

Abstract
Genetic experiments have positioned the fgfr1 gene at the top of the gene hierarchy that governs gastrulation, as well as the subsequent development of the major body axes, nervous system, muscles, and bones, by affecting downstream genes that control the cell cycle, pluripotency, and differentiation, as well as microRNAs. Studies show that this regulation is executed by a single protein, the nuclear isoform of FGFR1 (nFGFR1), which integrates signals from development‐initiating factors, such as retinoic acid (RA), and operates at the interface of genomic and epigenomic information. nFGFR1 cooperates with a multitude of transcriptional factors (TFs), and targets thousands of genes encoding for mRNAs, as well as miRNAs in top ontogenic networks. nFGFR1 binds to the promoters of ancient proto‐oncogenes and tumor suppressor genes, in addition to binding to metazoan morphogens that delineate body axes, and construct the nervous system, as well as mesodermal and endodermal tissues. The discovery of pan‐ontogenic gene programming by integrative nuclear FGFR1 signaling (INFS) impacts our understanding of ontogeny, as well as developmental pathologies, and holds new promise for reconstructive medicine, and cancer therapy. J. Cell. Physiol. 231: 1199–1218, 2016. © 2016 The Authors. Journal of Cellular Physiology published by Wiley Periodicals, Inc.
Motto: Scientists and clinicians who study cancer (and other diseases—MKS) are caught up in the frantic and expensive search for an elusive cure but rarely ask why these diseases exist at all and what their place is in the grand story of life on Earth (paraphrased from Paul Davies, 2013, Physics World, vol. 26, pp 37–40).
Construction of a complex functional system, such as a living organism, requires not only raw building materials (genes encoding structural and other functional proteins), but also an assembly program, organized into flexible feedback and feed‐forward sub‐routines that can function within, and readily adapt to a non‐stable environment. Such a program is discussed below.
Unicellular organisms which appeared 3 billion years ago (Maynard Smith J, 1995) owed their ecological and evolutionary success to their ability to proliferate, invade new niches, and disseminate these abilities through their genetic material. The ancient genes that underwrite these functions are preserved in multicellular organisms and are often altered in cancer cells, leading to uncontrolled proliferation and migration. Thus, they are referred to as proto‐oncogenes, or tumor suppressor genes.
The ascent of multicellular organisms which begun 0.6 billion years ago, brought critical innovations. First, new categories of genes (morphogens) evolved which underwrote the cell speciation needed to form tissues and organs composed of different types of cells. Secondly, effective controls were imposed over the vestigial proto‐oncogenes/tumor suppressor genes as well as the new morphogens to coordinate their activities both in time and in space in developing multicellular organisms. In essence, the construction of a metazoan body engages thousands of genes and myriads of transcription factors. Their actions are orchestrated in the three‐dimensional space of occupied by chromatin, so as to allow the programmatic realization of the inherited genetic blueprint. How this immense task is accomplished is largely unknown.
In this article, we summarize experimental evidence outlining a new pan‐ontogenic paradigm, integrative nuclear fibroblast growth factor receptor 1 (FGFR1) signaling (INFS; Stachowiak et al., 2007, 2012b, 2015). The INFS operates at the interface of the genomic and epigenomic information. It computes and integrates diverse developmental signals, and elicits coordinate responses of thousands of genes to allow cell transitions between different developmental stages. INFS disruption is associated with cancer, and diverse developmental diseases like schizophrenia.
At the center of the INFS module are proteins that bear the historic name of fibroblast growth factors (FGF) and high affinity FGF receptors (FGFR). Neither FGFs nor FGFRs exist in single‐cell organisms, but are common to eumetazoans and essential for the generation of tissues with specialized cells (Stachowiak et al., 2015). Mutations within a single Fgfr1 gene disrupt gastrulation, as well as the development of the central and peripheral nervous systems, in addition to the development of mesodermal somites, muscles, bones, and endoderm. These developmental consequences of Fgfr1 mutations reflect changes in the expression of diverse genes (Ciruna et al., 1997; Partanen et al., 1998; Ciruna and Rossant, 2001; Dequeant and Pourquie, 2008) and microRNAs (Bobbs et al., 2012; Stuhlmiller and Garcia‐Castro, 2012) that control development, and thus, firmly place Fgfr1 at the top of the developmental hierarchy. But how can a single gene exert such global pan‐ontogenic function?
FGFs emerged early in the evolution of metazoans. One such metazoan, Caenorhabditis elegans, possesses an FGF ortholog, LET‐756, that is equipped with three nuclear localization signals (NLSs), which are functionally necessary, as its biological effects depend on nuclear accumulation (Popovici et al., 2006). Members of the mammalian FGF family similarly retain an NLS, and/or have acquired a cleavable secretion signal peptide (SP). The NLS‐containing FGFs, for example, the 23 kDa FGF‐2, act in the nucleus to promote differentiation, whereas secreted members of the FGF family, for example, 18 kDa FGF‐2, act on the cell surface, to carry out their mitogenic functions (Ornitz and Itoh, 2011; Stachowiak et al., 2015).
Individual FGFRs (in mammals, FGFR1‐4) have adaptations that direct them to different subcellular compartments (Myers et al., 2003; Stachowiak et al., 2007). An atypical transmembrane domain in FGFR1 allows the newly translated receptor to be released from the pre‐Golgi membrane into cytosol and to translocate into the nucleus, (Box 1; Fig. 1A). The accumulation of hypoglycosylated nuclear FGFR1 (nFGFR1) is highly regulated by diverse developmental signals, and is, thus, named as an integrative signaling (Fang et al., 2005; Stachowiak et al., 2007, 2015). In proliferating neural stem/progenitor cells (NS/PC) of the brain subventricular zone (SVZ), FGFR1 is cytoplasmic, while in differentiating cells FGFR1 localizes to the nucleus (Stachowiak et al., 2009, 2012a). Likewise, nuclear accumulation of FGFR1 is observed in developing midbrain substantia nigra (SN) neurons while they extend their telencephalic projections (Fang et al., 2005). When this developmental process reaches completion, FGFR1 re‐localizes, becoming predominantly cytoplasmic (Fang et al., 2005). Nuclear FGFR1 accumulation is observed also in vitro during the differentiation of diverse types of stem cells, as well as during the growth and differentiation of glial and neuronal cells, endothelial and mesodermal cells, and in cancer cells (Stachowiak et al., 1997b, 2007, 2015).
Figure 1.
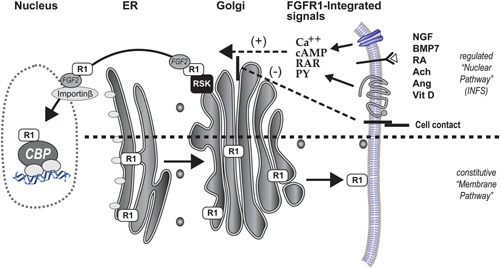
Newly synthesized FGFR1 can enter either the constitutive membrane pathway or regulated nuclear pathway in membrane pathway the receptor is processed and glycosylated in Golgi and accumulates in plasma membrane. In the nuclear pathway, an atypical transmembrane domain in FGFR1 allows newly translated immobile receptor to be released from the pre‐Golgi membrane into cytosol generating a highly mobile protein in a process that involves proteasomes and is facilitated by the FGF‐2 ligand, and ribosomal S6 kinase (Dunham‐Ems et al., 2006, 2009). The nuclear transport of FGFR1 is mediated by importin‐β (Reilly and Maher, 2001). The nuclear accumulation of the hypoglycosylated nuclear form of FGFR1 (nFGFR1) is stimulated by a variety of developmental signals, including various growth factors (i.e., EGF, NGF, BDNF, BMP), vitamins D and retinoids, hormones, and neurotransmitters, calcium, cyclic AMP, and is inhibited by cell contact receptors. This is the reason that this pathway is referred to as integrative signaling (Stachowiak et al., 2015). FRET and co‐immunoprecipitation assays show that the NLS containing 23 kDa FGF‐2 interacts with nFGFR1 during nuclear transport and in the nucleus while, 18 kDa FGF‐2, which lacks a bipartite NLS, interacts with FGFR1 only in the cytoplasm (Dunham‐Ems et al., 2009). Biophotonic assays, including FLIP and FRAP, have demonstrated that cytoplasmic FGFR1 exists in three separate populations: (1) an immobile, newly synthesized Endoplasmic Reticulum (ER) population; (2) a highly mobile, non‐glycosylated, cytosolic population; and (3) a slowly diffusing, membrane receptor population (Dunham‐Ems et al., 2006). Nuclear accumulation of FGFR1 in live cells is promoted by an accelerated cytoplasmic to nuclear import, as well by as a reduced nuclear to cytoplasmic export (Lee et al., 2012, 2013; Stachowiak et al., 2012b). In addition, activation of cell surface FGFR1 by FGF‐2 induces FGFR1 internalization, which is dependent upon the ARF6, Dynamin2 and Rab5 endocytic machinery, and is inhibited by cell surface E‐cadherin adhesion complexes (Bryant and Stow, 2005). Once internalized, FGFR1 may be released from endosomes, or trafficked in a retrograde fashion to the ER/Golgi for cytoplasmic release via the RSK1‐associated pathway.
Box 1. Constitutive plasma membrane and regulated nuclear targeting of FGFR1.
Dual plasma membrane and nuclear FGFR1 distribution is observed in developing cells (Stachowiak et al., 2015). For instance in proliferating brain stem cells FGFR1 is cytoplasmic whereas in differentiating neural cells FGFR1 accumulates in the cell nucleus (Stachowiak et al., 2012a). In agreement with this dual distribution, brain‐targeted FGFR1 knockout impairs both the cell proliferation and differentiation (Pirvola et al., 2002; Ohkubo et al., 2004) which may reflect the loss of FGFR1 signaling at the cell surface and the INFS, respectively (Stachowiak et al., 2012b).
Gene Transcription Is Regulated by nFGFR1 Acting Cell Nucleus
nFGFR1 concentrates in nuclear speckles, which are pre‐RNA co‐transcriptional processing factories (Peng et al., 2002; Lee et al., 2012, 2013; Somanathan et al., 2003; Stachowiak et al., 2003b, 2012b). Within these speckles nFGFR1 affects the rate of new RNA synthesis.
Studies focused on genes encoding for neural Tyrosine Hydroxylase (TH), enolase (NSE), neurofilament‐l (NF‐l), doublecortin, βIII‐tubulin, developmental fgf‐2, c‐Jun, cyclin D1, nurr1, and nur77 showed that they can be activated following transfection with a constitutively active nuclear FGFR1(SP‐/NLS), or by nuclear HMW‐FGF‐2 ligand (Peng et al., 2001; Reilly and Maher, 2001; Lee et al., 2013; Narla et al., 2013). In contrast, a dominant negative nuclear FGFR1(SP‐/NLS)(TK‐) blocked gene and promoter activation by neurotransmitters, hormones, growth factors, Ca++, cAMP or tyrosine kinase signaling. However, extracellular FGFR1 antagonists do not prevent such gene activations. Similarly, blocking extracellular ligand‐induced FGFR1 internalization does not affect gene activation by transfected FGFR1 and its nuclear ligand, HMW FGF‐2. Hence, the gene activation by nFGFR1 is not a consequence of ligand‐induced receptor internalization from the cell surface. These results indicate that instead, these gene activations are mediated by intracellular, nuclear nFGFR1, and that an increase in nFGFR1 content is sufficient, and essential for the activation of these specific genes (Stachowiak et al., 2003b, 2012b; Peng et al., 2001, 2002). DNA electrophoretic mobility shift assays (EMSAs) and cross‐linked chromatin immunoprecipitations (ChiPs) demonstrated that nFGFR1 binds to the promoter regions of nFGFR1 regulated genes (including nurr1 and nur77, fgfr1 and fgf‐2, dcx, th, βIII tubulin)) along with its binding partner, a common transcriptional coactivator, CREB binding protein (CBP; Peng et al., 2002; Fang et al., 2005; Narla et al., 2013; Lee et al., 2012, 2013). Another binding partner of CBP is ribosomal S6 kinase (RSK; Hu et al., 2004). nFGFR1 disassociates inactive RSK‐CBP complexes, replacing them with FGFR1‐CBP complexes that activate RNA Pol II and histone acetylation (Fang et al., 2005), as well as with FGFR1‐RSK1 complexes, in which FGFR1 augments RSK1 phosphorylation of TFs and, potentially, other chromatin proteins (Hu et al., 2004; Fang et al., 2005; Fig. 2A). The dynamic nature of gene regulation by nFGFR1 and its partner proteins (Dunham et al., 2004; Dunham‐Ems et al., 2009; Baron et al., 2012; Lee et al., 2012) is illustrated in Box 2 (Fig. 2B).
Figure 2.
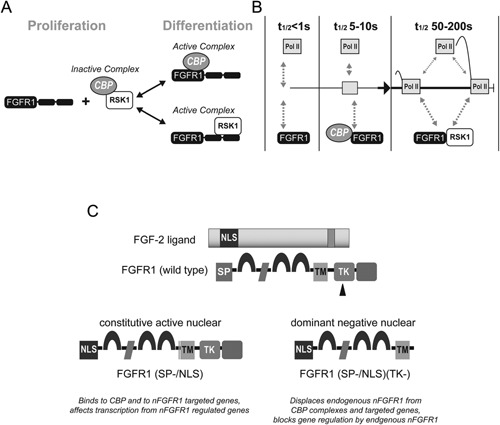
(A) The investigation has framed a threefold mechanism for nFGFR1 activation of CBP‐dependent transcription (Fang et al., 2005) in proliferating non‐differentiated cells, CBP is arrested in an inactive complex with RSK1 or RSK2. As FGFR1 accumulates in the nucleus, the TK domain binds to the N‐terminal kinase domain of RSK and disrupts the CBP‐RSK complex. The N‐terminal domain of another FGFR1 molecule interacts with the N‐terminal domain of CBP forming a CBP‐nFGFR1 complex. The nFGFR1‐CBP complex activates transcription by recruiting RNA Pol II and acetylating histones (Fang et al., 2005), while RSK1‐bound nFGFR1 phosphorylates TFs and, potentially, other chromatin proteins. (B) Kinetic model of INFS from FRAP mobility measurements (Dunham‐Ems et al., 2009; Stachowiak et al., 2012b). Bimodal analyses of the R1‐EGFP FRAP recovery demonstrate that nuclear FGFR1 contains a hyperkinetic component (“F” Fast recovering t1/2 < 1 sec; present in nucleoplasm), a hypokinetic component (“S” Slow‐recovering, t1/2 = 24 sec; chromatin‐associated), and an immobile component (non‐recovering, nuclear matrix‐associated). FGFR1 that is not engaged in transcription associates with nuclear matrix and is immobile. Activation of transcription by cAMP releases FGFR1 from matrix via an FGFR1 interaction with 23 kDa FGF‐2 generating an “F” R1, which engages in rapid (t1/2 < 1 sec) “non‐productive” molecular collisions and chromatin scanning. R1‐CBP binding converts “F” R1 into hypokinetic “S” R1 (t1/2 5–10 sec). We propose that the “S” FGFR1 represents FGFR1‐CBP oscillations that drive the formation of the RNA Pol II (Pol II) Preinitiation Complex (PIC). CBP binding to DNA‐associated transcription factors may extend the residence of CBP‐FGFR1 on chromatin, thereby promoting initiation of transcription. During transcriptional activation, the rate of oscillations is further reduced (“S” R1 converts into “ES”, (t/12 > 50 sec), possibly reflecting FGFR1 and RSK1 binding events and the formation of productive elongating complexes. The kinetics of RNA Polymerase II (RPII) are based on the methods of (Darzacq et al., 2007) and are similar to FGFR1. Close co‐localization of nFGFR1 and RNA Pol II is illustrated by immunostaining and super‐resolution microscopy in Video—Supplemental Material (collaborative experiment with Dr. Hari Shroff, NIH) l (Video 1). (C) Engineering constitutive active and dominant negative FGFR1. Testing function of nFGFR1 by gain and loss experiments—transfection of the constitutively active/nuclear variant FGFR1(SP‐/NLS), in which the cleavable SP is replaced with the NLS of FGF2, provides a means to generate INFS signals directly in the nucleus bypassing the afferent stimuli. Transfection of dominant‐negative variants of FGFR1(SP‐/NLS)(TK‐), which lack the tyrosine kinase (TK) domain, block nFGFR1 function specifically in the cell nucleus (Stachowiak et al., 2012b, 2015). The tyrosine kinase deleted (TK‐) mutant displaces endogenous nFGFR1 from the CBP complex and the gene promoter (Peng et al., 2002).
Box 2. Nuclear FGF receptor‐1 and CREB binding protein—an integrative signaling module (Stachowiak et al., 2015).
The interaction of CBP with hypo‐glycosylated nFGFR1 was indicated by GST‐CBP pull downs of FGFR1, coimmunoprecipitations of CBP and nFGFR1, and by biophotonics of live cells (Fang et al., 2005; Dunham‐Ems et al., 2009). CBP associates with diverse TFs during their binding to gene regulatory sequences. Together they recruit RNA Pol II and thus may promote the formation of preinitiation complexes (PICs) that include RNA Pol II as well as the enzymatic activities needed for the chromatin remodeling required for transcriptional initiation.
Transfection studies conducted with constitutively active nuclear FGFR1(SP‐/NLS), and its dominant‐negative variant FGFR1(SP‐/NLS)(TK‐) (Fig. 2C) in vitro, showed that (1) nFGFR1 alone is sufficient to induce changes in gene activities and neuronal differentiation in cultured neural progenitor cells, and diverse types of stem cells, and (2) that nRGFR1 is necessary for RA, NGF, BMP, BDNF, acetylcholine, and cAMP induced neuronal differentiation (Horbinski et al., 2002; Stachowiak et al., 2003a; Fang et al., 2005; Lee et al., 2013). In vivo transfection of FGFR1(SP‐/NLS), or activation of endogenous nFGFR1 was also shown to reactivate developmental‐like neuronogenesis by neural stem/progenitor cells in adult brain (Bharali et al., 2005; Stachowiak et al., 2009). These gain and loss of function experiments demonstrated that nFGFR1 and INFS broadly participate in developmental transitions, most commonly as a switch to differentiation and post‐mitotic development (Stachowiak et al., 2015).
nFGFR1 Programs Development of Pluripotent Stem Cells
In early embryogenesis, soon after conception, cells in the interior of the blastocyst proliferate rapidly to yield a growing mass of cells from which the organism will be built. These non‐differentiated embryonic stem cells (ESC) are defined as pluripotent, that is, give a rise to any of the three embryonic layers ecto‐, endo‐, or mesoderm as they form during gastrulation (Gluache, 2011). The exit of ESC from the pluripotent state is induced by retinoic acid (RA) which has broad ontogenic functions (Morriss‐Kay and Sokolova, 1996; Kam et al., 2012). RA triggers transcriptional cascades that cause cells to differentiate into neuronal, cardiac, myogenic, adipogenic, and vascular smooth muscle cells, with the exact outcome depending on ligand concentration. At high concentrations (1–10 μM) RA promotes development specifically along the neuronal lineage, while also inhibiting the generation of glial, mesodermal or endodermal cells (Guan et al., 2001; Okada et al., 2004; Akanuma et al., 2012; Kam et al., 2012; Lee et al., 2012).
Nuclear accumulation of FGFR1 is a common response to RA in human ESCs and mESC. Furthermore, loss‐ and gain‐of‐function experiments have demonstrated that nFGFR1 is both necessary for RA‐induced differentiation of neurons, as well as sufficient to induce neuronal differentiation in the absence of RA stimulation (Lee et al., 2012). How can a single nuclear protein program the neural development of ESCs—a process that involves the coordinated regulation of thousands of genes that are located on different chromosomes and contain diverse regulatory elements? Given that CBP interacts with a large variety of TFs, nFGFR1 can potentially act as a global master regulator that delivers the RA signal to a large number of genes, including those that lack an RXR or Nur‐related site (Fig 3B, Box 3). Indeed nFGFR1‐xdCBP complexes form even larger complexes with RXR, RAR, and orphan Nur receptors, as well as with CREB and other TFs (Forthmann et al., 2015; Stachowiak et al., 2015). nFGFR1 synergistically regulates transcription by these TFs at their targeted sequences: RARE, NBRE, NurRE, CRE, etc. (Stachowiak et al., 2015).
Figure 3.
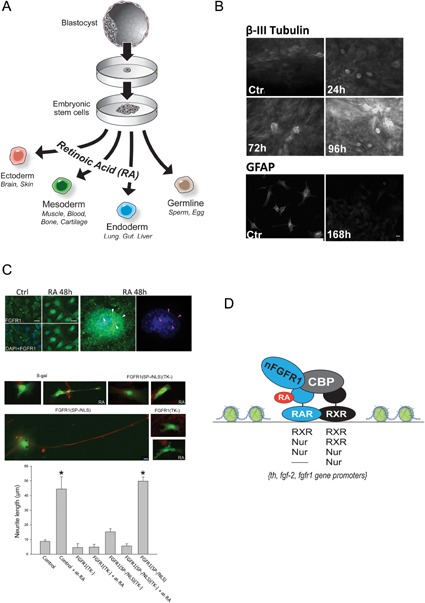
(A) During early embryogenesis embryonic stem cells (ESCs) appear in the inner cell mass of the blastocyst. Each of ESC is capable of developing into an organism with all of its tissues and thus is referred to as pluripotent. Retinoic acid appears in the primitive streak during gastrulation when three germ layers are formed, and has broad regulatory functions in ontogenesis including axis development in the vertebrate embryo (Morriss‐Kay and Sokolova, 1996). RA stimulates pluripotent cells to differentiate into neuronal, cardiac, myogenic, adipogenic, and vascular smooth muscle cells, depending on ligand concentration. (B) In vitro, at high concentrations (1–10 μM), RA promotes the exit of ESCs from the pluripotent state and their development specifically into the neuronal lineages. Within a few hours, nFGFR1 accumulates in the nuclei of ESC,s as the cells exit from the cell cycle, and neurogenic and neuronal genes are upregulated. By 24 h, the cells display a neuronal morphology (including long neurites and growth‐cone endings), and express neuron‐specific β‐III tubulin (B), MAP2, neurofilament L, TH (Lee et al., 2012), as well as glutamate and acetylcholine receptors (Guan et al., 2001; Okada et al., 2004; Akanuma et al., 2012; Lee et al., 2012). After 96 h of at‐RA treatment, only single cells displayed characteristic glial morphology and GFAP immunoreactivity (B), (Lee et al., 2012). Bar size—20 µm. With additional time and appropriate conditions the ESCs develop complex neuronal networks and 3D brain‐like organoids (not shown). Reprinted with permission from J Cell Biochem 113:2920–2936. (C) RA acid stimulates the nuclear accumulation of nFGFGR1 in ESCs which is both necessary and sufficient for RA‐induced neuronal differentiation. Top—ESC were incubated with or without (control) 1 µM at‐RA for 48 h and immunolabeled with N‐terminal αFGFR1 (ABCAM) (+goat‐anti mouse‐Alex488). On the enlarged nucleus arrowheads point to weakly stained (DAPI) euchromatin regions with high FGFR1‐IR after at‐RA stimulation. Center and bottom—the at‐RA‐induced outgrowth of β‐III‐tubulin containing neurites in mESC is inhibited by transfected dominant negative nuclear FGFR1 (FGFR1(SP‐/NLS)(TK‐) and by cytoplasmic/nuclear FGFR1(TK‐). In the absence of at‐RA, the average neurite outgrowth induced by active nuclear FGFR1(SP‐/NLS) is similar to the at‐RA induced outgrowth. Bar size—20 µm (Lee et al., 2012). (D) RA signaling is mediated by both retinoic acid receptors (RARs) and retinoid X receptors (RXRs), which can act as homo or heterodimers on RA‐responsive elements (RAREs) within RA‐regulated genes (Lefebvre et al., 2010). Additionally, RXR is highly versatile with respect to its heterodimerization; among the many other nuclear receptors with which it can interact are two members of the orphan nuclear subfamily, Nur77 and Nurr1. These factors also function independently by binding Nur‐response elements, as monomers (NBRE) and dimers (NurRE) (Maira et al., 1999; Maira et al., 2003; Lefebvre et al., 2010). nFGFR1 forms CBP‐containing low mobility chromatin complexes with RXR, RAR, and Nurs. These complexes bind to RARE, NBRE, and NurRE sequences within RA‐activated Fgfr1, Fgf‐2, and Th genes, and synergistically activate isolated RA‐ and Nur‐responsive elements (Baron et al., 2012; Lee et al., 2012, 2013).
Box 3. Ontogenic programing of ESC—role of RA.
Global Genome Targeting by nFGFR1
We speculated that nFGFR1 mediates RA‐induced neural programming of ESCs by targeting “master developmental” genes and/or interacting directly with multiple sets of genes within diverse development pathways. The ascent of next generation DNA sequencing and its applications to chromatin immunoprecipitation (ChIP) and global RNA analyses enabled us to identify gene networks that are directly targeted by FGFR1 and its partners, RXR and Nur77, and to characterize the associated gene regulation during RA‐induced neuronal programing of mESCs.
Experiments were performed on nondifferentiated female mouse (m)ESCs maintained in the pluripotent state with leukemia inhibitory factor (LIF) on LIF‐free monolayers treated with 1 µM RA for 2 days to induce neuronal cell (NC) differentiation (Terranova et al., 2015). ChIPseq revealed that nFGFR1, RXR, and Nur77 bind to thousands of sites nonrandomly distributed on all mouse chromosomes (Box 4, Fig. 4A). There are 30,586 RXR binding sites in ESCs but only 15,224 in NCs. The number of Nur77 sites are similar in ESCs (22,651) and in NCs (25,995). In contrast, the number of nFGFR1‐targeted sites in pluripotent ESC is 11,378, much smaller than the number of sites for either RXR or Nur77. However the number of nFGFR1 sites in NCs (46,137) is fourfold higher than in pluripotent ESCs. Thus, during neuronal differentiation, nFGFR1 targeted sites become more abundant than either the RXR or Nur77 targeted sites (Terranova et al., 2015).
Figure 4.
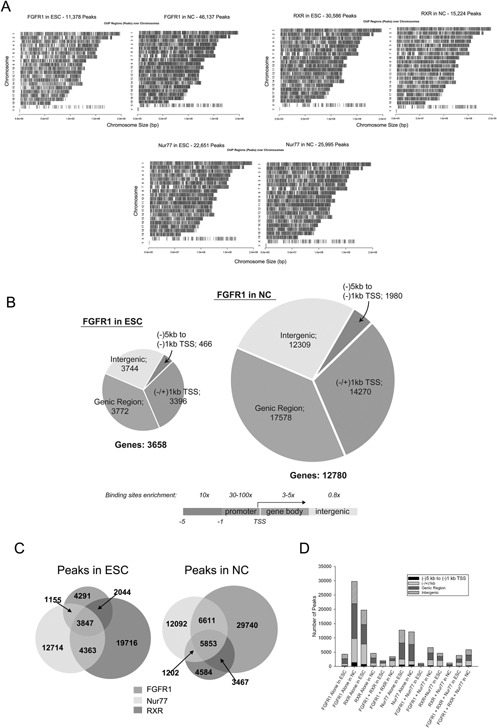
(A) nFGFR1, RXR, and Nur77 peaks are heterogeneously distributed on all chromosomes in both ESCs and NCs. The density of binding sites on Ch. X was approximately fivefold lower than on other chromosomes (chromosome Y is absent). (reprinted from PLoS ONE 10:e0123380). (B) In mESC and NC genomes 2/3 of targeted sites are within promoter and genic regions. Relative to their global genomic representation, one observes an enrichment of FGFR1, RXR or Nur77 peaks within 5'UTR and exon regions and lack enrichment in the intron or 3'UTR regions. Within the promoter, the highest enrichment was found in the bidirectional promoter (50‐ to 100‐fold). In unidirectional promoters 10‐ to 30‐fold enrichment was observed −1/+1 kb from TSS and 5–10 fold in −1/−5 kb. (C) Genome‐wide colocalization of nFGFR1, RXR, and Nur77 peaks. Venn diagram illustrates the number of individual and overlapping nFGFR1, RXR, and Nur77 binding sites (Reprinted from PLoS ONE 10:e0123380). (D) nFGFR1, RXR, and Nur77 peaks co‐localize within all genomic regions. Specifically in the proximal promoter and NCs, the number of sites at which RXR or Nur77 bind together with nFGFR1 was markedly higher than the number of sites at which either RXR or Nur77 bind without nFGFR1. (Reprinted from PLoS ONE 10:e0123380).
Box 4. Genome‐wide binding of nFGFR1, RXR and Nur77 in pluripotent ESC and RA‐induced NC and enrichment of binding sites within promoter and genic regions (Terranova et al., 2015).
The binding sites for nFGFR1 are present throughout the genome, similar to those for RXS and Nur77. Of particular interest in these regards, nFGFR1 binding sites are most highly concentrated within gene regulatory regions. Similarly, the binding of all factors to genomic DNA is highly enriched within the upstream proximal promoters (−1 kb), the bidirectional promoter and the 5'UTR, but not in the 3′UTR, or introns (Box 4, Fig. 4B; Terranova et al., 2015).
In ESCs, 1/5th of all of the RXR and all the Nur77 binding sites colocalize with nFGFR1 binding sites, and nearly half (45%) of all sites co‐occupied by RXR+Nur77 co‐localize with nFGFR1 binding sites. What distinguishes NCs from ESCs, is that the abundance of all categories of sites that include nFGFR1 are markedly higher (two‐ to threefold), even though the numbers of RXR and RXR+Nur77 are reduced. Most notably, large numbers of binding sites for nFGFR1 alone are present in ESCs as well as in NCs (37% in ESCs and 64% in NCs; Box 4, Fig. 4C). Specifically, within the proximal and distal promoter regions, prominent increases are observed in nFGFR1 binding, regardless of whether nFGFR1 binds alone, or co‐localizes with RXR or Nur77. However, the number of sites that bind RXR and RXR‐Nur77, but lack nFGFR1 is low, and the number of sites that bind Nur77 alone are relatively unchanged (Box 4, Fig. 4D).
Combined RNA‐seq and ChIP‐seq analyses (Box 5) reveal that >85% of genes with promoters targeted by nFGFR1, RXR, Nur77, individually or in combination, are in an active (mRNA expressing) state (Fig. 5A and B) including proximal promoters of both up‐ and down‐regulated genes (Fig. 5C). During the transition of ESCs to NCs, the numbers of expressed and regulated genes with promoters targeted by nFGFR1 (either alone or in combination with RXR, Nur77 or both), increases markedly. In contrast, the population of genes bound by RXR and/or Nur77, but not nFGFR1, diminishes. Likewise, the increased incorporation of the histone variant H3.3 (a marker of gene activation) into nFGFR1 containing peaks is observed both in expressed and differentially regulated genes, further relating nFGFR1 binding to gene regulation (Box 5, Fig. 5D; Terranova et al., 2015). Thus, nFGFR1, acting either alone or together with RXR and Nur77, emerges as an active mediator of RA‐induced gene programing, consistent with the instructive function of nFGFR1 in the differentiation of ESCs into NCs (Lee et al., 2012; Terranova et al., 2015).
Figure 5.
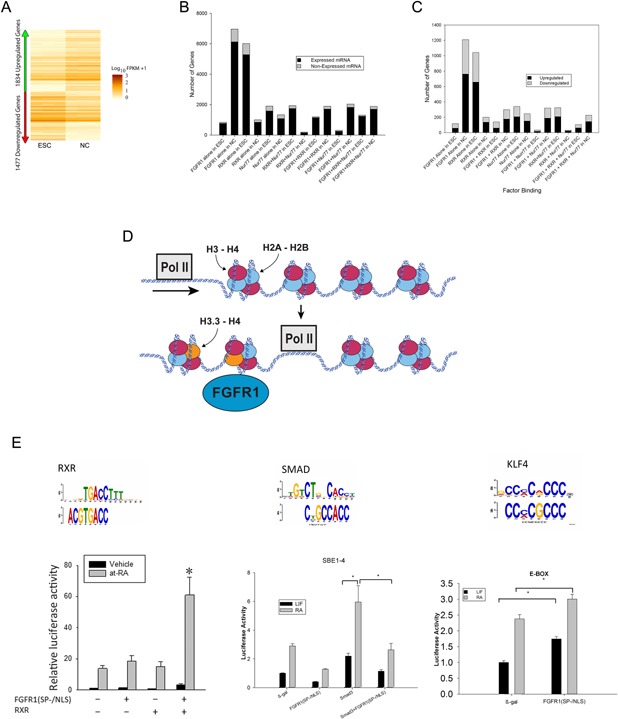
(A) Heatmap representation of genes that are differentially expressed in pluripotent ESCs and RA‐induced NCs from three biological replicates. Out of 14,443 expressed genes, 1,834 were up‐regulated and 1,477 were down‐regulated in NCs (fold change [FC] >±2.0 and P‐value <0.035 were considered significant). Values are displayed as fragments per kb of transcript per million fragments mapped (FPKM). (B) >85% of promoter targeted sites are on active, expressed, genes. (C) Approximately 60% of genes with proximal promoters targeted by nFGFR1, RXR, and Nur77 or their combinations were upregulated and approximately 40% of genes were downregulated. In NCs, the population of regulated genes targeted by RXR was reduced, the number of regulated genes targeted by Nur77 was not changed, while the number of regulated genes targeted by nFGFR1 alone, nFGFR1+RXR, nFGFR1+Nur77, or nFGFR1+RXR+Nur77 increased several fold. In NCs, the population of regulated genes that were targeted by nFGFR1 (2,058 genes) constituted over 62% of all differentially regulated genes; that is, it was noticeably larger than the population of regulated genes that did not bind nFGFR1 (480 genes). (D) Increased incorporation of histone variant H3.3 (marker of gene activation), into nFGFR1 peaks on gene promoters links even more nFGFR1 binding to gene regulation. (E) nFGFR1 targeting of different promoter elements confers gene activation or inhibition. (A‐E reprinted or modified from PLoS ONE 10:e0123380), E—left part reprinted with permission from J Cell Biochem 113:2920–2936.
Box 5. Binding of nFGFR1, RXR and Nur77, and gene regulation (Terranova et al., 2015).
The role of nFGFR1, either individually, or in combination with RXR and/or Nur77, is further illustrated by the overlapping lists of recognition motifs in the target DNAs of these proteins (Box 5, Table I). nFGFR1 associates with sequences targeted by the classical nuclear receptors Nur77 and/or RXR, as well as others that are not. nFGFR1‐targeted sites encompass the consensus sequences of CREB and other diverse TFs, all of which interact with CBP, and thus may engage in nFGFR1‐CBP mediated transcriptional regulation. The nFGFR1 regulation of the identified elements has been verified in several genes, using isolated, luciferase‐linked elements. These experiments illuminated a correlation between nFGFR1 binding with gene activation, as well as inhibition, as nFGFR1 targeting of different promoter elements can produce opposing transcriptional effects (Box 5, Fig. 5E; Fang et al., 2005; Lee et al., 2012; Stachowiak et al., 2012b; Terranova et al., 2015).
Table I.
The DNA sequences targeted by nFGFR1, as with RXR and Nu77 targeted sequences, displayed a high evolutionary conservation across vertebrate species corroborating their importance as genomic regulators (ref)
| Factor | Motif (bold are unique to factor) |
|---|---|
| nFGFR1 | ARNT, ATF1, CTCF, ERG/ELK4, KLF4, MAX, MZF1, NRF1, Nurr1, Pou2f3, Pou5f1:Sox2, RARα, RFX1, RXRα, SMAD, Sox8, SP1, STAT, TCF3, TP53, YY1, ZBTB33, ZFP161 |
| RXR | ATF1, CTCF, Irx4, Mycn, MZF1, Nurr1, Nr1h3:RXRα, Pitx2, Pou3f3, PPARG, Prrx2, RARα, RFX1, RXRα, Sox8, SP1, YY1, ZEB1, ZFP161 |
| Nur77 | ATF1, CTCF, Hic1, MAX, Mycn, NRF1, Nurr1, Pax6, Prrx2, Six6, SP1, STAT, RXRα, YY1, ZFP161 |
Table shows the results of motif analyses of sequences targeted by nFGFR1, RXR and/or Nur77. MEME‐ChiP and other methods (Terranova et al., 2015) verified earlier findings (Lee et al., 2012) that nFGFR1 associates not only with the core AGGTCA sequences targeted by the classical nuclear receptors Nur77 and/or RXR, but also to sequences that are not targeted by Nur77 and/or RXT. nFGFR1‐targeted sites encompass the consensus sites for CREB, STAT, Sp1, and Smad as well other diverse TF that interact with CBP, and thus may engage in the nFGFR1‐CBP mediated transcriptional regulation. These observations substantiated earlier findings that FGFR1 which lacks its DNA binding domain, can associate with gene promoters via CBP (Fang et al., 2005; reprinted from PLoS ONE 10:e0123380).
The extensive list of nFGFR1‐targeted motifs highlights how widespread the use of the mechanism underlying RA‐initiated gene regulation is, and the vast size of the population of responsive genes. The discovery that this regulation involves a plethora of TFs beyond RAR/RXR and Nurs, and reveals that RA‐induced transfer and retention of nFGFR1 in the nucleus (Lee et al., 2012), and nFGFR1 gene targeting represent a global paradigm of gene programing (Terranova et al., 2015).
nFGFR1 Binds to and Controls Genes of Pluripotency Core
Core networks of interconnected TFs control the ability of embryonic stem cells (ESCs) to maintain their pluripotent state, or to differentiate into three germ layers (Ivanova et al., 2006; Chen et al., 2008; Box 6, Fig. 6A and B). Research efforts have focused primarily on the identification of pluripotency genes in ESCs, and on the reconstitution of their activities in induced pluripotent stem cells (iPSCs). Much less is known about the mechanisms that turn off pluripotency genes, so that cells may abandon their nondifferentiated state, and develop into a multitude of lineages.
Figure 6.
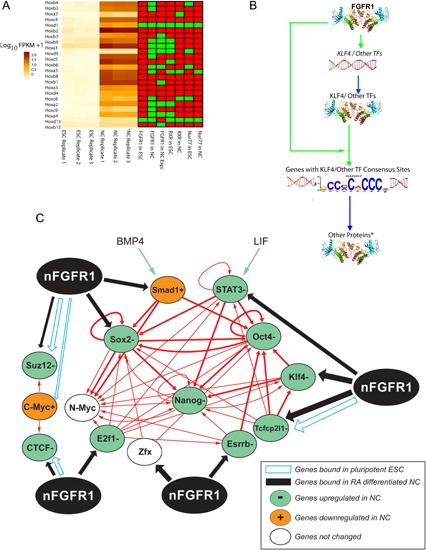
(A) Heat‐map illustrating the expression patterns of core pluripotent genes (left) and associated proximal promoter binding of nFGFR1, RXR and Nur77 (right; Terranova et al., 2015; reprinted from PLoS ONE 10:e0123380). (B) The pluripotent gene network is based on Chen et al. (2008) (Ivanova et al., 2006). nFGFR1 binding to gene promoters is marked by blue and black arrows. Genes expressed at high levels in ESC and repressed in NC are marked with green ovals, genes activated in NC with orange ovals and genes expressed in ESC and NC at the same levels with white ovals (Terranova et al., 2015). These pluripotency TFs are wired to the ES genome in two major ways. The first cluster includes the self‐assembling Oct4 and Sox2 complex, their partner Nanog. The second cluster consists of c‐Myc, n‐Myc, Zfx, and E2f1, which with additional self‐renewal regulators Esrrb, Tcfcp21, and Klf4 wired to and control the first cluster as shown in Fig. B. Among those genes, important OSKM group (Oct4, Sox2,Klf4, and Myc) has been distinguished by the ability to induce pluripotent state in fibroblast and other differentiated cells (Tanabe et al., 2014). Maintenance of the self‐renewing state of the mouse embryonic stem cells (ESCs), also requires the environmental factor cytokine, LIF, which activates STAT3 through phosphorylation and serum derived BMP4 which triggers Smad1 phosphorylation. BMP4, acts in conjunction with LIF to maintain the self‐renewal and pluripotency of mESC by activating the first cluster Oct4, Sox2, and Nanog cluster genes (Ying et al., 2003). Closely related to the pluripotency networks are Polycomb repressor SUZ12 and CTCF an insulator binding protein. Suz12 protein binds to diverse genes, which direct cell development and differentiation, but which are repressed by SUZ12 in ESC. As the SUZ12 is replaced by E2f1 or other coactivators, the developmental genes become activated, cells exit from the pluripotent state and begin lineage development. The CTCF is the vertebrate insulator and chromatin architectural protein. CTCF forms multi‐subunit protein complexes with cohesion, which differentially co‐localizes in the vicinity of specific CTCF binding sites (Balakrishnan et al., 2012). The CTCF protein controls expression of gene multi‐gene programs including the self‐renewal network. In the pluripotent ESC (+LIF), Suz12, Myc, Tcfcp2l1, and Stat3 are under direct inhibition by nFGFR1 (Terranova et al., 2015). In RA‐differentiated NC the Klf4, Sox2, Stat3, E2f1, Esrrb, Suz12, Smad1, Zfx, Tcfcp2l1, and Ctcf pluripotency genes become down‐regulated in a process that involves direct promoter binding by nFGFR1 and accompanied by a loss of RXR binding. The Oct4 and Nanog genes, which do not bind nFGFR1 are repressed by nFGFR1 indirectly in ESC and NC. The repression of Klf4, Sox2, Stat3, E2f1, Esrrb, Suz12, Smad1, Zfx, Tcfcp2l1, Ctcf, Oct4, and Nanog genes in differentiating NC by endogenous nFGFR1 was demonstrated by transfection of dominant negative nuclear FGFR1 (SP‐/NLS)(TK‐). FGFR1 (SP‐/NLS)(TK‐) profoundly increased expression of the pluripotency genes and prevented cell differentiation. Moreover, the same pluripotency genes were turned off by constitutively active nuclear FGFR1 (SP‐/NLS) which stopped cell self‐renewal and induced differentiation. Figure is based on Terranova et al. (2015) and Chen et al. (2008). (C) Dual level of transcriptional regulation by nFGFR1. In the case of Klf4 and several other pluripotent TFs (i.e., TP53, SMAD, CTCF, MYC, OCT4, SOX2, STAT3, RXR, Nurr1, and Nur77) nFGFR1 interacts with TFs encoding genes as well as with the consensus sequences to which they bind. This dual level regulation implies a feed‐forward mechanism, in which nFGFR1 controls both the generation of TFs and their downstream function to finely tune the pluripotency and other ontogenetic gene networks (modified from PLoS ONE 10:e0123380).
Box 6. nFGFR1 targets and regulates pluripotent core genes and motifs targeted by pluripotent transcription‐factors (TFs; (Terranova et al., 2015) and linked database).
In the center of the pluripotency network resides a self‐assembling TF complex of Oct4 and Sox2, and their partner Nanog. Genes encoding these factors are regulated by c‐Myc, n‐Myc, Zfx, and E2F1 as well as additional self‐renewal regulators genes: Esrrb, Tcfcp21, and Klf4 (Fig. 6B). Environmental cytokines BMP4 and LIF activate the Oct4, Sox2 and Nanog cluster genes (Ying et al., 2003), so as to maintain their capacity for self‐renewal and pluripotency. In addition, the self‐renewal network is controlled by chromatin architectural protein CTCF, while cellular differentiation is prevented by SUZ12, a component of Polycomb Repressive Complex II that binds to developmental regulatory genes. In ESCs, nFGFR1 has the capacity to bind to the promoter of SUZ12, so as to repress SUZ12‐mediated repression. Similarly, nFGFR1 binds within the promoters of Myc and Tcfcp2l1, so as to act as a partial repressor (Fig. 6A and B). These repressive effects of nFGFR1 are only a small proportion of the broad categories of the pluripotency genes targeted by nFGFR1 in ESCs (Terranova et al., 2015).
The RA induced differentiation of pluripotent ESCs into NCs was accompanied by nFGFR1 binding to promoters of the Klf4, Sox2, Stat3, E2f1, Esrrb, Suz12, Smad1, Zfx, Tcfcp2l1, and Ctcf genes, and nFGFR1‐mediated their inactivation (Fig. 6A and B; Terranova et al., 2015). Transfection of the constitutively active nuclear variant FGFR1(SP‐/NLS) into ESCs was sufficient to repress these pluripotency genes, even in the absence of RA treatment (Terranova et al., 2015), and to induce cellular differentiation, similar to that induced by RA does (Lee et al., 2012). These experiments have established nFGFR1 is a repressor of the pluripotency core during cellular differentiation. The inactivation of the Klf4, Sox2, Stat3, E2f1, Esrrb and Suz12, Smad1, Zfx, Tcfcp2l1, and Ctcf genes following the recruitment of nFGFR1 to their proximal promoters, was accompanied by the disassociation of RXR and Nur77 from many of the same sites (Fig. 6A). These findings suggest that while RXR and Nur77 bind and regulate core pluripotency genes in undifferentiated cells, nFGFR1 binds to and down‐regulates the same genes during neuronal differentiation (Fig. 6B; Terranova et al., 2015). In addition, loss and gain of function experiments demonstrate that nFGFR1 also represses Oct4 and Nanog, although these genes do not bind nFGFR1. This indirect inhibition could involve the binding and inhibition of the Tcfcp2l1 and Klf4 genes by nFGFR1. Normally, these two upstream genes, Tcfcp2l1 and Klf4, activate Oct4, and Nanog, as outlined in the pluripotency network (Box 6, Fig. 6B).
nFGFR1 binding and inhibition of the SUZ12 and Ctcf genes is an additional, new mechanism to coordinately switch off pluripotency genes, and turn on genes involved in cellular differentiation. The CTCF protein associates with the pluripotency genes NANOG, SOX2, and cMYC. In ESCs, the depletion of CTCF accelerates the loss of pluripotency, and disrupts the process of differentiation. In our studies, we found that nFGFR1 binds to the promoter of the Ctcf gene and inhibits its expression (Terranova et al., 2015). Moreover, nFGFR1 targets DNA sequences that are CTCF binding sites. Given these dual actions (Box 6, Fig. 6C), nFGFR1 may act as an effective regulator of high‐order chromatin structure. nFGFR1's global control over cellular pluripotency and lineage‐specific gene expression is likely to be related such mechanism.
A similar dual nFGFR1 action is observed in other genes that encode TFs affecting pluripotency and other gene networks (Box 6, Fig. 6C). Indeed, nFGFR1 binds to the promoters of the Myc, Smad, Klf4, and Sox2, Oct4, and Stat3 genes, as well as to the consensus DNA sequences recognized by their proteins. This dual‐level of regulation, with nFGFR1 controlling the generation of TFs, as well as their downstream actions, offers a feed‐forward mechanism for fine tuning complex ontogenic networks (Terranova et al., 2015).
nFGFR1 Binds to and Regulates Genes of Neural Development (Box 7, Fig. 7)
Figure 7.
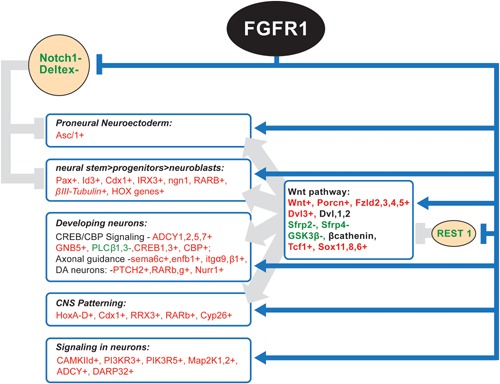
nFGFR1 targets and regulates key genes that program and execute different stages of neural development (based on the results of ChiPseq, ChiP, RNAseq, and RNA analyses). nFGFR1 removes “developmental road blocks” imposed by anti‐neural Notch1 and REST genes. nFGFR1 binds and inhibits the Notch1 gene in NC and vacates Deltex1 gene promoter in down‐regulated Deltex1 gene. nFGFR1 shows dynamic binding to the REST gene promoter and 3′ region indicating nFGFR1 role in the REST inactivation in differentiated NC. nFGFR1 targets and activates several master gene that instruct neural development. Those include proneural Ascl1, and multiple genes in the Wnt pathway. In general nFGFR1 binding correlates the activation of genes that stimulate or transduce WNT signals (red +) and with down‐regulation of the genes that inhibit Wnt receptors (green −). nFGFR1 binding activates neuronal developmental genes Pax, Id3, Cdx1, IRX3, CREB/CBP signaling genes, and the CNS patterning genes. nFGFR1 targets activated axonal guidance genes, and genes involved in synaptic plasticity and development of DA neurons.
Induction of neuroectoderm
nFGFR1 is both essential and sufficient for the programing of neuronal development by brain neural stem/progenitor cells in vivo, as well as by ESCs, neural progenitor cells (NPC), and neural tumor cell lines in vitro (Stachowiak et al., 2015). In agreement with these functions, nFGFR1 targets genes which govern the development of embryonic neuroectoderm, neural tubes, in addition to the patterning of the central nervous system, the development of neuronal cells and networks, the induction of the neural crest and the development of the peripheral nervous system (Box 7, Fig. 7; (Terranova et al., 2015) and linked database).
Box 7. nFGFR1 controls multiple stages in neural development ((Terranova et al., 2015) and linked database).
The event that immediately precedes the induction of neuroectoderm is the formation of a proneural cluster on the dorsal surface of vertebrate embryos. This latter event is initiated by the expression of the Ascl1 gene (achaete and scute, previously Mash1). Ascl1 encodes for a basic helix‐loop‐helix TF that causes epidermal cells to acquire neural competence, and later defines the neural lineages generated in the neurogenic ventricular zone of the brain (Bertrand et al., 2002). These Ascl1 actions are facilitated by cooperative Wnt, Frizzled (Fz), Disheveled (Dvl) signaling. The Dvl protein is recruited by Wnt activated Frizzled receptors, and relays signals downstream to β catenin, which is in turn liberated from an inactive complex with glycogen synthase kinase‐3β (GSK‐3β), permitting the β catenin‐activated transcription factors Tcf/LEF to stimulate neurodevelopmental genes (Gao and Chen, 2010).
Expression of the Ascl1 gene increases several‐fold during RA induced neuronal differentiation of mESCs, an event that occurs following the recruitment of nFGFR1, and the loss of the RXR from the Ascl1 promoter. In NCs nFGFR1 is recruited to a number of genes activating the Wnt pathway, including genes encoding for several Wnt ligands, Porcn involved in Wnt biogenesis and recycling, the receptor Fz 2–4 genes, as well as the upregulated FZ activator, proneural Dvl 3 gene (Box 7, Fig. 7). The related disheveled one and two genes, similarly have promoters that bind nFGFR1, and are constitutively expressed, both in ESCs and NCs. nFGFR1 binds to the β‐catenin gene, which is also expressed in ESCs and NCs. nFGFR1, along with RXR, also binds to the promoter of the GSK3β gene. Lastly, in RA differentiated NCs, nFGFR1 targets the upregulated Wnt TFs genes Tcf1, Sox11, 8, and 6. In addition to nFGFR1 activation of the WNT pathway genesnFGFR1 binds to the Sfrp2 and Sfrp4 genes, which encode for secreted antagonists of Frizzled. The binding of nFGFR1 to Sfrp2 and Sfrp4 correlates with the inactivation of both of these latter two genes ((Terranova et al., 2015) and linked database).
Generation of the proneural cluster is restricted to a group of dorsal ectodermal cells which lack Notch, a dual function protein which, like FGFR1, acts as a cell membrane receptor as well as a transcriptional regulator. Notch proteins block the proneural signals of the Ascl1 and Wnt pathway in adjacent cells. The anti‐neural action of Notch is enhanced by Deltex, which displaces Hairless from Hairless/Notch binding complexes, and in this manner prevents suppression of Hairless by Notch (Matsuno et al., 1995). Recent investigations of INFS have revealed that the Notch signaling is under two‐pronged control by nFGFR1 (Terranova et al., 2015). nFGFR1 binds to the Notch1 gene promoter both in ESCs and NCs. In addition an nFGFR1 binding site has been found within the Notch1 gene body in NCs. Both basal Notch1 gene activity in ESCs, and Notch1 downregulation in NCs are antagonized by a dominant negative nFGFR1, nuclear FGFR1(SP‐/NLS)(TK‐), thus revealing that nFGFR1 confers Notch1 gene inhibition. nFGFR1 also controls Notch 1 signaling indirectly, by targeting the Deltex1 gene. In ESCs, FGFR1 binds to the promoter of Deltex1, activating the gene (Terranova et al., 2015). The Deltex1 gene becomes inactivated in differentiating NCs, when nFGFR1 binding to Deltex1 declines. Hence, during neuronal development, the repression of Notch signaling by nFGFR1 very likely involves direct inhibition of the Notch1 gene by nFGFR1, as well as the loss of nFGFR1 binding and activation of the Deltex1 gene.
In summary, nFGFR1 binding results in the activation of proneural genes, and the inhibition anti‐proneural genes in differentiating NCs. These actions provide a mechanistic account for nFGFR1 neural programing of ESC.
Further neuronal development programing—nFGFR1 removes inhibitory signals
Development of neuronal cells and neural tissue requires the concerted action of a number of genes, many of which are targeted by nFGFR1. Notch1 and RE1‐silencing transcription factor (REST) constitute roadblocks that must be removed to allow for the development of the neuronal lineage of cells. Notch1 prevents neuronal differentiation of neural progenitors, while promoting glial development (Gaiano and Fishell, 2002). Thus, direct inhibition of the Notch1 gene by nFGFR1 allows for not only the initial induction of the neuroectoderm, but also enables neuronal development (as shown with brain stem/progenitor cells), while blocking gliogenesis (Stachowiak et al., 2009; Lee et al., 2012).
The REST gene facilitates the exit of ESCs from the pluripotent state by blocking the core Oct4, Sox2, and Nanog genes, but at the same time, prevents neural development by turning off Wnt signaling (Johnson et al., 2008). However, as RA‐treated ESCs undergo the process of differentiation into NCs, their REST mRNA becomes markedly depleted (Terranova et al., 2015). During this time, nFGFR1 displays a complex pattern of binding to the 5′ region of the mouse REST gene (comprising the promoter), as well as to the 3′ region of the gene. This pattern of binding changes during cellular differentiation in a manner suggesting that nFGFR1 directly downregulates the REST gene.
nFGFR1 augments proneuronal signals
Following the initial formation of the neural plate, Wnt signaling continues to be involved in the development and patterning of the embryonic nervous system. Wnt signals are essential for the formation of the neural crest, the proliferation of hippocampal neuronal precursors, as well as neuronal maturation, synapse formation and synaptic plasticity (Gao and Chen, 2010; Bengoa‐Vergniory and Kypta, 2015). Thus, nFGFR1 targeting of the genes in the Wnt pathway may very well underlie the established role of the Fgfr1 gene in all of these processes.
Other important proneuronal genes induced during ESC neuronal differentiation which are regulated by nFGFR1 encode for TFs, including Pax‐3, Id3, IRX3, and Cdx1 (Terranova et al., 2015). Pax‐3, which regulates cell proliferation, migration, regional specification, and apoptosis, is expressed in the ventricular zone of developing spinal cord, the hindbrain, the midbrain and diencephalon, as well as in the neural crest cells of developing spinal ganglia (Thompson and Ziman, 2011). Pax3 is also involved in myogenesis, its mutations playing a role in cancer (Medic and Ziman, 2009). Pax3 gene expression is markedly increased during the RA induced differentiation of mESCs into NCs (Terranova et al., 2015). Of particular interest in these regards, is the evidence indicating that nFGFR1 targets the Pax3 promoter only in NCs, and not in ESCs. Consistent with the existence of this stage‐specific binding, is the observation that the blocking of endogenous nFGFR1 by FGFR1(SP‐/NLS)(TK‐) greatly diminished the upregulation of Pax3 mRNA in NCs, while having no effect on Pax3 mRNA in ESCs. (Terranova et al., 2015)
nFGFR1 binds and activates the proneuronal ID3 (inhibitor of DNA binding 3) gene that prevents helix‐loop‐helix TFs from binding to DNA. Also, nFGFR1 binds and activates the Irx3 (Iroquois homeobox 3) gene which in combination with the sonic hedgehog gene (Shh), defines specific groups of neurons in developing brain. nFGFR1 targets the caudal type homeobox 1 (Cdx1) master gene, which relays the proneural Wnt and retinoid signals to the Hox genes. The activation of the ID3, Irx3, and Cdx1 genes in NCs is accompanied by the binding of nFGFR1 to their promoters, and is prevented by dominant negative FGFR1(SP‐/NLS(TK‐) (Terranova et al., 2015). nFGFR1 also binds to an important neurodevelopmental gene, neurogenin 1 (ngn1), both in ESCs and in NCs. Ngn1 is active at defined times during development, and affecting the positions of different progenitor cell pools. In NCs, nFGFR1 binds to the promoter of the upregulated Ngn1 gene, but does not bind to the related nonregulated ngn3 gene (Terranova et al., 2015).
nFGFR1 promotes neuronal development via “CREB/CBP signaling” and “Axonal Guidance Pathway” genes
The IPA analysis of mESC genes with promoters bound by nFGFR1 identified “CREB signaling in neurons” as a major nFGFR1‐targeted pathway (Terranova et al., 2015) (Supplemental Fig. S1A). This substantiates earlier findings that nFGFR1 nuclear accumulation was essential and sufficient for the activation of neuronal genes and neuronal differentiation induced by diverse receptors and second messengers that converge on CREB and CBP (Fang et al., 2005; Stachowiak et al., 2003a, 2007). Neuronal differentiation by nFGFR1 is executed directly via the promoters of the cAMP, PKC, BMP7, dopamine, and Wnt/β‐catenin signaling genes. The top differentially regulated genes targeted by nFGFR1 include CamkII, Adenylate cyclase (AC), Phospholipase C (PLC), and G‐protein β (Gβ), all of which provide converging inputs to the CREB and CBP effectors (Terranova et al., 2015).
Transfection of human and mouse neural progenitor cells with a constitutively active nuclear FGFR1(SP‐/NLS) or with HMW FGF‐2 (which signals via endogenous nFGFR1) produces a striking neuronal morphology (Fang et al., 2005; Stachowiak et al., 2012a). Similar result were obtained with ESCs, and in developing brain neural progenitors (Lee et al., 2012). Furthermore, nFGFR1 mediates BMP7 and NGF initiated neuronal development and regeneration (Horbinski et al., 2002; Lee et al., 2013). The mechanisms of nFGFR1 action were substantiated by an IPA identified class of Axonal Guidance Pathway genes targeted by nFGFR1 (Terranova et al., 2015; Supplemental Fig. S1B). nFGFR1 binds to Semaphorin 6D, Ephrins, the Ephrin receptor and α3‐integrin genes, as well as the Smad effector genes, all of which are engaged in axonal development, guidance and neuronal network formation (Terranova et al., 2015). Other genes important in neuronal growth which have promoters bound by nFGFR1, and which are regulated by nFGFR1 include Nestin, Doublecortin and βIII tubulin. nFGFR1 binding was verified in developing brain tissues and in cultured PC12 cells (Baron et al., 2012; Lee et al., 2012, 2013; Narla et al., 2013).
nFGFR1 regulates development of dopamine (DA) neurons
DA neurons located in the ventral midbrain (substantia nigra and ventral tegmental area) are involved in motor, sensory, cognitive and emotional functions, and in a variety of neurologic and psychiatric disorders. Development of these neurons is controlled by nFGFR1 and its nuclear partner proteins (Klejbor et al., 2006; Smidt and Burbach, 2009). nFGFR1, CBP, and Nurr1 accumulate in the nuclei of developing DA progenitors, all three proteins colocalizing in the nuclear bodies (Fang et al., 2005; Baron et al., 2012). The knock down of Nurr1 or the blocking of endogenous FGFR1 in DA progenitors attenuates neuronal development in transgenic mice, thus verifying their ontogenic function (Klejbor et al., 2006; Smidt and Burbach, 2009). nFGFR1 directly targets the Patch2, Ngn, RAR, and Nurr1 genes (Terranova et al., 2015), which are instrumental in DA neuronal development, and nFGFR1, together with Nurr1, binds and co‐activates the TH gene involved in DA synthesis (Lee et al., 2012). nFGFR1 may also play a role in the synaptic plasticity of DA transmission mediated by phosphatase inhibitors, effectors of DA receptors, and DARPP‐32. nFGFR1 binds the promoter of the Darpp‐32 gene as it becomes upregulated during RA induced differentiation of ESCs to NCs (Terranova et al., 2015).
CNS and Body Patterning by nFGFR1 Control of Hox and Related Genes
The layout of body axes and body dimensions are programmed by a phylogenetically conserved superfamily of “morphogenic” transcriptional factors many of which contain a 60 amino acid, helix‐turn‐helix, DNA binding Homeobox domain and thus are called homeobox genes. Homologues of these genes appear in radially symmetric pre‐Paleozoic cnidarians and evolve into Hox genes present in all bilaterian metazoans (Larroux et al., 2007). The Drosophila genome has 8 Hox genes organized into a single cluster that outlines the embryo's anterior–posterior axis, patterning and segmentation. The invertebrate Hox cluster has evolved into four (A–D) vertebrate clusters, which include 11 HoxA, 10 HoxB, 9 HoxC, and 9 HoxD genes within the mouse and human genomes (Box 8, Fig. 8A). In both fruit flies and mice, the deletion of a single Hox gene leads to altered axial identities and the transformation of specific embryonic structures into more anterior ones (Montavon and Soshnikova, 2014). In addition the Hox genes are essential for the outgrowth and patterning of limbs along both the anterior–posterior and proximal–distal axes (Zakany and Duboule, 2007). To execute these complex functions, both the pattern and time of Hox gene expression must be tightly controlled. Hox genes located at the 3′ end of the clusters are transcribed first, and generally are in more anterior regions of the embryo, as compared to the genes situated at the 5′ end, which are expressed later and in more posterior areas (Montavon and Soshnikova, 2014). This spatial and temporal collinearity is especially evident in development of the mesodermal musculature, and bones as well as in the segmentation of the central nervous system. The initial state of the neural tube is forebrain like. The midbrain, hindbrain, and spinal cord arise through a progressive transformation to more posterior fates by the Hox paralog groups 1–4 in the hindbrain and 5–13 in the segmentally‐restricted domains of the spinal cord (Mallo and Alonso, 2013; Pascual‐Anaya et al., 2013). Furthermore, Hox genes shape development and synaptic specificity of motoneurons and underwrite their selective segmental muscle innervation. Altogether, Hox genes have been designated as the architects of the neural system and its developmental choreographers (Philippidou and Dasen, 2013).
Figure 8.
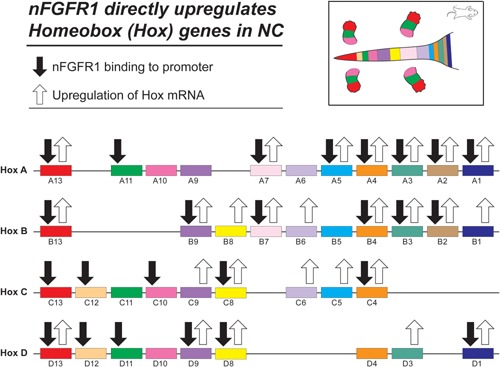
(A) Activation of Hox genes during RA induced ESC transition to NC is accompanied by nFGFR1 binding to their promoters. The analyses of the HoxA cluster showed that nFGFR1 binding plays an essential role in the activation HoxA genes by RA. Furthermore, nFGFR1 binding is sufficient to activate several of the HoxA gene in ESC in the absence RA stimulation (Terranova et al., 2015). Hox genes are organized based on information from (P. Turpenny). (B) nFGFR1‐dependent mechanism may promote the sequential expression of Hox genes during ontogeny. During development Hox genes at the 3′ end of the clusters are activated first followed by Hox genes located at more 5′ positions. The mechanism of 3′–5′ time co‐linearity could involve an initial direct 1 activation of the 3′ Hox genes (i.e., Hox A1 and A2) by nFGFR1 followed a delayed co‐activation of the Hox 5′ genes by accumulating HoxA1, A2, and Cdx1 transcriptional factors along with nFGFR1.
Box 8. nFGFR1 controls CNS and body patterning Hox genes ((Terranova et al., 2015) and linked database).
The layouts of the metazoan bodies are governed by phylogenetically conserved transcription factors that contain 60 amino acid DNA binding Homeobox domain. Vertebrate Hox genes are organized into four A‐D clusters that underwrite body axes, dimensions, and body and limbs segmentation, patterning of the nervous system and mesoderm‐derived muscles and bones. Mutations of the individual Hox genes disrupt the body and CNS segmentation and the borders between the CNS regions. Hox genes are transcriptionally inactive in blastocyst and in pluripotent ESC. During gastrulation and further development Hox genes are activated by RA, Wnts, and Cdx1 in a time order that reflects their location within the clusters (3' to 5').
Regulation of Hox genes is achieved mostly at the transcriptional level (Kondrashov et al., 2011), and is dictated by their clustered organization (Duboule, 2007). In vertebrate embryos, prior to gastrulation, Hox clusters are silenced by multiprotein complexes encoded by Polycomb group (PcG) genes (Schuettengruber and Cavalli, 2009; Young and Deschamps, 2009). As gastrulation ensues, Hox genes become activated in the posterior part of the primitive streak in a time order that reflects their location within the clusters (Izpisua‐Belmonte et al., 1991; Tschopp and Duboule, 2011). The 3′–5′ time collinearity is governed by cis‐acting elements located within the clusters themselves. The search for regulators of Hox transcription in trans revealed a major role for RA, Cdx1, FGFs, and Wnts (Alexander et al., 2009; Young and Deschamps, 2009). In the developing central nervous system, RA activates the 3′ (1–5) Hox cluster, whereas the 5′ cluster (6–13) is activated by Cdx1, Wnt, and FGF8. Furthermore, 3′ Hox A1 is essential for the activation of downstream HoxA and Hox B genes. This results in a model in which the upstream genes activate the downstream genes, thus spreading the activation in the 3′–5′ direction (Montavon and Soshnikova, 2014).
In non‐differentiated mESCs, Hox genes are largely repressed, but many become upregulated during RA‐induced neural ESC differentiation (Terranova et al., 2015). In Hox A and C clusters, RA activated genes localize predominantly to the 3′ region, which during development is activated first. While many of the inactive Hox genes In ESCs are bound by RXR, a few, HoxD11 and HoxD13, are bound by nFGFR1 (Box 8, Fig. 8A; Terranova et al., 2015). In contrast, the RA‐induced upregulation of the HoxA1–5, HoxA7, and Hox A13 genes is accompanied by nFGFR1 binding to their promoters (Box 8, Fig. 8A). Also the upregulated Hox B2–4, B7, B9, B13, C4, C8, and the Hox D1, D8, and D13 genes have promoters targeted by nFGFR1. The role of nFGFR1 binding in regulation of the Hox genes was demonstrated by an analysis of the levels of the mRNAs of the 3′ cluster (HoxA1–7) (Terranova et al., 2015). RA‐induced differentiation of NCs was accompanied by the up‐regulation of HoxA1–5 and HoxA7 mRNAs. The up‐regulation of the HoxA mRNAs in the RA‐treated NCs was abolished by a dominant negative nuclear FGFR1(SP‐/NLS)(TK‐), as observed in RA induced ESC neuronal differentiation. These findings reveal the essential role of nFGFR1 in the programming of the HoxA genes, and are consistent with the repression of Hox genes by dominant negative FGFR1 mutant in Xenopus embryos (Isaacs et al., 1998; Pownall et al., 1998).
Notably, overexpression of active nuclear FGFR1(SP‐/NLS) is sufficient to activate Hox genes (Terranova et al., 2015), and neuronal development in mESCs (Lee et al., 2012), even in the absence of RA stimulation. These findings have established nFGFR1 as a factor that programs stem‐cell development, and highlights the importance of nFGFR1‐mediated Hox gene activation. In pluripotent ESCs, low‐level HoxA activity is maintained by endogenous nFGFR1. However, the effect of endogenous nFGFR1 is indirect, as nFGFR1 does not bind to HoxA‐gene promoters in ESCs.
The HOX upstream regulators identified so far (i.e., RA) are able to activate rather large subsets of Hox genes, and are active in broad time windows, along the anterior–posterior axis. How can these “generic” factors trigger the collinear expression of Hox genes, in both space and time? We propose that nFGFR1 which activates the Hox A1, A2, and A3 genes to the largest degree, initiates the activation of downstream Hox by accumulating HoxA1 and A2 proteins (Box 8, Fig. 8B). Furthermore, nFGFR1 targeting and activation of the Cdx1 gene (Fig. 7) could lead to a gradual accumulation of the CDX1 protein at the promoters of downstream 5′ Hox genes, and their delayed activation as outlined on Figure 8B.
Developmental patterning and regional cell specification in the central nervous system are also directed by a functional concentration gradient of RA. The concentration gradient of RA is created by varying the expression of the RA‐generating enzyme, retinaldehyde dehydrogenase 2 (RALDH2; overexpressed posteriorly) and the RA‐metabolizing protein CYP26A (overexpressed anteriorly; White and Schilling, 2008; Rhinn and Dolle, 2012). Genetic experiments placed the FGFs upstream of the RALDH2 and Cyp26 genes (Rhinn and Dolle, 2012). ChIP‐seq experiments and independent ChIP assays suggest that this regulation is mediated at least in part directly by nFGFR1 binding to the Cyp26a1 promoter and, along with RXR, to the intragenic region of RALDH2 (Terranova et al., 2015).
In summary, genomic inquiries place nFGFR1 at the top of Notch and Hox controlled developmental patterning, and suggest an additional role for nFGFR1 in the formation of developmental RA gradients.
nFGFR1 Regulation of Endodermal and Mesodermal Genes
RA programs ESCs to differentiate into neuronal, endothelial or mesodermal cells depending on ligand concentration. In agreement with specific neuronal programing by 5 µM RA, genes encoding the early endodermal markers GDF‐1, GDF‐3, and HNF‐4 (https://www.rndsystems.com/research-area/early-endodermal-lineage-markers) are downregulated in RA treated ESCs (Terranova et al., 2015). Similarly, genes typically expressed by mature endodermal cells, Cytokeratin 19, EOMES, FABP1, HNF3α, are downregulated, or are not expressed in NCs. Gene downregulation is accompanied by increased nFGFR1 binding to the intragenic regions of the GDF‐1, GDF‐3, HNF‐4, HNF‐3α, and FaBP1 genes, or to the promoter region of the Cytokeratin 19 and eomesodermin (EOMES) gene (Terranova et al., 2015). The Jarid2 gene, which controls development of liver, spleen, thymus, and cardiovascular system (Landeira et al., 2015), is expressed in nondifferentiated ESCs, and is downregulated in NCs (Terranova et al., 2015). Jarid2 mRNA downregulation is accompanied by de novo nFGFR1 binding to the Jarid2 promoter, suggesting direct repression by nFGFR1. Thus, nFGFR1 targeting the promoters or intragenic loci of endothelial genes may underwrite their repression during neuronal development.
The Fgfr1 gene regulated FGF/Wnt/Notch‐based oscillatory mechanism is key to controlling cell differentiation and maturation in the CNS, as well as positioning segmental boundaries in the developing presomitic mesoderm (Yamaguchi et al., 1994; Wahl et al., 2007; Dequeant and Pourquie, 2008). The results of ChiPseq and independent ChiP analyses in ESCs and NCs are consistent with this model, in that nFGFR1 not only targets various neuronal genes in the Wnt/β‐catenin pathway (Fig. 7 and S4D Fig), but also binds within the promoters of key mesodermal genes, Dusp6, Perlecan (Hspg2), Porcn, Lfng, Nkd1, Nrarp, and within the promoter and gene body of the Mesp2 and Notch 1 genes (Terranova et al., 2015). Dominant negative nuclear FGFR1(SP‐/NLS)(TK‐) reversed the repression of the Mesp2, Notch1 and Hspg2 genes in ESCs and NCs, while reducing the RA‐induced upregulation of nFGFR1 targeted neurodevelopmental Pax3, Id3, Irx3, and Cdx1 genes (section above). Thus, endogenous nFGFR1 represses mesodermal genes, and activates neural genes as the ESCs progress towards a neuronal phenotype.
Fgfr1 gene ablation demonstrated its essential role in bone development and remodeling (Jacob et al., 2006). One of the bone development controlling genes is FGF‐23 (Han et al., 2015a). In mouse ESCs only trace levels of FGF23 mRNA are detected and are further diminished in NCs (Terranova et al., 2015). nFGFR1 binds to the FGF23 gene promoter in both ESCs and NCs, and additionally to the 3′UTR in NCs. Thus, during neuronal development, the FGF23 gene may very well be directly inhibited by nFGFR1. In contrast, in human osteoblastic cells which express FGF23, Quarles and colleagues have presented evidence indicating that the FGF‐23 gene is activated by INFS (Han et al., 2015b). Included amongst this evidence is the observation that (1) nFGFR1 binds to the FGF23 promoter along with CREB and CBP, and (2) transfection of human osteoblastic cells with FGFR1(SP‐/NLS) upregulated FGF23 promoter activity. Thus, the nature of the gene regulation by nFGFR1, whether inhibitory, or activating and instructive, may depend upon the context of the cell lineage controlling the signals and the cell phenotype.
nFGFR1 Targets Promoters of miRNA Genes ((Terranova et al., 2015) and Linked Database)
In mESCs and NCs nFGFR1 targets not only mRNA‐encoding genes, but also binds to the proximal promoters of several expressed miRNAs genes. Exemplary mir301 and mir9 are upregulated several fold during RA induced ESC differentiation (Box 9, Fig. 9). In ESCs, the mir301 and mir9 promoters interact with RXR but not with nFGFR1, nor with Nur77. In contrast, in NCs both nFGFR1 and Nur7, bind to the mir301 and mir9 promoters. This likely confers mir301 and mir9 upregulation, as Nur77a and nFGFR1 were shown to coactivate transcription from the Nur‐responsive elements. mir301, mir9 and several other nFGFR1 targeted miRNA genes are known to promote neurogenesis and are involved in diverse aspects of ontogenesis by targeting mRNAs of hundreds of developmental genes (Box 9, Fig. 9). This seminal finding offers an additional new mechanism for global genome regulation in ontogenesis by nFGFR1.
Figure 9.
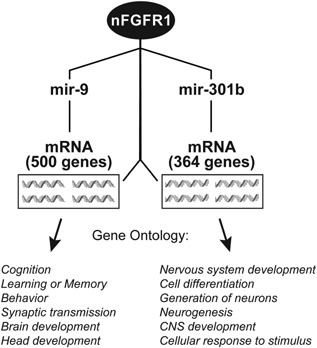
nFGFR1 targets promoters of miRNAs upregulated during RA induced mESC differentiation to NC. (A) nFGFR1 binds to promoters of mir301 and mir9 in NC but not ESC. Thus, nFGFR1 may indirectly control translation of mRNAs targeted by mir301 and mir9 in differentiating NC. Mir301 and mir9 have binding sequences in a great number of mRNAs (364 and 500, respectively; identified by mirtarbase data base analysis, Supplementary table). Gene Ontology identifies functions of mir301 and mir9 targeted genes as being related to cell development and differentiation, brain development and function.
Box 9. nFGFR1 targeting of miRNA genes that control ontogenetic gene expression ((Terranova et al., 2015) and linked database).
Role of nFGFR1 in Ontogenic Gene Regulations—Summary Notes
Sequencing of human, mouse, and other genomes has unlocked an unprecedented vast information which could be mined to unknot the tangled scores that underwrite life's evolution, and its abbreviated replay—ontogeny. Recent discoveries summarized in this article reveal basic components of the emerging paradigm for master ontogenic networks involved in cell pluripotency, tissue and organ development and axis specification being coordinated by a common mechanism, INFS (Box 10, Fig. 10). In this new paradigm, both ancient proto‐oncogenes and tumor suppressor genes, which assured developmental success of unilateral organisms, as well as metazoan “morphogens” are subjugated to a common integrating function of nFGFR1.
Figure 10.
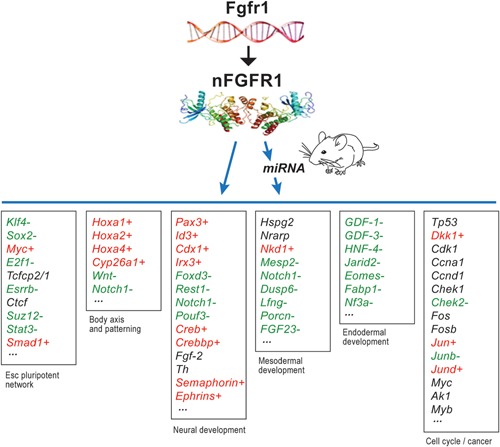
Genetic experiments position the Fgfr1 gene at the top of gene hierarchy that directs the development of multicellular animals. Fgfr1 governs gastrulation, as well as development of the major body axes, neural plate, central and peripheral nervous systems, and mesoderm by affecting the genes that control the cell cycle, pluripotency and differentiation (Ciruna et al., 1997; Partanen et al., 1998; Ciruna and Rossant, 2001; Dequeant and Pourquie, 2008) and microRNAs (Bobbs et al., 2012). This regulation is executed by nuclear protein, nFGFR1, which integrates a plethora of development controlling epigenomic signals (Stachowiak et al., 2015). nFGFR1 cooperates with RXR, Nurs and other CBP binding TFs, and targets thousands of genomic loci, including promoters of the mRNA and miRNA genes that reside at the top of the ontogenetic networks (Terranova et al., 2015). The dynamic nature of nFGR1 and its partner proteins illustrates how probabilistic molecular collisions (see Box 2, Fig. 2B) control the flow of information in structured genomic networks that underwrite development. The figure lists the discussed examples of nFGFR1 binding (ChiPseq) to promoters of different categories of developmental genes in which color denotes upregulation (red +) or downregulation (green −) during transition from ESCs to NCs (RNA‐seq). Black indicates nFGFR1 targeted genes that are not differentially expressed in ESC and NC (<2‐fold change). Figure is based on (Terranova et al., 2015) and linked database).
Box 10. Paradigm for global ontogenetic genome programming by nFGFR1 expression ((Terranova et al., 2015) and linked database).
nFGFR1‐mediated control of master developmental mRNA and miRNA gene networks and chromatin architectural factors emerges as an unprecedented central mechanism that integrates and orchestrates genome function in forming multicellular organisms. The overall importance of such mechanisms in animal development is supported by the conservation of nFGFR1 genomic targets, and the evolutionary emergence of nuclear FGFs (Popovici et al., 2006) and FGFR1 (Bertrand et al., 2009) in early metazoans.
INFS in Disease
The unprecedented Pan‐ontogenic nature of INFS indicates its role as a potential Pan‐pathological mechanism that contributes to a wide variety of disorders. In this article, we will discuss the role of INFS in cancer and a developmental disorder—schizophrenia.
Ancient protoncogenes and tumor suppressor genes are targeted by nFGFR1—role in cancer (Box 11, Fig. 11A)
Figure 11.
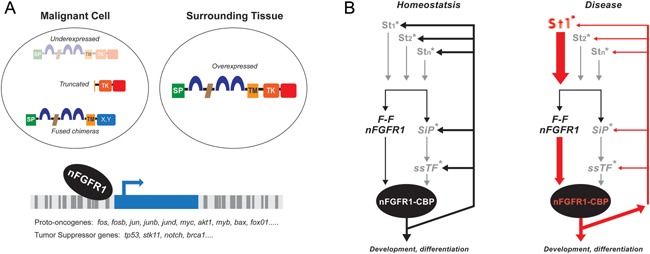
(A) Malignancy—in cancer cells nFGFR1 has been found depleted, truncated or fused with other genes. In cancer cell lines with reduced nFGFR1 expression, an up‐regulation of endogenous nFGFR1 or transfection of constitutively active nuclear FGFR1(SP‐/NLS) promote cell differentiation. Studies of non‐transformed ESC and NC show that nFGFR1 binds to promoters of key developmental proto‐oncogenes and tumor suppressor genes suggesting that the generation of cancerous phenotypes may involve altered structure/function of nFGFR1 and/or regulation of altered genes by nFGFR1. In cells surrounding pancreatic or glioma tumors nFGFR1 is found overexpressed and may promote cell migration and metastasis. (B) Developmental disease—disruption of INFS in schizophrenia. “Feed‐Forward‐And‐Gate” signaling by INFS in development. Neurogenic signals generated by diverse extracellular stimuli (St; neurotransmitters, hormones, growth factors, cell contact receptors) are propagated through signaling pathways (SiP; cAMP, Ca++/PKC, MAPK) to sequence specific transcription factors ([TF; CREB, AP1, NfkB, Smads, Klf4, Stat3, nuclear retinoid receptors {RXR/RAR} and orphan Nur receptors, etc.,]). In parallel, a newly synthesized FGFR1 translocates into the nucleus and “feeds forward” (F‐F) developmental signals directly to CREB binding protein (CBP), an essential transcriptional co‐activator and gene‐gating factor. The coupled activation of TFs and CBP by nFGFR1 allows genes to coordinately respond to developmental signals and cell development. In the proposed transcriptional circuit INFS co‐ordinates incoming developmental signals (St) through the Feed‐Forward (FF) module and reinforces or turns off those input signals via a feedback (FB) module (Lee et al., 2012). *marks signaling pathways in which schizophrenia‐linked genes a have been found, including cAMP, G‐protein signaling, PKC, MAPK, NfkB, CREB, RXR, and Nurr1 (Buervenich et al., 2000; Sun et al., 2010; Jablensky et al., 2011). In schizophrenia and other neurodevelopmental diseases, mutations of these individual genes, including “weak” copy variations could deregulate this auto‐regulated genomic circuit (red lines) and thus lead to broad molecular and developmental dysfunctions (Figure is based on information in (Stachowiak et al., 2013) and in (Terranova et al., 2015) and linked database).
An ontology of ESC expressed genes whose promoters are targeted by nFGFR1, but not by RXR or Nur77, identified the cell cycle, proliferation and cancer as the top biological functions and diseases (Fig. 4A). At the top of the cell cycle network, nFGFR1 targeted promoters of diverse genes, including checkpoint kinase 1/2, Dkk1, and Camk2d, which are often deregulated in various types of cancer (Terranova et al., 2015; Supplemental Fig. S1C). A prominent category of genes which have promoters targeted by nFGFR1 are phylogenetically old proto‐oncogenes which control normal cell growth and migration. Over 100 cancer‐causing proto‐oncogenes have already been discovered nested deep within the human genome and have homologues in ancient, highly conserved genes (Cline, 1986). In human cancers, proto‐oncogenes may generate qualitatively, or quantitatively abnormal oncogenic products that set‐off programs resulting in malignancies. The list of nFGFR1 targeted proto‐oncogenes include c‐fos and fos‐b expressed in ESCs and NCs, c‐jun, jun b, jun d, c‐myc, and Akt1, which are upregulated in NCs, and c‐myb, Bax, and FoxO1, which are downregulated in NCs. nFGFR1 binds also to the promoters of suppressor genes, including TP53, STK11, BRCA1 which are downregulated in NCs. While TP53 is the most commonly mutated gene in human cancers, other nFGFR1 targeted tumor suppressor genes also play a major role in breast, colon, brain, ovarian, and lung cancer, as well as in renal carcinomas (Brose MS and Weber, 2003).
Box 11. Role of nFGFR1 in disease.
The nFGFR1 regulated Notch1 gene, which acts by an ancient mechanism to regulate cell proliferation, may act as an oncogene, or less commonly, as a tumor suppressor gene in various malignancies. Epidermis‐derived melanoma cells display grossly overexpressed activated Notch‐1 (Nickoloff et al., 2005) in addition to changes in nuclear FGFs and FGFR signaling (Halaban et al., 1988; Katoh and Nakagama, 2014).
The binding of nFGFR1 to proto‐oncogenes and tumor suppressor genes in mESCs and mouse NCs is conserved in human ESCs and iPSCs (our unpublished data), and, thus is likely to be of major developmental importance. In general, studies have shown a reduced content of nFGFR1 in the nuclei of glioma, pheochromocytoma, neuroblastoma and medulloblastoma lines, as compared to non‐transformed cells (Stachowiak et al., 2015). Restoration of nuclear FGFR1 accumulation (i.e., by NGF treatment of PC12 cells, RA treatment of neuroblastoma cells, the transfection of constitutively active nuclear FGFR1(SP‐/NLS) in neuroblastoma, medulloblastoma and glioma cells) promote a normal cellular phenotype, suggesting that nFGFR1 has a tumor suppressor‐like action. This is contrasted by the mitogenic action of plasma membrane localized FGFRs, that occurs when cells are stimulated by extracellular FGFs.
The laboratory of Richard Grosse has demonstrated the nuclear accumulation of a truncated nFGFR1 (lacking the N‐terminal region) in human breast cancer cells, as well as the binding of this truncated nFGFR1 to EBI3, GRINA, KRTAP5‐6, POUF2, PRSS27, Stac3, and SFN genes, with a consequent increase in cell migration and, potentially, metastasis (Chioni and Grose, 2012). The wild‐type nFGFR1 was subsequently observed to bind to the same genes in mouse ESCs/NCs (Terranova et al., 2015) and/or in human NCs (our unpublished observations). Thus, the replacement of full‐length nFGFR1 with the truncated form distorts the regulation of genes which are normally targeted, and is an integral part of the biology of cancer cells. Likewise, several reports have described chromosomal transfers in cancer cells, which create diverse oncogenic fusions of FGFR1 with other proteins. For example, fusions of FGFR1 with the transcriptional regulator TIF1have been observed. These FGFR1/TIF1 fusion proteins, like nFGFR1, localize to nuclear bodies, where they interact with RA nuclear receptors (Belloni et al., 2005), even under cellular conditions where normal FGFR1 would interact with cytoplasmic membrane proteins, rather than accumulating in the nucleus. Oncogenic fusions of FGFR1 to the transforming acidic coiled–coil (TACC) coding domain of the tachykinin family TACC1 gene were found in human glioma cells (Singh et al., 2012). The impact of these and other fusions of nFGFR1 with proteins on genomic deprograming, and on the biology of cancer cells requires further investigation.
Recent studies have identified the presence of nFGFR1 in neoplasms, and have suggested an additional complexity to the role of nFGFR1 in cancer. Overexpression of nFGFR1 is a feature of pancreatic ductal adenocarcinomas (PDACs). Nuclear FGFR1 and FGF‐2 are found in activated pancreatic stellate cells (PSCs), the cells primarily responsible for desmoplasia in pancreatic carcinomas (Coleman et al., 2014). Nuclear translocation of FGFR1 is necessary for the invasion of PSCs into regions containing pancreatic carcinomas, and subsequently, the formation of desmoplasia around the cancer cells. Similarly, reactive astrocytes which surround human glioma tumors overexpress nFGFR1, an event that can potentially promote their migration towards glioma cells (Stachowiak et al., 1997a). Understanding the complex roles of FGF signaling in cancer biology holds a promise for novel treatment strategies.
Role of nFGFR1 in ontogenic dysregulation in Schizophrenia (Box 11, Fig. 11B)
Schizophrenia is a developmental disorder characterized by complex aberrations in the structure, wiring, and chemistry of multiple neuronal systems. The abnormal developmental trajectory of the brain appears to be established during gestation, long before clinical symptoms of the disease appear in early adult life. Over 200 genes selected by their linkage, association, and expression, as reported in the literature, have been proposed to contribute to the etiology of the disease. However, there is no single gene, whose expression is altered in a majority of schizophrenia patients (Sun et al., 2010; Rodriguez‐Murillo et al., 2012). These schizophrenia linked genes mediate a wide range of neurodevelopmental events including progenitor cell proliferation and differentiation, migration, cytoskeletal reorganization, axonal connectivity, and patterning of brain structures (Jaaro‐Peled et al., 2009; Moskvina et al., 2009; Potkin et al., 2010; Sun et al., 2010; Ayalew et al., 2012) and operate within 24 known pathways (Feng et al., 2005; Wong et al., 2011; Ayalew et al., 2012; Rodriguez‐Murillo et al., 2012), all of which converge on the INFS Feed Forward and Gate module (Box 11). Hence, changes in schizophrenia (SZ)‐linked genes affect INFS, and thereby lead to a common “watershed” dysregulation of developmental gene networks. Indeed, iPSCs derived neural cells from different SZ patients have several hundred of the same genes dysregulated, including FGFR1 and the nuclear FGF14 ligand. Our recent investigations suggest that alterations in nFGFR1 interactions with developmental gene networks, miRNA genes, and chromatin topology factors in schizophrenia may underlie the neurodevelopmental pathology of this disease. Furthermore, FGFR1 and its partner proteins bind to promoters of schizophrenia‐linked neurodevelopmental genes including: DISC1, ANK3, ZFP395, Ngn1, FZD3, FGF‐2, and FGFR1 and thus play a direct role in their regulation (Stachowiak et al., 2013; Terranova et al., 2015). These observations suggest a dysregulation of transcriptional circuitry in which INFS integrates incoming developmental signals including signals from altered schizophrenia‐linked gene, and affects functions of other schizophrenia genes via a feedback loop (Box 11B), mutations in individual patients may deregulate this self‐controlled genomic circuit, and result in common developmental dysfunctions.
Role of nFGFR1 in ontogenic diseases—summary notes and perspective
INFS dysregulation in neoplastic and developmental diseases induced by a limited genomic change may spread across the genomic networks and deconstruct biological homeostasis. Studies of the pan‐ontogenic INFS should help to understand the nature these diseases, how they develop and what information nodes may be targeted to restore stability of the ontogenic networks. Eventually, these efforts may help to combat cancer and developmental diverse disorders that plague humanity.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Ingenuity Pathway Analysis (IPA) of genes expressed differentially in ESCs and NCs and targeted by nFGFR1.
Table S1. Mir301 and mir9 have binding sequences in a great number of mRNAs (364 and 500, respectively; identified by mirtarbase data base analysis)
Video—Supplemental Material.
Acknowledgements
We are deeply indebted to Dr. Barbara Birkaya who helped to conceive and perform many of the described experiments. We gratefully acknowledge the experimental work and intellectual input of all our students including Christopher Terranova, Sridhar Narla, Yu‐Wei Lee, Xiaogong Fang, Courtney Benson, Brandon Decker, and Seerat Elahi. We thank Dr. Hari Shroff for allowing us to use the Super‐resolution Microscope in his laboratory at NIH, Dr. Mary Taub, Dr. Scott Doyle (SUNY at Buffalo), and Dr. John Aletta (Ch3 Biosystems) for extensive discussion and editing of our manuscript. We acknowledge grant support from the New York State Stem Cell Science (NYSTEM), Patrick P. Lee Foundation and the Esther Trachtman Foundation.
Literature Cited
- Akanuma H, Qin XY, Nagano R, Win‐Shwe TT, Imanishi S, Zaha H, Yoshinaga J, Fukuda T, Ohsako S, Sone H. 2012. Identification of stage‐specific gene expression signatures in response to retinoic acid during the neural differentiation of mouse embryonic stem cells. Front Genet 3:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander T, Nolte C, Krumlauf R. 2009. Hox genes and segmentation of the hindbrain and axial skeleton. Annu Rev Cell Dev Biol 25:431–456. [DOI] [PubMed] [Google Scholar]
- Ayalew M, Le‐Niculescu H, Levey DF, Jain N, Changala B, Patel SD, Winiger E, Breier A, Shekhar A, Amdur R, Koller D, Nurnberger JI, Corvin A, Geyer M, Tsuang MT, Salomon D, Schork NJ, Fanous AH, O'Donovan MC, Niculescu AB. 2012. Convergent functional genomics of schizophrenia: From comprehensive understanding to genetic risk prediction. Mol Psychiatry 17:887–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan SK, Witcher M, Berggren TW, Emerson BM. 2012. Functional and molecular characterization of the role of CTCF in human embryonic stem cell biology. PLoS ONE 7:e42424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron O, Forthmann B, Lee YW, Terranova C, Ratzka A, Stachowiak EK, Grothe C, Claus P, Stachowiak MK. 2012. Cooperation of nuclear fibroblast growth factor receptor 1 and Nurr1 offers new interactive mechanism in postmitotic development of mesencephalic dopaminergic neurons. J Biol Chem 287:19827–19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni E, Trubia M, Gasparini P, Micucci C, Tapinassi C, Confalonieri S, Nuciforo P, Martino B, Lo‐Coco F, Pelicci PG, Di Fiore PP. 2005. 8p11 myeloproliferative syndrome with a novel t(7;8) translocation leading to fusion of the FGFR1 and TIF1 genes. Genes Chromosomes Cancer 42:320–325. [DOI] [PubMed] [Google Scholar]
- Bengoa‐Vergniory N, Kypta RM. 2015. Canonical and noncanonical Wnt signaling in neural stem/progenitor cells. Cell Mol Life Sci 72:4157–4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. 2002. Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3:517–530. [DOI] [PubMed] [Google Scholar]
- Bertrand S, Somorjai I, Garcia‐Fernandez J, Lamonerie T, Escriva H. 2009. FGFRL1 is a neglected putative actor of the FGF signalling pathway present in all major metazoan phyla. BMC Evol Biol 9:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharali DJ, Klejbor I, Stachowiak EK, Dutta P, Roy I, Kaur N, Bergey EJ, Prasad PN, Stachowiak MK. 2005. Organically modified silica nanoparticles: a nonviral vector for in vivo gene delivery and expression in the brain. Proc Natl Acad Sci USA 102:11539–11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobbs AS, Saarela AV, Yatskievych TA, Antin PB. 2012. Fibroblast growth factor (FGF) signaling during gastrulation negatively modulates the abundance of microRNAs that regulate proteins required for cell migration and embryo patterning. J Biol Chem 287:38505–38514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose MS ST, Weber B. 2003. Genetic basis of cancer syndromes In: Kufe DW, Pollock RE, Weichselbaum RR, et al., editors. Holland‐Frei cancer medicine. 6th edition Hamilton (ON): BC Decker. [Google Scholar]
- Bryant DM, Stow JL. 2005. Nuclear translocation of cell‐surface receptors: Lessons from fibroblast growth factor. Traffic 6:947–954. [DOI] [PubMed] [Google Scholar]
- Buervenich S, Carmine A, Arvidsson M, Xiang F, Zhang Z, Sydow O, Jonsson EG, Sedvall GC, Leonard S, Ross RG, Freedman R, Chowdari KV, Nimgaonkar VL, Perlmann T, Anvret M, Olson L. 2000. NURR1 mutations in cases of schizophrenia and manic‐depressive disorder. Am J Med Genet 96:808–813. [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, Loh YH, Yeo HC, Yeo ZX, Narang V, Govindarajan KR, Leong B, Shahab A, Ruan Y, Bourque G, Sung WK, Clarke ND, Wei CL, Ng HH. 2008. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133:1106–1117. [DOI] [PubMed] [Google Scholar]
- Chioni AM, Grose R. 2012. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J Cell Biol 197:801–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruna B, Rossant J. 2001. FGF signaling regulates mesoderm cell fate specification and morphogenetic movement at the primitive streak. Dev Cell 1:37–49. [DOI] [PubMed] [Google Scholar]
- Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J. 1997. Chimeric analysis of fibroblast growth factor receptor‐1 (Fgfr1) function: A role for FGFR1 in morphogenetic movement through the primitive streak. Development 124:2829–2841. [DOI] [PubMed] [Google Scholar]
- Cline MJ. 1986. Oncogenes and the pathogenesis of human cancers. Ric Clin Lab 16:503–507. [DOI] [PubMed] [Google Scholar]
- Coleman SJ, Chioni AM, Ghallab M, Anderson RK, Lemoine NR, Kocher HM, Grose RP. 2014. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol Med 6:467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzacq X, Shav‐Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14:796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dequeant ML, Pourquie O. 2008. Segmental patterning of the vertebrate embryonic axis. Nat Rev Genet 9:370–382. [DOI] [PubMed] [Google Scholar]
- Duboule D. 2007. The rise and fall of Hox gene clusters. Development 134:2549–2560. [DOI] [PubMed] [Google Scholar]
- Dunham SM, Pudavar HE, Prasad PN, Stachowiak MK. 2004. Cellular signaling and protein–protein interactions studied using fluorescence recovery after photobleaching. J Phys Chem B 108:10540–10546. [Google Scholar]
- Dunham‐Ems SM, Pudavar HE, Myers JM, Maher PA, Prasad PN, Stachowiak MK. 2006. Factors controlling fibroblast growth factor receptor‐1's cytoplasmic trafficking and its regulation as revealed by FRAP analysis. Mol Biol Cell 17:2223–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham‐Ems SM, Lee YW, Stachowiak EK, Pudavar H, Claus P, Prasad PN, Stachowiak MK. 2009. Fibroblast growth factor receptor‐1 (FGFR1) nuclear dynamics reveal a novel mechanism in transcription control. Mol Biol Cell 20:2401–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Stachowiak EK, Dunham‐Ems SM, Klejbor I, Stachowiak MK. 2005. Control of CREB‐binding protein signaling by nuclear fibroblast growth factor receptor‐1: A novel mechanism of gene regulation. J Biol Chem 280:28451–28462. [DOI] [PubMed] [Google Scholar]
- Feng J, Chen J, Yan J, Jones IR, Craddock N, Cook EH, Jr. , Goldman D, Heston LL, Sommer SS. 2005. Structural variants in the retinoid receptor genes in patients with schizophrenia and other psychiatric diseases. Am J Med Genet B Neuropsychiatr Genet 133B:50–53. [DOI] [PubMed] [Google Scholar]
- Forthmann B, Aletta JM, Lee YW, Terranova C, Birkaya B, Stachowiak EK, Stachowiak MK, Claus P. 2015. Coalition of nuclear receptors in the nervous system. J Cell Physiol 230:2875–2880. [DOI] [PubMed] [Google Scholar]
- Gaiano N, Fishell G. 2002. The role of notch in promoting glial and neural stem cell fates. Annu Rev Neurosci 25:471–490. [DOI] [PubMed] [Google Scholar]
- Gao C, Chen YG. 2010. Dishevelled: The hub of Wnt signaling. Cell Signal 22:717–727. [DOI] [PubMed] [Google Scholar]
- Gluache IaR I. 2011. Hetergeneity and flexibility of stem cell fate decisions. New Jersey, London, Singapore, Bejing, Shanghai, Hong Kong, Taipei Chennai: World Scientific. [Google Scholar]
- Guan K, Chang H, Rolletschek A, Wobus AM. 2001. Embryonic stem cell‐derived neurogenesis. Retinoic acid induction and lineage selection of neuronal cells. Cell Tissue Res 305:171–176. [DOI] [PubMed] [Google Scholar]
- Halaban R, Kwon BS, Ghosh S, Delli Bovi P, Baird A. 1988. BFGF as an autocrine growth factor for human melanomas. Oncogene Res 3:177–186. [PubMed] [Google Scholar]
- Han X, Xiao Z, Quarles LD. 2015a. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF‐23. J Biol Chem 290:10447–10459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Xiao Z, Quarles LD. 2015b. Membrane and integrative nuclear fibroblastic growth factor receptor (FGFR) regulation of FGF‐23. J Biol Chem 290:20101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horbinski C, Stachowiak EK, Chandrasekaran V, Miuzukoshi E, Higgins D, Stachowiak MK. 2002. Bone morphogenetic protein‐7 stimulates initial dendritic growth in sympathetic neurons through an intracellular fibroblast growth factor signaling pathway. J Neurochem 80:54–63. [DOI] [PubMed] [Google Scholar]
- Hu Y, Fang X, Dunham SM, Prada C, Stachowiak EK, Stachowiak MK. 2004. 90‐kDa ribosomal S6 kinase is a direct target for the nuclear fibroblast growth factor receptor 1 (FGFR1): Role in FGFR1 signaling. J Biol Chem 279:29325–29335. [DOI] [PubMed] [Google Scholar]
- Isaacs HV, Pownall ME, Slack JM. 1998. Regulation of Hox gene expression and posterior development by the Xenopus caudal homologue Xcad3. EMBO J 17:3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. 2006. Dissecting self‐renewal in stem cells with RNA interference. Nature 442:533–538. [DOI] [PubMed] [Google Scholar]
- Izpisua‐Belmonte JC, Falkenstein H, Dolle P, Renucci A, Duboule D. 1991. Murine genes related to the Drosophila AbdB homeotic genes are sequentially expressed during development of the posterior part of the body. EMBO J 10:2279–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro‐Peled H, Hayashi‐Takagi A, Seshadri S, Kamiya A, Brandon NJ, Sawa A. 2009. Neurodevelopmental mechanisms of schizophrenia: Understanding disturbed postnatal brain maturation through neuregulin‐1‐ErbB4 and DISC1. Trends Neurosci 32:485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablensky A, Morar B, Wiltshire S, Carter K, Dragovic M, Badcock JC, Chandler D, Peters K, Kalaydjieva L. 2011. Polymorphisms associated with normal memory variation also affect memory impairment in schizophrenia. Genes Brain Behav 10:410–417. [DOI] [PubMed] [Google Scholar]
- Jacob AL, Smith C, Partanen J, Ornitz DM. 2006. Fibroblast growth factor receptor 1 signaling in the osteo‐chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev Biol 296:315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML, Volta M, Chan SS, Lipovich L, Pollard SM, Karuturi RK, Wei CL, Buckley NJ, Stanton LW. 2008. REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol 6:e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam RK, Deng Y, Chen Y, Zhao H. 2012. Retinoic acid synthesis and functions in early embryonic development. Cell Biosci 2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh M, Nakagama H. 2014. FGF receptors: Cancer biology and therapeutics. Med Res Rev 34:280–300. [DOI] [PubMed] [Google Scholar]
- Klejbor I, Myers JM, Hausknecht K, Corso TD, Gambino AS, Morys J, Maher PA, Hard R, Richards J, Stachowiak EK, Stachowiak MK. 2006. Fibroblast growth factor receptor signaling affects development and function of dopamine neurons—Inhibition results in a schizophrenia‐like syndrome in transgenic mice. J Neurochem 97:1243–1258. [DOI] [PubMed] [Google Scholar]
- Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, Ishijima J, Shiroishi T, Barna M. 2011. Ribosome‐mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell 145:383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeira D, Bagci H, Malinowski AR, Brown KE, Soza‐Ried J, Feytout A, Webster Z, Ndjetehe E, Cantone I, Asenjo HG, Brockdorff N, Carroll T, Merkenschlager M, Fisher AG. 2015. Jarid2 coordinates nanog expression and PCP/Wnt signaling required for efficient ESC differentiation and early embryo development. Cell Rep 12:573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larroux C, Fahey B, Degnan SM, Adamski M, Rokhsar DS, Degnan BM. 2007. The NK homeobox gene cluster predates the origin of Hox genes. Curr Biol 17:706–710. [DOI] [PubMed] [Google Scholar]
- Lee YW, Terranova C, Birkaya B, Narla S, Kehoe D, Parikh A, Dong S, Ratzka A, Brinkmann H, Aletta JM, Tzanakakis ES, Stachowiak EK, Claus P, Stachowiak MK. 2012. A novel nuclear FGF receptor‐1 partnership with retinoid and Nur receptors during developmental gene programming of embryonic stem cells. J Cell Biochem 113:2920–2936. [DOI] [PubMed] [Google Scholar]
- Lee YW, Stachowiak EK, Birkaya B, Terranova C, Capacchietti M, Claus P, Aletta JM, Stachowiak MK. 2013. NGF‐induced cell differentiation and gene activation is mediated by integrative nuclear FGFR1 signaling (INFS). PLoS ONE 8:e68931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P, Benomar Y, Staels B. 2010. Retinoid X receptors: Common heterodimerization partners with distinct functions. Trends Endocrinol Metab 21:676–683. [DOI] [PubMed] [Google Scholar]
- Maira M, Martens C, Philips A, Drouin J. 1999. Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol Cell Biol 19:7549–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maira M, Martens C, Batsche E, Gauthier Y, Drouin J. 2003. Dimer‐specific potentiation of NGFI‐B (Nur77) transcriptional activity by the protein kinase A pathway and AF‐1‐dependent coactivator recruitment. Mol Cell Biol 23:763–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallo M, Alonso CR. 2013. The regulation of Hox gene expression during animal development. Development 140:3951–3963. [DOI] [PubMed] [Google Scholar]
- Matsuno K, Diederich RJ, Go MJ, Blaumueller CM, Artavanis‐Tsakonas S. 1995. Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development 121:2633–2644. [DOI] [PubMed] [Google Scholar]
- Maynard Smith J SE. 1995. The major transitions in evolution. New York: Oxford University Press. [Google Scholar]
- Medic S, Ziman M. 2009. PAX3 across the spectrum: from melanoblast to melanoma. Crit Rev Biochem Mol Biol 44:85–97. [DOI] [PubMed] [Google Scholar]
- Montavon T, Soshnikova N. 2014. Hox gene regulation and timing in embryogenesis. Semin C Dev Biol 34C:76–84. [DOI] [PubMed] [Google Scholar]
- Morriss‐Kay GM, Sokolova N. 1996. Embryonic development and pattern formation. FASEB J 10:961–968. [DOI] [PubMed] [Google Scholar]
- Moskvina V, Craddock N, Holmans P, Nikolov I, Pahwa JS, Green E, Owen MJ, O'Donovan MC. 2009. Gene‐wide analyses of genome‐wide association data sets: Evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatry 14:252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JM, Martins GG, Ostrowski J, Stachowiak MK. 2003. Nuclear trafficking of FGFR1: A role for the transmembrane domain. J Cell Biochem 88:1273–1291. [DOI] [PubMed] [Google Scholar]
- Narla ST, Klejbor I, Birkaya B, Lee YW, Morys J, Stachowiak EK, Prokop D, Bencherif M, Stachowiak MK. 2013. Activation of developmental nuclear fibroblast growth factor receptor 1 signaling and neurogenesis in adult brain by alpha7 nicotinic receptor agonist. Stem Cells Transl Med 2:776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickoloff BJ, Hendrix MJ, Pollock PM, Trent JM, Miele L, Qin JZ. 2005. Notch and NOXA‐related pathways in melanoma cells. J Investig Dermatol Symp Proc 10:95–104. [DOI] [PubMed] [Google Scholar]
- Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM. 2004. Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci 24:6057–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Shimazaki T, Sobue G, Okano H. 2004. Retinoic‐acid‐concentration‐dependent acquisition of neural cell identity during in vitro differentiation of mouse embryonic stem cells. Dev Biol 275:124–142. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Itoh N. 2011. Fibroblast growth factors. Genome Biol 2:S3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- P. Turpenny SE. Elements of medical genetics. London, UK: Elsevier. [Google Scholar]
- Partanen J, Schwartz L, Rossant J. 1998. Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev 12:2332–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual‐Anaya J, D'Aniello S, Kuratani S, Garcia‐Fernandez J. 2013. Evolution of Hox gene clusters in deuterostomes. BMC Dev Biol 13:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Moffett J, Myers J, Fang X, Stachowiak EK, Maher P, Kratz E, Hines J, Fluharty SJ, Mizukoshi E, Bloom DC, Stachowiak MK. 2001. Novel nuclear signaling pathway mediates activation of fibroblast growth factor‐2 gene by type 1 and type 2 angiotensin II receptors. Mol Biol Cell 12:449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Myers J, Fang X, Stachowiak EK, Maher PA, Martins GG, Popescu G, Berezney R, Stachowiak MK. 2002. Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J Neurochem 81:506–524. [DOI] [PubMed] [Google Scholar]
- Philippidou P, Dasen JS. 2013. Hox genes: Choreographers in neural development, architects of circuit organization. Neuron 80:12–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirvola U, Ylikoski J, Trokovic R, Hebert JM, McConnell SK, Partanen J. 2002. FGFR1 is required for the development of the auditory sensory epithelium. Neuron 35:671–680. [DOI] [PubMed] [Google Scholar]
- Popovici C, Fallet M, Marguet D, Birnbaum D, Roubin R. 2006. Intracellular trafficking of LET‐756, a fibroblast growth factor of C. elegans, is controlled by a balance of export and nuclear signals. Exp Cell Res 312:1484–1495. [DOI] [PubMed] [Google Scholar]
- Potkin SG, Macciardi F, Guffanti G, Fallon JH, Wang Q, Turner JA, Lakatos A, Miles MF, Lander A, Vawter MP, Xie X. 2010. Identifying gene regulatory networks in schizophrenia. Neuroimage 53:839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pownall ME, Isaacs HV, Slack JM. 1998. Two phases of Hox gene regulation during early Xenopus development. Curr Biol 8:673–676. [DOI] [PubMed] [Google Scholar]
- Reilly JF, Maher PA. 2001. Importin beta‐mediated nuclear import of fibroblast growth factor receptor: Role in cell proliferation. J Cell Biol 152:1307–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, Dolle P. 2012. Retinoic acid signalling during development. Development 139:843–858. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Murillo L, Gogos JA, Karayiorgou M. 2012. The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annu Rev Med 63:63–80. [DOI] [PubMed] [Google Scholar]
- Schuettengruber B, Cavalli G. 2009. Recruitment of polycomb group complexes and their role in the dynamic regulation of cell fate choice. Development 136:3531–3542. [DOI] [PubMed] [Google Scholar]
- Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A. 2012. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 337:1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smidt MP, Burbach JP. 2009. Terminal differentiation of mesodiencephalic dopaminergic neurons: the role of Nurr1 and Pitx3. Adv Exp Med Biol 651:47–57. [PubMed] [Google Scholar]
- Somanathan S, Stachowiak EK, Siegel AJ, Stachowiak MK, Berezney R. 2003. Nuclear matrix bound fibroblast growth factor receptor is associated with splicing factor rich and transcriptionally active nuclear speckles. J Cell Biochem 90:856–869. [DOI] [PubMed] [Google Scholar]
- Stachowiak EK, Maher PA, Tucholski J, Mordechai E, Joy A, Moffett J, Coons S, Stachowiak MK. 1997a. Nuclear accumulation of fibroblast growth factor receptors in human glial cells‐association with cell proliferation. Oncogene 14:2201–2211. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Moffett J, Maher P, Tucholski J, Stachowiak EK. 1997b. Growth factor regulation of cell growth and proliferation in the nervous system. A new intracrine nuclear mechanism. Mol Neurobiol 15:257–283. [DOI] [PubMed] [Google Scholar]
- Stachowiak EK, Fang X, Myers J, Dunham S, Stachowiak MK. 2003a. CAMP‐induced differentiation of human neuronal progenitor cells is mediated by nuclear fibroblast growth factor receptor‐1 (FGFR1). J Neurochem 84:1296–1312. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Fang X, Myers JM, Dunham SM, Berezney R, Maher PA, Stachowiak EK. 2003b. Integrative nuclear FGFR1 signaling (INFS) as a part of a universal “feed‐forward‐and‐gate“ signaling module that controls cell growth and differentiation. J Cell Biochem 90:662–691. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Maher PA, Stachowiak EK. 2007. Integrative nuclear signaling in cell development—A role for FGF receptor‐1. DNA Cell Biol 26:811–826. [DOI] [PubMed] [Google Scholar]
- Stachowiak EK, Roy I, Lee YW, Capacchietti M, Aletta JM, Prasad PN, Stachowiak MK. 2009. Targeting novel integrative nuclear FGFR1 signaling by nanoparticle‐mediated gene transfer stimulates neurogenesis in the adult brain. Integr Biol 1:394–403. [DOI] [PubMed] [Google Scholar]
- Stachowiak EK, Roy I, Stachowiak MK. 2012a. Triggering neuronogenesis by endogenous brain stem cells with DNA nanoplexes In: Stachowiak EST MK, editor. Stem cells from mechanisms to technologies. Hakensack, NJ, USA/London, UK: World Scientific Publishing; pp 333–359. [Google Scholar]
- Stachowiak MK, Stachowiak EK, Aletta JM, Tzanakakis ES. 2012b. A common integrative nuclear signaling module for stem cell development In: Stachowiak EST MK, editor. Stem cells from mechanisms to technologies. World Scientific; pp 87–132. [Google Scholar]
- Stachowiak MK, Kucinski A, Curl R, Syposs C, Yang Y, Narla S, Terranova C, Prokop D, Klejbor I, Bencherif M, Birkaya B, Corso T, Parikh A, Tzanakakis ES, Wersinger S, Stachowiak EK. 2013. Schizophrenia: A neurodevelopmental disorder‐integrative genomic hypothesis and therapeutic implications from a transgenic mouse model. Schizophr Res 143:367–376. [DOI] [PubMed] [Google Scholar]
- Stachowiak MK, Birkaya B, Aletta JM, Narla ST, Benson CA, Decker B, Stachowiak EK. 2015. Nuclear FGF receptor‐1 and CREB binding protein: An integrative signaling module. J Cell Physiol 230:989–1002. [DOI] [PubMed] [Google Scholar]
- Stuhlmiller TJ, Garcia‐Castro MI. 2012. FGF/MAPK signaling is required in the gastrula epiblast for avian neural crest induction. Development 139:289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Jia P, Fanous AH, van den Oord E, Chen X, Riley BP, Amdur RL, Kendler KS, Zhao Z. 2010. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS ONE 5:e11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Takahashi K, Yamanaka S. 2014. Induction of pluripotency by defined factors. Proc Jpn Acad Ser B Phys Biol Sci 90:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terranova C, Narla ST, Lee YW, Bard J, Parikh A, Stachowiak EK, Tzanakakis ES, Buck MJ, Birkaya B, Stachowiak MK. 2015. Global developmental gene programing involves a nuclear form of fibroblast growth factor receptor‐1 (FGFR1). PLoS ONE 10:e0123380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JA, Ziman M. 2011. Pax genes during neural development and their potential role in neuroregeneration. Prog Neurobiol 95:334–351. [DOI] [PubMed] [Google Scholar]
- Tschopp P, Duboule D. 2011. A regulatory ‘landscape effect’ over the HoxD cluster. Dev Biol 351:288–296. [DOI] [PubMed] [Google Scholar]
- Wahl MB, Deng C, Lewandoski M, Pourquie O. 2007. FGF signaling acts upstream of the NOTCH and WNT signaling pathways to control segmentation clock oscillations in mouse somitogenesis. Development 134:4033–4041. [DOI] [PubMed] [Google Scholar]
- White RJ, Schilling TF. 2008. How degrading: Cyp26s in hindbrain development. Dev Dyn 237:2775–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Woon HG, Weickert CS. 2011. Full length TrkB potentiates estrogen receptor alpha mediated transcription suggesting convergence of susceptibility pathways in schizophrenia. Mol Cell Neurosci 46:67–78. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. 1994. Fgfr‐1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8:3032–3044. [DOI] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Chambers I, Smith A. 2003. BMP induction of Id proteins suppresses differentiation and sustains embryonic stem cell self‐renewal in collaboration with STAT3. Cell 115:281–292. [DOI] [PubMed] [Google Scholar]
- Young T, Deschamps J. 2009. Hox, Cdx, and anteroposterior patterning in the mouse embryo. Curr Top Dev Biol 88:235–255. [DOI] [PubMed] [Google Scholar]
- Zakany J, Duboule D. 2007. The role of Hox genes during vertebrate limb development. Curr Opin Genet Dev 17:359–366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher's web‐site.
Fig. S1. Ingenuity Pathway Analysis (IPA) of genes expressed differentially in ESCs and NCs and targeted by nFGFR1.
Table S1. Mir301 and mir9 have binding sequences in a great number of mRNAs (364 and 500, respectively; identified by mirtarbase data base analysis)
Video—Supplemental Material.