

Summary
Background
Exportin 1 (XPO1/CRM1) plays prominent roles in the regulation of nuclear protein export. Selective inhibitors of nuclear export (SINE) are small orally bioavailable molecules that serve as drug‐like inhibitors of XPO1, with potent anti‐cancer properties. Traumatic brain injury (TBI) presents with a secondary cell death characterized by neuroinflammation that is putatively regulated by nuclear receptors.
Aims and Results
Here, we report that the SINE compounds (KPT‐350 or KPT‐335) sequestered TBI‐induced neuroinflammation‐related proteins (NF‐kB, AKT, FOXP1) within the nucleus of cultured primary rat cortical neurons, which coincided with protection against TNF‐α (20 ng/mL)‐induced neurotoxicity as shown by at least 50% and 100% increments in preservation of cell viability and cellular enzymatic activity, respectively, compared to non‐treated neuronal cells (P's < 0.05). In parallel, using an in vivo controlled cortical impact (CCI) model of TBI, we demonstrate that adult Sprague‐Dawley rats treated post‐injury with SINE compounds exhibited significant reductions in TBI‐induced behavioral and histological deficits. Animals that received KPT‐350 orally starting at 2 h post‐TBI and once a day thereafter over the next 4 days exhibited significantly better motor coordination, and balance in the rotorod test and motor asymmetry test by 100–200% improvements, as early as 4 h after initial SINE compound injection that was sustained during subsequent KPT‐350 dosing, and throughout the 18‐day post‐TBI study period compared to vehicle treatment (P's < 0.05). Moreover, KPT‐350 reduced cortical core impact area and peri‐impact cell death compared to vehicle treatment (P's < 0.05).
Conclusions
Both in vitro and in vivo experiments revealed that KPT‐350 increased XPO1, AKT, and FOXP1 nuclear expression and relegated NF‐kB expression within the neuronal nuclei. Altogether, these findings advance the utility of SINE compounds to stop trafficking of cell death proteins within the nucleus as an efficacious treatment for TBI.
Keywords: Cell death protein trafficking, Neuroinflammation, Nuclear export inhibitor, Secondary cell death
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability in young people in the United States 1, 2. The estimated cost of TBI in the United States is $56 billion annually 1, 2, with over 1.7 million people yearly suffering from TBI, often resulting in undiagnosed pathology that can lead to chronic disability 3, 4. TBI involves a cascade of events, from the initial neural and vascular damage to the subsequent inflammation, ischemia, and bleeding that lead to progressive neurodegeneration 5, 6. After the initial insult, many brain cells continue to die due to the complex trafficking of cell death signals from primary injured cells to neighboring cells that eventually leads to “self‐destruction” and death of brain cells even in remote areas 7, 8. Moreover, although different mechanisms are involved in the pathogenesis of TBI, increasing evidence shows that secondary cell death mediated by inflammation plays a key role for its pathogenic progression 9, 10. Thus, treatments that curtail neuroinflammation after TBI may mitigate the evolution of secondary cell damage. The paradigm‐shifting TBI treatment modality being advanced in this project operates via a novel mechanism of “nuclear sequestration of cell death signals” (i.e., withholding cell death signals in the nucleus to inhibit their propagation to the cytoplasm), which could potentially enhance cell survival and rescue damaged neural cells in the TBI brain.
The nucleus is often referred to as the “brain” of the cell. Environmental damage to the cell leads to changes in gene expression as the cell reacts to the damage 11, 12, 13. Damage is communicated to the cell nucleus through the nuclear pore, a semi‐porous opening in the nuclear envelope that separates the nucleus from the cytoplasm 11, 12, 13 (Figure 1). The nuclear pore is thus a complex gate between the nucleus and cytoplasm that regulates the entry and exit of most macromolecules, including large proteins (≥30–40 kDa) which communicate cell damage and response to that damage 11, 12, 13. Transit of proteins (and RNAs) through the pore requires carrier proteins called importins and exportins. Exportin 1 (XPO1, also called CRM1) 14 is the major exportin, with over 200 cargo proteins 15, 16, including many that are known to play fundamental roles in the response of cells to injury 17. XPO1 is dramatically upregulated in neurons following diverse types of neuronal damage including in advanced multiple sclerosis (post‐mortem samples) 18, in neurons from rats following stroke 19 and TBI 20, involving a cascade of events, from the initial neuronal and vascular damage to the subsequent inflammation, ischemia, and bleeding that lead to increased intracranial pressure and further neuronal death. New treatments for TBI that target the cascade of injury‐induced pathology and that act on both the directly injured neurons in the core impacted area, as well as the peri‐impact area, are desperately needed. We have discovered and developed a class of potent, small molecule inhibitors of XPO1, termed selective inhibitor of nuclear export (SINE) compounds. The compound KPT‐330 (selinexor) is the most advanced SINE and is currently being tested in phase 1 and 2 clinical trials for hematology and solid tumor malignancies, while another SINE compound KPT‐335 (verdinexor) has received “minor use” designation by U.S. Food and Drug Administration (FDA) Center for Veterinary Medicine. This makes the product eligible for conditional approval similar to orphan drug/accelerated approvals used for submissions of human therapeutics. KPT‐350 is an orally available, brain penetrant SINE compound that shows good tolerability and excellent efficacy in animal models of neurodegenerative diseases and inflammatory conditions, including multiple sclerosis 21. The rationale for the use of XPO1 inhibitors for the treatment of TBI is based on their anti‐inflammatory, anti‐oxidant, and neuroprotective activities in vitro and in vivo.
Figure 1.
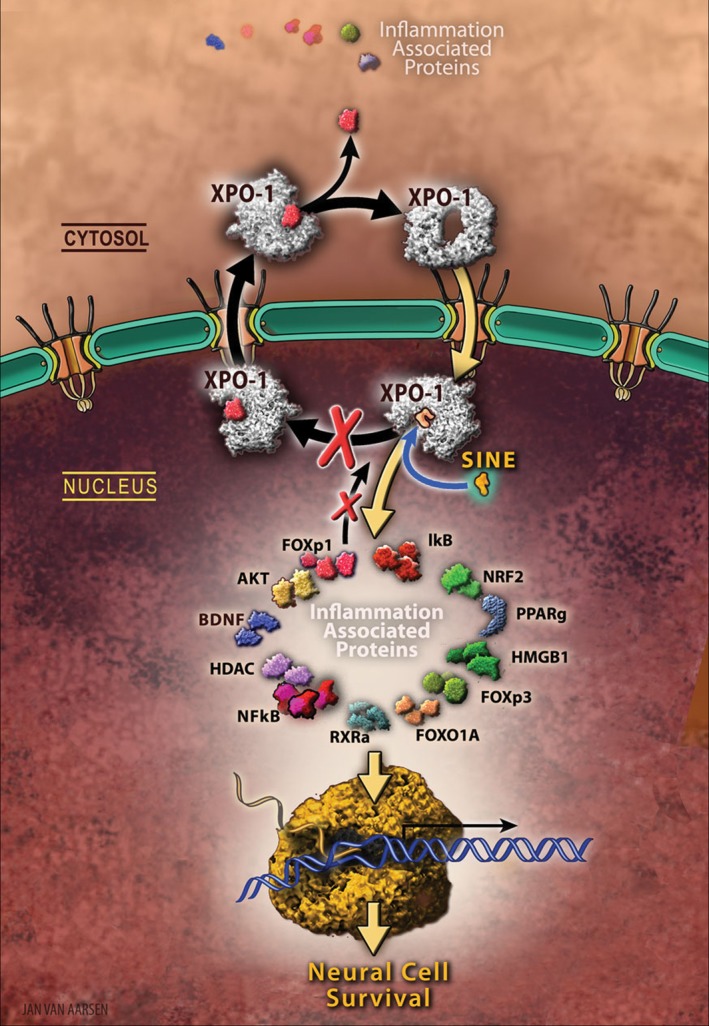
Nuclear sequestration of cell death. The nucleus acting as the “brain” of the cell directs how the cell reacts to injury. Damage is communicated to the cell nucleus through the nuclear pore, which serves as a critical gate between the nucleus and cytoplasm, and regulates the entry and exit of most proteins involved in the propagation of cell damage, as well as in mounting a response to the cell injury. Transit of proteins through the pore requires carrier proteins called importins and exportins. The major exportin is called Exportin 1 or XPO1 is dramatically upregulated in neural cells following diverse types of neural damage including Traumatic brain injury (TBI), suggesting that nuclear export underlies the pathology of neurodegeneration and inflammation associated with the disease. Therefore, nuclear inhibition of XPO1 would be beneficial for progressive forms of acute or chronic neuronal injury including those associated with TBI. The present study explored a novel class of potent, small molecule, brain‐penetrating inhibitors of XPO1, termed selective inhibitors of nuclear export (SINE), which can sequester cell surviving signals (FOXP1, AKT, among other therapeutic proteins), as well as cell death signals (NF‐kB) within the nucleus after TBI, resulting in reduced inflammation and improved behavioral recovery in SINE‐treated TBI animals. Because SINE is now currently being tested in the clinic for patients with cancer (i.e., SINE sequesters cell death signals within the nucleus, thereby inhibiting the spread of cancer to neighboring healthy cells), the proposed laboratory‐to‐clinic translational research will expedite the entry of SINE as a safe and effective drug, with evidence‐based mechanism of action, for TBI.
Because exaggerated nuclear export underlies the pathology of neurodegeneration, the export of inflammation‐associated components in these diseases may serve as a key cell death initiator. Therefore, in this study we examined whether inhibition of XPO1‐mediated nuclear export by SINE compounds would be beneficial for progressive forms of neuroinflammation associated with TBI.
Materials and Methods
These set of studies were divided into two, namely in vitro and in vivo studies, all designed to show the efficacy and mechanism of action of SINE compounds in experimental models of TBI.
For the in vitro studies, we assessed SINE compounds' regulation of nuclear exporter XPO1 in cultured neuronal cells exposed to an inflammation‐based model of TBI. This first pilot study was designed to show that SINE compounds protected against TBI‐mediated secondary cell death (albeit inflammation) and targeted the nucleus of neuronal cells in affording its therapeutic benefits against TBI. We also evaluated inhibition of nuclear factor‐kappaB (NF‐kB), blockade of AKT, and increases in forkhead box protein p1 (FOXP1) in cultured neuronal cells. This second pilot study was designed to reveal potential mechanism of action involving markers of inflammation and apoptosis, in tandem with demonstrating that SINE compounds produced these therapeutic effects against TBI by sequestration of cell death signals within the nucleus. We used the following in vitro methods.
TNF‐α In Vitro Model of Inflammation‐Mediated Injury
Details on the TNF‐α method are provided in our publications 22. Cultures of primary rat cortical neurons followed the supplier's protocol (BrainBits LLC, Springfield, IL, USA). Briefly, immediately after thawing, cells (4 × 104 cells/well) were seeded and grown in 96‐well plate coated by Poly‐L‐Lysine in 200 μL neural medium (NbActive 4, BrainBit NB4‐500) containing 2 mM l‐glutamine and 2% B27 in the absence of antibiotics for 4 days at 37°C in humidified atmosphere containing 5% CO2 23, 24. These cells were incubated initially for 2 h with SINE (using effective SINE dosing regimen (10 μM KPT‐350 or 10 μM KPT‐335) in vitro as per communication with Karyopharm) or vehicle. Human recombinant TNF‐α 20 ng/mL (R&D Systems, Minneapolis, MN, USA) was added to cell cultures for 48 h after 2 h incubation of SINE and vehicle treatment. All treatment conditions were performed in triplicate with investigators blinded to the treatment conditions.
Measurement of Cell Viability
Measurement of cell viability was performed by both fluorescent live/ dead cell assay and trypan blue exclusion method 25. A two‐color fluorescence cell viability assay was performed by Calcein‐AM (L3224; Invitrogen, Carisbad, CA, USA) to be retained within live cells, including an intense uniform green fluorescence and ethidium homodimer (EthD‐1) to bind the nuclei of damaged cells (bright red fluorescence). Briefly, the cells were incubated with 2 μM Calcein‐AM and 4 μM EthD‐1 for 45 min at room temperature in darkness according to the manufacturer's instructions. The green fluorescence of the live cells was measured by the Gemini EX florescence plate reader (Molecular Device), excitation at 485 nm and emission at 538 nm. In addition, trypan blue (0.2%) exclusion method was conducted and mean viable cell counts were calculated in four randomly selected areas (1 mm2, n = 10) to reveal the cell viability. To precisely calibrate the cell viability, the values were standardized form florescence intensity and trypan blue data 23, 24.
Measurement of Mitochondrial Activity
For quantification of TNF‐α‐induced inflammatory cell death, we conducted the 3‐(4, 5‐dimethyl‐2‐thiazoyl)‐2,5‐diphenyltetrazolium bromide (MMT) (Roche, 11465007001) by mitochondrial dehydrogenases, which was used as a measure of mitochondrial activity as previously described 23, 24. Briefly, the cells were treated with 0.5 mg/mL MTT solution at 37°C for 4 h and subsequently incubated with lysis buffer for 14 h in the incubator in a humidified atmosphere at 37°C and 5% CO2. The optical density of solubilized purple forzmazan was measured at 570 nm on a Synergy HT plate reader (BioTek, Winooski, VT, USA). Data are represented as relative percentage mean proliferation, defined as optical density reading number of each treated cells normalized to control cells (in the absence of treatment).
Immunocytochemistry
For nuclear export regulation and upstream and downstream inflammatory pathways, nuclear and cell death, including inflammatory, phenotypic markers were used for immunocytochemical analyses of cultured primary rat cortical neurons following treatment with SINE and vehicle.
Primary rat cortical neurons (8 × 104 cell/well) were cultured in 400 μL neural medium containing 2 mM l‐glutamine and 2% B27 in the absence of antibiotics in Poly‐L‐Lysine 8‐chamber (354632; BD Bioscience, Franklin Lakes, NJ, USA) for 4 days 23, 24. The cells were then incubated initially for 2 h with SINE (10 μM KPT‐350 or 10 μM KPT‐335) or vehicle. Human recombinant TNF‐α 20 ng/mL (R&D Systems) was added to cell cultures for 48 h after 2 h incubation of SINE and vehicle treatment and fixed in 4% paraformaldehyde for 20 min at room temperature (RT). The cells were washed 5 times for 10 min in PBST. Then, they were blocked by 5% normal goat serum (50‐062Z, Invitrogen) in PBS containing 0.1% Tween 20 (PBST) (Sigma‐Aldrich, St. Louis, MO, USA) for 60 min at RT. Primary antibodies, anti‐AKT1 mouse monoclonal antibody (1:100, ab124341, Abcam, Cambridge, MA, USA), anti‐XPO1 (CRM1) rabbit polyclonal antibody (1:100, ab24189, Abcam), anti‐NF‐kB p65 rabbit polyclonal antibody (1:100, ab16502, Abcam), and anti‐FOXP1 rabbit polyclonal antibody (1:100, ab16645, Abcam) were used. The cells were incubated overnight at 4°C with primary antibody with 5% normal goat serum in PBST. The cells were washed five times for 10 min in PBST and then soaked in 5% normal goat serum in PBST containing corresponding secondary antibodies, goat anti‐mouse IgG‐Alexa 488 (green; 1:1000, A11029, Invitrogen), and goat anti‐rabbit IgG‐Alexa 488 (green; 1:1000, A11034, Invitrogen) for 90 min in the dark. Finally, cells were washed 5 times for 10 min in PBST and 3 times for 5 min in PBS, then processed for Hoechst 33258 (bisBenzimideH 33258 trihydrochloride; Sigma) for 30 min, washed in PBS, and coverslipped with Fluoromount (Sigma). Immunofluorescent images were visualized using confocal microscope (FV1000; Olympus, Tokyo, Japan). Control experiments were performed with the omission of the primary antibodies yielding negative results.
For the in vivo studies, we tested the hypothesis that SINE compound exerted its “upstream” therapeutic benefits (i.e., sequestration of signaling molecules within the nucleus) against TBI by regulating the nuclear export function. For this first in vivo study, we performed acute studies to reveal direct action of SINE compound on nuclear exporter, specifically on XPO1. Based on pilot studies, we used the reported effective SINE compound dose and regimen in TBI. We exposed adult rats (2 months old) to TBI or sham surgery, and evaluated nuclear export function in the short‐term phase of injury (day 18, study period of the pilot study). Primary endpoints included assays of brain XPO1 and neuronal cell death. In addition, we conducted routine neurobehavioral outcomes to assess SINE compound efficacy in reducing TBI symptoms. Next, we tested the hypothesis that SINE attenuated the TBI pathological outcome through the inflammatory response “downstream”. We evaluated anti‐inflammation‐related pathways, including the inhibition of NF‐kB, enhancement of AKT, and elevation of FOXP1 expression. For this second in vivo study, we utilized the brain tissues from the acute studies of Aim 1. Using the pilot study employing the reported effective SINE compound dose and regimen, we exposed adult rats (2 months old) to TBI or sham surgery, and evaluated anti‐inflammation‐related pathways. Primary endpoints included inhibition of NF‐kB, enhancement of AKT, and elevation of FOXP1 expression. These phenotypic expressions detected via immunofluorescence staining provided insights on potential mechanisms of action underlying SINE compounds' therapeutic benefits against TBI. We used the following in vivo methods.
Subjects
All in vivo experiments were conducted in accordance with the National Institute of Health Guide and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use committee of the University of South Florida, Morsani College of Medicine. Rats were housed two per cage in a temperature‐ and humidity‐controlled room that was maintained on 12‐/12‐h light/dark cycles. They had free access to food and water.
TBI Model of Controlled Cortical Impact (CCI) Injury
Details on the CCI method are provided in our publications 26, 27. Rats (n = 12 per treatment condition) were deeply anesthetized with isoflurane in an induction chamber and then placed on a species‐specific nose cone apparatus in a stereotaxic apparatus. After exposing the skull, a 4.0 mm craniectomy was performed over the right frontoparietal cortex (−2.0 mm anteroposterior and +2.0 mm mediolateral to bregma). The pneumatically operated TBI device (with a convex tip diameter of 3.0) impacted the brain at a velocity of 6.0 m/s reaching a depth of 1.0 mm below dura for 150 ms to produce a moderate TBI model. The impactor rod was angled 15° to the vertical to maintain a perpendicular position in reference to the tangential plane of the brain curvature at the impact surface. A linear variable displacement transducer (Macrosensors, Pennsauken, NJ, USA), which was connected to the impactor, measured the velocity and duration to verify consistency. Control animals (n = 12) underwent the same surgical procedure (including craniectomy) except that the rats did not receive the impact injury.
Oral Drug Administration
In an animal model of moderate TBI, adult Sprague‐Dawley rats underwent CCI injury or sham surgery, followed by oral administration of KPT‐350 (5 mg/kg or 7 mg/kg 2 h later and once a day thereafter over the next 4 days) throughout the 18‐day post‐TBI study.
Neurobehavioral Tests
We employed behavioral tests that have been validated as sensitive for characterizing TBI in animal models, including the CCI TBI model. Animals were tested at baseline (before TBI), and at daily over the 18‐day post‐TBI survival period. These tests included rotorod and elevated body swing tests (EBST). Rotorod test of motor coordination involved placing the animal on an accelerating, revolving rod, and the latency to the first fall was recorded. There were 3 trials/days 28. EBST which measured motor activity involved lifting the animal by the tail ~10 cm above the surface of their home cage, and the number of times the animal moved its head and body to left or right recorded was recorded over 20 consecutive trials 29, 30.
Immunohistochemistry
Rats were anesthetized and transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (PB). Brains were removed and post‐fixed for 24 h followed by 30% sucrose in PB for 1 week and sectioned (30 μm) using a Leica cryostat 30. Sections were either processed for cell death (Nissl) and inhibition of nuclear exporter XPO1, or assayed for inflammation‐related markers including NF‐kB, AKT, and FOXP1. Details on the antibodies are provided in Immunocytochemistry Section. Densitometric analysis using Image Pro II (Image Pro II, Bioimager, Maple, ON, Canada) image analysis software was used.
Assessment of Lesion Volume
Six coronal slices from each brain between the anterior edge and posterior edge of the infarct were collected and processed for Nissl staining. The cavity area was defined at the damaged region. Brain sections were examined by using a light microscope (Olympus) and Keyence microscope. Lesion volume in each brain was calculated from the formula: ([area of the damaged region in each section] × 0.30) (mm3). Lesion volume was then expressed as a percentage of the ipsilateral hemisphere compared to the contralateral hemisphere 30.
Microarray
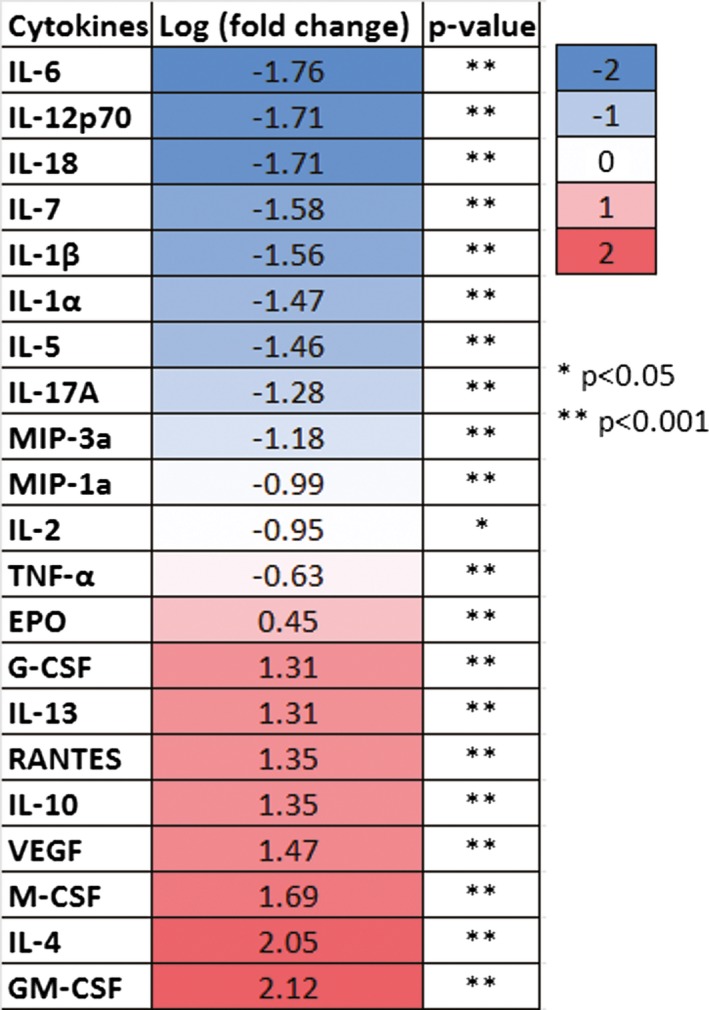
Rats were euthanized, and the brains were divided into the ipsilateral and contralateral hemispheres and then flash‐frozen. A Bio‐Rad 23‐Plex protein assay panel (Bio‐Rad, Hercules, CA, USA) was used to measure cytokine levels. Through the use of the 23‐Plex multiplex bead‐based suspension array system, coupled reactions of the specific beads isolated the desired biomarkers. The entire sample was then thoroughly washed to eliminate the unbounded, unwanted proteins. A promptly added biotinylated detection antibody formed the final detection complex. Analysis was performed according to system and panel instructions 31. Measurement of cytokines was conducted by comparing expression levels in CCI injury brains relative to sham brains, followed by KPT‐350 treatment.
Statistical Analyses
The data were evaluated statistically using ANOVA and subsequent post hoc Bonferroni's test. Statistical significance was preset at P < 0.05.
Results
The present experiments were designed to test the hypothesis whether nuclear sequestration of cell death afforded therapeutic benefits for TBI. To assess the ability of SINE compounds to protect against TBI‐mediated secondary cell death (albeit inflammation), we assessed the effect of KPT‐350 regulation of XPO1 on nuclear sequestration of inflammation‐related proteins cell viability in cultured primary rat cortical neuronal cells exposed to an inflammation‐based model of TBI. Results revealed that either SINE compound KPT‐350 or KPT‐335 reduced TNF‐α neurotoxicity and significantly increased both cell viability and mitochondrial activity compared to TNF‐α alone (P < 0.05) (Figure 2). In tandem, whereas XPO1 was detected in nuclei and cytosol in untreated cells, SINE compounds increased XPO1 nuclear expression under both normal cell culture condition and TNF‐α exposure (Figure 2). Similarly, nuclear factor‐kappaB (NF‐kB), which normally resides in its unactivated form in the cytoplasm, remained in the cytoplasm following TNF‐α treatment, but SINE compounds induced NF‐kB translocation to the nucleus without TNF‐α and enhanced its nuclear expression with TNF‐α exposure (Figure 2). Moreover, although the anti‐apoptotic AKT normally resides in the cytosol, and its cytoplasmic expression is unchanged in the presence of TNF‐α, SINE compounds increased AKT nuclear expression with or without TNF‐α treatment (Figure 2). Anti‐inflammatory and anti‐apoptotic forkhead box protein p1 (FOXP1) is also normally expressed in the cytoplasm, but SINE increased nuclear expression of FOXP1 with or without TNF‐α exposure (Figure 2). Consistent with its action as a selective XPO1 inhibitor, KPT‐350 has been found to induce the nuclear retention of a wide variety of XPO1 cargo proteins that are known to have anti‐inflammatory functions, including IκBα, FOXP3, FOXO1A and Nrf2 transcription factors, RXRγ and PPARγ nuclear receptors, and the chromatin binding protein HMGB1 (data not shown). This KPT‐350‐induced nuclear retention was similarly demonstrated in human monocytic leukemia THP‐1 and in human cervical cancer HeLa cells with or without prior TNF‐α exposure 32, 33, 34, 35, 36. These observations imply a potential for broad spectrum anti‐inflammatory activity of KPT‐350, based upon activation of a wide array of anti‐inflammatory mechanisms. These in vitro results, altogether, demonstrate that SINE compounds rescued cell viability and preserved mitochondrial activity against TNF‐α‐induced neurotoxicity possibly via sequestration of SINE's potent action on the nucleus as evidenced by increased nuclear expression of XPO1, as well as nuclear sequestration of pro‐survival and pro‐death cell signaling molecules, such as NF‐kB, AKT, and FOXP1.
Figure 2.
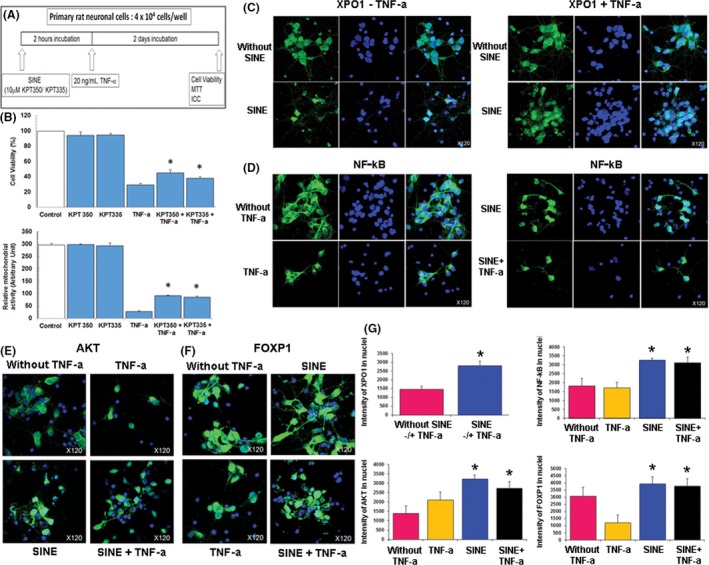
Selective inhibitors of nuclear export (SINE) therapeutic benefits and mechanism of action in vitro. (A) Overall experimental design is shown in schematic diagram. (B) SINE dosing regimen (10 μM KPT‐350 or 10 μM KPT‐335) reduces TNF‐α neurotoxicity. ANOVA revealed significant treatment effect of SINE on cell viability and mitochondrial activity (P's < 0.01). Post hoc pairwise comparisons revealed that treatment with either 10 μM KPT‐350 or 10 μM KPT‐335 reduced TNF‐α neurotoxicity in both cell viability and mitochondrial activity assays (*P's < 0.05). (C) SINE increases XPO1 nuclear expression under normal cell culture condition. The nuclear transporter XPO1 is normally detected in nuclei and cytosol. Without SINE compounds, XPO1 expression was found to be present in both nuclei and cytosol. Interestingly, SINE compound treatment increased XPO1 nuclear localization. In addition, SINE compound also increased XPO1 nuclear expression under TNF‐α exposure. Without SINE treatment, XPO1 expression was found to be present in both nuclei and cytosol following treatment with TNF‐α. In contrast, SINE treatment increased XPO1 nuclear expression under TNF‐α exposure. (D) NF‐kB is expressed and activated by TNF‐α in the cytosol, but translocates to the nucleus with SINE treatment. NF‐kB is a transcription factor which resides in its unactivated form in the cytoplasm. Following activation subsequent to TNF‐α treatment, NF‐kB is elevated and may induce the expression of anti‐apoptotic genes (e.g., AKT). In addition, NF‐kB translocates to the nucleus following SINE treatment under normal condition or TNF‐α exposure. SINE contained NF‐kB expression within the nuclei in both normal cell culture condition and TNF‐α treatment, again demonstrating the ability of SINE to sequester signaling molecules to the nucleus. (E) Anti‐apoptotic AKT is normally expressed in the cytosol, but SINE increases AKT's nuclear expression under normal condition or TNF‐α exposure. Under normal cell condition, the anti‐apoptotic AKT is expressed in the cytosol and its expression reduced by TNF‐α treatment. Treatment with SINE increased AKT nuclear expression and importantly with TNF‐α treatment, providing further support for SINE's robust ability to target the nucleus in sequestering cell death‐related, but also cell survival‐associated molecules. (F) Anti‐inflammatory and anti‐apoptotic FOXP1 is also normally expressed in the cytosol, but SINE increases FOXP1's nuclear expression under normal condition or TNF‐α exposure. Under normal cell condition, the anti‐inflammatory and anti‐apoptotic FOXP1 is detected in the cytosol and its expression reduced by TNF‐α treatment. Treatment with SINE increased FOXP1 nuclear expression under normal condition and more importantly with TNF‐α treatment, providing further evidence of SINE capacity to sequester pro‐cell survival‐associated molecules within the nucleus. (G) Quantification of XPO1, NF‐kB, AKT, and FOXP1 was obtained from three independent experiments, and each experiment was conducted in triplicates. Data are mean ± SEM and significance preset at *P < 0.05. These in vitro results, altogether, demonstrate that SINE rescued cell viability and preserved mitochondrial activity against TNF‐α‐induced neurotoxicity possibly via sequestration of SINE's potent action on the nucleus as evidence by increased nuclear expression of the nuclear transporter XPO1, as well as sequestration of pro‐survival and pro‐death cell signaling molecules, NF‐kB, AKT, and FOXP1.
Based on the positive in vitro effects of SINE compounds against inflammation‐induced injury, we next evaluated effects of KPT‐350 in an in vivo model of TBI (see Figure 3A for overall experimental design). Adult Sprague‐Dawley male rats (2 months old) underwent TBI (unilateral controlled cortical impact model) or sham surgery, then received oral KPT‐350 (5 or 7 mg/kg) 2 h later and once a day thereafter over the next 4 days. ANOVA revealed significant treatment effect of KPT‐350 in attenuating TBI‐induced motor deficits (P < 0.01) (Figure 3B), based upon improved performance in elevated body swing test (EBST) either 5 mg/kg KPT‐350 or 7 mg/kg KPT‐350 compared to those TBI animals that received vehicle alone (P's < 0.05). ANOVA also revealed significant treatment effect of KPT‐350 on motor coordination as revealed by rotorod test (P's < 0.01), with either 5 mg/kg KPT‐350‐ or 7 mg/kg KPT‐350‐treated animals demonstrating amelioration of TBI‐induced dysfunctions in motor coordination compared to those TBI animals that received vehicle alone (P < 0.05). Next, we embarked in histological/immunohistochemical assessments of KPT‐350 therapeutic benefits. KPT‐350 (5 or 7 mg/kg) reduced TBI‐induced lesions in core impact (P < 0.01) and peri‐impact area (P < 0.05) based on Nissl staining (Figure 3C). In addition, either 5 or 7 mg/kg KPT‐350 led to increased nuclear expression of XPO1 in TBI brain, based on immunofluorescent imaging (Figure 3D, quantified in Figure 3E). In contrast, expression of XPO1 in vehicle‐treated TBI brain was very low and predominantly outside of the nucleus. In addition, brains from animals that received sham surgery displayed no detectable XPO1 signals. KPT‐350 at 7 mg/kg, but not 5 mg/kg, led to sequestration of NF‐kB expression within nuclei in TBI brain (Figure 3D, quantified in Figure 3E). In contrast, expression of NF‐kB vehicle‐treated TBI brain showed extranuclear NF‐kB localization. In addition, brains from animals that received sham surgery exhibited no detectable NF‐kB signals. In contrast, expression of NF‐kB in vehicle‐treated TBI brain was primarily cytoplasmic. Furthermore, both doses of KPT‐350 forced nuclear retention of AKT expression in TBI brain compared to vehicle‐treated TBI brain or sham group (Figure 3D, quantified in Figure 3E). Moreover, brains from animals that received sham surgery exhibited no detectable AKT. However, overall expression of AKT appeared to be elevated in vehicle‐treated TBI brain, which may indicate an ongoing endogenous repair mechanism. Nonetheless, the nuclear expression of AKT signals was increased in TBI brain of KPT‐350‐treated animals. Furthermore, KPT‐350 increased FOXP1 nuclear expression in TBI brain compared to vehicle‐treated TBI brain or sham group (Figure 3D, quantified in Figure 3E). Quantification using immunofluorescence intensity for each phenotypic marker revealed (i.e., XPO1, NF‐kB, AKT, and FOXP1) significant treatment effects across treatment groups (ANOVA, P's < 0.01). Post hoc pairwise comparisons revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) resulted in increased expression of XPO1, NF‐kB, AKT, and FOXP1 compared to vehicle‐treated TBI brain. Additionally, microarray profiling of different cytokines revealed specific upregulation and downregulation of anti‐inflammatory and pro‐inflammatory cytokines, respectively (Table 1).
Figure 3.
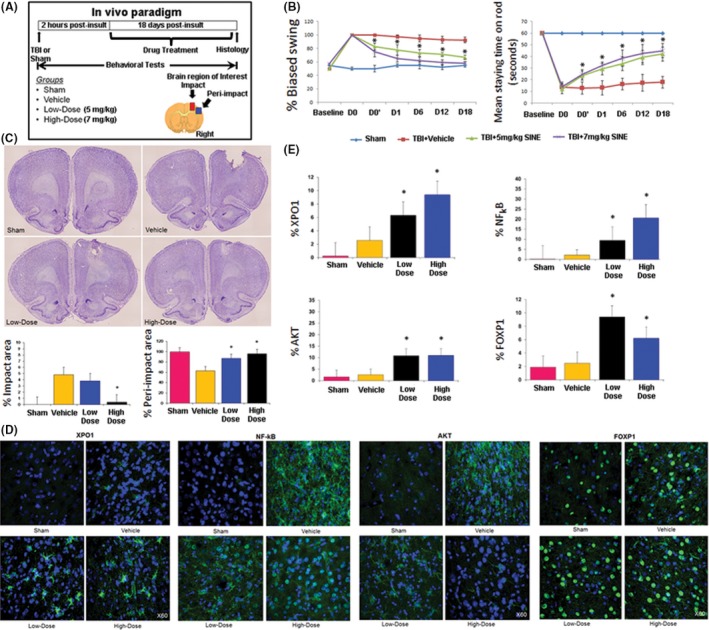
Selective inhibitors of nuclear export (SINE) therapeutic benefits and mechanism of action in vivo. (A) Overall experimental design is shown in a schematic diagram, with brain region of interest for histological and immunohistochemical assessment also indicated (B) SINE attenuates TBI‐induced motor deficits. ANOVA revealed significant treatment effect of SINE on motor activity as revealed by EBST (P's < 0.01). Post hoc pairwise comparisons revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) reduced TBI‐induced motor deficit compared to those TBI animals that received vehicle alone (*P's < 0.05). In addition, SINE attenuates TBI‐induced dysfunctions in motor coordination. ANOVA revealed significant treatment effect of SINE on motor coordination as revealed by rotorod test (P's < 0.01). Post hoc pairwise comparisons revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) reduced TBI‐induced dysfunctions in motor coordination compared to those TBI animals that received vehicle alone (*P's < 0.05; Legend—D0: Day 0, immediately after surgery but prior to drug treatment; D0': Day 0, 4 h after drug treatment; D1–D18: Day 1–Day 18 postsurgery). Next, we embarked on histological/ immunohistochemical assessments. (C) SINE reduces TBI‐induced lesions in core impact and peri‐impact areas. ANOVA revealed significant treatment effects of SINE on TBI‐induced lesion as revealed by Nissl staining (P's < 0.01). Post hoc pairwise comparisons revealed that treatment with 7 mg/kg KPT‐350 (high dose) reduced TBI‐induced impact area (A), while either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) reduced the TBI‐induced peri‐impact area (B) compared to TBI animals that received vehicle alone (*P's < 0.05). (D) Representative images of immunostained TBI brains for XPO1, NF‐kB, AKT, and FOXP1. SINE increases XPO1 nuclear expression in TBI brain. Immunofluorescent imaging revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) led to increased nuclear expression of XPO1 in TBI brain. In contrast, expression of XPO1 in vehicle‐treated TBI brain was very low and predominantly showed extranuclear signals. In addition, brain from animal that received sham surgery displayed negligible XPO1 signals. SINE sequesters NF‐kB expression within the nucleus in TBI brain. Immunofluorescent imaging revealed that treatment with 7 mg/kg KPT‐350 (high dose) resulted in increased nuclear expression of NF‐kB in TBI brain. Treatment with 5 mg/kg KPT‐350 (low dose) also led to increased nuclear expression of NF‐kB but to a lesser extent compared to 7 mg/kg KPT‐350 (high dose) treatment. In contrast, expression of NF‐kB in vehicle‐treated TBI brain showed extranuclear NF‐kB signals. In addition, brain from animals that received sham surgery exhibited no detectable NF‐kB signals. SINE limits AKT expression within the nucleus in TBI brain. Immunofluorescent imaging revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) resulted in increased sequestration of AKT signals within the nucleus of the TBI brain. In contrast, expression of AKT in vehicle‐treated TBI brain showed extranuclear AKT signals. In addition, brain from animal that received sham surgery exhibited only trace levels of AKT signals. SINE increases FOXP1 nuclear expression in TBI brain. Immunofluorescent imaging revealed that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) resulted in increased expression of FOXP1 within the nucleus of the TBI brain compared to vehicle‐treated TBI brain or sham group. (E) Quantification of XPO1, NF‐kB, AKT, and FOXP1 expression in TBI brain. Quantification involved unbiased stereology of immunofluorescent cells with nuclear expression of each phenotypic marker within the peri‐impact cortex. Analyses revealed significant treatment effects (ANOVA, P's < 0.01), with post hoc pairwise comparisons showing that treatment with either 5 mg/kg KPT‐350 (low dose) or 7 mg/kg KPT‐350 (high dose) resulted in increased expression of XPO1, AKT, NF‐kB, and FOXP1 compared to vehicle‐treated TBI brain or sham (P's < 0.05).
Table 1.
Microarray profiling of different cytokines in TBI brain treated with SINE. Upregulation and downregulation of anti‐inflammatory and pro‐inflammatory cytokines, respectively, were detected when comparing TBI brains treated with SINE compared to those treated with vehicle
Discussion
These studies reveal that SINE compounds treatment affords therapeutic benefits in both in vitro and in vivo paradigms of TBI. We recognize that XPO1/CRM1 plays prominent roles in the regulation of nuclear protein export and was recently shown to be overexpressed in CNS lesions in rats following TBI and to regulate cell death following brain injury 20. Here, we provide evidence that SINE compounds acting on XPO1 exerts both histological and behavioral effects on TBI. SINE compounds were found to significantly counteract TNF‐α‐induced loss of cell viability and mitochondrial activity compared to vehicle‐treated primary rat neuronal cells. We saw similar SINE neuroprotective effects against an MS model of glutamate toxicity in cultured rat neurons 21.
In the present animal model of moderate TBI, TBI animals treated orally with KPT‐350 exhibited significantly better motor coordination and balance in the rotorod test, general motor activity as revealed by the motor asymmetry test, and cognitive performance as revealed by MWM test. Up to twofold improvement was observed as early as 4 h after initial treatment, and was sustained over the 18‐day post‐injury assessment period, compared to vehicle treatment. Histological data showed reduction of cell death in the core impact and peri‐impact cortical areas in KPT‐350‐treated animals, in particular with the high dose. Both in vitro and in vivo studies revealed that SINE compounds sequestered inflammation‐related proteins within the nucleus. Cytokine profiling offered insights on potential “inflammation” targets of SINE compounds in abrogating TBI‐induced inflammation. Microarray analysis identified that certain inflammation‐related cytokines, in particular IL‐6 and GM‐CSF, were significantly downregulated or upregulated by SINE treatment. In summary, our data provided evidence of nuclear sequestration of cell death signals, as well as cell survival molecules, as SINE compound's primary therapeutic target in abrogating the neuroinflammatory response in TBI.
This study consisted of a set of basic science‐ and mechanism‐driven hypotheses, and translational research experiments designed to assess the safety and efficacy of KPT‐350 in an animal model of TBI. The scientific concept of cell death inhibition by nuclear sequestration of that we advanced here is based on patented findings demonstrating that a family of closely related, oral SINE showed potent and selective anti‐inflammatory activities in multiple preclinical models [e.g. 32, 33, 34, 35, 36]. Of note, selinexor, an oral SINE compound, has advanced to phase 2 human clinical trials for solid tumors and hematologic malignancies. In addition, KPT‐335 (verdinexor) oral SINE compound in phase 3 trial in dogs with non‐Hodgkin's lymphoma showed clear activity and good tolerability 35. Altogether, these studies provide solid guidance on feasibility, efficacy, and safety profiles of SINE compounds for clinical application in cancer, as well as in inflammation‐associated diseases. Here, we endeavored to explore the therapeutic benefits of oral SINE compounds against TBI, a neurological disorder that shows a major secondary cell death accompanied by an inflammatory response 37, 38, 39. To this end, we showed that nuclear sequestration of inflammatory cell death by SINE compounds retarded secondary pathological manifestations of TBI.
Selective inhibitors of nuclear export compounds comprise a class of potent, small molecule, orally available, brain‐penetrating inhibitors of XPO1. In contrast with other XPO1/CRM1 inhibitors (e.g., Leptomycin B, ratjadone, anguinomycin, etc.) 12, 17, 34, 40 which bind irreversibly to XPO1, SINE compounds inhibit XPO1‐mediated nuclear‐cytoplasmic transport by transient (i.e., 12–24 h) covalent binding to the XPO1 cargo binding site, corresponding to inhibition period sufficient for retention of specific proteins in the nucleus 34, 41, 42, 43, 44, 45, 46, 47. Of note, irreversible binding to XPO1 by other XPO1/CRM1 inhibitors (e.g. Leptomycin B) has been assumed to induce significant systemic toxicity in both animals and humans 48, 49. Furthermore, SINE compounds have been demonstrated to be slowly reversible and orally bioavailable, an advantage of SINE over other existing XPO1 inhibitors which require intravenous delivery, limiting frequency of drug administration 34, 50. Recent studies have shown good tolerability and excellent efficacy of SINE analog KPT‐350 in neurodegenerative diseases and inflammatory conditions including spinal cord injury, multiple sclerosis, amyotrophic lateral sclerosis, rheumatoid arthritis, and systemic lupus erythematosus 51, 52, as well as in animal models of TBI 53, 54. Moreover, SINE KPT‐350 demonstrates high CNS penetration and drug‐like features, making it an excellent candidate for further drug development 34, 55. A closely related SINE compound, KPT‐330 (selinexor), has been tested in >1000 human cancer patients to date, which was well tolerated and highly efficacious 45, 46, 47. Selinexor, applied topically, significantly improved wound healing in model of ischemic wounds by improving blood flow and reducing inflammation (clinical studies later this year). Our present data found that KPT‐350 reduces the impact of CCI by increasing the expression of neuroprotectant proteins (e.g., AKT and FOXP1) and reducing the inflammatory response (NF‐kB), leading to accelerated and improved recovery of motor functions after TBI. These exciting efficacy and mechanistic readouts of SINE compounds in experimental TBI studies provide the impetus to pursue a preclinical development plan via a mechanistic and translational pathway to advance the clinical application of SINE compounds to TBI patients. The present study showed (1) the use of an oral anti‐inflammatory agent (SINE), with robust inhibitory effect on nuclear export, in attenuating the secondary cell death associated with TBI; (2) the elucidation of mechanistic interaction between inflammation and SINE compounds, through nuclear sequestration of cargo molecules (NF‐kB, AKT, and FOXP1), altogether improving our understanding of pathological outcome and treatment of TBI, which may be relevant to other inflammation‐plagued diseases.
Conflict of Interest
Karyopharm Inc. holds patents and intellectual property rights on SINE compounds.
Acknowledgment
This study was funded by Karyopharm Inc. The authors thank the excellent technical assistance of Mr. Quinn Colburn in final preparation of the manuscript.
The copyright line for this article was changed on 25 March, 2016 after original online publication.
References
- 1. Asemota AO, George BP, Bowman SM, Haider AH, Schneider EB. Causes and trends in traumatic brain injury for united states adolescents. J Neurotrauma 2013;30:67–75. [DOI] [PubMed] [Google Scholar]
- 2. Kou Z, VandeVord PJ. Traumatic white matter injury and glial activation: From basic science to clinics. Glia 2014;62:1831–1855. [DOI] [PubMed] [Google Scholar]
- 3. Sundman MH, Hall EE, Chen NK. Examining the relationship between head trauma and neurodegenerative disease: A review of epidemiology, pathology and neuroimaging techniques. J Alzheimers Dis Parkinsonism 2014;4:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tashlykov V, Katz Y, Volkov A, et al. Minimal traumatic brain injury induce apoptotic cell death in mice. J Mol Neurosci 2009;37:16–24. [DOI] [PubMed] [Google Scholar]
- 5. Hadass O, Tomlinson BN, Gooyit M, et al. Selective inhibition of matrix metalloproteinase‐9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS ONE 2013;8:e76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lo EH. A new penumbra: Transitioning from injury into repair after stroke. Nat Med 2008;14:497–500. [DOI] [PubMed] [Google Scholar]
- 7. Fang Y, Bonini NM. Axon degeneration and regeneration: Insights from drosophila models of nerve injury. Annu Rev Cell Dev Biol 2012;28:575–597. [DOI] [PubMed] [Google Scholar]
- 8. Osterloh JM, Yang J, Rooney TM, et al. dSarm/Sarm1 is required for activation of an injury‐induced axon death pathway. Science 2012;337:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campolo M, Esposito E, Ahmad A, et al. Hydrogen sulfide‐releasing cyclooxygenase inhibitor ATB‐346 enhances motor function and reduces cortical lesion volume following traumatic brain injury in mice. J Neuroinflammation 2014;11:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shojo H, Kaneko Y, Mabuchi T, Kibayashi K, Adachi N, Borlongan CV. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury‐induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience 2010;171:1273–1282. [DOI] [PubMed] [Google Scholar]
- 11. Cooper GM. The Nuclear Envelope and Traffic between the Nucleus and Cytoplasm In: Geoffrey MC, editor. The Cell: A Molecular Approach, 2nd edn Sunderland, MA: Sinauer Associates, 2000;1–689. [Google Scholar]
- 12. Nguyen KT, Holloway MP, Altura RA. The CRM1 nuclear export protein in normal development and disease. Int J Biochem Mol Biol 2012;3:137–151. [PMC free article] [PubMed] [Google Scholar]
- 13. Wang W, Budhu A, Forgues M, Wang XW. Temporal and spatial control of nucleophosmin by the ran‐CRM1 complex in centrosome duplication. Nat Cell Biol 2005;7:823–830. [DOI] [PubMed] [Google Scholar]
- 14. Kudo N, Khochbin S, Nishi K, et al. Molecular cloning and cell cycle‐dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J Biol Chem 1997;272:29742–29751. [DOI] [PubMed] [Google Scholar]
- 15. Guttler T, Gorlich D. Ran‐dependent nuclear export mediators: A structural perspective. EMBO J 2011;30:3457–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu D, Farmer A, Chook YM. Recognition of nuclear targeting signals by karyopherin‐beta proteins. Curr Opin Struct Biol 2010;20:782–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Turner JG, Sullivan DM. CRM1‐mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem 2008;15:2648–2655. [DOI] [PubMed] [Google Scholar]
- 18. Kim JY, Shen S, Dietz K, et al. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat Neurosci 2010;13:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen Y, Samal B, Hamelink CR, et al. Neuroprotection by endogenous and exogenous pacap following stroke. Regul Pept 2006;137:4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li A, Zou F, Fu H, et al. Upregulation of CRM1 relates to neuronal apoptosis after traumatic brain injury in adult rats. J Mol Neurosci 2013;51:208–218. [DOI] [PubMed] [Google Scholar]
- 21. Haines JD, Herbin O, de la Hera B, et al. Nuclear export inhibitors avert progression in preclinical models of inflammatory demyelination. Nat Neurosci 2015;18:511–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bachstetter AD, Jernberg J, Schlunk A, et al. Spirulina promotes stem cell genesis and protects against lps induced declines in neural stem cell proliferation. PLoS ONE 2010;5:e10496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaneko Y, Tajiri N, Shojo H, Borlongan CV. Oxygen‐glucose‐deprived rat primary neural cells exhibit dj‐1 translocation into healthy mitochondria: A potent stroke therapeutic target. CNS Neurosci Ther 2014;20:275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kaneko Y, Sullivan R, Dailey T, Vale FL, Tajiri N, Borlongan CV. Kainic acid‐induced golgi complex fragmentation/dispersal shifts the proteolysis of reelin in primary rat neuronal cells: An in vitro model of early stage epilepsy. Mol Neurobiol 2015; doi: 10.1007/s12035-015-9126-1 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bell E, Cao X, Moibi JA, et al. Rapamycin has a deleterious effect on min‐6 cells and rat and human islets. Diabetes 2003;52:2731–2739. [DOI] [PubMed] [Google Scholar]
- 26. Acosta SA, Diamond DM, Wolfe S, et al. Influence of post‐traumatic stress disorder on neuroinflammation and cell proliferation in a rat model of traumatic brain injury. PLoS ONE 2013;8:e81585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Acosta SA, Tajiri N, Shinozuka K, et al. Long‐term upregulation of inflammation and suppression of cell proliferation in the brain of adult rats exposed to traumatic brain injury using the controlled cortical impact model. PLoS ONE 2013;8:e53376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi K, Yasuhara T, Shingo T, et al. Embryonic neural stem cells transplanted in middle cerebral artery occlusion model of rats demonstrated potent therapeutic effects, compared to adult neural stem cells. Brain Res 2008;1234:172–182. [DOI] [PubMed] [Google Scholar]
- 29. Borlongan CV, Sanberg PR. Elevated body swing test: A new behavioral parameter for rats with 6‐hydroxydopamine‐induced hemiparkinsonism. J Neurosci 1995;15:5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tajiri N, Acosta SA, Shahaduzzaman M, et al. Intravenous transplants of human adipose‐derived stem cell protect the brain from traumatic brain injury‐induced neurodegeneration and motor and cognitive impairments: Cell graft biodistribution and soluble factors in young and aged rats. J Neurosci 2014;34:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tajiri N, Hernandez D, Acosta S, et al. Suppressed cytokine expression immediately following traumatic brain injury in neonatal rats indicates an expeditious endogenous anti‐inflammatory response. Brain Res 2014;1559:65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng Y, Holloway MP, Nguyen K, et al. XPO1 (CRM1) inhibition represses stat3 activation to drive a survivin‐dependent oncogenic switch in triple‐negative breast cancer. Mol Cancer Ther 2014;13:675–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kojima K, Kornblau SM, Ruvolo V, et al. Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood 2013;121:4166–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. London CA, Bernabe LF, Barnard S, et al. Preclinical evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT‐335 in spontaneous canine cancer: Results of a phase I study. PLoS ONE 2014;9:e87585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendonca J, Sharma A, Kim HS, et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014;5:6102–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perwitasari O, Johnson S, Yan X, et al. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza a virus replication in vitro and in vivo. J Virol 2014;88:10228–10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Simard JM, Woo SK, Schwartzbauer GT, Gerzanich V. Sulfonylurea receptor 1 in central nervous system injury: A focused review. J Cereb Blood Flow Metab 2012;32:1699–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gatson JW, Liu MM, Abdelfattah K, et al. Estrone is neuroprotective in rats after traumatic brain injury. J Neurotrauma 2012;29:2209–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das M, Leonardo CC, Rangooni S, Pennypacker KR, Mohapatra S, Mohapatra SS. Lateral fluid percussion injury of the brain induces CCL20 inflammatory chemokine expression in rats. J Neuroinflammation 2011;8:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Turner JG, Dawson J, Sullivan DM. Nuclear export of proteins and drug resistance in cancer. Biochem Pharmacol 2012;83:1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Azmi AS, Aboukameel A, Bao B, et al. Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology 2013;144:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Draetta GF, Shacham S, Kauffman M, et al. Cytotoxicity of novel, small molecule, CRM1‐selective inhibitors of nuclear export (SINE) in colorectal cancer (CRC) cells. J Clin Oncol 2011;29:suppl: abstract #e14091 [Google Scholar]
- 43. Etchin J, Sanda T, Mansour MR, et al. KPT‐330 inhibitor of CRM1 (XPO1)‐mediated nuclear export has selective anti‐leukaemic activity in preclinical models of t‐cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol 2013;161:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood 2012;120:4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McCauley D, Landesman Y, Senapedis W, et al. Preclinical evaluation of selective inhibitors of nuclear export (SINE) in basal‐like breast cancer (BLBC). J Clin Oncol 2012;30:suppl: abstract #1055 [Google Scholar]
- 46. Ranganathan P, Yu X, Na C, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012;120:1765–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang K, Wang M, Tamayo AT, et al. Novel selective inhibitors of nuclear export CRM1 antagonists for therapy in mantle cell lymphoma. Exp Hematol 2013;41:67 e64–78 e64 [DOI] [PubMed] [Google Scholar]
- 48. Tai YT, Landesman Y, Acharya C, et al. CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: Molecular mechanisms and therapeutic implications. Leukemia 2014;28:155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer 1996;74:648–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mutka SC, Yang WQ, Dong SD, et al. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res 2009;69:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Owen T, Wang W, McCauley D, et al. Novel nuclear export inhibitors deplete autoreactive plasma cells and protect mice with lupus‐like disease from nephritis. Arthritis Rheum 2012;64:suppl: abstract #2678 [Google Scholar]
- 52. Owen T, Rangel‐Moreno J, Klebanov B, et al. Novel selective inhibitors of nuclear export (SINE) decrease type I interferon activation and deplete autoreactive plasma cells in the kidney in murine lupus. In: 13th ACR/ARHP Annual Meeting; October 26–30, 2013; San Diego, CA, USA, Abstract #2890. [Google Scholar]
- 53. Tajiri N, Acosta SA, Pabon MM, et al. Selective inhibitors of nuclear export (SINE) exert robust therapeutic benefits in traumatic brain injury models. In: Tenth World Congress on Brain Injury; March 19‐22, 2014, San Francisco, CA, Abstract #0635 [Google Scholar]
- 54. Tajiri N, Acosta SA, Pabon MM, et al. A nuclear attack: Sequestration of traumatic brain injury‐induced cell death at the nuclear level via selective inhibitors of nuclear export (SINE). Cell Transplant 2014;23:suppl: abstract #785 [Google Scholar]
- 55. Landesman Y, Senapedis W, Saint‐Martin JR, et al. Pharmacokinetic (PK)/pharmacodynamic (PD) and efficacy relationship of selective inhibitors of nuclear export (KPT‐SINE). Cancer Res 2012;72:suppl: abstract #3775 22710432 [Google Scholar]