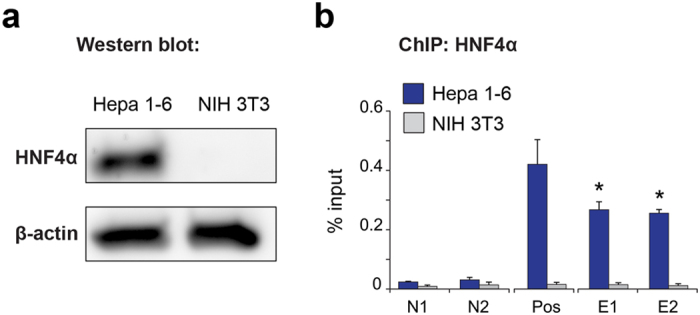
(a) Western blotting demonstrates HNF4α protein expression in the murine hepatocyte cell line Hepa 1–6, but not in the murine fibroblast cell line NIH 3T3. (b) ChIP analyses of HNF4α binding to DNA sequences of interest. Dark blue–Hepa 1–6 cells, which express both Hnf4a and Agxt2, Gray–NIH 3T3 fibroblasts, which express neither Hnf4a, nor Agxt2 (negative control). N1 and N2 (negative controls) -sequences 1 and 5 kb downstream of Agxt2 promoter, lacking known HNF4A consensus binding site. Pos (positive control) –sequence comprising previously shown HNF4α binding site in Hnf1a gene promoter region. E1 and E2 (experiment) -two different primer pairs, both centered around the predicted HNF4α binding site in the Agxt2 promoter. Average of 3 (for N2 and P) or 4 (for N1, E1 and E2) independent biological replicates, error bars indicate SEM, Mann-Whitney test *P = 0.02866.
A qPCR analysis of the chromatin precipitated with anti-HNF4A antibodies from Hepa 1–6 cell lysates showed enrichment of the DNA fragments containing the putative HNF4α binding site in the Agxt2 promoter, when compared to the negative control regions. The positive control region, containing known HNF4α binding site, also demonstrated enrichment compared to the negative control regions. The negative control experiment performed with the lysate from NIH 3T3 cells demonstrated no enrichment in any of the tested regions (Fig. 3b).
siRNA-mediated knockdown of Hnf4a reduces Agxt2 expression
Next we downregulated expression of Hnf4a in cultured Hepa 1–6 cells by siRNA-mediated knockdown to determine whether HNF4α is required for endogenous Agxt2 expression. Four different siRNAs against Hnf4a mRNA were used with the following negative controls: two different scrambled siRNAs, the mock control without siRNA and intact cells. Efficiency of Hnf4a knockdown was confirmed at the protein level by Western blotting (Fig. 4a). mRNA levels of Agxt2 and Hnf4a were measured by RT-qPCR. Glutamate-cysteine ligase (Gclc) mRNA level was assessed as an additional positive control for Hnf4a knockdown, since this gene is known to be regulated by HNF4α35. 60S ribosomal protein L13 (RpL13) mRNA level was assessed as an additional negative control for global effects of Hnf4a knockdown, since we did not expect regulation of RpL13 expression by HNF4α.
Figure 4. siRNA-mediated knockdown of Hnf4a reduces Agxt2 expression in cultured murine hepatocytes.

(a) Validation of knock-down efficiency of Hnf4a in Hepa 1–6 cell line. Western blotting analysis with anti-HNF4α antibodies in whole cell lysates after treatment with four distinct siRNAs against Hnf4a and four negative controls: two scramble siRNAs, transfection reagent alone and untreated cells. Beta-actin is used as loading control. (b) Reduced Agxt2 mRNA accumulation in Hepa 1–6 cells after Hnf4a knock-down. qPCR was performed in four siRNA-treated and four negative control cell lines, with ΔΔCt quantification relative to scramble 1 control. Gclc serves as positive control. RpL13 serves as negative control. Average of 9 independent biological replicates, error bars indicate SEM, one-way ANOVA, Dunnett’s multiple comparison test: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (Precise P-values, Hnf4a: Hnf4a siRNA 1–0.0001, Hnf4a siRNA 2–0.003, Hnf4a siRNA 4–0.0137; Agxt2: Hnf4a siRNA 1–0.0001, Hnf4a siRNA 2–0.0089, Hnf4a siRNA 3 − <0.0001, Hnf4a siRNA 4− <0.0001; Gclc: Hnf4a siRNA 1–0.0032, Hnf4a siRNA 3 − <0.0001, Hnf4a siRNA 4–0.0005).
Transfection of Hepa 1–6 cells with siRNAs against Hnf4a led to a 40% decrease in Hnf4a mRNA levels and about 55% decrease in Agxt2 mRNA levels compared to the control scrambled siRNA. The levels of Gclc mRNA were decreased by approximately 40% in the cells expressing anti-HNF4α siRNAs 1, 3 and 4 indicating efficiency of HNF4α knockdown. The levels of Gclc mRNA were increased by 19% (n.s.) in the cells expressing anti-HNF4α siRNA 2 likely due to nonspecific activity of this particular Hnf4a siRNA (Fig. 4b). No difference in RpL13 gene expression between the samples was observed (Fig. 4b).
Agxt2 mRNA levels are decreased in Hnf4a knockout mice
We took advantage of the previously characterized inducible liver-specific Hnf4a knockout mice27 to determine whether HNF4α regulates Agxt2 expression in vivo. No HNF4α protein was detected by Western blot in the liver of knockout animals after induction of Cre recombinase expression by tamoxifen. Deletion of Hnf4a led to greater than 90% decrease in Agxt2 mRNA levels in the liver. No significant changes in the Agxt2 mRNA levels were observed in the kidneys, where HNF4α protein levels remained intact (Fig. 5a). Partial deletion of Hnf4a in heterozygous Hnf4a knockout mice led to respective decrease in Agxt2 mRNA levels in a dose-dependent manner (Fig. 5c). Neither the liver, nor the kidneys showed any compensatory changes in Ddah1 expression. The levels of Ddah2 remained unchanged in the liver, but were increased by about 50% in the kidney (p < 0.05) (Fig. 5b).
Figure 5. Expression of Agxt2 in inducible liver-specific Hnf4a knockout mice.

(a) Western blotting of liver and kidney lysates from liver-specific Hnf4a knockout mice (Hnf4a KO) and the wild type littermates (wild type). Beta-actin was used as loading control. (b) Agxt2, Ddah1 and Ddah2 mRNA levels in the liver and kidneys of liver-specific Hnf4a knockout mice and littermates determined by qPCR. n = 7 for WT, n = 6 for Hnf4a knockout mice, error bars indicate SEM, Mann-Whitney test: *P = 0.04; ***P < 0.0001. (c) Agxt2 mRNA levels in the liver of homozygous and heterozygous liver-specific Hnf4a knockout mice and littermates determined by qPCR. n = 7 for both heterozygous and homozygous Hnf4a knockout mice, n = 8 for WT, error bars indicate SEM, one-way ANOVA, Tukey’s multiple comparison test: Hnf4a level: Hnf4a+/+ vs. Hnf4a+/−, **P = 0.0012; Hnf4a+/+ vs. Hnf4a−/−, ***P < 0.0001; Hnf4a+/− vs. Hnf4a−/−, ***P = 0.0009; Agxt2 level: Hnf4a+/+ vs. Hnf4a+/−, **P = 0.0006; Hnf4a+/+ vs. Hnf4a−/−, ***P < 0.0001; Hnf4a+/− vs. Hnf4a−/−, ***P < 0.0001.
AGXT2 enzymatic activity is decreased and metabolites levels are changed in plasma of Hnf4a knockout mice
Next, we determined, whether liver-specific Hnf4a deficiency affects tissue AGXT2 activity and systemic levels of AGXT2-related metabolites. We observed an 85% decrease in tissue AGXT2 activity in the liver and no changes in AGXT2 activity in the kidneys of the liver-specific Hnf4a knockout mice (Fig. 6a). The plasma levels of the AGXT2 substrates ADMA and SDMA were increased by 18.3% and 23.3% (p < 0.05), while the plasma levels of the corresponding AGXT2 products ADGV and SDGV were decreased by 67.9% and 74.8%, respectively (p < 0.05) (Fig. 6b,c). Further, a dramatic increase in the plasma levels of the AGXT2 substrate BAIB was observed in Hnf4a knockout mice, whereas no BAIB was detected in the plasma of the wild-type littermates (Fig. 6b).
Figure 6. AGXT2 activity and metabolites levels in plasma of inducible liver-specific Hnf4a knockout mice.

(a) AGXT2 activity was assessed as a production rate of AGXT2-specific metabolite of ADMA–asymmetric dymethylguanovaleric acid (ADGV) per mg of tissue after incubation with isotope-labeled ADMA. ADGV concentrations were determined by HPLC-MS-MS. n = 7 for WT, n = 6 for Hnf4a knockout mice, error bars indicate SEM, Mann-Whitney test: **P = 0.0012. (b) AGXT2 substrates levels in plasma of liver specific Hnf4a knockout and wild type mice. ADMA, SDMA, and BAIB concentrations determined by HPLC-MS-MS. In case of ADMA and SDMA n = 6 for WT and n = 7 for Hnf4a knockout mice, in case of BAIB n = 7 for both WT and Hnf4a knockout mice; error bars indicate SEM, Mann-Whitney test: ADMA *P = 0.0291; SDMA *P = 0.0231. Levels of BAIB in all wild type samples were below quantification limit of 0.2 μmol/L (N.D.–non detectable). (c) AGXT2 products levels in plasma of liver-specific Hnf4a knockout mice and wild type littermates. ADGV and SDGV concentrations were determined by HPLC-MS-MS. n = 7 for both WT and Hnf4a knockout mice. For SDGV culture-derived isotope labelled [2H6]-SDGV was used as the internal standard, therefore, only relative units (area ratios) are provided. Error bars indicate SEM, Mann-Whitney test: **P = 0.0014; ***P = 0.0006.
Discussion
In this study we applied several complementary in vitro and in vivo approaches to demonstrate that HNF4α is the major regulator of AGXT2 gene expression. First, in silico analysis predicted the presence of a functionally active HNF4α binding site in the mammalian AGXT2 promoter. The role of this site during murine Agxt2 promoter activation was demonstrated in vitro by mutational analysis and subsequent detection of the resulting decrease in promoter activity in luciferase reporter assays. Direct binding of HNF4α to the predicted consensus motif in the Agxt2 promoter was confirmed by ChIP assay. The role of HNF4α in regulation of Agxt2 expression in cultured hepatocytes was demonstrated by detection of a significant decrease in Agxt2 mRNA levels after siRNA-mediated Hnf4a knockdown. In the in vivo part of our study we showed that inducible liver-specific Hnf4a knockout led to a dramatic downregulation of AGXT2 expression and activity in the murine liver. Furthermore, deletion of Hnf4a in the murine liver led to an elevation of plasma levels of the AGXT2 substrates ADMA, SDMA and BAIB and a decrease in plasma levels of the AGXT2 products ADGV and SDGV, thus replicating the previously reported biochemical phenotypes of patients and experimental animals with reduced or absent AGXT2 activity.
AGXT2 is a mitochondrial pyridoxal phosphate-dependent amino transferase. It is encoded by a single gene located on the chromosome 5 in humans and on the chromosome 15 in mice. We found that the sequence −211 to +1 relative to the translation start site of murine Agxt2 was sufficient for driving Agxt2 promoter activity in the Agxt2 promoter/luciferase reporter assay (Fig. 2b). This region is 66% homologous between mice and humans with the longest conserved sequence containing the HNF4α consensus binding site. Our study demonstrated the essential role of this HNF4α binding site in regulation of Agxt2 expression in the murine liver. Presence of a homologous HNF4α binding site in the 5′ UTR of human AGXT221,39 suggests that HNF4α also regulates hepatic AGXT2 expression in humans. This is consistent with high RNA polymerase II (Pol2) occupancy of the AGXT2 promoter region in both mouse and human liver (Fig. 1c). Interestingly, both HNF4A and AGXT2 are coexpressed in the renal proximal tubules, raising the possibility that HNF4α might also be involved in regulation of AGXT2 expression in the kidneys (GEO GDS339740). This possibility is also supported by high Pol2 occupancy of the predicted AGXT2 core promoter region in the kidneys (Fig. 1c).
While outside the scope of this study, our analyses indicate that additional factors may be involved in transcriptional regulation of AGXT2. Indeed, while levels of AGXT2 and HNF4a mRNAs generally correlate in both mouse and human tissues, small intestine represents a peculiar exception, expressing only HNF4a, but not AGXT241. This is likely explained by either tissue-specific mechanism for negative regulation of Agxt2 expression, such as DNA methylation, or local chromatin modifications that may prevent HNF4α binding or activity. Alternatively, additional cofactor(s), absent in small intestine, may be required for efficient HNF4α-dependent activation of Agxt2. Another possibility is that only certain isoforms of HNF4α are involved in regulation of Agxt2 expression. Indeed, HNF4a gene contains two promoters, one with kidney and liver expression (P1) and one with pancreatic-specific expression (P2), and has 13 exons, which encode twelve distinct isoforms as the result of alternate promoter usage and differential splicing. The expression profile of these isoforms varies with development, differentiation, and tissue origin42,43,44. The P1-driven isoforms are known to have stronger transcriptional potentials, compared to the P2-originated ones, due to presence of AF-1 activation function domain in their structure45. Moreover, the downstream targets of HNF4α include the genes, expression of which is strictly or mainly dependent on the presence of a functional AF-1 domain in the HNF4α protein (CAR, apoAIV, apoCII), and the genes, expression of which is independent of this motif, although dependent upon HNF4α (OTC and apoAII)46. The experimental approaches, utilized in the current manuscript, were targeting all isoforms of HNF4α, so determination of the role of the specific isoforms of HNF4α in regulation of Agxt2 expression in different tissues lies beyond the scope of the current work and will be examined in our future studies.
Interestingly, we did not observe complete loss of Agxt2 expression in the liver of the liver-specific Hnf4a knockout mice (Fig. 5b) suggesting that the basal Agxt2 transcription is independent of HNF4α. This is consistent with the proposed mechanism of HNF4α action, wherein it interacts with and augments transcriptional activation by the Mediator complex47. Additionally, HNF4α has been demonstrated to facilitate recruitment of histone acetyltransferase coactivators and to increase promoter accessibility for transcription factors via interaction with ATP-dependent chromatin remodelers47.
An almost 90% decrease in hepatic Agxt2 expression, which we detected in the liver-specific Hnf4a knockout mice in the current study, did not result in compensatory changes in expression of Ddah1 or Ddah2 genes in the liver or compensatory changes in Agxt2 and Ddah1 expression in the kidneys. The levels of Ddah2 in the kidneys were slightly elevated (Fig. 5b) possibly suggesting presence of an inducible tissue-specific enhancer, which still remains to be identified. Potential trigger for upregulation of Ddah2 expression in the kidney could be increased circulating levels of ADMA, as a part of a possible negative feedback loop. Interestingly, upregulation of Ddah2 expression in the kidneys was not capable of compensating the elevation of systemic ADMA levels, which was observed in our experiment, suggesting insufficiency of the potential endogenous DDAH2-mediated compensatory mechanisms regulating systemic ADMA levels.
Downregulation of Agxt2 expression in the liver led to elevation of systemic levels of both ADMA and SDMA, which mimicked the metabolic phenotype observed by Caplin and colleagues in mice with global Agxt2 deficiency14. These findings emphasize the essential role of hepatic AGXT2 in regulation of plasma levels of endogenous methylarginines and argue against the initial hypothesis of Lee and colleagues that AGXT2 is primarily a renal enzyme48. The major contribution of hepatic AGXT2 to total AGXT2 activity, however, is consistent with the previous reports from our and other groups, which showed strong expression of AGXT2 in the liver15,30,32,49.
Hepatic AGXT2 deficiency in response to liver-specific Hnf4a knockout resulted in appearance of micromolar levels of BAIB in plasma, whereas plasma levels of BAIB in the wild type littermates remained below the detection limit. Hence, Hnf4a knockouts are mimicking the biochemical phenotype of the patients with hyper-β-aminoisobutyric aciduria caused by AGXT2 deficiency50. Our data suggest that hepatic AGXT2 not only plays a major role in metabolism of endogenous methylarginines, but is also essential for systemic BAIB homeostasis. It is possible, however, that dramatic elevation of systemic BAIB concentration in liver-specific Hnf4a knockout mice is due not only to deficiency of AGXT2, but also to alterations in expression of some other genes involved in BAIB metabolism or transport. One such gene may be solute carrier family 6 member 13 (Slc6a13, Gat2), a known transporter of BAIB that is associated with hyper-β-aminoisobutyric aciduria in humans17. Supporting this hypothesis, gene expression analyses suggest that this transporter is almost completely depleted in the liver of Hnf4α knockout mice23.
Our finding that even partial downregulation of HNF4α either in cultured hepatocytes after siRNA-mediated Hnf4α knockdown (Fig. 4b) or in vivo in heterozygous Hnf4a knockout mice (Fig. 5c) still leads to a statistically significant proportional decrease in Agxt2 expression suggests that the effect of HNF4a on Agxt2 transcription is dose-dependent, which makes our study relevant to the large number of patients with mild HNF4α deficiency. Indeed, while severe inborn HNF4α deficiency is rare and leads to maturity-onset diabetes of the young 1 (MODY1)24, mild HNF4α deficiency due to HNF4A polymorphisms in either intronic or promoter regions is common and is associated with increased risk of type 2 diabetes mellitus25,51, gestational diabetes mellitus52, metabolic syndrome, dyslipidemia53 and arterial hypertension54. For instance, the allele frequency for common HNF4A polymorphisms rs2144908, rs3818247 and rs1884614, which are associated with higher susceptibility to type 2 diabetes, is approximately 20% in healthy population and increases up to 29% in type 2 diabetes patients55,56.
The finding that HNF4α deficiency and subsequent downregulation of AGXT2 leads to an increase in plasma ADMA concentration might also be clinically relevant, as the epidemiological studies have shown that even modest elevation in plasma ADMA levels is associated with increased risk of cardiovascular events and mortality in people with cardiovascular disease57,58. Furthermore, the pathophysiological significance of even modest changes in plasma ADMA concentration has been demonstrated in experimental studies59. A potential explanation for this phenomenon is that ADMA accumulates in endothelial cells in vivo at the levels 5–10 folds higher than the levels of ADMA in plasma, so the magnitude of ADMA changes in plasma might only partially reflect the levels in the intracellular compartments60. What further supports the potential clinical relevance of our findings is that in the in vivo part of our study we downregulated HNF4α only in one tissue, i.e. in the liver, and this alone led to significant changes in plasma levels of dimethylarginines. We speculate that downregulation of HNF4α levels in multiple tissues would lead to even more profound changes in metabolism of ADMA and SDMA.
Changes in AGXT2 activity can affect several metabolites, such as endogenous methylarginines13, BAIB31,61, NO13 and lipids9,62, which are all known to be dysregulated in diabetes. In addition, studies in animal models have demonstrated that AGXT2 deficiency leads to hypertension14, a classic feature of the metabolic syndrome. Furthermore, AGXT2 polymorphisms have been linked to increased carotid intima-media thickness63, and, in diabetic patients, to increased risk of coronary artery disease64. These observations raise the possibility that HNF4α-related diabetes may represent a metabolically-distinct subset of diabetes characterized by impairment in AGXT2 activity and a higher risk for development of cardiovascular complications due to the resulting metabolic abnormalities. Published data regarding the association between diabetes and circulating levels of methylarginines such as ADMA are contradictory65,66, perhaps because HNF4α-related diabetes represents only a subset of patients. Testing this hypothesis by assessing ADMA- and BAIB-related metabolic changes as well as the spectrum and prevalence of cardiovascular complications in the diabetic patients with HNF4A polymorphisms may lead to development of novel targeted therapeutic approaches for this subgroup of patients.
Additional Information
How to cite this article: Burdin, D. V. et al. Diabetes-linked transcription factor HNF4α regulates metabolism of endogenous methylarginines and β-aminoisobutyric acid by controlling expression of alanine-glyoxylate aminotransferase 2. Sci. Rep. 6, 35503; doi: 10.1038/srep35503 (2016).
Supplementary Material
Acknowledgments
We thank Carmen Friebel, Isabell Wenzel and Annette Rexin for their help with cell culture and Western blot experiments. We thank Theresa Reetz for her help with the luciferase experiments and the expression analysis in the in vitro part of the project. We thank Max Plank Institute Dresden and personally Martin Stoeter and Marc Bickle for help with set-up and optimization of siRNA experiments. We thank Vladimir Todorov for his help with the luciferase experiments set-up. This work was supported by the grant from Else Kröner-Fresenius-Stiftung 2011_A168 to R.N.R., by the Erasmus Mundus Action 2 MULTIC scholarship (award 11-664) to A.K. and by the DAAD scholarship (award 50024759) to D.V.B.; A.A.S. is HHMI Fellow of the Damon Runyon Cancer Research Foundation (DRG-2185-14).
Footnotes
Author Contributions D.V.B. and A.A.K. –research design, experimental work, data analysis, manuscript preparation; C.B., S.B., R.M., S.B.B., J.M.-L. and M.M. –experimental work, data analysis; N.S. –research design, computational analysis; S.R.B.–data analysis, manuscript preparation; A.V.D., A.A.S., N.J., F.G., N.W. and R.N.R. –research design, data analysis, manuscript preparation.
References
- Schwedhelm E. & Boger R. H. The role of asymmetric and symmetric dimethylarginines in renal disease. Nature reviews. Nephrology 7, 275–285, 10.1038/nrneph.2011.31 (2011). [DOI] [PubMed] [Google Scholar]
- Kittel A. & Maas R. Pharmacology and clinical pharmacology of methylarginines used as inhibitors of nitric oxide synthases. Current pharmaceutical design 20, 3530–3547 (2014). [DOI] [PubMed] [Google Scholar]
- Antoniades C. et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. European heart journal 30, 1142–1150, 10.1093/eurheartj/ehp061 (2009). [DOI] [PubMed] [Google Scholar]
- Cooke J. P. Asymmetrical dimethylarginine: the Uber marker? Circulation 109, 1813–1818, 10.1161/01.CIR.0000126823.07732.D5 (2004). [DOI] [PubMed] [Google Scholar]
- Schwedhelm E. et al. Incidence of all-cause and cardiovascular mortality predicted by symmetric dimethylarginine in the population-based study of health in pomerania. PloS One 9, e96875, 10.1371/journal.pone.0096875 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore M. O. et al. Symmetrical dimethylarginine predicts mortality in the general population: observations from the Dallas heart study. Arteriosclerosis, thrombosis, and vascular biology 33, 2682–2688, 10.1161/ATVBAHA.113.301219 (2013). [DOI] [PubMed] [Google Scholar]
- Strobel J. et al. Transport of asymmetric dimethylarginine (ADMA) by cationic amino acid transporter 2 (CAT2), organic cation transporter 2 (OCT2) and multidrug and toxin extrusion protein 1 (MATE1). Amino acids 45, 989–1002, 10.1007/s00726-013-1556-3 (2013). [DOI] [PubMed] [Google Scholar]
- Closs E. I., Basha F. Z., Habermeier A. & Forstermann U. Interference of L-arginine analogues with L-arginine transport mediated by the y+ carrier hCAT-2B. Nitric oxide: biology and chemistry/official journal of the Nitric Oxide Society 1, 65–73, 10.1006/niox.1996.0106 (1997). [DOI] [PubMed] [Google Scholar]
- Speer T. et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 38, 754–768, 10.1016/j.immuni.2013.02.009 (2013). [DOI] [PubMed] [Google Scholar]
- Leiper J. & Nandi M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nature reviews. Drug discovery 10, 277–291, 10.1038/nrd3358 (2011). [DOI] [PubMed] [Google Scholar]
- Martens-Lobenhoffer J., Rodionov R. N., Drust A. & Bode-Boger S. M. Detection and quantification of alpha-keto-delta-(N(G),N(G)-dimethylguanidino)valeric acid: a metabolite of asymmetric dimethylarginine. Analytical biochemistry 419, 234–240, 10.1016/j.ab.2011.08.044 (2011). [DOI] [PubMed] [Google Scholar]
- Ogawa T., Kimoto M., Watanabe H. & Sasaoka K. Metabolism of NG,NG-and NG,N’G-dimethylarginine in rats. Archives of biochemistry and biophysics 252, 526–537 (1987). [DOI] [PubMed] [Google Scholar]
- Rodionov R. N., Murry D. J., Vaulman S. F., Stevens J. W. & Lentz S. R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. The Journal of biological chemistry 285, 5385–5391, 10.1074/jbc.M109.091280 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplin B. et al. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arteriosclerosis, thrombosis, and vascular biology 32, 2892–2900, 10.1161/ATVBAHA.112.254078 (2012). [DOI] [PubMed] [Google Scholar]
- Luneburg N. et al. Genome-wide association study of L-arginine and dimethylarginines reveals novel metabolic pathway for symmetric dimethylarginine. Circulation. Cardiovascular genetics 7, 864–872, 10.1161/CIRCGENETICS.113.000264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno E., Minatogawa Y. & Kido R. Co-purification of alanine-glyoxylate aminotransferase with 2-aminobutyrate aminotransferase in rat kidney. Biochimica et biophysica acta 715, 97–104 (1982). [DOI] [PubMed] [Google Scholar]
- Rhee E. P. et al. A genome-wide association study of the human metabolome in a community-based cohort. Cell metabolism 18, 130–143, 10.1016/j.cmet.2013.06.013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begriche K. et al. Beta-aminoisobutyric acid prevents diet-induced obesity in mice with partial leptin deficiency. Obesity 16, 2053–2067, 10.1038/oby.2008.337 (2008). [DOI] [PubMed] [Google Scholar]
- Roberts L. D. et al. beta-Aminoisobutyric acid induces browning of white fat and hepatic beta-oxidation and is inversely correlated with cardiometabolic risk factors. Cell metabolism 19, 96–108, 10.1016/j.cmet.2013.12.003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, 10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome research 22, 1798–1812, 10.1101/gr.139105.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic acids research 41, D171–D176, 10.1093/nar/gks1221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle M. A. et al. Hepatocyte nuclear factor 4alpha orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proceedings of the National Academy of Sciences of the United States of America 103, 8419–8424, 10.1073/pnas.0600246103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajans S. S., Bell G. I. & Polonsky K. S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. The New England journal of medicine 345, 971–980, 10.1056/NEJMra002168 (2001). [DOI] [PubMed] [Google Scholar]
- Cho Y. S. et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nature genetics 44, 67–72, 10.1038/ng.1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson S. et al. Studies in 3,523 Norwegians and meta-analysis in 11,571 subjects indicate that variants in the hepatocyte nuclear factor 4 alpha (HNF4A) P2 region are associated with type 2 diabetes in Scandinavians. Diabetes 56, 3112–3117, 10.2337/db07-0513 (2007). [DOI] [PubMed] [Google Scholar]
- Bonzo J. A., Ferry C. H., Matsubara T., Kim J. H. & Gonzalez F. J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. The Journal of biological chemistry 287, 7345–7356, 10.1074/jbc.M111.334599 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzolino A. M. et al. TGFbeta overrides HNF4alpha tumor suppressing activity through GSK3beta inactivation: implication for hepatocellular carcinoma gene therapy. Journal of hepatology 58, 65–72, 10.1016/j.jhep.2012.08.023 (2013). [DOI] [PubMed] [Google Scholar]
- Martens-Lobenhoffer J. & Bode-Boger S. M. Quantification of L-arginine, asymmetric dimethylarginine and symmetric dimethylarginine in human plasma: a step improvement in precision by stable isotope dilution mass spectrometry. Journal of chromatography. B, Analytical technologies in the biomedical and life sciences 904, 140–143, 10.1016/j.jchromb.2012.07.021 (2012). [DOI] [PubMed] [Google Scholar]
- Martens-Lobenhoffer J., Rodionov R. N. & Bode-Boger S. M. Probing AGXT2 enzyme activity in mouse tissue by applying stable isotope-labeled asymmetric dimethyl arginine as substrate. Journal of mass spectrometry: JMS 47, 1594–1600, 10.1002/jms.3125 (2012). [DOI] [PubMed] [Google Scholar]
- Kittel A. et al. Alanine-glyoxylate aminotransferase 2 (AGXT2) polymorphisms have considerable impact on methylarginine and beta-aminoisobutyrate metabolism in healthy volunteers. PloS One 9, e88544, 10.1371/journal.pone.0088544 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittel A. et al. In vivo evidence that Agxt2 can regulate plasma levels of dimethylarginines in mice. Biochemical and biophysical research communications 430, 84–89, 10.1016/j.bbrc.2012.11.008 (2013). [DOI] [PubMed] [Google Scholar]
- Frazer K. A., Pachter L., Poliakov A., Rubin E. M. & Dubchak I. VISTA: computational tools for comparative genomics. Nucleic acids research 32, W273–W279, 10.1093/nar/gkh458 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B., Mane-Padros D., Bolotin E., Jiang T. & Sladek F. M. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic acids research 40, 5343–5356, 10.1093/nar/gks190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M. & Lash A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic acids research 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorrez L. et al. Using ribosomal protein genes as reference: a tale of caution. PloS One 3, e1854, 10.1371/journal.pone.0001854 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijnen M. J., Peerlinck K., Maasdam D., Bertina R. M. & Reitsma P. H. Hemophilia B Leyden: substitution of thymine for guanine at position -21 results in a disruption of a hepatocyte nuclear factor 4 binding site in the factor IX promoter. Blood 82, 151–158 (1993). [PubMed] [Google Scholar]
- Reijnen M. J., Sladek F. M., Bertina R. M. & Reitsma P. H. Disruption of a binding site for hepatocyte nuclear factor 4 results in hemophilia B Leyden. Proceedings of the National Academy of Sciences of the United States of America 89, 6300–6303 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein M. B. et al. Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100, 10.1038/nature11245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright J. et al. Transcriptional adaptation to Clcn5 knockout in proximal tubules of mouse kidney. Physiological genomics 33, 341–354, 10.1152/physiolgenomics.00024.2008 (2008). [DOI] [PubMed] [Google Scholar]
- Petryszak R. et al. Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic acids research 44, D746–D752, 10.1093/nar/gkv1045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Padilla M. E., Fougere-Deschatrette C. & Weiss M. C. Expression of HNF4alpha isoforms in mouse liver development is regulated by sequential promoter usage and constitutive 3′ end splicing. Mechanisms of development 109, 183–193 (2001). [DOI] [PubMed] [Google Scholar]
- Suaud L., Joseph B., Formstecher P. & Laine B. mRNA expression of HNF-4 isoforms and of HNF-1alpha/HNF-1beta variants and differentiation of human cell lines that mimic highly specialized phenotypes of intestinal epithelium. Biochemical and biophysical research communications 235, 820–825, 10.1006/bbrc.1997.6888 (1997). [DOI] [PubMed] [Google Scholar]
- Dean S., Tang J. I., Seckl J. R. & Nyirenda M. J. Developmental and tissue-specific regulation of hepatocyte nuclear factor 4-alpha (HNF4-alpha) isoforms in rodents. Gene expression 14, 337–344 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eeckhoute J. et al. Hepatocyte nuclear factor 4 alpha isoforms originated from the P1 promoter are expressed in human pancreatic beta-cells and exhibit stronger transcriptional potentials than P2 promoter-driven isoforms. Endocrinology 144, 1686–1694, 10.1210/en.2002-0024 (2003). [DOI] [PubMed] [Google Scholar]
- Briancon N. & Weiss M. C. In vivo role of the HNF4alpha AF-1 activation domain revealed by exon swapping. The EMBO journal 25, 1253–1262, 10.1038/sj.emboj.7601021 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik S., Wallberg A. E., Kang Y. K. & Roeder R. G. TRAP/SMCC/mediator-dependent transcriptional activation from DNA and chromatin templates by orphan nuclear receptor hepatocyte nuclear factor 4. Molecular and cellular biology 22, 5626–5637 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I. S., Nishikimi M., Inoue M., Muragaki Y. & Ooshima A. Specific expression of alanine-glyoxylate aminotransferase 2 in the epithelial cells of Henle’s loop. Nephron 83, 184–185, 45507 (1999). [DOI] [PubMed] [Google Scholar]
- Rodionov R. N. et al. Role of alanine:glyoxylate aminotransferase 2 in metabolism of asymmetric dimethylarginine in the settings of asymmetric dimethylarginine overload and bilateral nephrectomy. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association-European Renal Association 29, 2035–2042, 10.1093/ndt/gfu236 (2014). [DOI] [PubMed] [Google Scholar]
- Kakimoto Y., Taniguchi K. & Sano I. D-beta-aminoisobutyrate:pyruvate aminotransferase in mammalian liver and excretion of beta-aminoisobutyrate by man. The Journal of biological chemistry 244, 335–340 (1969). [PubMed] [Google Scholar]
- Kooner J. S. et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nature genetics 43, 984–989, 10.1038/ng.921 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy V. S., Diaz C. A., Trenado L. M., Peralta J. M. & Soto S. M. Thr130Ile polymorphism of HNF4A gene is associated with gestational diabetes mellitus in Mexican population. Journal of investigative medicine: the official publication of the American Federation for Clinical Research 62, 632–634, 10.2310/JIM.0000000000000045 (2014). [DOI] [PubMed] [Google Scholar]
- Weissglas-Volkov D. et al. Common hepatic nuclear factor-4alpha variants are associated with high serum lipid levels and the metabolic syndrome. Diabetes 55, 1970–1977, 10.2337/db06-0035 (2006). [DOI] [PubMed] [Google Scholar]
- Saif-Ali R., Harun R., Al-Jassabi S. & Wan Ngah W. Z. Hepatocyte nuclear factor 4 alpha P2 promoter variants associate with insulin resistance. Acta biochimica Polonica 58, 179–186 (2011). [PubMed] [Google Scholar]
- Silander K. et al. Genetic variation near the hepatocyte nuclear factor-4 alpha gene predicts susceptibility to type 2 diabetes. Diabetes 53, 1141–1149 (2004). [DOI] [PubMed] [Google Scholar]
- Love-Gregory L. D. et al. A common polymorphism in the upstream promoter region of the hepatocyte nuclear factor-4 alpha gene on chromosome 20q is associated with type 2 diabetes and appears to contribute to the evidence for linkage in an ashkenazi jewish population. Diabetes 53, 1134–1140 (2004). [DOI] [PubMed] [Google Scholar]
- Meinitzer A. et al. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (The Ludwigshafen Risk and Cardiovascular Health study). Clinical chemistry 53, 273–283, 10.1373/clinchem.2006.076711 (2007). [DOI] [PubMed] [Google Scholar]
- Valkonen V. P. et al. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 358, 2127–2128, 10.1016/S0140-6736(01)07184-7 (2001). [DOI] [PubMed] [Google Scholar]
- Leiper J. et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nature medicine 13, 198–203, 10.1038/nm1543 (2007). [DOI] [PubMed] [Google Scholar]
- Masuda H., Goto M., Tamaoki S. & Azuma H. Accelerated intimal hyperplasia and increased endogenous inhibitors for NO synthesis in rabbits with alloxan-induced hyperglycaemia. British journal of pharmacology 126, 211–218, 10.1038/sj.bjp.0702298 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhre K. et al. A genome-wide association study of metabolic traits in human urine. Nature genetics 43, 565–569, 10.1038/ng.837 (2011). [DOI] [PubMed] [Google Scholar]
- Moore K. J. & Fisher E. A. Dysfunctional HDL takes its toll in chronic kidney disease. Immunity 38, 628–630, 10.1016/j.immuni.2013.03.006 (2013). [DOI] [PubMed] [Google Scholar]
- Yoshino Y. et al. Missense variants of the alanine: glyoxylate aminotransferase 2 gene correlated with carotid atherosclerosis in the Japanese population. Journal of biological regulators and homeostatic agents 28, 605–614 (2014). [PubMed] [Google Scholar]
- Zhou J. P. et al. Association of the AGXT2 V140I polymorphism with risk for coronary heart disease in a Chinese population. Journal of atherosclerosis and thrombosis 21, 1022–1030 (2014). [DOI] [PubMed] [Google Scholar]
- Boger R. H. et al. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 119, 1592–1600, 10.1161/CIRCULATIONAHA.108.838268 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaciragic A. et al. An assessment of correlation between serum asymmetric dimethylarginine and glycated haemoglobin in patients with type 2 diabetes mellitus. Bosnian journal of basic medical sciences/Udruzenje basicnih mediciniskih znanosti = Association of Basic Medical Sciences 14, 21–24 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane T. M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294, 10.1038/nature10413 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K., Yang C., Sladek F. M. & Jiang T. Identifying transcription factor binding sites through Markov chain optimization. Bioinformatics 18 Suppl 2, S100–S109 (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.