

ABSTRACT
Whole‐exome sequencing (WES) is increasingly applied to research and clinical diagnosis of human diseases. It typically results in large amounts of genetic variations. Depending on the mode of inheritance, only one or two correspond to pathogenic mutations responsible for the disease and present in affected individuals. Therefore, it is crucial to filter out nonpathogenic variants and limit downstream analysis to a handful of candidate mutations. We have developed a new computational combinatorial system UMD‐Predictor (http://umd‐predictor.eu) to efficiently annotate cDNA substitutions of all human transcripts for their potential pathogenicity. It combines biochemical properties, impact on splicing signals, localization in protein domains, variation frequency in the global population, and conservation through the BLOSUM62 global substitution matrix and a protein‐specific conservation among 100 species. We compared its accuracy with the seven most used and reliable prediction tools, using the largest reference variation datasets including more than 140,000 annotated variations. This system consistently demonstrated a better accuracy, specificity, Matthews correlation coefficient, diagnostic odds ratio, speed, and provided the shortest list of candidate mutations for WES. Webservices allow its implementation in any bioinformatics pipeline for next‐generation sequencing analysis. It could benefit to a wide range of users and applications varying from gene discovery to clinical diagnosis.
Keywords: pathogenicity prediction, mutation, bioinformatics, NGS, substitution, synonymous, nonsynonymous, nonsense
Introduction
The human genome is composed of approximately 3.2 billion nucleotides and contains at least 25,000 genes. Despite very efficient systems to ensure the accurate replication of this genome, it has been estimated that the germ‐line base substitution rate ranges from 1.1 to 3 × 10−8 per base per generation [Roach et al., 2010]; therefore, each individual carries at least 10 de novo variations. In addition, early whole genome sequencing (WGS) experiments have revealed that every human carries about 3 million single‐nucleotide polymorphisms (SNP) [Levy et al., 2007; Wang et al., 2008; Wheeler et al., 2008; Pushkarev et al., 2009], these data being consistently confirmed. The analysis of the dbSNP build 142 database reveals that 15% of identified SNPs are localized in genes, from which about 86% map to introns and 2.75% to exons. Considering these exonic variations, more than 50% correspond to missense mutations and about 37% to synonymous changes. Nevertheless, these data do not provide information about the potential pathogenicity of these variations and may thus be biased for nonpathogenic events. However, the Human Gene Mutation Database [Stenson et al., 2003] indicates that the vast majority of human pathogenic variations correspond to missense mutations (50%) corroborating that these mutations represent a key element in human genetics as their interpretation is challenging as next‐generation sequencing (NGS) technologies are widely used both for research and clinical diagnosis. Thus, performing a whole‐exome sequencing (WES) or a WGS will result in about 23,000 exonic SNPs with about 11,500 missense variations and 8,500 synonymous changes from which only one to two mutations, based on the mode of inheritance, are responsible for the Mendelian genetic disease.
The identification of human disease‐causing mutations is critical in human medicine as more than 7,000 rare human genetic diseases have been characterized [Amberger et al., 2015]. Only 55% of disease‐causing genes have been identified and international initiatives are organized to speed up genes identification and drug development such as the International Rare Disease Research Consortium (IRDIRC ‐ http://www.irdirc.org). In parallel, the identification of disease‐causing mutations in known genes is still a challenge in clinical practice as no in vitro functional assay is usually available and most families harbor private mutations. This challenge is exemplified in the NGS context and the genetically heterogeneous diseases where candidate disease‐causing mutations are present in various genes. Currently, bioinformatics pipelines are required to handle the “data deluge” [Schatz and Langmead, 2013] and facilitate the identification of pathogenic events. These cascade of tools combine multiple layers allowing run quality analysis, alignment of filtered sequences against the human reference genome GRCh37 (hg19), variant calling, annotation, and filtration [Pabinger et al., 2014]. A key step of the annotation process is the integration of pathogenicity predictions from various tools, which aim to efficiently predict substitutions effects on protein structure and function.
Multiple tools have been designed using three main approaches: sequence and evolutionary conservation‐based methods, protein sequence and structure‐based methods, and supervised‐learning methods. The first approach relies on the observation that disease‐causing missense mutations mainly occur at evolutionarily conserved positions that have an essential role in the structure and/or function of the encoded protein. The most widely used systems of this type are SIFT and Mutations assessor [Reva et al., 2011; Sim et al., 2012]. The second approach takes into account the degree of similarity between amino acids as well as physical disruption of protein domains. Archetypes for this approach are the Polyphen‐2, and SNPeffect systems [Adzhubei et al., 2010; De Baets et al., 2012]. The third approach includes various semisupervised learning methods trained with positive (disease‐causing mutations) and negative (nonpathogenic mutations) datasets from which rules have been established based on various features. The most widely used systems of this type are MutationTaster2, SNAP, and CADD [Bromberg and Rost, 2007; Kircher et al., 2014; Schwarz et al., 2014]. Nevertheless, none of these approaches could be considered as a gold standard as they have limitations: a high sensitivity to multiple sequence alignments that could drastically alter predictions for the first type; availability of 3D structures that are not accessible for many proteins or only partially available for parts of a given‐protein for the second type, and the requirement of very large experimentally validated datasets to train systems of the third type. To solve these issues, PON‐P and CONDEL have been developed as metapredictors as they combine predictions from other tools to build consensus scores [González‐Pérez and López‐Bigas, 2011; Olatubosun et al., 2012]. If they give very good results for a subset of variations, they have limitations, especially for situations where individual systems give conflicting results. Unfortunately, it corresponds to variations difficult to evaluate and for which predictions are very helpful. Finally, a combinatorial approach has been proposed. It is based on the assumption that single‐nucleotide variations could not only alter the protein sequence but also impact mRNA [Frédéric et al., 2009]. In fact, coding‐sequence mutations could affect splicing signals such as donor and acceptor splice sites or auxiliary sequences such as exonic splicing enhancers and exonic splicing silencers. Thus, mutations affecting the ultimate base of exons are now well recognized to affect the donor splice site while often resulting in a synonymous change [Desmet et al., 2009]. The purpose of this work was to develop this combinatorial approach through the addition of new elements such as the conservation over 100 species and the variation frequency in the general population. In addition, as WES and WGS experiments result in annotated variations at the nucleotide level, this system was designed to handle large datasets, to predict the pathogenicity of missense and synonymous changes and make it available through Webservices allowing integration in any bioinformatics pipeline. To evaluate its efficiency, we compared our system with the seven most used and reliable prediction tools: SIFT 5.1.1 [Sim et al., 2012], Polyphen 2.2.2 [Adzhubei et al., 2010], Provean 1.1.3 [Choi et al., 2012], Mutation Assessor 2 [Reva et al., 2011], CONDEL 1.5 [González‐Pérez and López‐Bigas, 2011], MutationTaster 2 [Schwarz et al., 2014], and CADD [Kircher et al., 2014]. To avoid any bias linked to a specific dataset or mutation type, we selected four widely used different datasets containing pathogenic and nonpathogenic variations: Varibench [Sasidharan Nair and Vihinen, 2013] combined with dbSNP [Sherry et al., 2001] (19,335 pathogenic, 7,897 nonpathogenic), Uniprot [UniProt Consortium, 2014] (20,821 pathogenic, 36,825 nonpathogenic), Clinvar [Landrum et al., 2014] (10,669 pathogenic, 1,817 nonpathogenic), and PredictSNP [Bendl et al., 2014] (24,082 pathogenic, 19,800 nonpathogenic).
Materials and Methods
UMD‐Predictor Ecosystem
We designed the UMD‐Predictor ecosystem as a stand‐alone platform that contains all predictions for all substitutions from any human transcript. It was conceptualized as a three‐tier architecture. The Presentation Tier was developed in PHP, javascript, and html. This interface integrates the d3.js (http://d3js.org/) and jquery (http://jquery.com/) libraries to ensure easy access to the system (http://umd‐predictor.eu). The Application Tier integrates the prediction algorithm itself and the data‐tier integrates the Ensembl v71 human genome reference sequence and transcripts. All theoretical substitutions of protein‐coding transcripts (280,315,899 mutations) have then been modeled, computed, and stored in a PostgreSQL (http://www.postgresql.org/) database allowing rapid analysis of large datasets.
To facilitate integration into bioinformatics pipelines, we developed a Webservice to access UMD‐Predictor database. Various parameters can be used to search for a single or multiple mutations, all mutations from a specific gene, transcript, or chromosome and a full VCF file. Additionally, the Website provides educational materials (tutorials, help sections, and screencasts) to facilitate user experience.
Combinatorial Pathogenicity Prediction Algorithm
The new combinatorial algorithm was derived from the previously described one [Frédéric et al., 2009]. Three major modifications have been made, notably the replacement of the amino acids conservation previously extracted from the SIFT system by a new conservation score; the automatic extraction of key residues information from Swissprot to replace manual annotation, and the addition of variation frequency in the general population.
The new UMD‐Predictor algorithm combines the following features: Blosum62 conservation matrix, the Yu's biochemical substitution matrix, protein key residues, impact on splicing signals (splice sites and auxiliary sequences), the variation frequency at the population level, and the conservation score in 100 species with Grantham's substitution matrix. To normalize the predictions on a scale from 0 to 100, the formula also includes a ‘‘C’’ constant value. The pathogenicity of a given variation is thus given by the formula:
X(i, j) refers to a matrix table with ‘‘i’’ corresponding to the feature and ‘‘j’’ to the UMD‐Predictor value associated with the original element's value. For example, BLOSUM62 original values range from ‘‘−4’’ to ‘‘11.’’ The X(i, j) for original value ‘‘−4’’ is ‘‘−15,’’ whereas the X(i, j) value for ‘‘11’’ is ‘‘+15.” Value range for each element has been determined based on expert knowledge to avoid any bias induced by a trial and error approach using limited sets of data. Thus, the conservation among species was given the strongest impact (−40 to +60) on prediction followed by the annotation of protein key residues that reflect either key structural or functional elements (+30) and the potential activation of cryptic splice sites (+30). Other features have been given a lower value as they either correspond to more general information such as the Yu's biochemical substitution matrix (−20 to +20), the BLOSUM62 matrix (−15 to +15), or either to more difficult to interpret data such as the effect on auxiliary splicing sequences (0 to +10). The variation frequency in the general population, which could be considered as a conservation score among humans, was used to penalize frequent events most probably associated to polymorphisms (−50 to 0). Finally, when a wild‐type splice site is abolished, the value is set to +90 to significantly impact the prediction. As for the original UMD‐Predictor algorithm [Frédéric et al., 2009], score ranges from 0 to 100 and is divided into four classes: (i) <50 polymorphism; (ii) 50–64 probable polymorphism; (iii) 65–74 probably pathogenic mutation; and (iv) >74 pathogenic mutation.
Conservation Score from 100 Species
The conservation is a critical element to assess pathogenicity of mutations. We therefore built a new feature able to quantify the impact of a given amino acid substitution based on conservation. We used the Vertebrate Multiz Alignment & Conservation (phastCons100way) data from UCSC [Rosenbloom et al., 2015]. We clustered the species into five groups: primates; Euarchontoglires; Laurasiatheria, Afrotheria, and mammals; birds and Sarcopterygii; and Fish. The conservation of a specific residue was assessed using a window of ±3 residues. All species with more than one substitution were excluded for the score computation. For each position, we counted the number of alternative residues present in each group and a conservation score was then computed depending on the residue position and the group. It was subsequently normalized for the number of selected species in each group. For each mutation, we compared the Grantham score of the mutation itself and the highest Grantham score of natural variants of each group. If this variation is positive, the conservation score increases, otherwise it is decreased. The conservation score is thus translated into a new matrix ranging from −40 to +60.
For example, if we consider a Glutamic acid residue (Glu) at position 1,003 of the FBN1 gene (ENSEMBL gene transcript ENST00000316623), the conservation displayed a natural variation to an Alanine residue (Ala ‐ Grantham score of 107) in the Armadillo species (Supp. Fig. S1). A Glu>Lys mutation will result in a conservation score of 66.8 as the Grantham score of the mutant residue is lower than the Grantham score of the natural variant. A Glu>Val mutation will result in a conservation score of 73.3 as the Grantham score of the mutant residue is higher than the Grantham score of the natural variant.
Automatic Extraction of Key Residues Information from Swissprot
In the original combinatorial algorithm, each database curator manually annotated a key residue parameter accounting for a 3D structure key element (Proline‐induced hinges) or a functional key element (glycosylation site or disulfide bound). With the development of a global system, an automatic annotation system was required. We therefore used Uniprot/SwissProt data for the following terms: "CARBOHYD," "DISULFID," "ZN_FNG," "MOD_RES," "TRANSMEM," "CA_BIND," "DNA_BIND," "NP_BIND," "MOTIF," "ACT_SITE," "METAL," "BINDING," "SITE," "SE_CYS," "LIPID," and "CROSSLNK."
Variation Frequency in the General Population
As Mendelian‐inherited human genetic diseases are considered rare, a high frequency of a specific variation in the human population is indicative of a nonpathogenic mutation also named polymorphism. Thus, we collected frequency information from the Ensembl database [Cunningham et al., 2015], which include dbSNP data (build 139) [NCBI Resource Coordinators, 2015]. If a variation frequency is available, a penalty score ranging from −50 to 0 was used for mutations with frequencies above 0.001 with −50 for frequency above 0.05.
Prediction Assessment
Each predictor system was run using subsets of known variants as input and predictions were assessed in terms of true positive (TP), true negative (TN), false positive (FP), and false negative (FN). The sensitivity (TPR), specificity (TNR), Matthews correlation coefficient (MCC) [Matthews, 1975], and diagnostics odds ratio (DOR) [Glas et al., 2003] were calculated.
The MCC scores range from +1 (a perfect prediction) to −1 (an inverse prediction) where 0 represents an average random prediction. MCC interpretation can be done using the following classes: MCC = +0.70 or higher, very strong positive relationship; MCC = +0.40 to +0.69, strong positive relationship; MCC = +0.30 to +0.39, moderate positive relationship; MCC = +0.20 to +0.29, weak positive relationship; MCC = +0.01 to +0.19, no or negligible relationship; MCC = −0.01 to −0.19, no or negligible relationship; MCC = −0.20 to −0.29, weak negative relationship; MCC = −0.30 to −0.39, moderate negative relationship; MCC = −0.40 to −0.69, strong negative relationship; and MCC = −0.70 or lower, very strong negative relationship. This measurement has been favored over “accuracy,” as it is less sensitive to the different numbers of pathogenic and nonpathogenic variant classes in each gene.
The DOR ranges from zero to infinity. A higher DOR value indicates a better test performance. The log(DOR) is used to evaluate the trade‐off between sensitivity and specificity and ranges from −10 to 10. A color‐coded graph is usually used to display the efficiency of various tests.
Evaluation Datasets and Systems
To allow unbiased comparison of tools, we selected large available datasets that contain annotated pathogenic and nonpathogenic mutations. These datasets are freely available and have already been used for tools evaluation. Note that only pathogenic mutations have been selected from the Varibench dataset and combined to the dbSNP dataset in order to reach a significant number of both mutation types. We therefore used the four following datasets: Varibench [Sasidharan Nair and Vihinen, 2013] with dbSNP [Sherry et al., 2001] (19,335 pathogenic, 7,897 nonpathogenic); Uniprot [UniProt Consortium, 2014] (20,821 pathogenic, 36,825 nonpathogenic); Clinvar [Landrum et al., 2014] (10,669 pathogenic, 1,817 nonpathogenic); and PredictSNP [Bendl et al., 2014] (24,082 pathogenic, 19,800 nonpathogenic). Due to discrepancies in datasets genomic variations description (genomic or protein coordinates), not all predictors were able to perform analysis for all variants; therefore, accuracy was evaluated both on the overall dataset and on a common dataset corresponding to mutations for which all tools were able to perform predictions.
The seven most used and reliable prediction tools were used for comparison: SIFT 5.1.1 [Sim et al., 2012], Polyphen 2.2.2 [Adzhubei et al., 2010], Provean 1.1.3 [Choi et al., 2012], Mutation Assessor 2 [Reva et al., 2011], CONDEL 1.5 [González‐Pérez and López‐Bigas, 2011], MutationTaster 2 [Schwarz et al., 2014], and CADD [Kircher et al., 2014]. For each system, the most recent version was used with default parameters.
Other Comparison Assessment Parameters
The efficiency of prediction systems is mainly based on the prediction themselves but other parameters were also taken into account. They include: the time required for NGS datasets annotation; the ability to identify true pathogenic mutations; and the number of predicted pathogenic mutations. These parameters were evaluated using three clinical WES datasets from unrelated patients provided by the molecular genetics laboratory of the “La Timone Children Hospital” (Marseille, France). The molecular diagnostic of recessive diseases was confirmed by Sanger sequencing and family study for each proband (data not shown). The WES datasets contained 58,145, 54,006, and 57,936 variants. The assessment was performed using the on‐line version of each system and the Webservices of the UMD‐Predictor system.
Results
UMD‐Predictor Ecosystem
The UMD‐Predictor was designed as a stand‐alone platform that contains all predictions for single‐nucleotide substitutions (280,315,899) of human transcripts from the human reference genome GRCh37. It is freely available for noncommercial use at http://umd‐predictor.eu. It can be queried either directly on‐line through a user‐friendly Web interface or through Webservices to facilitate its integration in any bioinformatics pipeline. The Web interface allows: (i) single analysis of all mutations from a gene selected by gene symbol, Ensembl gene ID, Ensembl transcript ID, RefSeq peptide ID, or Uniprot ID; (ii) multiple analysis of mutations from different loci or chromosomal positions; or (iii) a VCF file analysis. A predictions table containing advanced filtration and sorting options allow rapid access to single predictions. It contains prediction values and color‐coded classes (from green for polymorphisms to red for pathogenic mutations). A distribution graph is displayed for each transcript for single analysis. Additionally, all results can be exported into xml, csv, tsv, or json formats.
Pathogenicity Predictions Evaluation
To assess the pathogenicity predictions of the UMD‐Predictor system, we collected data from 141,246 annotated variations, either pathogenic or nonpathogenic, using four reference datasets: Varibench [Sasidharan Nair and Vihinen, 2013] with dbSNP [Sherry et al., 2001] (19,335 pathogenic, 7,897 nonpathogenic); Uniprot [UniProt Consortium, 2014] (20,821 pathogenic, 36,825 nonpathogenic); Clinvar [Landrum et al., 2014] (10,669 pathogenic, 1,817 nonpathogenic), and PredictSNP [Bendl et al., 2014] (24,082 pathogenic, 19,800 nonpathogenic). These datasets were then used for prediction assessment using the seven most used and reliable prediction tools: SIFT 5.1.1 [Sim et al., 2012], Polyphen 2.2.2 [Adzhubei et al., 2010], Provean 1.1.3 [Choi et al., 2012], Mutation Assessor 2 [Reva et al., 2011], CONDEL 1.5 [González‐Pérez and López‐Bigas, 2011], MutationTaster 2 [Schwarz et al., 2014], and CADD [Kircher et al., 2014]. For each tool, default parameters were used. While the UMD‐Predictor results are divided into four classes, for this evaluation, data were combined in two classes only: “polymorphism” and “probable polymorphism” were combined in a “nonpathogenic” class, whereas “probably pathogenic” mutations and “pathogenic” mutations were combined in a “pathogenic” class. For tools comparisons, we used statistical measures of the performance of a binary classification test: sensitivity (true‐positive rate), specificity (true‐negative rate), positive predictive value (PPV), negative predictive value (NPV), accuracy, MCC, DOR, and its logarithm, log(DOR), to study the trade‐off between sensitivity and specificity.
Data from the Varibench/dbSNP dataset are presented in Table 1, whereas data from other datasets are available in the Supporting Information (Supp. Figs. S2–S15; Supp. Tables S1–S14). As shown for this dataset, UMD‐Predictor provided with more accurate results for PPV, NPV, sensitivity, and MCC, whereas MutationTaster 2 was more sensitive. The DOR and log(DOR) data also demonstrated a better efficiency for UMD‐Predictor with a DOR of 86.6 versus 12.6 for the second most efficient predictor system. The log(DOR) measures consistently demonstrated an almost 2‐logs improvement compared with other predictors as exemplified for this dataset (4.46 vs. 2.53). As shown in Figure 1, this high difference is driven by a strong improvement in specificity with a very good sensitivity. It is important to note that MutationTaster2 automatically annotates variations as disease causing when marked as pathogenic in ClinVar. Even in the situation of the ClinVar dataset, MutationTaster2 did not perform better than UMD‐Predictor (Supp. Figs. S8–S11; Supp. Tables S7–S10).
Table 1.
Comparison Between UMD‐Predictor and Other Predictors Using the Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] Dataset (n = 17,329)
| SIFT | PPH2 | Provean | Mutation assessor | CONDEL | MutationTaster | CADD | UMD‐Predictor | |
|---|---|---|---|---|---|---|---|---|
| TP | 9,596 | 10,290 | 9,638 | 9,775 | 8,797 | 11,174 | 10,182 | 10,727 |
| TN | 2,805 | 3,045 | 3,088 | 3,162 | 3,287 | 2,937 | 3,214 | 4,024 |
| FP | 1,229 | 1,189 | 1,147 | 1,073 | 948 | 1,298 | 1,021 | 211 |
| FN | 3,498 | 2,803 | 3,456 | 3,319 | 4,297 | 1,920 | 2,912 | 2,367 |
| PPV | 0.89 | 0.90 | 0.89 | 0.90 | 0.90 | 0.90 | 0.91 | 0.98 |
| NPV | 0.45 | 0.52 | 0.47 | 0.49 | 0.43 | 0.60 | 0.52 | 0.63 |
| Sensitivity | 0.73 | 0.79 | 0.74 | 0.75 | 0.67 | 0.85 | 0.78 | 0.82 |
| Specificity | 0.70 | 0.72 | 0.73 | 0.75 | 0.78 | 0.69 | 0.76 | 0.95 |
| Accuracy | 0.72 | 0.77 | 0.73 | 0.75 | 0.70 | 0.81 | 0.77 | 0.85 |
| MCC | 0.38 | 0.46 | 0.41 | 0.44 | 0.39 | 0.52 | 0.48 | 0.69 |
| DOR | 6.3 | 9.7 | 7.7 | 9.0 | 7.2 | 12.6 | 11.2 | 86.6 |
| log(DOR) | 1.84 | 2.27 | 2.04 | 2.20 | 1.97 | 2.53 | 2.42 | 4.46 |
TP, true positives; TN, true negatives; FP, false positives; FN, false negatives; PPV, positive predictive value; NPV, negative predictive value; MCC, Matthews correlation coefficient; DOR, diagnostic odds ratio.
Figure 1.
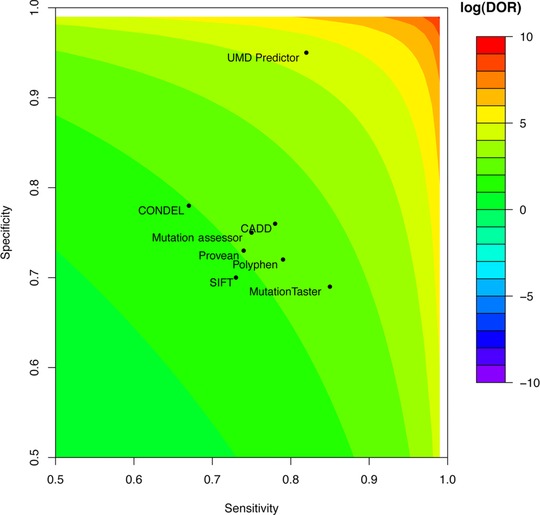
log(DOR) comparison between predictors using the Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] dataset (n = 17,329). X‐axis, sensitivity; Y‐axis, specificity; color‐coded scale, log(DOR).
In parallel with global statistical parameters, we evaluated the sensitivity of the various methods to distinguish pathogenic and nonpathogenic mutations using the receiver operating characteristic (ROC) curve that represents the sensitivity as a function of false‐positive rate. As shown in Figure 2 and Table 2, the UMD‐Predictor ROC‐AUC (area under curve) shows a significant improvement compared with other systems (0.954 vs. 0.834 for the second most efficient tool).
Figure 2.

Sensitivity of methods in distinguishing pathogenic and nonpathogenic variants. Receiver operating characteristics (ROCs) curves including AUC for seven predictors using the Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] dataset (n = 17,329).
Table 2.
Receiver Operating Characteristics (ROCs) Area for Seven Predictors Using the Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] Dataset (n = 17,329)
| Confidence | ROC area | Standard error | Min ROC area | Max ROC area | |
|---|---|---|---|---|---|
| UMD‐Predictor | 0.95 | 0.954 | 0.002 | 0.950 | 0.957 |
| SIFT | 0.95 | 0.784 | 0.005 | 0.774 | 0.794 |
| PPH2_VAR | 0.95 | 0.826 | 0.004 | 0.819 | 0.834 |
| PROVEAN | 0.95 | 0.789 | 0.004 | 0.781 | 0.797 |
| CONDEL | 0.95 | 0.778 | 0.004 | 0.771 | 0.786 |
| MUT‐ASS | 0.95 | 0.813 | 0.004 | 0.806 | 0.820 |
| CADD | 0.95 | 0.834 | 0.004 | 0.827 | 0.841 |
Min ROC area, lower bound for the confidence interval of a vector of length two; Max ROC area, upper bound for the confidence interval of a vector of length two. All data were generated using the “ci.cvAUC” function of the “cvAUC” package (https://github.com/ledell/cvAUC) for the ROCR R‐package [Sing et al., 2005].
Impact of Mutations Frequency in the General Population
As indicated in Material and Methods section, the new UMD‐Predictor algorithm integrates a penalty score when a mutation has been described at a high frequency in the human population. Such information has already been used by other systems such as MutationTaster, which automatically annotates mutations as polymorphisms if they have been reported more than four times at a homozygous state in the 1000 genomes or HapMap projects. To assess the benefit of this new parameter, we removed its value from the algorithm and compared the sensitivity of methods in distinguishing pathogenic and nonpathogenic variants. Even without adjustment of the other parameters, the UMD‐Predictor still gives the most accurate predictions (0.79 vs. 0.77 for PPH2 and CADD), whereas it is outscored by CADD for AUC of 0.828 vs. 0.834 (Fig. 3; Table 3).
Figure 3.
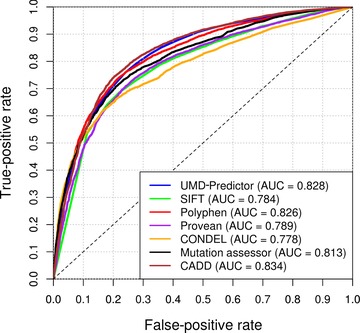
Sensitivity of methods in distinguishing pathogenic and nonpathogenic variants. Receiver operating characteristics (ROCs) are shown discriminating pathogenic mutations from nonpathogenic mutations defined by Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] dataset (n = 17,329). UMD‐Predictor values were obtained without the mutations’ frequency information.
Table 3.
Receiver Operating Characteristics (ROCs) Area for Seven Predictors Using the Varibench–dbSNP [Sherry et al., 2001; Sasidharan Nair and Vihinen, 2013] Dataset (n = 17,329)
| Confidence | ROC area | Standard error | Min ROC area | ax ROC area | |
|---|---|---|---|---|---|
| UMD‐Predictor | 0.95 | 0.828 | 0.004 | 0.821 | 0.836 |
| SIFT | 0.95 | 0.784 | 0.005 | 0.774 | 0.794 |
| PPH2_VAR | 0.95 | 0.826 | 0.004 | 0.819 | 0.834 |
| PROVEAN | 0.95 | 0.789 | 0.004 | 0.781 | 0.797 |
| CONDEL | 0.95 | 0.778 | 0.004 | 0.771 | 0.786 |
| MUT‐ASS | 0.95 | 0.813 | 0.004 | 0.806 | 0.820 |
| CADD | 0.95 | 0.834 | 0.004 | 0.827 | 0.841 |
Min ROC area, lower bound for the confidence interval of a vector of length two; Max ROC area, upper bound for the confidence interval of a vector of length two. All data were generated using the “ci.cvAUC” function of the “cvAUC” package (https://github.com/ledell/cvAUC) for the ROCR R‐package [Sing et al., 2005]. UMD‐Predictor values were obtained without the mutations’ frequency information.
WES Annotation
One of the main applications of global pathogenicity prediction systems is the annotation of NGS data, either WES or WGS. To assess the ability of the various systems in this context, we used data from three clinical diagnostic exomes. Three criteria were analyzed: the annotation processing‐time (PT), the correct annotation of disease‐causing mutations, and the number of annotated potential pathogenic variations (NV).
As shown in Table 4, the UMD‐Predictor system was able to process a VCF file of about 56,700 variations in 180 sec (from 93 to 240 sec), whereas other systems did it on average in 2,991 sec (from 240 to 11,160sec). SIFT 5.1.1, Polyphen 2.2.2, Provean 1.1.3, CONDEL 1.5, and CADD required a preprocessing of the VCF file that was not taken into account in this evaluation. The availability of Webservices reduced the analysis time to 69 sec (± 4 sec) for UMD‐Predictor when annotating extracted SNPs from VCF files, and allowed batch submission of variants including direct analysis of a full VCF file. In this condition, three VCF files were annotated in 66, 68, and 74 sec.
Table 4.
Comparison Between UMD‐Predictor and Other Prediction Tools for VCF Processing Using Three Files That Contained 58,145, 54,006, and 57,936 Variants, Respectively
| SIFTa | PPH2a | Proveana | Mutation assessora , b | CONDELa , c | Mutation Taster | CADDa | UMD‐Predictor | |
|---|---|---|---|---|---|---|---|---|
| PT1 (s) | 1,200 | 420 | 3,240 | 540 | 3,000 | 2,100 | 8,700 | 93 |
| PT2 (s) | 240 | 420 | 8,100 | 960 | 1,500 | 2,340 | 9,360 | 206 |
| PT3 (s) | 540 | 420 | 4,140 | 600 | 1,500 | 2,340 | 11,160 | 240 |
| NV1 | 1,958 | 2,881 | 1,540 | 1,339 | 1,376 | 2,677 | 3,241 | 871 |
| NV2 | 1,341 | 2,350 | 1,332 | 1,049 | 1,111 | 2,437 | 2,555 | 540 |
| NV3 | 1,842 | 2,781 | 1,542 | 1,350 | 1,376 | 3,401 | 3,098 | 807 |
All tests have been performed using the Web interface of each system. PT1‐3, processing time in seconds for VCF files 1–3; NV1‐3, number of variants predicted to be pathogenic for VCF files 1–3.
A preprocessing of the VCF file is required before analysis (this time has not been included in the tests).
VCF analysis was not possible on‐line; therefore, data have been generated through downloaded data.
VCF files cannot include more than 1,000 rows; therefore, the original VCF files have been split (this time has not been included in the tests).
All systems have successfully annotated six disease‐causing mutations found in the three patients. Nevertheless, they provided a wide range of candidate pathogenic mutations from 540 to 3,401. The UMD‐Predictor system gave for each case the shortest list with an average of 739 variations (from 540 to 871), whereas other predictors gave an average of 2,027 variations (from 1,049 to 3,401). This corresponds to a reduction of 63.5% of candidate variations.
Discussion
The now widely used WES and WGS technologies produce huge amount of variations from which only a handful are useful for genetic counseling or gene discovery. Therefore, it is critical to access variant annotations to select candidate pathogenic mutations. Unfortunately, this cannot be achieved through variants annotations from central‐ or locus‐specific databases as their content is still limited and of heterogeneous quality. In addition, disease‐causing mutations are often private and therefore not reported. In this situation, it is necessary to rely on bioinformatics prediction systems to annotate all variants and facilitate the critical filtration process that should ideally result in a limited set of candidate pathogenic mutations, as they need to be individually validated through familial segregation and in vitro or animal models.
The UMD‐Predictor system was developed to predict the pathogenicity of all cDNA substitutions from human genes (human reference genome GRCh37), therefore allowing the pathogenicity assessment of missense and synonymous mutations. To do so, we developed a combinatorial approach that pools information at the nucleotide level (conservation, variation frequency in the general population), at the protein level (physicochemical properties, global BLOSUM62 substitution matrix, conservation of the specific residue over 100 species, and involvement in functional or structural domains), and at the mRNA level (disruption or creation of splicing signals). This system was optimized to handle large sets of variations resulting from NGS studies thanks to Webservices that could be accessed in any NGS bioinformatics pipeline. It can also be used for any variant evaluation through a user‐friendly interface at http://umd‐predictor.eu.
To improve the predictions accuracy, we developed three new features. The first one is able to quantify the impact of a given amino acid substitution based on conservation between 100 species. To do so, it clusters species into five groups and the conservation of a specific residue is assessed using a window of ±3 residues. The second one corresponds to an automatic key residue annotation system able to extract relevant information from the Uniprot/SwissProt dataset. Finally, the third one takes into account the frequency information from the dbSNP data (build 138) (NCBI Resource Coordinators, 2015).
To evaluate the efficiency of this new pathogenicity prediction system, we collected data from 141,246 annotated variations, either pathogenic or nonpathogenic, using four reference datasets. These datasets were then used for prediction assessment using the seven most used and reliable prediction tools: SIFT 5.1.1 [Sim et al., 2012], Polyphen 2.2.2 [Adzhubei et al., 2010], Provean 1.1.3 [Choi et al., 2012], Mutation Assessor 2 [Reva et al., 2011], CONDEL 1.5 [González‐Pérez and López‐Bigas, 2011], MutationTaster 2 [Schwarz et al., 2014], and CADD [Kircher et al., 2014]. All these predictors are considered as standards in silico predictive algorithms for missense predictions by the American College of Medical Genetics and Genomics [Richards et al., 2015]. All evaluations demonstrate better accuracy (0.85), specificity (0.95), Matthews’ correlation coefficient (0.69), and DOR (86.6) for the UMD‐Predictor system. If it is well accepted that conservation between species is an important element to highlight key residues, we believe that allele frequency among humans could be considered as a complementary “conservation score” in a specific species. The simple removal of this criterion demonstrates that, even if UMD‐Predictor still ranks among the best systems, this parameter is important as it may account for up to 7% of the accuracy (0.79 vs. 0.85). This underlines that frequency information in humans should be considered to improve other pathogenicity predictions systems as already done for Mutation Taster. We also believe that allele frequency in other species might be important information to consider in the future when it will be available.
In order for prediction systems to be efficiently integrated into bioinformatics pipelines [Sherry et al., 2001; Pabinger et al., 2014], they need to include the following features: compliant with the variant call format (VCF), as accurate as possible, rapid, and programmatically accessible. To evaluate the ability of the various predictors to match these criteria, we used VCF datasets resulting from three WES performed in a clinical diagnostic context and for which pathogenic mutations have been confirmed. We demonstrated that all systems were able to accurately annotate the identified pathogenic mutations as candidate pathogenic mutations. Nevertheless, these pathogenic mutations were always part of a list of candidate pathogenic mutations that ranged from 540 to 3,401. In each situation, UMD‐Predictor provided the shortest list of candidate mutations, therefore being the most accurate. These results impacted the downstream filtration and validation processes that were reduced by approximately 64%.
Overall, we demonstrated that a single system, using a combinatorial prediction algorithm, could perform better than other prediction systems. This pinpoints that it is not required to develop expert systems aggregating predictions [González‐Pérez and López‐Bigas, 2011; Olatubosun et al., 2012] from various primary tools to improve efficiency. In addition, machine‐learning systems also prove to be limited, most probably because of the current training datasets quality/completeness. Moreover, we believe that such combinatorial approach could be applied for the prediction of mutations impact on other signals such as transcription factors binding sites, enhancers, miRNA, or ultraconserved regions.
As underlined by Pabinger et al. (2014], legal issues might arise when annotating lists of variants through on‐line systems as they do not guarantee data confidentiality. To solve this issue, once batch analyses have been performed, corresponding files are automatically deleted from the UMD‐Predictor system and no data is stored.
As described by the American College of Medical Genetics and Genomics [Richards et al., 2015], bioinformatics pipelines play a pivotal role in NGS analysis. They include a multistep processing that could result from many different programs and databases combinations and involves handling large amounts of data. They typically include mapping and assembly, sequence alignments, and variant calling (including SNV, structural variations, and copy‐number variations). Once SNVs have been identified, a second layer of multistep processing is initiated to annotate each variant and use several filtration processes depending on the mode of inheritance, the disease frequency, the phenotype, and the related tissue expression pattern. A key element of this layer is the pathogenicity prediction step that could be obtained from different systems. UMD‐Predictor is now one of them and could rapidly become a reference system according to its efficiency, rapidity, and easy implementation in existing NGS pipelines through Webservices.
UMD‐Predictor only predicts pathogenicity for coding SNVs. It would be of interest to develop pathogenicity predictions for in‐frame deletions and insertions. Nevertheless, because of the paucity of data, this remains a challenge. For exhaustive pathogenicity prediction of all variation types, UMD‐Predictor should be combined with other specific systems such as the Human Splicing Finder (HSF – http://www.umd.be/HSF3/) [Desmet et al., 2009] that is able to predict the pathogenicity of intronic variants.
Supporting information
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1: Multiple alignments of the Glutamic acid residue (E) at position 1003 of the FBN1 gene transcript ENST00000316623. Red square: surrounding windows of 3 residues used to remove species with more than one substitution per window. Thus Mouse, Rat, and all species below Armadillo were excluded. The remaining 55 species were used to compute the conservation score of 97.68. This score was subsequently normalized based on the number and categories of remaining species to 71.9. In a second step, the conservation score is adjusted based on the Grantham substitution matrix in comparison to natural observed variants if any are available. In this example, one natural variant was observed for the Armadillo species: Glu>Ala with a Grantham score of 107. Therefore, all potential substitutions were thus compared to this natural variant. Thus a Glu>Lys mutation (Grantham score of 56) will result in a conservation score of 66.8, while a Glu>Val mutation (Grantham score of 121) will result in a conservation score of 73.3.
Figure S2: log(DOR) comparison between predictors using the full Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233).
X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S3: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the full Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233).
Figure S4: log(DOR) comparison between predictors using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S5: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Uniprot (UniProt Consortium 2014) dataset (n=18.401).
Figure S6: log(DOR) comparison between predictors using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S7: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646).
Figure S8: log(DOR) comparison between predictors using the common Clinvar (Landrum et al. 2014) dataset (n=10.023). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations, but rather directly integrates conclusion from ClinVar.
Figure S9: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Clinvar (Landrum et al. 2014) dataset (n=10.023). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Clinvar (Landrum et al. 2014) dataset (n=10.023).
Figure S10: log(DOR) comparison between predictors using the Clinvar (Landrum et al. 2014) full dataset (n=12.486). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
Figure S11: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the Clinvar (Landrum et al. 2014) full dataset (n=12.486). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the Clinvar (Landrum et al. 2014) full dataset (n=12.486).
Figure S12: log(DOR) comparison between predictors using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
Figure S13: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the PredictSNP (Bendl et al. 2014) full dataset (n=43.882).
Figure S14: log(DOR) comparison between predictors using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
False positive rateTrue positive rate0.00.20.40.60.81.00.00.20.40.60.81.00.00.10.20.30.40.50.60.70.80.91.00.00.10.20.30.40.50.60.70.80.91.0UMD Predictor (AUC = 0.851)SIFT (AUC = 0.77)Polyphen (AUC = 0.799)Provean (AUC = 0.787)CONDEL (AUC = 0.755)Mutation Assessor (AUC = 0.801)CADD (AUC = 0.771
Figure S15: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common PredictSNP (Bendl et al. 2014) dataset (n=24.300).
Table S1: Comparison between UMD‐Predictor and other prediction tools using the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233)
Table S2: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233)
Table S3: Comparison between UMD‐Predictor and other prediction tools using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401)
Table S4: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401)
Table S5: Comparison between UMD‐Predictor and other prediction tools using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646)
Table S6: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Uniprot (UniProt Consortium 2014) full dataset (n=57.646)
Table S7: Comparison between UMD‐Predictor and other prediction tools using the common Clinvar (Landrum et al. 2014) dataset (n=10.023)
Table S8: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common Clinvar (Landrum et al. 2014) dataset (n=10.023)
Table S9: Comparison between UMD‐Predictor and other prediction tools using the Clinvar (Landrum et al. 2014) full dataset (n=12.486)
Table S10: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Clinvar (Landrum et al. 2014) full dataset (n=12.486)
Table S11: Comparison between UMD‐Predictor and other prediction tools using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882)
Table S12: Receiver Operating Characteristics (ROCs) area for 7 predictors using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882)
Table S13: Comparison between UMD‐Predictor and other prediction tools using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300)
Table S14: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300)
Contract grant sponsors: The European Union Seventh Framework Program (grant no. 305444); “An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research” (RD‐CONNECT; www.rd‐connect.eu; grant no. 200754); "Genotype‐To‐Phenotype Databases: A Holistic Solution” (GEN2PHEN; www.gen2phen.org); The FHU A*MIDEX project MARCHE n.ANR‐11‐IDEX‐001‐02 funded by the “Investissement d'avenir” French government program, managed by the French National Research Agency (ANR).
Communicated by Sean V. Tavtigian
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. 2015. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res 43(Database issue):D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. 2014. PredictSNP: robust and accurate consensus classifier for prediction of disease‐related mutations. PLoS Comput Biol 10:e1003440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg Y, Rost B. 2007. SNAP: predict effect of non‐synonymous polymorphisms on function. Nucleic Acids Res 35:3823–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acid substitutions and indels. PloS One 7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho‐Silva D, Clapham P, Coates G, Fitzgerald S, Gil L, Girón CG, et al., 2015. Ensembl 2015. Nucleic Acids Res 43:D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baets G, Van Durme J, Reumers J, Maurer‐Stroh S, Vanhee P, Dopazo J, Schymkowitz J, Rousseau F. 2012. SNPeffect 4.0: on‐line prediction of molecular and structural effects of protein‐coding variants. Nucleic Acids Res 40:D935–D939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet F‐O, Hamroun D, Lalande M, Collod‐Beroud G, Claustres M, BEROUD C. 2009. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frédéric MY, Lalande M, Boileau C, Hamroun D, Claustres M, BEROUD C, Collod‐Beroud G. 2009. UMD‐predictor, a new prediction tool for nucleotide substitution pathogenicity—application to four genes: FBN1, FBN2, TGFBR1, and TGFBR2. Hum Mutat 30:952–959. [DOI] [PubMed] [Google Scholar]
- Glas AS, Lijmer JG, Prins MH, Bonsel GJ, Bossuyt PMM. 2003. The diagnostic odds ratio: a single indicator of test performance. J Clin Epidemiol 56:1129–1135. [DOI] [PubMed] [Google Scholar]
- González‐Pérez A, López‐Bigas N. 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88:440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42:D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, Lin Y, MacDonald JR, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5:e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BW. 1975. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta 405:442–451. [DOI] [PubMed] [Google Scholar]
- NCBIResourceCoordinators . 2015. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 43:D6–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olatubosun A, Väliaho J, Härkönen J, Thusberg J, Vihinen M. 2012. PON‐P: integrated predictor for pathogenicity of missense variants. Hum Mutat 33:1166–1174. [DOI] [PubMed] [Google Scholar]
- Pabinger S, Dander A, Fischer M, Snajder R, Sperk M, Efremova M, Krabichler B, Speicher MR, Zschocke J, Trajanoski Z. 2014. A survey of tools for variant analysis of next‐generation genome sequencing data. Brief Bioinform 15:256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pushkarev D, Neff NF, Quake SR. 2009. Single‐molecule sequencing of an individual human genome. Nat Biotechnol 27:847–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reva B, Antipin Y, Sander C. 2011. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 39:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, et al., 2010. Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science 328:636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, et al. 2015. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43:D670–D81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan Nair P, Vihinen M. 2013. VariBench: a benchmark database for variations. Hum Mutat 34:42–49. [DOI] [PubMed] [Google Scholar]
- Schatz MC, Langmead B. 2013. The DNA Data deluge: fast, efficient genome sequencing machines are spewing out more data than geneticists can analyze. IEEE Spectr 50:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Cooper DN, Schuelke M, Seelow D. 2014. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 11:361–362. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim N‐L, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. 2012. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40:W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. 2005. ROCR: visualizing classifier performance in R. Bioinformatics 21:3940–3941. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN. 2003. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21:577–581. [DOI] [PubMed] [Google Scholar]
- Consortium UniProt. 2014. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res 42:D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, Guo Y, Feng B, et al. 2008. The diploid genome sequence of an Asian individual. Nature 456:60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen Y‐J, Makhijani V, Roth GT, Gomes X, Tartaro K, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452:872–876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclaimer: Supplementary materials have been peer‐reviewed but not copyedited.
Figure S1: Multiple alignments of the Glutamic acid residue (E) at position 1003 of the FBN1 gene transcript ENST00000316623. Red square: surrounding windows of 3 residues used to remove species with more than one substitution per window. Thus Mouse, Rat, and all species below Armadillo were excluded. The remaining 55 species were used to compute the conservation score of 97.68. This score was subsequently normalized based on the number and categories of remaining species to 71.9. In a second step, the conservation score is adjusted based on the Grantham substitution matrix in comparison to natural observed variants if any are available. In this example, one natural variant was observed for the Armadillo species: Glu>Ala with a Grantham score of 107. Therefore, all potential substitutions were thus compared to this natural variant. Thus a Glu>Lys mutation (Grantham score of 56) will result in a conservation score of 66.8, while a Glu>Val mutation (Grantham score of 121) will result in a conservation score of 73.3.
Figure S2: log(DOR) comparison between predictors using the full Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233).
X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S3: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the full Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233).
Figure S4: log(DOR) comparison between predictors using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S5: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Uniprot (UniProt Consortium 2014) dataset (n=18.401).
Figure S6: log(DOR) comparison between predictors using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR).
Figure S7: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646).
Figure S8: log(DOR) comparison between predictors using the common Clinvar (Landrum et al. 2014) dataset (n=10.023). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations, but rather directly integrates conclusion from ClinVar.
Figure S9: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common Clinvar (Landrum et al. 2014) dataset (n=10.023). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common Clinvar (Landrum et al. 2014) dataset (n=10.023).
Figure S10: log(DOR) comparison between predictors using the Clinvar (Landrum et al. 2014) full dataset (n=12.486). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
Figure S11: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the Clinvar (Landrum et al. 2014) full dataset (n=12.486). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the Clinvar (Landrum et al. 2014) full dataset (n=12.486).
Figure S12: log(DOR) comparison between predictors using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
Figure S13: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the PredictSNP (Bendl et al. 2014) full dataset (n=43.882).
Figure S14: log(DOR) comparison between predictors using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300). X‐axis: sensitivity; Y‐axis: specificity; color‐coded scale: log(DOR). Note that the MutationTaster2 tool does not perform any prediction on ClinVar pathogenic variations but rather directly integrates conclusion from ClinVar.
False positive rateTrue positive rate0.00.20.40.60.81.00.00.20.40.60.81.00.00.10.20.30.40.50.60.70.80.91.00.00.10.20.30.40.50.60.70.80.91.0UMD Predictor (AUC = 0.851)SIFT (AUC = 0.77)Polyphen (AUC = 0.799)Provean (AUC = 0.787)CONDEL (AUC = 0.755)Mutation Assessor (AUC = 0.801)CADD (AUC = 0.771
Figure S15: Sensitivity of methods in distinguishing pathogenic and non‐pathogenic variants using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300). Receiver Operating Characteristics (ROCs) are shown discriminating pathogenic mutations from non‐pathogenic mutations defined by the common PredictSNP (Bendl et al. 2014) dataset (n=24.300).
Table S1: Comparison between UMD‐Predictor and other prediction tools using the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233)
Table S2: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Varibench – dbSNP (Sherry et al. 2001; Sasidharan Nair and Vihinen 2013) full dataset (n=27.233)
Table S3: Comparison between UMD‐Predictor and other prediction tools using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401)
Table S4: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common Uniprot (UniProt Consortium 2014) dataset (n=18.401)
Table S5: Comparison between UMD‐Predictor and other prediction tools using the common Uniprot (UniProt Consortium 2014) full dataset (n=57.646)
Table S6: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Uniprot (UniProt Consortium 2014) full dataset (n=57.646)
Table S7: Comparison between UMD‐Predictor and other prediction tools using the common Clinvar (Landrum et al. 2014) dataset (n=10.023)
Table S8: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common Clinvar (Landrum et al. 2014) dataset (n=10.023)
Table S9: Comparison between UMD‐Predictor and other prediction tools using the Clinvar (Landrum et al. 2014) full dataset (n=12.486)
Table S10: Receiver Operating Characteristics (ROCs) area for 7 predictors using the Clinvar (Landrum et al. 2014) full dataset (n=12.486)
Table S11: Comparison between UMD‐Predictor and other prediction tools using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882)
Table S12: Receiver Operating Characteristics (ROCs) area for 7 predictors using the PredictSNP (Bendl et al. 2014) full dataset (n=43.882)
Table S13: Comparison between UMD‐Predictor and other prediction tools using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300)
Table S14: Receiver Operating Characteristics (ROCs) area for 7 predictors using the common PredictSNP (Bendl et al. 2014) dataset (n=24.300)