

Summary
Aims
Long noncoding RNAs (lncRNAs) play a key role in regulating immunological functions. Their impact on the chronic inflammatory disease multiple sclerosis (MS), however, remains unknown. We investigated the expression of lncRNAs in peripheral blood mononuclear cells (PBMCs) of patients with MS and attempt to explain their possible role in the process of MS.
Methods
For this study, we recruited 26 patients with MS according to the revised McDonald criteria. Then, we randomly chose 6 patients for microarray analysis. Microarray assays identified outstanding differences in lncRNA expression, which were verified through real‐time PCR. LncRNA functions were annotated for target genes using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses, and regulatory relationships between lncRNAs and target genes were analyzed using the “cis” and “trans” model.
Results
There were 2353 upregulated lncRNAs, 389 downregulated lncRNAs, 1037 upregulated mRNAs, and 279 downregulated mRNAs in patients with MS compared to healthy control subjects (fold change >2.0). Real‐time PCR results of six aberrant lncRNAs were consistent with the microarray data. The coexpression network comprised 864 lncRNAs and 628 mRNAs. Among differentially expressed lncRNAs, 10 lncRNAs were predicted to have 10 cis‐regulated target genes, and 33 lncRNAs might regulate their trans target genes.
Conclusions
We identified a subset of dysregulated lncRNAs and mRNAs. The differentially expressed lncRNAs may be important in the process of MS. However, the specific molecular mechanisms and biological functions of these lncRNAs in the pathogenesis of MS need further study.
Keywords: Long noncoding RNAs, Microarray, Multiple sclerosis
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system (CNS) mediated by CD4+ T cells; it is characterized by demyelinating lesions and progressive axon loss 1. The pathogenesis of MS is thought to be complex and not well understood, and genetic and environmental risk factors are reported to be involved. However, our current level of genetic knowledge can explain only about 25% of the overall risk of MS, depending on ethnicity and environment 2, 3, 4. Epigenetic changes, such as altered DNA methylation, histone modifications, and microRNA‐mediated posttranscriptional gene silencing may affect the initiation and progression of MS 5, 6. Emerging evidence shows that long noncoding RNAs (lncRNAs) play a key role in the regulation of immunological functions 7, suggesting that they might also be involved in MS. However, the precise role of lncRNAs in the pathogenesis of MS remains elusive.
LncRNAs that are >200 nucleotides in length represent a new class of noncoding RNA 8, 9, 10. They contribute to a variety of biological cascades and are reported to be involved in neurodegenerative diseases, diabetic mellitus, cancer, and cardiovascular diseases 11, 12, 13, 14. Noncoding RNAs are emerging as a new regulatory layer that affects both the development of the immune system and its function 15, 16. Although thousands of long intergenic noncoding RNAs (lincRNAs) have been identified in the mammalian genome by bioinformatics analyses of transcriptomic data, their functional characterization is still largely incomplete. Recent studies show widespread changes in the expression of lncRNAs during the activation of the innate immune response and T‐cell development, differentiation, and activation 17. These lncRNAs control important aspects of immunity, such as production of inflammatory mediators, differentiation, and cell migration by regulating protein–protein interactions or via their ability to base pair with RNA and DNA 15, 16, 18. Although several lncRNAs have been implicated in diverse processes and diseases 19, 20, only a few examples of their regulation of the autoimmune diseases have been described 21, 22, 23.
In the present study, we performed an array of lncRNA chip assays on peripheral blood mononuclear cells (PBMCs) of patients with MS. Outstanding lncRNA functions were annotated based on coexpression genes and a gene ontology (GO) biological analysis process. The relationships among lncRNAs and mRNAs were revealed through “cis” and “trans” analyses.
Materials and Methods
Study Population and Trial Design
During the open enrollment, a total of 26 relapsing–remitting patients with MS in the acute stage of disease were recruited at Tianjin Medical University General Hospital from May 2014 to August 2015 (Table 1). These patients met the McDonald criteria for MS, as revised in 2010, and all met the criteria of the disease being disseminated in space (i.e., involvement of multiple areas of the CNS) and time (i.e., ongoing disease activity over time). We also verified MS by oligoclonal bands observed in the cerebrospinal fluid (CSF) of all patients with MS. Exclusion criteria were the following: 1 the presence of other diseases of the CNS in addition to MS 2 the presence of tumor(s) and systemic hematologic diseases 3 the presence of recent infection, and 4 concomitant use of antineoplastic or immune‐modulating therapies prior to blood sampling. The Tianjin Medical University General Hospital institutional review boards approved the trial protocol and supporting documentation. We also recruited 20 age‐ and gender‐matched healthy volunteers (control subjects) for the comparative study. Informed consent was obtained at enrollment from all patients or legally acceptable surrogates.
Table 1.
Baseline characteristics
| Control (n = 26) | MS (n = 26) | P‐value | |
|---|---|---|---|
| Gender, M/F | 8/18 | 8/18 | 1.00 |
| Age at onset, years, median (range) | – | 34 (19–64) | – |
| Annual relapse rate, median (range) | – | 0.6 (0.04–2.7) | – |
| OCBs positive/tested (%) | – | 21/26 (81) | – |
| Brain MRI abnormalities (%) | – | 26/26 (100) | – |
| Spinal MRI abnormalities (%) | – | 17/26 (65) | – |
| EDSS score, median (range) | – | 4 (1–10) | – |
| Poor neurological outcome (%) | – | 11/26 (42) | – |
MS, multiple sclerosis; OCBs, oligoclonal bands; MRI, magnetic resonance imaging; EDSS, Expanded Disability Status Scale.
Isolation of human peripheral blood mononuclear cells (PBMCs)
Peripheral blood anticoagulated by ethylenediaminetetraacetic acid (EDTA) was obtained from all patients with MS and healthy controls. Human PBMCs were isolated using Ficoll–Hypaque gradients.
RNA Extraction and Chip Arrays
Total RNA was extracted from PBMCs using Trizol® reagent (Invitrogen, Grand Island, NY, USA). Approximately 200 ng of total RNA from each sample was used for the lncRNA microarray analyses. LncRNA expression was analyzed using OE_Biotech Human lncRNA chip software, containing 41,000 lncRNAs and 34,000 mRNAs. Those lncRNA and mRNA target sequences were merged from multiple databases: 23,898 from GENCODE/ENSEMBL; 14,353 from the Human lincRNA Catalog 24; 7760 from RefSeq; 5627 from USCS; 13,701 from ncRNA Expression Database (NRED); 21,488 from LNCipedia; 1038 from H‐InvDB; 3019 from lncRNAs‐a (Enhancer‐like); 1053 from the Antisense ncRNA pipeline; 407 Hox ncRNAs; 962 UCRs; and 848 from the Chen Ruisheng laboratory (Institute of Biophysics, Chinese Academy of Sciences, Beijing, China).
Each RNA was detected by probes; this was repeated twice. The array also contained 4974 Agilent control probes. The lncRNA chip experiments were conducted at Capitalbio Corporation in Beijing, China. Different lncRNAs and mRNAs were analyzed using Cluster 3.0 software. The results were further analyzed using Tree View software. Green indicates low expression, and red indicates high expression in the output for these analyses.
Quantitative Real‐time PCR Validation
Total RNA was extracted from PBMCs by Trizol® reagent (Invitrogen) following the manufacturer's instructions. RNA quantity and quality were assessed using a Nanodrop ND‐100 Spectrophotometer (Nanodrop Technologies, Wilmington, USA) and a 2100 Bioanalyzer (Agilent RNA 6000 Nano Kit, Waldbronn, Germany), with a 260:280 ratio of ≥1.5 and an RNA integrity number of ≥7 for the majority of the samples. For the reverse transcriptase (RT) reaction, SYBR Green RT reagents (Bio‐Rad, Indianapolis, USA) were used.
In brief, the RT reaction was performed for 60 min at 37°C, followed by 60 min at 42°C, using oligo (dT) and random hexamers. PCR amplifications were performed using SYBR Green Universal Master Mix. In brief, reactions were performed in duplicate containing 2 × concentrated Universal Master Mix, 1 μL of template cDNA, and 100 nM of primers in a final volume of 12.5 μL, followed by analysis in a 96‐well optical reaction plate (Bio‐Rad). The lncRNA PCR results were quantified using the 2ΔΔct method against β‐actin for normalization. The data represent the means of three experiments.
We used the following real‐time PCR primers:
XLOC 010881F CCTCTGGGCTTCCTGATAAA
XLOC 010881R AGACCTCCATCCTCAAACCA
ENSG00000231898.3 F CCTCTGGGCTTCCTGATAAA
ENSG00000231898.3 R GGCACCTAACTATGGGAGGAG
XLOC 009626 F CATCGGCAGTGATTTCCTCT
XLOC 009626 R GACTATGTGCTTCTTCCCTTGG
ENSG00000233392.1 F ACCTGTGTGGGCTGACCTAT
ENSG00000233392.1 R CTTCCTGTGGCTGTTCTTCC
ENSG00000259906.1 F GAGCACTGGATGATTTGGAA
ENSG00000259906.1 R TGATGAGCCTTCTTGGTCA
XLOC 010931 F CTCGGCAAATGACTGAACCT
XLOC 010931 R CTCTCTGTGAAGCCCACCAC
LncRNA Coexpression Analysis and Gene Function Annotation
Volcano plot filtering was used to identify lncRNAs and mRNAs with statistically significant differences in expression. Hierarchical clustering was applied to present the diacritical lncRNA and mRNA expression patterns among the samples. LncRNA classification was carried out to explore the potential function of the differentially expressed lncRNAs. GO analysis and pathway analyses were also performed to describe more fully the roles of the differentially expressed mRNAs 25. Furthermore, a coding–noncoding gene coexpression network (CNC network) was drawn using Cytoscape 26, with Pearson coefficient >0.98. The microarray analysis was performed by Capitalbio Corporation.
Target Prediction
For each lncRNA, we calculated the Pearson correlation of its expression value with that of each mRNA. The mRNAs that were coexpressed with the lncRNA were defined as having a Pearson coefficient that exceeded 0.98 and a P‐value <0.05. We identified the mRNAs as “cis‐regulated target genes” when 1 the mRNA loci were within a 10‐kb window up‐ or downstream of the given lncRNA; and 2 the Pearson correlation of lncRNA–mRNA expression was significant (r > 0.98 and P < 0.05). We then determined which mRNAs were likely to be transregulated by the lncRNA of interest. For each lncRNA, we calculated the overlap of the coexpressed mRNA set with transcriptional factor (TF) target genes and used hypergeometric distribution to calculate the significance of this overlap. If the mRNAs coexpressed with a given lncRNA significantly overlapped with the target genes of a given TF, it suggested that this TF might interact with the lncRNA and that these mRNAs might be the transregulated target genes of that particular lncRNA 27.
Results
LncRNA and mRNA Expression Profiles in MS
To determine the expression levels of lncRNAs associated with MS, we performed lncRNA and mRNA microarray analyses on the PBMCs of patients with MS. These data were then compared to those obtained from the PBMCs of age‐ and gender‐matched healthy controls. After separating signal from noise and performing t‐tests, we observed significant differences in lncRNAs and mRNA expression of up to 2‐fold (P < 0.05) (Figure 1A,B,D,E). These results are summarized in Table 2. The lncRNA and mRNA expression data were clustered using Cluster 3.0 and are plotted in Figure 1C and F.
Figure 1.
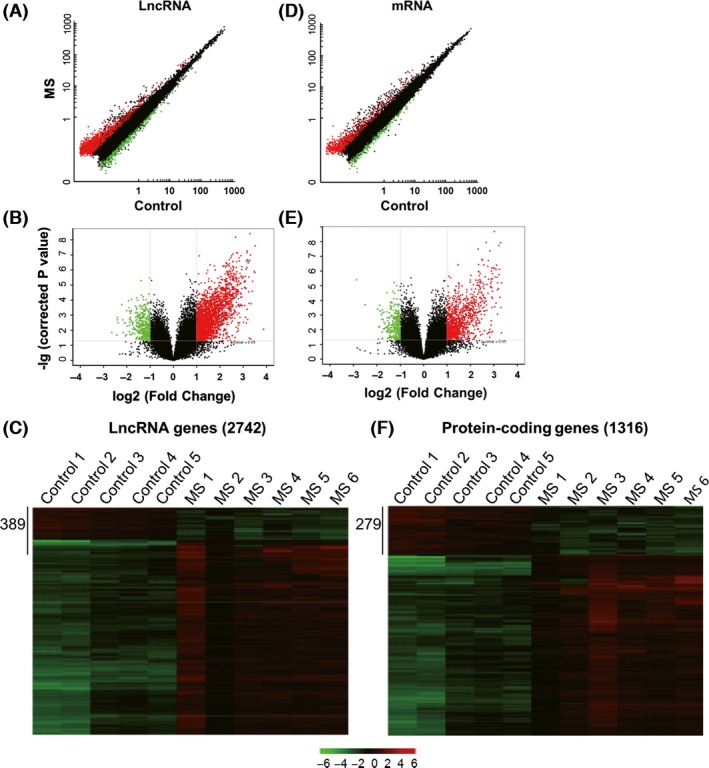
LncRNA and mRNA profiles of microarray data. (A, D). Log–log scatter plots of lncRNA and mRNA expression. The red and green points in the plot indicate more than 2‐fold change of lncRNAs and mRNAs between multiple sclerosis (MS) and healthy control samples. (B, E). Volcano plots of the differentially expressed lncRNAs and mRNAs. The red and green points in the plot represent the differentially expressed lncRNAs and mRNA having statistical significance. (C, F). Hierarchical clustering shows a distinguishable lncRNA and mRNA expression profile between the two groups. Plots here represent analysis of RNA extracted from peripheral blood mononuclear cells (PBMCs) obtained from 6 patients with MS and 5 healthy control subjects.
Table 2.
Dysregulated lncRNAs and mRNAs in multiple sclerosis (MS)
| mRNA | lncRNAs | |
|---|---|---|
| Upregulation | 1037 | 2353 |
| Downregulation | 279 | 389 |
| Total | 1316 | 2742 |
lncRNAs, long noncoding RNAs.
Validation of Disrupted lncRNA Expression in MS
To verify the disruption of lncRNA expression in patients with MS, we performed real‐time PCR to determine the up‐ and downregulation of lncRNAs in each group. As shown in Figure 2, differences in the expression of 6 lncRNAs were detected in patients with MS compared with healthy control subjects. LncRNA ENSG00000231898.3 was the most elevated (12.7‐fold higher expression), followed by lncRNA XLOC_009626 (5.78‐fold higher expression) and lncRNA XLOC_010881 (4.57‐fold higher expression). LncRNA ENSG00000233392.1, lncRNA ENSG00000259906.1, and lncRNA XLOC_010931 exhibited 5.38‐, 2.99‐, and 4.13‐fold lower expression, respectively. These results were consistent with the results obtained from the microarray chip analyses.
Figure 2.
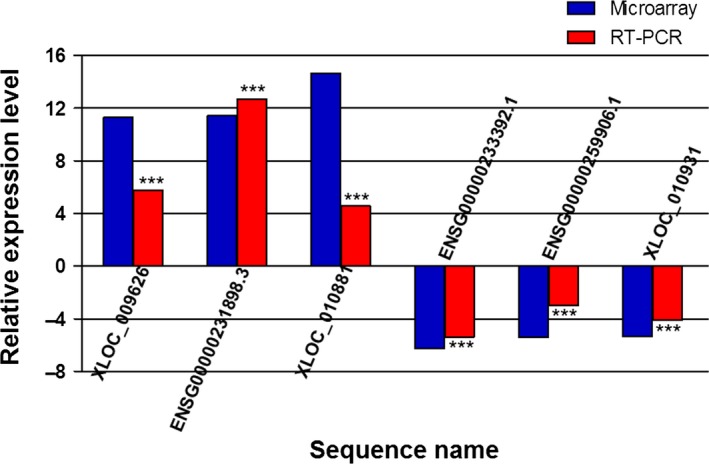
Comparison of lncRNA expression levels as determined by microarray and real‐time PCR analyses. Three upregulated and three downregulated differentially expressed lncRNAs were validated by real‐time PCR of RNA extracted from PBMCs from 20 patients with MS and 20 healthy control subjects. Each sample was analyzed in triplicate. Column heights represent mean fold changes in expression of the MS group. Real‐time PCR results are consistent with microarray data. ***P < 0.001: MS group versus healthy control group in real‐time PCR validation.
LncRNA Function Annotation
To further explore the function of lncRNAs in MS, we subjected the results of the lncRNA and mRNA chip analyses to Pearson's correlation coefficient analysis, in which coexpression was considered at P > 0.98. LncRNA function was annotated using GO and KEGG pathway analyses. LncRNA XR_427427.1 was associated with regulation of the apoptotic process, positive regulation of chemotaxis, and positive regulation of focal adhesion assembly (GO:0043066, GO:0050927, GO:0051894). ENST00000433734.1 was associated with neuron migration, axonal growth cone, dendritic spines, and stem cell differentiation (GO:0001764, GO:0044295, GO:0043197, GO:0048863). ENST00000453199.1 was associated with regulation of axonogenesis (GO:0050770). ENST00000559402.1 was associated with myelination and dendrites (GO:0042552, GO:0030425). XR_428585.1 was associated with the immune response, regulation of the acute inflammatory response, cytokine activity, and regulation of the apoptotic signaling pathway (GO:0006955, GO:0002675, GO:0005125, GO:2001235). XR_428553.1 was associated with the immune response, inflammatory response, regulation of immunoglobulin secretion, and cytokine activity (GO:0006955, GO:0006954, GO:0051024, GO:0005125). ENST00000447907.1 was associated with the innate immune response, positive regulation of the innate immune response, positive regulation of the cytokine biosynthetic process, and positive regulation of protein kinase activity (GO:0045087, GO:0045089, GO:0042108, GO:0045860). ENST00000524824.1 was associated with the inflammatory response (GO:0006954). ENST00000432148.1 was associated with leukocyte chemotaxis involved in the inflammatory response, regulation of the acute inflammatory response, positive regulation of cytokine production involved in the immune response, positive regulation of the acute inflammatory response to nonantigenic stimuli, and positive regulation of chemokine production (GO:0002232, GO:0002673, GO:0002720, GO:0002879, GO:0032722). Selecting the reliability prediction terms (according to P‐value and enrichment) produced a total of 30 enrichment GO terms (Figure 3A).
Figure 3.
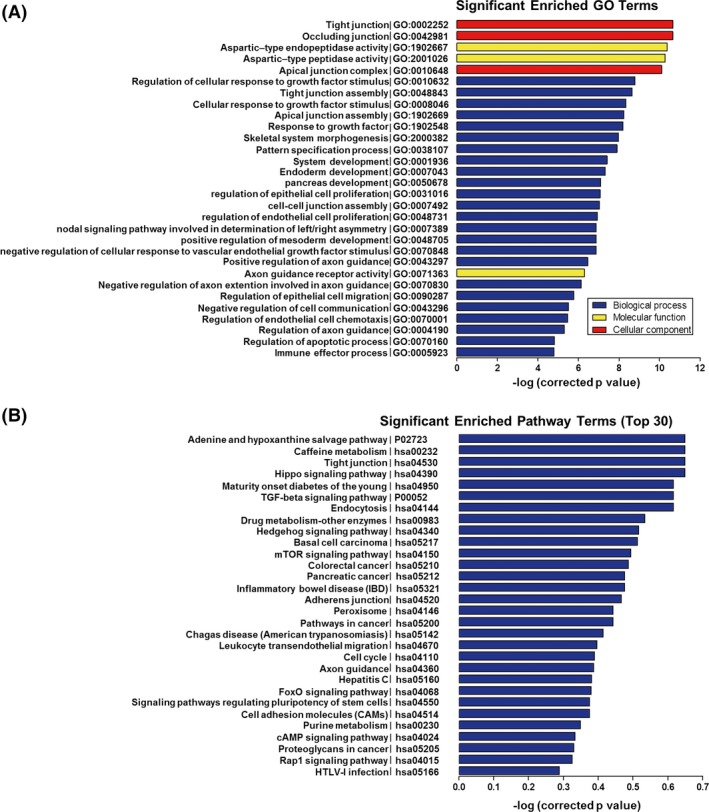
(A) Top 30 gene ontology (GO) terms for the difference in lncRNA coexpressed genes between patients with MS and healthy control subjects. (B) KEGG pathways analysis. Top 30 pathways for the difference in lncRNA coexpressed genes between the patients with MS and healthy control subjects.
LncRNA KEGG pathways are listed in Figure 3B, which included pathways for axon guidance, leukocyte transendothelial migration, mTOR signaling, tight junctions, and cell adhesion molecules (CAMs). All of these pathways are associated with immune and inflammatory responses.
The results of the GO and KEGG pathway analyses confirmed that lncRNAs might play important roles in the immune system, such as inflammation, cell differentiation, and proliferation.
“Cis” Analysis of the Expression of lncRNAs and Adjacent Coexpression Genes
Evidence suggests that several lncRNAs regulate their own transcription in cis‐regulatory fashion, as well as that of nearby genes, by recruiting remodeling factors to local chromatin 28. Prompted by this self‐regulation evidence, we identified chromosomal coexpression genes 10 kbp upstream and downstream of the differentially expressed lncRNAs to determine potential lncRNA “cis” genes. Comparing patients with MS to healthy controls, we found 10 “cis” genes (Table 3). Among these “cis” genes, PAX9 is associated with cellular processes occurring during neuronal differentiation, and DDIT4 is associated with oxidative stress.
Table 3.
“Cis” genes of aberrant lncRNAs
| lncRNA ID | mRNA Gene symbol | Cis regulation | Correlation | P‐value |
|---|---|---|---|---|
| TCONS_00027541 | SELV | Intergenic | 0.9672103 | 1.17E‐06 |
| ENST00000518861.1 | FLJ46284 | Intronic | 0.97479155 | 3.62E‐07 |
| ENST00000555107.1 | PAX9 | Antisense | 0.96835932 | 9.97E‐07 |
| ENST00000457076.1 | ASPRV1 | Intergenic | 0.98362009 | 5.27E‐08 |
| ENST00000418564.1 | ASPRV1 | Intergenic | 0.98044654 | 1.16E‐07 |
| ENST00000596259.1 | ASPRV1 | Intergenic | 0.9749509 | 3.52E‐07 |
| ENST00000415222.1 | ASPRV1 | Intergenic | 0.97408246 | 4.1E‐07 |
| uc.341‐ | HOXC9 | Intergenic | 0.95476031 | 4.89E‐06 |
| ENST00000411560.1 | STRC | Antisense | 0.95672734 | 4.01E‐06 |
| ENST00000491934.2 | DDIT4 | Antisense | 0.96485108 | 1.59E‐06 |
lncRNAs, long noncoding RNAs.
“Trans” Mechanism of Aberrant lncRNAs and Construction of TF–lncRNA–target Gene Network
Currently, known “trans” regulation mechanisms involve factors that mediate chromatin regulation and transcription 29. We calculated the lncRNA coexpression genes of chromatin regulators and transcription factors (TFs) in ENCODE to identify common genes involved in lncRNA regulation. Compared to healthy controls, in patients with MS, 33 “trans” genes were identified (Table 4). The “TF–lncRNA” two‐element network was generated using Cytoscape software. Figure 4A shows the “TF–lncRNA” core network map for patients with MS versus healthy controls (Pearson coefficient >0.997). The transcription factor NKx2‐5 modulated the expression of 9 lncRNAs, whereas the TF USF modulated the expression of 2 lncRNAs. This map provides a vivid picture of the relationship between lncRNAs and transcription factors and generates additional information for future studies. Based on the results of the lncRNA coexpression analyses, we added the target genes into the “TF–lncRNA” network to determine the “TF–lncRNA–target genes” three‐element network relationship. Figure 4B shows the core TF–lncRNA–target gene relationship map for patients with MS versus healthy controls, which contain 9 lncRNAs that have disrupted expression, 10 target genes, and 1 core TF NKx2‐5 in this core map. As observed for “NKx2‐5‐lncRNA XR_132575.3‐A_33‐P3368203” in this map, target genes, such as A_33‐P3368203, were coexpressed genes for lncRNA XR_132575.3. The transcription factor NKx2‐5 may regulate the expression of lncRNA XR_132575.3 and target genes, such as A_33‐P3368203. Thus, these maps provide valuable information concerning transcription factors, lncRNAs, and target genes.
Table 4.
“Trans” mechanism of the aberrant lncRNAs
| lncRNA ID | mRNA Gene symbol | Trans regulation | Correlation | P‐value |
|---|---|---|---|---|
| ENST00000601148.1 | NUFIP2 | miRNA sequestration | −0.9558084 | 4.4E‐06 |
| ENST00000567465.1 | LYRM7 | miRNA sequestration | −0.9535807 | 5.48E‐06 |
| TCONS_00025982 | LYRM7 | miRNA sequestration | −0.964201 | 1.73E‐06 |
| ENST00000443243.1 | EPHA10 | miRNA sequestration | 0.97663759 | 2.58E‐07 |
| ENST00000414562.1 | XDH | miRNA sequestration | 0.96781534 | 1.08E‐06 |
| TCONS_00026226 | MARVELD3 | miRNA sequestration | 0.96781534 | 1.08E‐06 |
| ENST00000432733.1 | MARVELD3 | miRNA sequestration | 0.96781534 | 1.08E‐06 |
| ENST00000467082.2 | CLDN11 | miRNA sequestration | 0.97835954 | 1.83E‐07 |
| ENST00000432733.1 | CLDN11 | miRNA sequestration | 0.95006245 | 7.57E‐06 |
| ENST00000609697.1 | CLDN11 | miRNA sequestration | 0.95533271 | 4.62E‐06 |
| ENST00000610021.1 | PARD6G | miRNA sequestration | 0.95328554 | 5.63E‐06 |
| ENST00000414562.1 | SCN2B | miRNA sequestration | 0.96415387 | 1.74E‐06 |
| ENST00000521021.1 | SCN2B | miRNA sequestration | 0.97187191 | 5.9E‐07 |
| ENST00000601293.1 | SCN2B | miRNA sequestration | 0.97470081 | 3.68E‐07 |
| ENST00000498967.2 | DCDC5 | miRNA sequestration | 0.97131936 | 6.44E‐07 |
| TCONS_00000838 | IAPP | miRNA sequestration | 0.95489628 | 4.82E‐06 |
| ENST00000500502.1 | IAPP | miRNA sequestration | 0.9505913 | 7.22E‐06 |
| ENST00000355837.4 | HHIP | miRNA sequestration | −0.9666635 | 1.26E‐06 |
| ENST00000589259.1 | HHIP | miRNA sequestration | 0.95894487 | 3.18E‐06 |
| ENST00000454681.2 | HHIP | miRNA sequestration | 0.95511078 | 4.72E‐06 |
| ENST00000558208.1 | HHIP | miRNA sequestration | 0.96053375 | 2.67E‐06 |
| TCONS_00009539 | SEMA5A | miRNA sequestration | 0.95524331 | 4.66E‐06 |
| ENST00000447907.1 | SEMA5A | miRNA sequestration | 0.95833933 | 3.39E‐06 |
| ENST00000593140.1 | GPR26 | miRNA sequestration | 0.96568018 | 1.43E‐06 |
| ENST00000568708.1 | NRIP3 | miRNA sequestration | 0.96221068 | 2.2E‐06 |
| ENST00000454681.2 | SMAD2 | miRNA sequestration | −0.9778117 | 2.05E‐07 |
| TCONS_00026226 | LRRC2 | miRNA sequestration | 0.97118335 | 6.57E‐07 |
| ENST00000593588.1 | DNAJC22 | miRNA sequestration | 0.95631307 | 4.18E‐06 |
| ENST00000413842.1 | DNAJC22 | miRNA sequestration | 0.96370422 | 1.84E‐06 |
| ENST00000549013.1 | SLC14A2 | miRNA sequestration | 0.95246178 | 6.09E‐06 |
| ENST00000423568.1 | SLC14A2 | miRNA sequestration | 0.96232625 | 2.17E‐06 |
| ENST00000602319.1 | YIPF4 | miRNA sequestration | 0.96315692 | 1.96E‐06 |
| ENST00000589259.1 | SLC30A7 | miRNA sequestration | −0.9550162 | 4.76E‐06 |
LncRNAs, long noncoding RNA.
Figure 4.
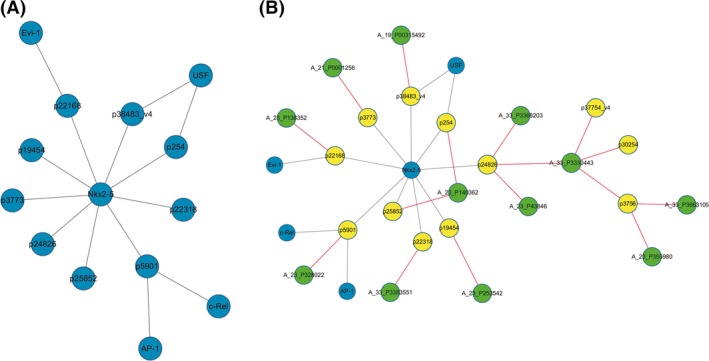
(A) “TF–lncRNA” network comparison between the patients with MS and healthy control subjects. (B) “TF–lncRNA–target gene” core network of disrupted lncRNA expression in patients with MS versus healthy control subjects.
Conclusion
LncRNAs play important roles in regulating gene expression. Abnormal expression of lncRNAs is often involved in the pathogenesis and progression of many diseases. Recent studies demonstrate that lncRNAs are closely related to the immune system 7, 30, 31. However, the expression pattern, potential targets, and functions of lncRNAs in terms of the development and pathogenesis of the MS are still unknown. Therefore, in this study, we systematically screened the genomewide expression pattern of lncRNAs as well as mRNAs in PBMCs obtained from patients with MS and healthy controls. We identified 2353 upregulated and 389 downregulated lncRNAs, and 1037 upregulated and 279 downregulated mRNAs. Among them, 137 lncRNAs had a >6‐fold difference, and 37 lncRNAs had a >8‐fold difference in expression level between the MS and control groups, suggesting that these lncRNAs may play important roles in the process of MS. Several candidate lncRNAs that we identified were chosen for real‐time PCR validation. Real‐time PCR revealed the same direction of regulation and significant differences in lncRNAs expression in MS and healthy controls. Therefore, our results from the real‐time PCR analysis confirmed the microarray data to some extent.
MS is a chronic inflammatory disease of the CNS mediated by CD4+ T cells and is characterized by demyelinating lesions and progressive axon loss 1. The pathogenesis of MS is traditionally thought to be complex and not well understood, and genetic and environmental risk factors are reported to be involved 2, 3, 4. Genetic susceptibility and environmental factors prime the immune response in MS that targets the CNS. Plasma cells arise in the periphery but can accumulate in the brain, where they can locally release antibodies that target both myelin sheaths and glial cells. Next, released inflammatory mediators open the blood–brain barrier and attract monocytes and additional lymphocytes, causing an influx of these cells and leading to the formation of phagocytic lesions 32, 33. Chronic oxidative injury, microglia activation, and accumulating mitochondrial damage in axons are critical factors driving neurodegeneration 34.
Here, we drew conclusions about lncRNA functions based on coexpressed gene relationships identified in GO and pathway analyses. In a comparison between patients with MS and healthy control subjects, the most enriched GO terms in the predicted target genes of the lncRNAs were tight junction, regulation of axon guidance, axon guidance receptor activity, and regulation of endothelial cell chemotaxis. Many of them were associated with the immune pathology of MS. Therefore, these results provide significant new information for further studies on MS.
Using a bioinformatics approach, we predicted that the differentially expressed lncRNAs were likely able to execute their functions by regulating gene expression in both a cis and trans fashion 29. Cis regulation is identified as occurring when the transcription of an lncRNA affects the expression of its neighbor genes. By screening the coexpressed genes located near the differentially expressed lncRNAs, we discovered some possible cis‐regulatory target genes. For instance, HOXC9 might be the target of lncRNA uc.341‐, which is upregulated by 2.9‐fold in PBMCs of patients with MS. Another example is DDIT4, which is upstream of lncRNA ENST00000491934.2 (2.87‐fold upregulation) and is a likely candidate for the cis‐regulated target of ENST00000491934.2. HOXC9 has been reported to regulate distinct sets of genes to coordinate diverse cellular processes during neuronal differentiation 35, 36. In addition, DDIT4, an oxidative gene, localizes to mitochondria, where it plays an important role in reducing reactive oxidative species (ROS) production and release by mitochondria 37. For example, excessive ROS production by mitochondria is related to lower sperm quality 38. These data suggest that expression of DDIT4 is important for cell survival during germ cell development, because only cells showing high expression of these genes seem to be sufficiently protected against oxidative stress and to be capable of reaching the ejaculate. Cells that do not sufficiently express these genes probably die before reaching a mature state, and as a result, sperm count is significantly decreased. Therefore, we hypothesize that lncRNAs ENST00000491934.2 and HSP90AA4P might be involved in the pathophysiology of MS by regulating HOXC9 and DDIT4 gene expression, respectively, in a cis fashion.
LncRNAs can also act on their target genes through long‐range trans regulation in conjunction with other TFs. For example, the lncRNA XR_132575.3 can affect the transportation of NKx2‐5, thereby influencing the expression of NKx2‐5 target genes. In our study, to identify which TFs exert coregulatory effects on differentially expressed lncRNAs, we overlapped the coding genes coexpressed with lncRNAs and the genes targeted by TFs.
In summary, this study described the expression profile of lncRNAs in patients with MS using a RNA microarray method. Bioinformatics approaches were used to predict the target genes and potential functions of the differentially expressed lncRNAs. These findings suggest that the differentially expressed lncRNAs may be important in the process of MS. However, the specific molecular mechanisms and biological functions of these lncRNAs in the pathogenesis of MS need further study.
Funding Sources
The authors disclose receipt of the following financial support for the research, authorship, and/or publication of this article. This work was financially supported by the National Natural Science Foundation of China (81571600, 81322018, 81273287, and 81100887 to J. W. H.); the Youth Top‐notch Talent Support Program; and the National Key Clinical Specialty Construction Project of China.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgment
We thank our patients for participating in this study. We also thank R. R. Han and C. J. Wei for facilitating recruitment of the patients, and S. W. Zhang for technical support.
References
- 1. McFarland HF, Martin R. Multiple sclerosis: A complicated picture of autoimmunity. Nat Immunol 2007;8:913–919. [DOI] [PubMed] [Google Scholar]
- 2. Sawcer S, Franklin RJ, Ban M. Multiple sclerosis genetics. Lancet Neurol 2014;13:700–709. [DOI] [PubMed] [Google Scholar]
- 3. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann Neurol 2007;61:288–299. [DOI] [PubMed] [Google Scholar]
- 4. Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: noninfectious factors. Ann Neurol 2007;61:504–513. [DOI] [PubMed] [Google Scholar]
- 5. Calabrese R, Valentini E, Ciccarone F, et al. TET2 gene expression and 5‐hydroxymethylcytosine level in multiple sclerosis peripheral blood cells. Biochim Biophys Acta 2014;1842:1130–1136. [DOI] [PubMed] [Google Scholar]
- 6. Tegla CA, Azimzadeh P, Andrian‐Albescu M, et al. SIRT1 is decreased during relapses in patients with multiple sclerosis. Exp Mol Pathol 2014;96:139–148. [DOI] [PubMed] [Google Scholar]
- 7. Zhang Y, Cao X. Long noncoding RNAs in innate immunity. Cell Mol Immunol 2016;13:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song X, Cao G, Jing L, et al. Analysing the relationship between lncRNA and protein‐coding gene and the role of lncRNA as ceRNA in pulmonary fibrosis. J Cell Mol Med 2014;18:991–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Necsulea A, Soumillon M, Warnefors M, et al. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 2014;505:635–640. [DOI] [PubMed] [Google Scholar]
- 10. Wan G, Hu X, Liu Y, et al. A novel non‐coding RNA lncRNA‐JADE connects DNA damage signalling to histone H4 acetylation. EMBO J 2013;32:2833–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carrieri C, Forrest AR, Santoro C, et al. Expression analysis of the long non‐coding RNA antisense to Uchl1 (AS Uchl1) during dopaminergic cells' differentiation in vitro and in neurochemical models of Parkinson's disease. Front Cell Neurosci 2015;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu JY, Yao J, Li XM, et al. Pathogenic role of lncRNA‐MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis 2014;5:e1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi Q, Yang X. Circulating MicroRNA and long noncoding RNA as biomarkers of cardiovascular diseases. J Cell Physiol 2015;231:751–755. [DOI] [PubMed] [Google Scholar]
- 14. Chang YN, Zhang K, Hu ZM, et al. Hypoxia‐regulated lncRNAs in cancer. Gene 2016;575:1–8. [DOI] [PubMed] [Google Scholar]
- 15. Ranzani V, Rossetti G. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc‐MAF‐4. Nat Immunol 2015;16:318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang P, Xue Y, Han Y, et al. The STAT3‐binding long noncoding RNA lnc‐DC controls human dendritic cell differentiation. Science (New York, NY) 2014;344:310–313. [DOI] [PubMed] [Google Scholar]
- 17. Spurlock CF 3rd, Tossberg JT, Guo Y. Expression and functions of long noncoding RNAs during human T helper cell differentiation. Nat Commun 2015;6:6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roux BT, Lindsay MA. LincRNA signatures in human lymphocytes. Nat Immunol 2015;16:220–222. [DOI] [PubMed] [Google Scholar]
- 19. Sauvageau M, Goff LA, Lodato S, et al. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2013;2:e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol 2011;21:354–361. [DOI] [PubMed] [Google Scholar]
- 21. Li Z, Chao TC, Chang KY, et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci USA 2014;111:1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carpenter S, Aiello D, Atianand MK, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science (New York, NY) 2013;341:789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu G, Tang Q, Sharma S, et al. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat Immunol 2013;14:1190–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Orom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer‐like function in human cells. Cell 2010;143:46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Z, Bai J, Wu A, et al. Co‐LncRNA: Investigating the lncRNA combinatorial effects in GO annotations and KEGG pathways based on human RNA‐Seq data. Database 2015. doi: 10.1093/database/bav082. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao F, Qu Y, Liu J, et al. Microarray profiling and co‐expression network analysis of LncRNAs and mRNAs in neonatal rats following hypoxic‐ischemic brain damage. Sci Rep 2015;5:13850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gendrel AV, Heard E. Noncoding RNAs and epigenetic mechanisms during X‐chromosome inactivation. Annu Rev Cell Dev Biol 2014;30:561–580. [DOI] [PubMed] [Google Scholar]
- 28. Guenzl PM, Barlow DP. Macro lncRNAs: A new layer of cis‐regulatory information in the mammalian genome. RNA Biol 2012;9:731–741. [DOI] [PubMed] [Google Scholar]
- 29. Bassett AR, Akhtar A, Barlow DP, et al. Considerations when investigating lncRNA function in vivo. Elife 2014;3:e03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu GC, Pan HF, Leng RX, et al. Emerging role of long noncoding RNAs in autoimmune diseases. Autoimmun Rev 2015;14:798–805. [DOI] [PubMed] [Google Scholar]
- 31. Heward JA, Lindsay MA. Long non‐coding RNAs in the regulation of the immune response. Trends Immunol 2014;35:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Henderson AP, Barnett MH, Parratt JD, Prineas JW. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann Neurol 2009;66:739–753. [DOI] [PubMed] [Google Scholar]
- 33. Hemmer B, Kerschensteiner M, Korn T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol 2015;14:406–419. [DOI] [PubMed] [Google Scholar]
- 34. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol 2015;14:183–193. [DOI] [PubMed] [Google Scholar]
- 35. Mao L, Ding J, Zha Y, et al. HOXC9 links cell‐cycle exit and neuronal differentiation and is a prognostic marker in neuroblastoma. Cancer Res 2011;71:4314–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stoll SJ, Kroll J. HOXC9: A key regulator of endothelial cell quiescence and vascular morphogenesis. Trends Cardiovasc Med 2012;22:7–11. [DOI] [PubMed] [Google Scholar]
- 37. Horak P, Crawford AR, Vadysirisack DD, et al. Negative feedback control of HIF‐1 through REDD1‐regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA 2010;107:4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koppers AJ, De Iuliis GN, Finnie JM, McLaughlin EA, Aitken RJ. Significance of mitochondrial reactive oxygen species in the generation of oxidative stress in spermatozoa. J Clin Endocrinol Metab 2008;93:3199–3207. [DOI] [PubMed] [Google Scholar]