

Abstract
Objective
Anti–tumor necrosis factor (anti‐TNF) agents are frequently used in combination with methotrexate (MTX) to treat rheumatoid arthritis (RA). We investigated the effect of a background MTX dose, in combination with anti‐TNF certolizumab pegol (CZP), on treatment efficacy and safety in RA patients.
Methods
A pre‐specified subgroup analysis comparing 2 MTX dosage categories (<15 mg/week and ≥15 mg/week) was carried out using data pooled from phase III clinical trials, Rheumatoid Arthritis Prevention of Structural Damage 1 (RAPID 1) and RAPID 2, according to treatment group: CZP 200 mg, CZP 400 mg, or placebo, every 2 weeks. Inclusion criteria required MTX dosage ≥10 mg/week. Efficacy end points included week 24 American College of Rheumatology criteria for 20%, 50%, and 70% improvement (ACR20/50/70) responses analyzed by logistic regression, and changes from baseline in the Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28‐ESR) and the modified Sharp/van der Heijde score (SHS) were analyzed by analysis of covariance. Incidence rates of treatment‐emergent adverse events (TEAEs) were categorized by baseline MTX dose. Post hoc sensitivity analysis investigated 3 MTX dose categories: ≤10 mg/week, >10 and ≤15 mg/week, and >15 mg/week.
Results
A total of 638, 635, and 325 patients received CZP 200 mg, CZP 400 mg, and placebo, respectively. At week 24, treatment responses in both CZP groups were uninfluenced by baseline MTX dose category, and were superior to the placebo group for all investigated end points: ACR20/50/70, DAS28‐ESR, and SHS. TEAE incidence rates were higher in patients receiving MTX ≥15 mg/week for most TEAE types across treatment groups.
Conclusion
CZP efficacy was not affected by background MTX dose category. It can be hypothesized that to minimize TEAEs, background MTX doses could be tailored to individual patient tolerance without affecting CZP efficacy.
INTRODUCTION
Methotrexate (MTX), the most commonly used synthetic disease‐modifying antirheumatic drug (DMARD) 1, 2, 3, was introduced for the treatment of rheumatoid arthritis (RA) more than 30 years ago. It is generally administered once weekly at dosages ranging from 7.5 to 30 mg/week 3, 4. MTX has one of the best benefit to risk ratios of any DMARD used in the management of RA 3, 5. However, some RA patients are intolerant to MTX. Moreover, many patients fail to respond completely so the dose needs to be increased or MTX is combined with other DMARDs. Furthermore, dose escalation may be necessary over time in some patients treated with MTX because of loss of response 6, which increases the risk of related adverse events (AEs). Most AEs associated with MTX, notably gastrointestinal (GI) AEs, are dose dependent 4, 6, 7, 8, 9. Patients receiving high doses may eventually require reduction to potentially less effective MTX doses or discontinuation of therapy because of AEs.
Box 1. Significance & Innovations.
Compared to placebo, certolizumab pegol (CZP) reduces the signs and symptoms of rheumatoid arthritis and inhibits progression of joint damage to a similar extent regardless of the baseline methotrexate (MTX) dose category.
Higher‐dose MTX was associated with an increase in the incidence of most treatment‐emergent adverse events (TEAEs) across all treatment groups, but TEAEs were generally mild to moderate.
The fact that CZP efficacy remains high across a range of baseline MTX dose categories may offer a benefit to patients who are unable to tolerate high doses of MTX.
Combining DMARDs is a widely used therapeutic approach to improve RA disease control 10, 11, 12, 13, 14. Tumor necrosis factor (TNF) plays a central role in the pathogenesis of RA 15, and anti‐TNFs have demonstrated efficacy in reducing the signs and symptoms of RA, as well as inhibiting structural damage, especially when used in combination with MTX 10, 11, 12, 13, 14, 16. Indeed, the combination of an anti‐TNF and MTX is more effective than monotherapy with either an anti‐TNF or MTX 17, 18.
Certolizumab pegol (CZP) is a PEGylated anti‐TNF consisting of a Fab′ fragment attached to a 40 kDa polyethylene glycol (PEG) moiety. In 2 phase III, placebo‐controlled clinical trials (Rheumatoid Arthritis Prevention of Structural Damage [RAPID]1 and 2), CZP significantly reduced the signs and symptoms of active RA and inhibited progression of structural joint damage when administered as add‐on therapy to MTX in patients with an inadequate response to MTX therapy alone 19, 20.
Because dose‐response and dose‐toxicity relationships exist for MTX therapy in RA and the optimal MTX dose is determined on an individual basis 4, 6, 7, 8, 9, it is important to establish whether an anti‐TNF agent will be effective across a wide dose range of background MTX doses. The objective of this subgroup analysis of data from the RAPID 1 and 2 trials was to assess the efficacy and safety/tolerability of CZP compared to placebo in RA patients receiving different background MTX doses.
MATERIALS AND METHODS
Study design
RAPID 1 (NCT00152386) and RAPID 2 (NCT00160602) study designs have been described previously 19, 20. Briefly, RAPID 1 and 2 were phase III, randomized, double‐blind placebo‐controlled trials, with durations of 52 weeks (RAPID 1) and 24 weeks (RAPID 2), respectively. Patients were eligible for inclusion in the trials if they met the following criteria: age ≥18 years with a diagnosis of RA, defined by American College of Rheumatology (ACR) 1987 criteria 21, of ≥6 months but <15 years’ duration; had received MTX (≥10 mg/week) for ≥6 months, with a stable dose for ≥2 months prior to baseline and had active RA at screening and baseline, defined as ≥9 tender joints and ≥9 swollen joints; and had an erythrocyte sedimentation rate (ESR; Westergren) ≥30 mm/hour or a C‐reactive protein concentration >15 mg/liter.
Patients were randomized 2:2:1 to 1 of 2 CZP treatment regimens (400 mg loading dose at weeks 0, 2, and 4, followed by 200 mg or 400 mg every 2 weeks) or placebo, added to stable‐dose MTX. All patients continued MTX therapy at the same dose as at study entry (≥10 mg/week). Patients failing to achieve ACR criteria for 20% improvement (ACR20) response at both weeks 12 and 14 were withdrawn from the study at week 16 (as per study protocol).
The primary efficacy end points were an ACR20 response rate at week 24 (RAPID 1 and RAPID 2) and change from baseline in the modified Sharp/van der Heijde score (SHS) at week 52 (RAPID 1) 19, 20. Sample size was determined based on expected differences between the CZP groups and placebo for both primary efficacy end points.
Analysis by baseline MTX dose
A pre‐specified subgroup analysis of the primary end point (i.e., ACR20 response at week 24) was performed in both RAPID 1 and 2 to assess the efficacy of CZP according to MTX dose category at enrollment (<15 mg/week or ≥15 mg/week).
Subsequently, a further subgroup analysis pooled data from RAPID 1 and 2 studies by treatment group (CZP 200 mg, CZP 400 mg, or placebo) and excluded all patients with unknown MTX dose at baseline. Efficacy and safety/tolerability were assessed in each pooled treatment group according to baseline MTX dose category: <15 mg/week or ≥15 mg/week. A post hoc sensitivity analysis was also undertaken in which patients were subdivided into 3 MTX dose categories as follows: ≤10 mg/week, >10 and ≤15 mg/week, and >15 mg/week.
Efficacy analyses
The efficacy end points examined in the intent‐to‐treat (ITT) population were week 24 ACR20/50/70 response rates, change from baseline in the Disease Activity Score in 28 joints using the ESR (DAS28‐ESR) assessment, DAS28‐ESR remission (defined as DAS28‐ESR <2.6), DAS28‐ESR low disease activity (LDA; defined as DAS28‐ESR ≤3.2), and change from baseline in the SHS.
Safety analyses
Safety/tolerability end points included treatment‐emergent adverse events (TEAEs), which were defined as AEs occurring after the first administration of the study drug and up to 12 weeks after the last dose for patients not continuing into the open‐label extension study. TEAEs were classified by system organ class and preferred term, according to the Medical Dictionary for Regulatory Activities (MedDRA), version 9.0. Tuberculosis (TB) was classified according to the MedDRA high‐level term, which included both latent TB and active TB.
Analysis of TEAEs (incidence rate per 100 patient‐years ± 95% confidence intervals) was conducted by baseline MTX dose category in the safety population, defined as any patient who received at least 1 dose of study drug. Due to a lack of TEAEs occurring in ≥10% of patients, “most frequent” TEAEs were defined as those occurring in ≥5% of patients in any one treatment group.
Statistical analysis
To explore differences in the CZP treatment effect compared to placebo between the 2 or 3 MTX dose categories, a factor for MTX category and an interaction term between MTX category and treatment were added to the logistic regression models, which originally had treatment and geographic region as factors, previously used to assess treatment effect on responder rates in the RAPID 1 and RAPID 2 trials 19, 20. For change from baseline in SHS and DAS28‐ESR scores, an analysis of covariance model, with treatment and region as factors, and baseline as covariate, was utilized 19, 20. All reported P values can only be interpreted in an exploratory manner, i.e., are nominal.
Following withdrawal or the use of rescue medication, missing patient data were imputed using nonresponder imputation for ACR20/50/70 responses and DAS28‐ESR remission and LDA, linear extrapolation for SHS, and last observation carried forward for the DAS28‐ESR change from baseline. Missing SHS baseline values were imputed by the median value of all patients within the treatment group.
RESULTS
Patients
Of the 1,601 patients originally randomized into RAPID 1 and RAPID 2, the MTX dose was available for 1,598 (638, 635, and 325 patients in the 200 mg CZP plus MTX, 400 mg CZP plus MTX, and placebo plus MTX groups, respectively) and were included in these analyses. Baseline demographics and disease activity were similar across the 3 treatment groups (CZP 200 mg, CZP 400 mg, or placebo) regardless of baseline MTX dose category (data not shown). Overall, more than 90% of patients were of Caucasian descent. Three patients were receiving MTX under 10 mg/week at study baseline (study protocol deviators), and 792 patients received MTX at a dosage of 10 mg/week (see Table 1 and Supplementary Table 1, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract. One patient randomized to the CZP 200 mg group did not receive treatment, and 2 patients randomized to the placebo group received CZP 200 mg [and were therefore included in the placebo group for ITT analyses and in the CZP 200 mg group for safety analyses]). Overall, 963 patients (60.3%) received MTX dosages of <15 mg/week and 635 (39.7%) received dosages of ≥15 mg/week.
Table 1.
Key efficacy end points at week 24 evaluated in subgroups of patients based on 3 categories of MTX doses at baseline (intent‐to‐treat population, sensitivity analysis)a
| MTX ≤10 mg/week | MTX >10 and ≤15 mg/week | MTX >15 mg/week | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PBO (n = 164) | CZP 200 mg (n = 326) | CZP 400 mg (n = 305) | PBO (n = 119) | CZP 200 mg (n = 201) | CZP 400 mg (n = 221) | PBO (n = 42) | CZP 200 mg (n = 111) | CZP 400 mg (n = 109) | Interaction P | |
| ACR response, % | ||||||||||
| ACR20 | 10.4 | 59.9 | 59.2 | 10.2 | 58.3 | 63.6 | 21.4 | 53.6 | 52.3 | 0.095 |
| ACR50 | 5.5 | 30.9 | 34.9 | 4.2 | 40.7 | 42.7 | 11.9 | 39.1 | 33.0 | 0.201 |
| ACR70 | 2.4 | 16.0 | 13.2 | 0.8 | 21.6 | 21.4 | 4.8 | 24.5 | 17.4 | 0.518 |
| Change from baseline in DAS28‐ESR, mean ± SD | −0.5 ± 1.2 | −2.2 ± 1.4 | −2.5 ± 1.4 | −0.6 ± 1.1 | −2.5 ± 1.5 | −2.6 ± 1.4 | −1.0 ± 1.4 | −2.5 ± 1.5 | −2.3 ± 1.5 | 0.083 |
| DAS28‐ESR remission, % | 1.2 | 8.0 | 8.9 | 0.9 | 11.6 | 14.7 | 2.4 | 17.1 | 11.0 | 0.902 |
| LDA, % | 1.8 | 15.1 | 18.4 | 1.8 | 22.1 | 27.5 | 7.1 | 29.7 | 19.3 | 0.597 |
| Mean change in radiographic end points | ||||||||||
| SHS, mean ± SD | 1.1 ± 4.0 | 0.3 ± 3.4 | −0.2 ± 2.6 | 1.6 ± 4.2 | 0.2 ± 2.7 | 0.3 ± 4.8 | 1.0 ± 1.9 | 0.1 ± 2.5 | −0.2 ± 2.7 | 0.708 |
| Erosion score, mean ± SD | 0.6 ± 2.5 | 0.0 ± 1.8 | −0.3 ± 1.8 | 0.7 ± 2.3 | 0.0 ± 1.5 | 0.1 ± 2.9 | 0.9 ± 1.8 | 0.2 ± 1.7 | 0.0 ± 0.9 | 0.530 |
| JSN score, mean ± SD | 0.5 ± 2.2 | 0.2 ± 2.3 | 0.0 ± 1.6 | 0.9 ± 2.9 | 0.2 ± 1.9 | 0.2 ± 2.4 | 0.1 ± 1.5 | −0.1 ± 2.1 | −0.1 ± 2.0 | 0.751 |
MTX = methotrexate; PBO = placebo; CZP = certolizumab pegol; ACR20/50/70 = American College of Rheumatology criteria for 20%/50%/70% improvement in disease activity; DAS28‐ESR = Disease Activity Score 28‐joint assessment using the erythrocyte sedimentation rate; LDA = low disease activity; SHS = modified Sharp/van der Heijde score; JSN = joint space narrowing.
In the sensitivity analysis (MTX 3‐dose categorization), baseline demographics and disease activity were also found to be similar across the treatment groups regardless of baseline MTX dose category (data not shown).
Clinical efficacy
In the MTX 2‐dose categorization (pre‐specified analysis), ACR20 response rates at week 24 were greater in patients receiving CZP 200 mg or 400 mg than in patients receiving placebo (P < 0.001 by logistic regression), and this effect was similar regardless of baseline MTX dose category (MTX <15 mg/week: 60.1%, 60.2%, and 9.8%; MTX ≥15 mg/week: 55.6%, 58.7%, and 15.1%) (Figure 1A). The baseline MTX dose category did not seem to have an impact on the treatment effect (Figure 1A).
Figure 1.
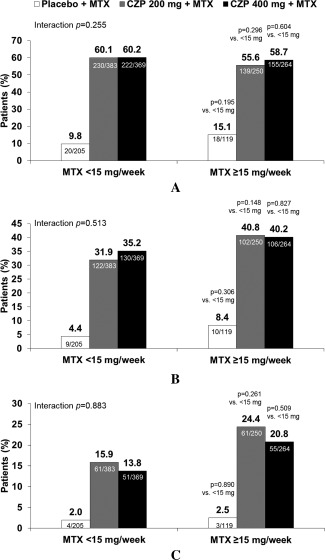
American College of Rheumatology criteria for 20% (A), 50% (B), and 70% (C) improvement (ACR20/50/70) response rates at week 24 by baseline methotrexate (MTX) dose category (intent‐to‐treat population; MTX 2‐dose categorization). Interaction P value between MTX <15 mg/week and MTX ≥15 mg/week across treatment groups based on logistic regression. Individual group P values based on logistic regression without interaction factor. CZP = certolizumab pegol.
ACR20 response rates at week 24 in the MTX 3‐dose categorization sensitivity analysis (≤10 mg/week, >10 and ≤15 mg/week, and >15 mg/week) were also greater in patients receiving CZP 200 mg or CZP 400 mg than in patients receiving placebo (Table 1). Response rates in the CZP groups appeared unaffected by baseline MTX dose category, ranging from 53.6% to 59.9% across the 3 MTX dose categories at week 24 in the CZP 200 mg group. Similar results were observed for patients in the CZP 400 mg group. In contrast, in patients receiving placebo, ACR20 response rates were higher in those receiving MTX >15 mg/week compared with those receiving MTX ≤10 or >10 to ≤15 mg/week at week 24 (21.4% versus 10.2% and 10.4%, respectively) (Table 1).
ACR50 and ACR70 responses at week 24 were greater in patients receiving CZP 200 mg or CZP 400 mg than in patients receiving placebo (Figure 1B and C). Numerically, slightly higher ACR50 and ACR70 response rates were observed in the higher MTX dose category (≥15 mg/week) compared with the lower MTX category (<15 mg/week) across all treatment groups (Figure 1B and C).
In the MTX 3‐dose categorization, ACR50 and ACR70 response rates at week 24 in all 3 MTX dose categories were greater in patients receiving CZP 200 mg or 400 mg than in patients receiving placebo, and response rates were only slightly higher in those receiving MTX >15 mg/week and >10 and ≤15 compared with those receiving MTX ≤10 mg/week (Table 1).
The mean change from baseline in DAS28‐ESR at week 24 was greater in patients receiving CZP 200 mg or CZP 400 mg than in patients receiving placebo (Figure 2) 19, 20. Although a numerically greater mean change was observed in the higher dose category for the CZP 200 mg group (Figure 2B), similar treatment effects were seen across baseline MTX dose categories in the placebo and CZP 400 mg groups. DAS28‐ESR remission rates at week 24 were also higher in patients receiving CZP 200 mg and CZP 400 mg compared to patients receiving placebo (MTX <15 mg/week: 7.6%, 9.8%, and 1.0%; MTX ≥15 mg/week: 15.6%, 13.4%, and 1.8%) (see Supplementary Figure 1A, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). Patients receiving CZP 200 mg or CZP 400 mg were more likely to report LDA at week 24 compared to placebo patients (MTX <15 mg/week: 15.1%, 19.8%, and 1.5%; MTX ≥15 mg/week: 27.2%, 24.4%, and 4.4%) with treatment effect being similar across baseline MTX dose categories (Supplementary Figure 1B, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). In the MTX 3‐dose categorization, treatment effect appeared unaffected by baseline MTX dose category for change from baseline in DAS28‐ESR, DAS28‐ESR remission, and LDA (Table 1).
Figure 2.
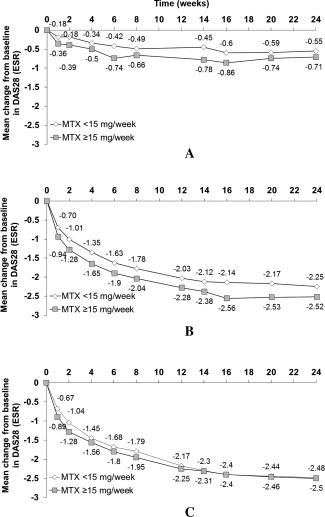
Mean change from baseline in the Disease Activity Score in 28 joints using the erythrocyte sedimentation rate (DAS28[ESR]) over time by baseline methotrexate (MTX) dose category (intent‐to‐treat population; MTX 2‐dose categorization). A, placebo plus MTX, B, certolizumab pegol (CZP) 200 mg plus MTX, and C, CZP 400 mg plus MTX. Week 24 interaction P value = 0.119 (MTX <15 mg/week vs. MTX ≥15 mg/week across treatment groups, based on analysis of covariance).
Inhibition of progression of structural damage
Mean change from baseline in SHS at week 24 was lower in patients receiving CZP 200 mg or CZP 400 mg versus those receiving placebo, regardless of baseline MTX dose category (<15 mg/week and ≥15 mg/week) (Figure 3). Baseline MTX dose category did not seem to have an impact upon treatment effect (Figure 3). In patients receiving placebo, mean changes from baseline in SHS at week 24 were similar in those receiving MTX <15 mg/week and ≥15 mg/week (1.2 versus 1.4). Mean changes in erosion (see Supplementary Figure 2A, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract) and joint space narrowing scores (see Supplementary Figure 2B, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract) at week 24 were markedly lower in patients receiving CZP 200 mg or CZP 400 mg compared with those receiving placebo (see Supplementary Figure 2).
Figure 3.
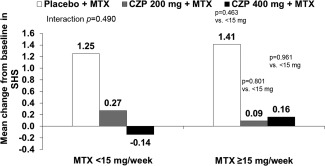
Mean change from baseline in the modified Sharp/van der Heijde score (SHS) at week 24 by baseline methotrexate (MTX) dose category (intent‐to‐treat population; MTX 2‐dose categorization). Interaction P value between MTX <15 mg/week and MTX ≥15 mg/week across treatment groups based on analysis of covariance (ANCOVA). Individual group P values based on ANCOVA. CZP = certolizumab pegol.
Safety and tolerability
All safety data from the 2 trials have previously been reported in detail 19, 20. In all treatment groups, the overall incidence of TEAEs appeared to be higher with increasing baseline dose of MTX (see Supplementary Table 1, available on the Arthritis Care & Research web site at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). Incidence rates of withdrawal due to TEAEs were low across the treatment groups and similar in all MTX dose categories (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract) with the most common TEAEs leading to withdrawal being infections (22 patients).
The most frequently reported infectious TEAEs were urinary tract infections, nasopharyngitis, and upper respiratory tract infections, with incidence rates of any infection slightly increased in patients in the higher MTX dose category compared to those in the lower dose category (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). The overall incidence rate of serious infections was similar across MTX categories, with the most frequently reported serious infections being pneumonia, erysipelas, and disseminated tuberculosis (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract).
The most frequent noninfectious TEAEs by primary system organ class, which occurred in ≥5% of patients in any one treatment group and MTX dose category, were back pain, rheumatoid arthritis, headache, pyrexia, rash, contusion, and cough (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). In all randomized treatment groups, the incidence rate of most categories of TEAEs was higher in patients receiving MTX ≥15 mg/week compared with those receiving <15 mg/week (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). In particular, GI disorder events were reported less frequently in those receiving <15 mg/week MTX compared to ≥15 mg/week MTX (12 [4.9%], 76 [11.1%], and 41 [8.1%] versus 38 [18.5%], 76 [16.3%], and 93 [19.3%] in CZP 200 mg, CZP 400 mg, and placebo groups, respectively). Noninfectious serious TEAEs were relatively rare, with the most common events reported being RA and cerebrovascular accident (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/acr.22676/abstract). The MTX 3‐dose categorization sensitivity analysis also reported increasing incidence rates of most categories of TEAEs at higher MTX dose categories.
Malignancies, including nonmelanoma skin cancer, affected 17 patients overall in the RAPID 1 and RAPID 2 studies 19, 20. When analyzed by baseline MTX dose category, malignancies were reported in 9 patients in the MTX <15 mg/week group and in 8 patients in the MTX ≥15 mg/week group.
Nine patients died during treatment, the details of which have been previously reported 19, 20. Of these, 7 received MTX <15 mg/week (all of which received MTX ≤10 mg/week), and 2 received MTX ≥15 mg/week. In all cases, events leading to death were judged as being unrelated to or unlikely to be related to study medication by the investigator.
DISCUSSION
The results of this subgroup analysis demonstrate that CZP reduces the signs and symptoms of RA and inhibits progression of joint damage with respect to placebo to a similar extent regardless of the baseline dose category of MTX.
ACR20 response rates at week 24 in the CZP treatment groups were unaffected by baseline MTX dose category (MTX 2‐ and 3‐dose categorizations). Regarding ACR50 and ACR70 response rates, there seemed to be a trend for numerically slightly higher response rates in higher MTX dose categories.
In the CZP 200 mg group, a slightly greater DAS28‐ESR mean change from baseline was observed with increasing baseline MTX dose. This finding was primarily driven by the lower changes from baseline DAS28‐ESR observed in patients receiving the lowest dose of MTX (≤10 mg/week). However, no such trend was observed for the placebo or CZP 400 mg groups.
A number of systematic reviews have evaluated the efficacy and safety of MTX as monotherapy or as therapy with other DMARDs in RA, but provide no insight into the impact of different background MTX dose categories on the efficacy and safety of anti‐TNF regimens 4, 22, 23. Data from open‐label studies have demonstrated that addition of an anti‐TNF (etanercept or infliximab) to background MTX therapy may allow MTX dose reduction or discontinuation without detrimental effects on efficacy 24, 25, 26.
The type of analyses described in the current paper, in which efficacy and safety have been assessed over a range of baseline MTX dose categories, has also been performed for infliximab and golimumab 27, 28. In the Japanese infliximab study, there was comparable efficacy of infliximab in RA patient subgroups receiving concomitant MTX at low (≤4 mg/week) or high (≥6 mg/week) doses over 54 weeks 27. Despite the use of lower MTX doses in Japan compared with those commonly utilized in the US and Europe, these data are largely consistent with our current findings in that the study drug was effective regardless of MTX dosage. Similar findings were reported for golimumab when the European League Against Rheumatism response rate over 6 months was assessed by baseline MTX dose category (<10 mg/week, ≥10 and <15 mg/week, or ≥15 mg/week) 28.
In a prospective early RA trial investigating the impact of MTX dose category on the efficacy of adalimumab, similar results were reported for patients receiving MTX 10 mg/week and MTX 20 mg/week, but efficacy was lower in patients receiving adalimumab in combination with MTX 5 or 2.5 mg/week 29. Additionally, a trial investigating MTX dose reduction in RA patients receiving adalimumab demonstrated similar outcomes for patients receiving MTX 7.5 mg/week compared to those receiving MTX 20 mg/week 30.
In our study, higher MTX doses seemed to be associated with an increased incidence of TEAEs. This was observed for infections and serious TEAEs, and was particularly the case for GI TEAEs. These observations are consistent with previous reports that describe MTX dose‐dependent increases in TEAEs in patients with RA 6, 8, 9, 29, 30. In an analysis of the safety of adalimumab in combination with different MTX doses in early RA, the incidence of infections appeared to be MTX dose category dependent 30, although the incidence rates of other TEAEs did not differ substantially between doses. This finding should be interpreted with caution, however, as MTX dose was not randomly assigned in the present study, and patients on higher MTX doses may be more prone to specific TEAEs due to more active disease.
The mean baseline MTX dose was approximately 13.5 mg/week in RAPID 1 and approximately 12.5 mg/week in RAPID 2 19, 20. These doses are low compared with current clinical practice recommendations for the use of MTX in RA, which suggest starting on MTX 10–15 mg/week and then escalating by 5 mg every 2–4 weeks to 25–30 mg/week or the highest tolerated dose, with a subsequent switch to subcutaneous administration in the event of an inadequate response 4, 23. Although all enrolled patients were required to have active RA despite at least 6 months of MTX therapy, incomplete MTX response was at the discretion of the investigator, and therefore the MTX dose may have not been optimized in enrolled patients receiving the lower doses of MTX (≤10 mg/week). Nevertheless the doses reflect those used in local clinical practice at the time of study, and the results from this analysis show similar efficacy for patients treated with CZP regardless of baseline MTX dose category. Furthermore, it should be noted that approximately 40% of the patients included in the pre‐specified 2‐dose analysis received MTX doses of ≥15 mg/week at baseline.
This analysis has several limitations. First, it is a subgroup analysis, and neither RAPID 1 nor RAPID 2 was powered to detect clinically relevant MTX dose‐related differences among the treatment effects. Second, mandatory withdrawal of ACR20 nonresponders at week 16 and the 2:2:1 randomization ratio in the trials resulted in longer mean exposure to the study drug in the CZP groups versus the placebo group 19, 20. This limitation, however, was somewhat mitigated in the AE analysis by reporting data as the number of cases per 100‐patient years. However, this method has limitations in itself, especially with low‐incidence TEAEs. For GI disorders, we therefore additionally reported the number of events and percentage of patients affected. Third, patients receiving the different baseline MTX doses may not have been similar in terms of the underlying disease process; physicians often prescribe higher MTX doses to better control the disease process, resulting in patients with poorer disease control receiving higher MTX doses. However, the baseline disease activity was similar across all dose categories, which may suggest that differences in dose reflect differences in local practice in the countries in which the studies were conducted. It is also not known whether patients’ baseline MTX dose represented the optimum dose to control their disease. Furthermore, no change or tailoring of the MTX dose was permitted during the study so it is not known whether a change in the MTX dose would lead to a difference in efficacy. Finally, these analyses were conducted in a clinical trial setting in patients with high disease activity at baseline, from a wide range of geographies, and who had already tolerated at MTX doses of at least 10 mg/week for over 6 months. These factors need to be considered when generalizing the results to patients in clinical practice, particularly those who are unable to tolerate even low doses of MTX.
In conclusion, this subgroup analysis suggests that CZP in combination with MTX reduces the signs and symptoms of RA and inhibits radiographic progression compared with placebo, irrespective of the baseline MTX dose category. Higher‐dose MTX was associated with an increase in the incidence of most TEAEs across all treatment groups, but TEAEs were generally mild to moderate. There was also a trend for numerically higher ACR50 and ACR70 rates in higher‐dose MTX patients. However, the fact that the efficacy of CZP remains high across a range of baseline MTX dose categories may offer a particular benefit to patients who are unable to tolerate high doses of MTX, hypothetically allowing physicians to tailor the MTX dose to the individual patient's tolerance without significantly compromising the efficacy of CZP.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be submitted for publication. Dr. Combe had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Emery.
Acquisition of data
Combe, Emery.
Analysis and interpretation of data
Furst, Keystone, van der Heijde, Luijtens, Ionescu, Goel.
ROLE OF THE STUDY SPONSOR
UCB Pharma sponsored the study and the development of this manuscript. In addition to content approval by the authors, UCB approved the manuscript following a full review to ensure the publication did not contain any information that had the potential to damage the intellectual property of UCB. UCB Pharma had no role in the study design or in the collection, analysis, or interpretation of the data, the writing of the manuscript, or the decision to submit the manuscript for publication. Publication of this article was not contingent upon approval by UCB Pharma.
Supporting information
Supplementary Figure 1: DAS28(ESR) A. Remission and B. LDA rates at Week 24 by baseline MTX dose category ITT population; MTX 2‐dose categorization)
Supplementary Figure 2: Mean change from baseline in radiographic endpoints at Week 24 by baseline MTX dose category (ITT population; MTX 2‐dose categorization). A. Erosion score, B. Joint space narrowing score
Supplementary Table 1. Treatment‐emergent AEs (TEAEs), including serious AEs, and most frequent TEAEs by primary system organ class (ie, occurring in ≥5% patients in any one treatment group), categorized by baseline MTX dose category (Safety populationc)
ACKNOWLEDGMENTS
The authors acknowledge ‘Matladi N. Ndlovu, PhD, UCB Pharma, Brussels, Belgium for publication coordination, and Owen Davies, UCB Pharma, Brussels, Belgium, for critically reviewing the manuscript. The authors would also like to thank all the investigators who participated in this study. Editorial assistance for the development of this manuscript was provided by Shelley Lindley and Andrew Richardson from PAREXEL, and by Costello Medical Consulting.
ClinicalTrials.gov identifier: NCT00152386 and NCT00160602.
REFERENCES
- 1. Braun J, Rau R. An update on methotrexate. Curr Opin Rheumatol 2009;21:216–23. [DOI] [PubMed] [Google Scholar]
- 2. Jobanputra P, Wilson J, Douglas K, Burls A. A survey of British rheumatologists’ DMARD preferences for rheumatoid arthritis. Rheumatology (Oxford) 2004;43:206–10. [DOI] [PubMed] [Google Scholar]
- 3. Swierkot J, Szechinski J. Methotrexate in rheumatoid arthritis. Pharmacol Rep 2006;58:473–92. [PubMed] [Google Scholar]
- 4. Visser K, van der Heijde D. Optimal dosage and route of administration of methotrexate in rheumatoid arthritis: a systematic review of the literature. Ann Rheum Dis 2009;68:1094–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pincus T, Yazici Y, Sokka T, Aletaha D, Smolen JS. Methotrexate as the “anchor drug” for the treatment of early rheumatoid arthritis. Clin Exp Rheumatol 2003;21 Suppl 31:S179–85. [PubMed] [Google Scholar]
- 6. Furst DE, Koehnke R, Burmeister LF, Kohler J, Cargill I. Increasing methotrexate effect with increasing dose in the treatment of resistant rheumatoid arthritis. J Rheumatol 1989;16:313–20. [PubMed] [Google Scholar]
- 7. Seideman P. Methotrexate: the relationship between dose and clinical effect. Br J Rheumatol 1993;32:751–3. [DOI] [PubMed] [Google Scholar]
- 8. Schnabel A, Reinhold‐Keller E, Willmann V, Gross WL. Tolerability of methotrexate starting with 15 or 25 mg/week for rheumatoid arthritis. Rheumatol Int 1994;14:33–8. [DOI] [PubMed] [Google Scholar]
- 9. Verstappen SM, Bakker MF, Heurkens AH, van der Veen MJ, Kruize AA, Geurts MA, et al. Adverse events and factors associated with toxicity in patients with early rheumatoid arthritis treated with methotrexate tight control therapy: the CAMERA study. Ann Rheum Dis 2009;69:1044–8. [DOI] [PubMed] [Google Scholar]
- 10. Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, et al. Etanercept therapy in rheumatoid arthritis: a randomized, controlled trial. Ann Intern Med 1999;130:478–86. [DOI] [PubMed] [Google Scholar]
- 11. Weinblatt ME, Keystone EC, Furst DE, Moreland LW, Weisman MH, Birbara CA, et al. Adalimumab, a fully human anti–tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum 2003;48:35–45. [DOI] [PubMed] [Google Scholar]
- 12. Maini RN, Breedveld FC, Kalden JR, Smolen JS, Furst D, Weisman MH, et al. Sustained improvement over two years in physical function, structural damage, and signs and symptoms among patients with rheumatoid arthritis treated with infliximab and methotrexate. Arthritis Rheum 2004;50:1051–65. [DOI] [PubMed] [Google Scholar]
- 13. Genovese MC, Bathon JM, Martin RW, Fleischmann RM, Tesser JR, Schiff MH, et al. Etanercept versus methotrexate in patients with early rheumatoid arthritis: two‐year radiographic and clinical outcomes. Arthritis Rheum 2002;46:1443–50. [DOI] [PubMed] [Google Scholar]
- 14. Keystone EC, Kavanaugh AF, Sharp JT, Tannenbaum H, Hua Y, Teoh LS, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti–tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo‐controlled, 52‐week trial. Arthritis Rheum 2004;50:1400–11. [DOI] [PubMed] [Google Scholar]
- 15. Kavanaugh AF. Anti–tumor necrosis factor‐α monoclonal antibody therapy for rheumatoid arthritis. Rheum Dis Clin North Am 1998;24:593–614. [DOI] [PubMed] [Google Scholar]
- 16. Castro‐Rueda H, Kavanaugh A. Biologic therapy for early rheumatoid arthritis: the latest evidence. Curr Opin Rheumatol 2008;20:314–9. [DOI] [PubMed] [Google Scholar]
- 17. Klareskog L, van der Heijde D, de Jager JP, Gough A, Kalden J, Malaise M, et al. Therapeutic effect of the combination of etanercept and methotrexate compared with each treatment alone in patients with rheumatoid arthritis: double‐blind randomised controlled trial. Lancet 2004;363:675–81. [DOI] [PubMed] [Google Scholar]
- 18. Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: a multicenter, randomized, double‐blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 2006;54:26–37. [DOI] [PubMed] [Google Scholar]
- 19. Keystone E, van der Heijde D, Mason D Jr, Landewe R, van Vollenhoven R, Combe B, et al. Certolizumab pegol plus methotrexate is significantly more effective than placebo plus methotrexate in active rheumatoid arthritis: findings of a fifty‐two–week, phase III, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐group study. Arthritis Rheum 2008;58:3319–29. [DOI] [PubMed] [Google Scholar]
- 20. Smolen J, Landewe RB, Mease P, Brzezicki J, Mason D, Luijtens K, et al. Efficacy and safety of certolizumab pegol plus methotrexate in active rheumatoid arthritis: the RAPID 2 study. A randomised controlled trial. Ann Rheum Dis 2009;68:797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 22. Katchamart W, Trudeau J, Phumethum V, Bombardier C. Efficacy and toxicity of methotrexate (MTX) monotherapy versus MTX combination therapy with non‐biological disease–modifying antirheumatic drugs in rheumatoid arthritis: a systematic review and meta‐analysis. Ann Rheum Dis 2009;68:1105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salliot C, van der Heijde D. Long‐term safety of methotrexate monotherapy in patients with rheumatoid arthritis: a systematic literature research. Ann Rheum Dis 2009;68:1100–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kremer JM, Weinblatt ME, Bankhurst AD, Bulpitt KJ, Fleischmann RM, Jackson CG, et al. Etanercept added to background methotrexate therapy in patients with rheumatoid arthritis: continued observations. Arthritis Rheum 2003;48:1493–9. [DOI] [PubMed] [Google Scholar]
- 25. Fleischmann RM, Cohen SB, Moreland LW, Schiff M, Mease PJ, Smith DB, et al, and the iRAMT Study Group . Methotrexate dosage reduction in patients with rheumatoid arthritis beginning therapy with infliximab: the Infliximab Rheumatoid Arthritis Methotrexate Tapering (iRAMT) trial. Curr Med Res Opin 2005;21:1181–90. [DOI] [PubMed] [Google Scholar]
- 26. Van Riel PL, Taggart AJ, Sany J, Gaubitz M, Nab HW, Pedersen R, et al, and the Add Enbrel or Replace Methotrexate Study Investigators. Efficacy and safety of combination etanercept and methotrexate versus etanercept alone in patients with rheumatoid arthritis with an inadequate response to methotrexate: the ADORE study. Ann Rheum Dis 2006;65:1478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wakabayashi H, Sudo A, Hasegawa M, Oka H, Uchida A, Nishioka K. Retrospective clinical study of the efficacy of lower‐dose methotrexate and infliximab therapy in patients with rheumatoid arthritis. Clin Rheumatol 2010;29:671–5. [DOI] [PubMed] [Google Scholar]
- 28. Combe B, Dasgupta B, Louw I, Pal S, Wollenhaupt J, Zerbini CA, et al. Efficacy and safety of golimumab as add‐on therapy to disease‐modifying antirheumatic drugs: results of the GO‐MORE study. Ann Rheum Dis 2014;73:1477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Burmester GR, Kivitz AJ, Kupper H, Arulmani U, Florentinus S, Goss SL, et al. Efficacy and safety of ascending methotrexate dose in combination with adalimumab: the randomised CONCERTO trial. Ann Rheum Dis 2015;74:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaeley GS, Evangelisto AM, Nishio, Midori J, Liu S, Kupper H. Impact of methotrexate dose reduction upon initiation of adalimumab on clinical and ultrasonographic parameters in patients with moderate to severe rheumatoid arthritis [abstract]. Arthritis Rheum 2013;65:S1147. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: DAS28(ESR) A. Remission and B. LDA rates at Week 24 by baseline MTX dose category ITT population; MTX 2‐dose categorization)
Supplementary Figure 2: Mean change from baseline in radiographic endpoints at Week 24 by baseline MTX dose category (ITT population; MTX 2‐dose categorization). A. Erosion score, B. Joint space narrowing score
Supplementary Table 1. Treatment‐emergent AEs (TEAEs), including serious AEs, and most frequent TEAEs by primary system organ class (ie, occurring in ≥5% patients in any one treatment group), categorized by baseline MTX dose category (Safety populationc)