

Abstract
Technologies that can visualize, capture, and identify subsets of biomolecules that are not encoded by the genome in the context of healthy and diseased cells will offer unique opportunities to uncover the molecular mechanism of a multitude of physiological and disease processes. We describe here a chemical reporter strategy for labeling of cell surface glycoconjugates that takes advantage of recombinant glycosyltransferases and a corresponding sugar nucleotide functionalized by biotin. The exceptional efficiency of this method, termed one-step selective exoenzymatic labeling, or SEEL, greatly improved the ability to enrich and identify large numbers of tagged glycoproteins by LC–MS/MS. We further demonstrated that this labeling method resulted in far superior enrichment and detection of glycoproteins at the plasma membrane compared to a sulfo-NHS-activated biotinylation or two-step SEEL. This new methodology will make it possible to profile cell surface glycoproteomes with unprecedented sensitivity in the context of physiological and disease states.
Graphical abstract
INTRODUCTION
The bioorthogonal chemical reporter strategy offers exciting possibilities to interrogate biomolecules of living cells that are not encoded by the genome.1 In this approach, the biosynthetic machinery of a cell is hijacked by feeding a metabolic precursor functionalized with a chemical reporter for incorporation into a target class of biomolecules, such as lipids or glycoconjugates. Next, a bioorthogonal reaction is performed to modify the reporter with a biophysical probe for visualization or enrichment. To date, azido modification is the most widely employed bioorthogonal chemical reporter because of its small size and inertness to most components in a biological environment. It can for example be tagged by Staudinger ligation using modified phosphines,2 copper(I)-catalyzed cycloaddition with terminal alkynes (CuAAC),3 or strain-promoted alkyne–azide cycloaddition (SPAAC).4 In particular, SPAAC is attractive because it avoids the cellular toxicity associated with copper ions without compromising alkyne reactivity.
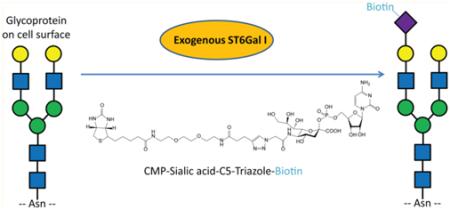
In an alternative approach, chemical reporters can be introduced into biomolecules of living cells by performing glycosyltransferase reactions at the cell surface.5 This process, for which we coined the term SEEL (selective exoenzymatic labeling), takes advantage of recombinant glycosyltransferases and a corresponding functionalized nucleotide sugar to install chemical reporters on cell surface acceptor glycans. For example, we have demonstrated that recombinant ST6Gal1 sialyltransferase and CMP-Neu5Ac9N3 (1) can be exploited for the selective labeling of N-linked glycans of living cells with azido-modified sialic acid. 5g,6 The attraction of the SEEL approach is that it only labels a specific class of cell surface molecules (e.g., N- vs O-glycans) and does not rely on feeding with metabolic substrates that must compete with natural sugar precursor pools. In light of the direct nature of this method, we explored whether SEEL can be accomplished in a single step by employing CMP-Neu5Ac modified by biotin.7 Such an approach would circumvent possible limitations associated with bioorthogonal reactions, such as side reactions,8 and decrease the time needed for labeling.
We have found that a one-step SEEL procedure, employing exogenously administered CMP-Neu5Ac analogues modified at C-5 or C-9 with biotin (compounds 2 and 3, Figure 1),5d,7a not only is feasible but dramatically improves cell surface labeling of glycoconjugates compared to two-step SEEL or metabolic labeling. The new methodology offers exciting possibilities to track, capture, and identify subsets of cell surface glycoconjugates with unprecedented sensitivity in whole cells. We have exploited the high efficiency labeling with compound 2 to enrich and identify tagged glycoproteins by LC–MS/MS, and could monitor the internalization and degradation of labeled cell surface glycoproteins over time, and identify a subset of glycoproteins that are subject to accumulation upon lysosomal disruption by chloroquine. Lastly, the new labeling strategy was compared with a commonly employed kit for cell surface biotinylation and was found to be vastly superior for enrichment of cell surface glycoproteins. Many diseases exhibit lysosomal dysfunction and altered recycling of cell surface glycoproteins.9 For example, Niemann-Pick type C (NPC) disease is characterized by impaired cholesterol efflux from late endosomes and lysosomes and secondary accumulation of lipids. Previously, we employed the chemical reporter strategy to demonstrate an unrecognized accumulation of glycoconjugates in endocytic compartments of NPC1-null and NPC2-deficient fibroblasts.9c The endosomal accumulation of sialylated glycoproteins was attributed to impaired recycling as opposed to altered fusion of vesicles. It is our expectation that identification of altered cell surface residency of glycoproteins in diseases such as NPC using the new SEEL methodology will uncover the molecular mechanisms that cause the phenotypes of these diseases, which in turn may lead to the design of new therapeutic strategies.
Figure 1.

Reagents for one- and two-step SEEL.
RESULTS
Synthesis of Modified Sugar Nucleotides and Enzymology
It is known that sialyltransferases tolerate modifications at C-5 and C-9,7b and it has for example been demonstrated that a CMP-sialic acid derivative having biotin at C-9 can be transferred to LacNAc.5d,7a Therefore, CMP-sialic acid derivatives 2 and 3 were prepared having biotin at the C-5 and C-9 positions. Thus, condensation of C-5 and C-9 azido modified sialic acid 5a and 5b with CTP in the presence of the recombinant CMP-sialic acid synthetase from Neisseria meningitis10 and the inorganic pyrophosphatase from Saccharomyces cerevisiae gave readily CMP-Neu5Ac derivatives 6 and 1, respectively. A CuAAC3a of 6 and 1 with alkyne modified biotin 7 in the presence of CuSO4, ascorbic acid, and TBTA gave, after purification by size exclusion column chromatography over Biogel P2, target compounds 2 and 3, respectively (Scheme 1). An alternative approach in which compounds 5a and 5b were modified by 7 followed by condensation with CTP in the presence of the recombinant CMP-sialic acid synthetase did not give the required products because the enzyme did not tolerate the bulky substituents at C-5 or C-9.
Scheme 1. Synthesis of Compounds 2 and 3a.
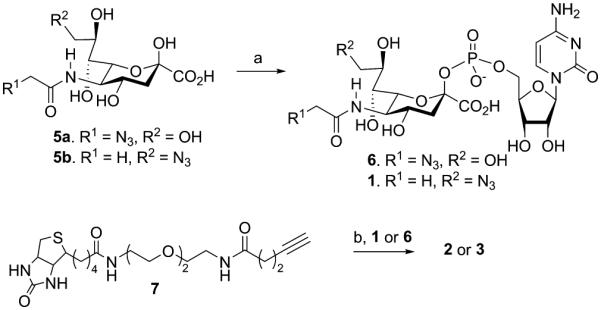
aReagents and conditions: (a) CTP, 100 mM Tris buffer (20 mM MgCl2, pH 8.9), inorganic pyrophosphatase CMP-sialic acid synthetase; (b) CuSO4, Na ascorbate, DMF/Tris-HCl buffer (pH = 7.5), TBTA.
Incubation of N-acetyllactosamine and CMP-sialic acid derivatives 1, 2, or 3 in the presence of ST6Gal1 led to almost quantitative formation of Neu5AcRα(2,6)Galβ(1,4)GlcNAc 8S, 9S, and 10S, respectively highlighting that, in addition to azide, the C-5 and C-9 triazole linked biotin moieties are tolerated by the transferase. Kinetic analysis of the enzymatic transformation showed that the modifications of 2 and 3 had no or a marginal impact on the Km value (2, 3, and CMP-Neu5Ac, Km = 53, 108, and 54 μM, respectively) with no appreciable influence on Vmax.
Labeling and Trafficking of Cell Surface Glycoproteins of Living Cells
HeLa cells were incubated with CMP-Neu5Ac derivatives 1, 2, and 3 (compounds shown in Figure 1) in the presence of ST6Gal1 for 2 h at 37 °C to examine the efficiency of one-step SEEL in comparison to the previously reported5g two-step SEEL method. The length of labeling and the chosen enzyme and substrate donor concentrations were optimized in order to achieve maximal labeling with minimal effects on cell viability. The labeling was performed with and without prior treatment with Vibrio cholerae sialidase. In addition, cells were metabolically labeled with azido containing sialosides by feeding peracetylated N-azidoacetylmannosamine (Ac4ManNAz).2a The cells modified by azido-containing glycoconjugates were biotinylated by exposure to sulfated dibenzocyclooctynylamide containing biotin (S-DIBO-biotin 4, 30 μM, Figure 1)11 for 1 h. The efficiency of the various surface-labeling procedures was determined by SDS–PAGE of cell lysates followed by Western blotting using an anti-biotin antibody conjugated with HRP. These experiments demonstrated that prior neuraminidase treatment of the cells increased the efficiency of labeling for both one- and two-step SEEL (consistent with the presence of abundant highly sialylated N-glycan structures in these cells which reduce the abundance of SEEL acceptors) (Figure 2a). Remarkably, the one-step labeling procedure using 2 and 3 gave a robust biotin signal even after exposure of the blot for only seconds, whereas the two-step protocol with compound 1 or metabolic labeling with ManNAz gave a faint biotin signal and longer exposure time was required to make the glycoproteins visible. Furthermore, labeling with 2 was more efficient than with 3 probably because it is a better substrate for ST6Gal1. The different labeling methods (metabolic, two-step, and one-step SEEL) were compared in three other cell lines (Figure S1) to gauge the generality of this striking improvement in labeling with one-step SEEL. In each case, the one-step SEEL procedure gave by far more robust labeling.
Figure 2.

One-step SEEL with compounds 2 and 3 greatly improved labeling efficiency of sialylated glycoproteins. (a) Western blot analysis of lysates from SEEL-labeled HeLa cells with ST6Gal1 using compound 1 (CMP-Neu5Ac9N3), compound 2 (C-5 biotinylated), or compound 3 (C-9 biotinylated) with or without pretreatment with Vibrio cholerae sialidase. SEEL labeling with 1 and metabolic labeling with Ac4ManNAz (met) was followed by incubation with 4. The above image is representative of three independent labeling experiments. The β-actin loading control is shown below the biotin blot. (b) Venn diagram for the proteomic analysis of HeLa cells subjected to two-step (compound 1) or one-step (compound 2) SEEL with ST6Gal1. (c) The spectral counts of the 20 most abundant glycoproteins detected using one-step SEEL are plotted relative to the counts detected using two-step SEEL (values for two-step SEEL set to 100%).
Next, we performed proteomic analysis on HeLa cells labeled by two-step SEEL or one-step SEEL (with ST6Gal1) to compare the labeling efficiency of the two methods (Table S1). 294 proteins were assigned at <1% false-discovery rate using one-step SEEL, whereas 174 proteins were assigned with two-step SEEL (Figure 2b). One-step SEEL detected 140 glycoproteins not observed using the two-step method, while two-step SEEL identified only 20 unique glycoproteins. The total spectral count of all assigned proteins from one-step SEEL was 2.1 times higher than that from two-step SEEL. Out of the 154 commonly found proteins in both methods, 85 proteins showed more than 2-fold increase in their spectral counts, while 57 proteins showed more than 3-fold increase and 25 proteins showed more than 5-fold increase in one-step SEEL. The relative spectral counts of the top 20 most abundant commonly assigned glycoproteins were compared in Figure 2c. Among these 20 proteins, PTPRF (752%), BSG (423%), ROBO1 (359%), and IGF2R (342%) showed at least a 3-fold improvement in assigned spectral counts. These data demonstrate the remarkably superior labeling eficiency of sialoglycoproteins at the cell surface when one-step SEEL is utilized.
Next, we examined whether the biotin containing sialosides at the cell surface are desialylated by neuraminidase. If the neuraminidase hydrolyzes only the natural form of sialosides but not the biotinylated sialosides, the one-step SEEL procedure can be done in 2 h with concurrent treatment of neuraminidase. Fibroblasts labeled with 2 and 3 were treated with Vibrio cholerae (VC) sialidase or Arthrobacter ureafaciens (AU) sialidase, and the results were compared with untreated cells by Western blotting. Only the C-9 compound was sensitive to treatment with VC sialidase whereas the C-5 analogue was resistant (Figure S2, panel A). In contrast, both modifications were resistant to AU sialidase treatment (panel B and data not shown). Treatment of the C-9 azide-modified sialyllactosamine (8S) with VC sialidase led to the removal of the modified sialic acid moiety within a period of 24 h whereas a similar treatment of C-9 biotin modified sialoside 9S resulted only in partial cleavage of the sialoside, and in this case an extended period of time was required for full removal. A sialoside modified at C-5 with biotin (10S) was resistant to VC sialidase treatment. None of the sialosides could be cleaved by AU sialidase. These findings confirm the cell-based results and also suggest that the AU sialidase can be used concurrently with SEEL labeling as a means to remove existing natural sialic acids without affecting the addition of the biotin-modified sialic acids.12
Next, attention was focused on the use of the new compounds to visualize trafficking of N-linked glycoconjugates in normal and chloroquine-treated cells. HeLa cells were enzymatically labeled with ST6Gal1 and CMP-sialic acid derivative 2, and the resulting biotin-modified N-glycoproteins were visualized by confocal microscopy following incubation with a fluorophore conjugated anti-biotin antibody (Figure 3a). As expected, robust staining at the cell surface was observed. Labeled cells were then incubated with or without chloroquine for 16 h to allow labeled glycoproteins to be internalized from the cell surface. Chloroquine is known to disrupt lysosomal pH and prevent eficient catabolism within this compartment. In the absence of chloroquine, a clear decrease in the intensity of labeling at the cell surface was detected, indicating that the SEEL-tagged glycoproteins can be internalized and/or degraded. In the chloroquine-treated cells, the labeled glycoconjugates instead accumulated within intracellular vesicles, consistent with late endosomes/lysosomes.
Figure 3.

(a) Sialoglycoproteins labeled with one-step SEEL can be internalized and are subject to intracellular accumulation in the presence of chloroquine. HeLa cells labeled by one-step SEEL using compound 2 incubated in the presence or absence of 50 μM chloroquine for 16 h following by confocal analysis. Anti-biotin antibody was applied to cells before internalization (without permeabilization). (b) Western blot analysis of SEEL-labeled HeLa cells under the three conditions tested and silver stain of anti-biotin immunoprecipitates from the same samples. (c) Quantification of normalized spectral counts for SEEL-labeled glycoproteins following analysis by tandem mass spectrometry. (d) Western blot analysis of IGF2R from anti-biotin immunoprecipitates under the three different conditions. 30 μg of protein was loaded in the input and flow through lanes whereas 180 μg of total protein was loaded for the eluted fraction in order to facilitate detection of this protein following the internalization period.
Proteomic Analysis of Labeled Glycoproteins Following Lysosomal Disruption
The high labeling efficiency achieved by the one-step SEEL procedure provides opportunities to enrich and identify cell surface glycoproteins and investigate how different glycoproteins respond to biological processes such as lysosomal disruption. For enrichment of tagged glycoproteins, immunoprecipitation was performed with an anti-biotin antibody, which was followed by SDS–PAGE and silver staining (Figure 3b). The results clearly show a loss of labeled N-linked glycoproteins following incubation in the absence of chloroquine (indicative of endocytosis and degradation of these glycoproteins) but a substantial reduction in the degradation of many labeled proteins in the presence of chloroquine. This observation was confirmed by Western blotting analysis of the same samples using an anti-biotin antibody (Figure 3b). Next, immunoprecipitated glycoproteins were subjected to tryptic digestion followed by shotgun proteomics (LC–MS/MS). The efficiency of labeling and enriching cell surface proteins is demonstrated by the identification of 255 membrane proteins out of the 282 proteins assigned (>90%) in the initial time zero sample (Table S2). Alterations in protein abundance were then examined in samples incubated with or without chloroquine for 16 h. A total of 173 proteins (161 of which are membrane proteins) were present in all three incubation conditions with less than a 1% false-discovery rate (Table S2).
Quantification via spectral counts revealed that 40 glycoproteins decreased in abundance by at least 5-fold (or became undetectable) after incubation of untreated cells for 16 h, but were detected in at least a 3-fold greater abundance when chloroquine was present during the 16 h incubation period (Figure 3c, Table S2). These glycoproteins, which include many receptors, such as the cation-independent mannose 6-phosphate receptor (IGF2R) and ephrin B receptor (EPH2A), likely represent those that internalize and turn over readily when lysosomal function is normal but will accumulate inside cells when the function of this organelle is compromised by chloroquine treatment. The data had been analyzed using stringent conditions for the quantification removing from consideration those glycoproteins that were detected in non-SEEL labeled control samples. Relaxation of this criterion, such as also considering proteins which were enriched at least 10-fold compared to control, captured additional proteins that exhibit the above-described internalization behavior such as CD44 and EGFR (Table S2). Interestingly, another set of glycoproteins including caveolin-1, mucin-1, and several cell surface channel proteins were not or only marginally affected in abundance after the chase period in the presence or absence of chloroquine (Figure 3c, Table S3).
To validate the proteomic findings, the steady-state level of one identified glycoprotein (IGF2R) was assessed by immunoprecipitation and Western blotting. IGF2R, also known as the cation-independent mannose 6-phosphate receptor, is primarily localized to post-Golgi compartments but also cycles to the plasma membrane where it can bind a variety of ligands.13 It is estimated that approximately 10–15% of the total IGF2R pool in cells localizes to the plasma membrane at steady state. Glycoproteins labeled with 2 were immunoprecipitated using an anti-biotin antibody and the resulting samples resolved by SDS–PAGE and Western blotting using an IGF2R protein-specific antibody. When the 6-fold higher protein load in the eluted fraction is taken into account, about 15% of the total IGF2R was labeled by SEEL, consistent with prior estimates of its cell surface distribution under steady state conditions. It can be seen in Figure 3d that incubation for 16 h resulted in a significant decrease in the abundance of the tagged glycoprotein whereas incubation in the presence of chloroquine showed less degradation of the labeled IGF2R. This closely mirrors the proteomic findings and supports the fidelity of the approach to monitor cell surface glycoprotein levels and dynamics, in particular for proteins that actively cycle between the plasma membrane and the endosomal system.
Proteomic Analysis of Cell Surface Proteins Labeled by SEEL and Amino-Reactive Reagents
Existing strategies for the enrichment of cell surface glycoproteins utilize reagents such as sulfo-NHS-activated biotin to tag lysine residues on available proteins.14 These reagents suffer from lack of selectivity and often isolate cytosolic proteins. Using non-adherent HEK293F cells, a direct comparison of the SEEL and amino labeling method was performed. As shown in Figure 4a, labeling with the sulfo-NHS-biotin reagent resulted in the detection of numerous proteins across a broad molecular weight. On the other hand, a more restricted profile of proteins was visualized by one-step SEEL labeling with ST6Gal1. Analysis of the total proteins detected (vs total cell surface glycoproteins detected), however, revealed a clear enrichment in the detection of cell surface glycoproteins using SEEL. Nearly 95% of the total proteins detected following SEEL with ST6Gal1 (367) were bona fide cell surface glycoproteins whereas only 18% of the total proteins detected using the sulfo-NHS-biotin reagent were cell surface glycoproteins (Figure 4b, top panel). Analysis of total spectral counts recovered using the two methodologies further enforced the obvious advantage of using SEEL-based labeling to enrich and detect cell surface glycoproteins (Figure 4b, bottom panel).
Figure 4.

(a) Western blot analysis of HEK293F cells labeled using sulfo-NHS-biotin or SEEL with ST6Gal1. (b) Quantification of the total proteins identified (top panel) and their spectral counts (bottom panel) under these conditions compared to the total cell surface glycoproteins identified.
DISCUSSION
The findings reported here demonstrate a remarkable labeling efficiency of cell surface glycoproteins by employing CMP-sialic acid modified by biotin and an appropriate sialyltransferase. The approach made it possible to track, capture, and identify subsets of cell surface glycoconjugates with unprecedented sensitivity in whole cells. The efficiency of the enzymatic transfer by ST6Gal1 is not substantially altered by the presence of the biotin (or azide) tag, and therefore we believe that the inferior labeling with the two-step SEEL method lies in the bioorthogonal reaction step. It is possible that the alkyne-containing compounds are prone to side reactions,8 but very little background is observed when cells are incubated with DIBO or S-DIBO alone. This observation would suggest instead that steric hindrance or electrostatic effects impact the efficiency of labeling at the cell surface. The use of the biotin conjugated CMP-sialic acid derivatives also decreases the total labeling time which makes it suitable for SEEL on cell types that may be sensitive to the conditions needed for this type of labeling. We did note that metabolic labeling most efficiently labeled a different subset of glycoproteins than SEEL (Figure 2). This may reflect the ability of metabolic labeling to label glycoproteins with high rates of turnover and biosynthesis. Likewise, it is anticipated that metabolic labeling can label intracellular glycoproteins (e.g., Golgi enzymes, etc.). The SEEL approach requires galactosyl acceptors at the cell surface, which in situ can be generated by performing the reactions in the presence of a bacterial neuraminidase that is capable of removing the natural sialic acids but not the biotin-bearing sialic acids. The ability to effectively label in the presence of the neuraminidase is advantageous since it may limit the effects that desialylation would have on the cell surface residence or endocytosis of glycoproteins.
Immunopurification of labeled glycoconjugates followed by tandem mass spectrometry demonstrated the power of the one-step SEEL approach to efficiently enrich and confidently identify cell surface glycoproteins, as approximately 300 known cell surface glycoproteins were detected. Almost twice as many proteins were identified compared to the two-step SEEL method (Figure 2b), and the new approach allows for many lower abundance glycoproteins to be uniquely identified. The 2.0-fold increase in spectral counts with one-step SEEL is far less dramatic than the increased labeling efficiency of glycoproteins detected by Western blotting. We believe that this difference arises from the fact that only one glycan of a glycoprotein that may have multiple glycans needs to be labeled for enrichment and subsequent proteomic analysis. In contrast, labeling multiple glycans of a single glycoprotein will greatly influence Western blot detection of a given glycoprotein as each biotin contributes independently to the overall signal.
A commonly performed procedure for analyzing cell surface proteomes is by biotinylation with reagents such as sulfo-NHS activated biotin followed by affinity capture and mass spectrometric analysis.14 This widely applied approach lacks selectivity and often leads to the isolation of cytosolic proteins likely due to cell permeability of the reagent. A number of alternative methods have been explored;15 however, all lack specificity required for comprehensive analysis of the surface membrane proteome.16 A more selective approach relies on mild oxidation of glycans with NaIO4 to install aldehyde functions, followed by tagging with a hydrazine containing bifunctional linker, proteolysis, capture of glycopeptides, enzymatic release of the N-linked glycans, and identification of the resulting peptides by LC–MS/MS.17 Although elegant, the approach requires a large number of steps and relies on rather inefficient hydrazone formation, and protein identification is based on the characterization of mainly one peptide.
The SEEL methodology described here is highly selective for cell surface proteins because the employed enzymes and reagents cannot cross the cell membrane. The high labeling efficiency and the selectivity for cell surface proteins achieved is best underscored by the impressive enrichment of cell surface glycoproteins vs total proteins detected (95% using SEEL; 18% using NHS-biotin). Moreover, the procedure is technically simple and allows glycoprotein trafficking and turnover to be easily investigated though of course it is limited to the glycoproteins that can be substrates for the enzyme. Furthermore, the one-step SEEL approach can readily be expanded to the labeling of specific classes of glycoproteins by employing alternative glycosyltransferases that have unique glycosyl acceptor specificities.18
The new strategy allowed the identification of glycoproteins whose steady state levels decrease following internalization as well as those that respond differently to incubation in the absence or presence of chloroquine. Many proteins such as IGF2R and EPHA2 were greatly reduced following the 16 h chase period for untreated cells but were more stable to degradation when chloroquine was present during the chase period. This profile indicates that these glycoproteins are subject to internalization and turnover or loss of label within the endolysosomal system. The ability of the new methodology to monitor glycoprotein dynamics in the context of lysosomal dysfunction will be particularly advantageous for the investigation of lysosomal storage disorders and the mechanisms that might explain the pathophysiology of these diseases. Abnormal recycling of glycoproteins is a known feature of these disorders that may lead to the intracellular accumulation of proteins that are required at the cell surface for normal health of cells such as neurons.9b,19 Methods that provide an accurate assessment of the cell surface glycoproteins have potential to uncover new targets for investigation and will no doubt find numerous applications in other areas of biology and biomedicine. For example, the global characterization of glycoproteins expressed on a cell surface and how this proteome responds to different stimuli can offer a better understanding of many disease processes, lead to new biomarkers for diagnosis and early detection of disease, and accelerate drug development.
EXPERIMENTAL SECTION
Methods
Methods, associated references, and additional information and data are available in the Supporting Information.
Materials
Recombinant rat α-(2,6)-sialyltransferase (ST6Gal1) was prepared as reported.20 CMP-Neu5Ac9N3, 4, and Ac4ManNAz were synthesized as previously described.5g,11 Vibrio cholerae neuraminidase was purchased from Sigma-Aldrich (N6514). Arthrobacter ureafaciens neuraminidase was purchase from NEB (P0722). Alkaline phosphatase (FastAP) was purchased from Thermo Scientific (EF0651). HRP conjugated anti-biotin antibody (200-032-211), Alexa Fluor 488 conjugated anti-biotin antibody (200-542-211), and unconjugated anti-biotin antibody (200-002-211) were purchased from Jackson ImmunoResearch Laboratories. HRP conjugated β-actin antibody was from Abcam (ab20272). Anti-CI-MPR (IGF2R) polyclonal antibody was a kind gift of Dr. Peter Lobel (CABM-Rutgers). Protease inhibitor cocktail tablet (88666), EZ-Link Sulfo-NHS-LC-Biotin (21335), and mass spectrometry compatible silver staining kit (24600) were from Thermo Scientific. Protein G beads were from Sigma-Aldrich (Protein G sepharose, Fast Flow, P3296).
Cell Lines and Culture
HeLa, human fibroblast (CRL-1509; Coriell Cell Repository, Camden, New Jersey), HepG2, or HEK293T cells were cultured in DMEM medium with high glucose (4.5 g/L) and l-glutamine, Jurkat cells were cultured in RPMI 1640 medium with l-glutamine, and all those media were supplemented with 10% fetal bovine serum (FBS, BenchMark) and penicillin (100 IU/mL)/ streptomycin (100 μg/mL, MediaTech). HEK293F cells were cultured in suspension with serum-free Freestyle 293 expression medium. All cell lines were cultured in a 5% CO2 atmosphere, 37 °C humid incubator.
Metabolic Labeling, Two-Step SEEL, or One-Step SEEL
Metabolic labeling of HeLa cells was done with 50–70% confluent cells in 12 well dishes by incubating with 30 μM Ac4ManNAz in DMEM supplemented with 10% FBS for 24 h in the cell culture incubator. Metabolic labeling of HepG2 and HEK293T cells was carried out by culturing 50–70% confluent cells in 10 cm culture dishes with 60 μM Ac4ManNAz for 24 h. Jurkat cells (~2.0 × 107 cells in suspension in a 10 cm dish) were also treated with 60 μM Ac4ManNAz for 24 h. Cells were washed with DPBS, further labeled with 4 (30 μM) in 2% FBS containing DPBS for 1 h at room temperature, and then lysed and analyzed by Western blot.
Two-step SEEL or one-step SEEL was done with confluent HeLa cells in 12 well dishes. Cells were pretreated with (+) or (−) Vibrio cholerae (VC) neuraminidase (50 mU/mL) for 2 h at 37 °C in serum free DMEM. After cells were washed with DPBS three times, cells were incubated in SEEL reaction mixture in 37 °C for 2 h. The SEEL reaction mixture (300 μL) was prepared using serum free DMEM with ST6Gal1 (42 μg/mL), CMP-sialic acid derivative (100 μM), 2 μL of BSA (2 mg/mL), alkaline phosphatase (2 μL), and 46 μL of 3 M sucrose. After cells were washed with DPBS, the cells with two-step SEEL were further labeled with 4 (30 μM) in 2% FBS containing DPBS for 1 h at room temperature and then lysed and analyzed by Western blot. Following one-step SEEL reaction, cells were directly lysed and analyzed by Western blot.
Two-step or one-step SEEL of Jurkat, HepG2, or HEK293T cells was carried out similarly except that they were labeled in suspension in Eppendorf tubes. In the case of HepG2 or HEK293T, cells confluent in 10 cm dishes were detached by pipetting after brief incubation (5–10 min) with DPBS and collected in Eppendorf tubes. For Jurkat cells, ~3.0 × 107 cells in Eppendorf tubes were prepared. Cells were pretreated with Arthrobacter ureafaciens (AU) neuraminidase (2 μL, 1:1 diluted in 50% glycerol) in 300 μL of serum free medium containing BSA (2 μL) for 90 min. Next, two-step SEEL, one-step SEEL, or 4 labeling was carried out similarly in 300 μL total volume in Eppendorf tubes.
SEEL and Chase Experiment of HeLa Cells with Chloroquine Treatment
For chase experiment, HeLa cells were labeled with the one-step SEEL method without sucrose since the sucrose-treated HeLa cells did not grow well over the chase period. The labeled cells were either (1) directly lysed for the following Western blot or immunoprecipitation or fixed and then stained with anti-biotin-488 for confocal imaging (0 h), or (2) further incubated in DMEM containing 10% FBS with or without chloroquine (50 μM) treatment (16 h). After the chase, the resulting cells were lysed or fixed depending on the desired analysis.
Cell Staining and Imaging
For staining after SEEL, HeLa cells were cultured on glass cover slides (gelatin coated) in 12 well dishes. For staining, one-step SEEL reaction (300 μL) was performed with ST6Gal1 and compound 2 (C-5 tetrazole) with cotreatment of Arthrobacter ureafaciens (AU) neuraminidase (2 μL/mL, 1:1 diluted in 50% glycerol) in the absence of sucrose. For 0 h analysis, cells were directly fixed with 3.7% formaldehyde for 15 min and then stained with anti-biotin-Alexa Fluor 488 (1:500) for 1 h. For chase, cells labeled by the SEEL reaction were further cultured with or without 50 μM chloroquine in DMEM containing 10% FBS for 16 h, followed by the fixing and staining with anti-biotin-Alexa Fluor 488. Stained cells were visualized using an Olympus FV1000 laser scanning confocal microscope.
Cell Surface Labeling with EZ-Link Sulfo-NHS-LC-Biotin
We followed the manufacturer’s protocol for the Sulfo-NHS-LC-Biotin reagent with modification in labeling temperature (4 °C instead of room temperature) in an attempt to reduce internalization of the compound. HEK293F cells (2.2 × 107 cells each for biotinylation or control) cultured in suspension were collected in Eppendorf tubes and pelleted. The cells were then washed three times with cold DPBS (1.0 mL, pH 8.0) and then resuspended in 500 μL of cold DPBS containing (+) or (−) 2 mM EZ-Link Sulfo-NHS-LC-Biotin. The resulting mixture was rotated at 4 °C for 30 min. Next, the cells were then washed three times with DPBS containing 100 mM glycine followed by three washes with DPBS at 4 °C. These biotinylated cells were lysed in the RIPA buffer for further analysis.
Immunoblotting, Immunoprecipitation, and Silver Staining
Labeled HeLa cells were lysed on plate by scraping in RIPA buffer (50 mM Tris-HCl buffer pH 8.0, 150 mM NaCl, 1.0% NP-40, 0.1% SDS, 0.5% sodium deoxycholate) supplemented with protease inhibitor cocktail on ice and collected in an Eppendorf tube. For the HEK293F cells labeled in suspension, cells were pelleted and lysed in an Eppendorf tube with the same lysis buffer. After vortexing for 20 s followed by incubation for 30 min on ice, the resulting tubes were spun down at 20000g for 10 min to clear nuclei. Protein concentration was determined by using BCA Protein Assay Kit (Thermo Science Pierce). Lysates were analyzed by SDS–PAGE and immunoblot using anti-biotin antibody conjugated with HRP typically at 1:50,000–1:100,000 dilution.
For immunoprecipitation, protein G beads (Sigma-Aldrich, St. Louis, MO) coated with anti-biotin antibody were prepared by incubating anti-biotin antibody (unconjugated) with protein G beads (protein G beads: antibody = 3:2 volume ratio) in the immunoprecipitation buffer (= RIPA buffer without protease inhibitor cocktail). We found that the anti-biotin antibody gave less background and higher specificity for enrichment compared to streptavidin-bound agarose. Cell lysates were precleared by incubating with protein G beads for 2 h at 4 °C. The precleared lysate was collected and then incubated with the antibody-coated protein G beads overnight at 4 °C. Next, the beads were washed 5 times with the RIPA buffer and then eluted with 2× sample loading buffer containing 10 mM dithiotheitol by boiling for 10 min. For proteomic analysis, one-step SEEL was performed in large scale without using sucrose for both HeLa and HEK293F cells, and the concentration of recombinant sialyltransferase and biotinylated CMP-sialic acid was the same as in the smaller scale reactions. HeLa cells were cultured confluent (~1.0 × 107 cells) in a 10 cm dish, and SEEL reaction was made on the same dish in 2 mL total reaction volume after 2 h of pretreatment of VC neuraminidase. In the case of HEK293F cells, cells cultured in suspension (1.7 × 107 cells) were collected in an Eppendorf tube and then SEEL reaction was performed with 530 μL total reaction volume in the presence of AU neuraminidase as concurrent treatment. 1.0 mg of each lysate was incubated with 50 μL of the protein G-beads (precoated with anti-biotin antibody) for complete pull-down of cell surface sialoglycoproteins. Eluted proteins were resolved by SDS–PAGE (8%), and the resulting gel was silver stained for in-gel trypsin digestion followed by proteomic MS analysis.
Proteomic Analysis
Each lane of the silver-stained SDS–PAGE gel was cut into 4 parts above 50 kDa and subsequently processed for in-gel digestion. Briefly, destained gel bands were denatured by incubating with 10 mM dithiothreitol at 56 °C for 1 h and alkylated by 55 mM iodoacetamide for 45 min in the dark prior to digestion with trypsin overnight. The resulting peptides were extracted, dried, and reconstituted in 0.1% formic acid. The peptides were separated on a 75 μm (i.d.) × 15 cm C18 capillary column (packed in house, YMC GEL ODS-AQ120 ÅS-5, Waters) and eluted into the nano-electrospray ion source of an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific) with a 180 min linear gradient consisting of 0.5–100% solvent B over 150 min at a flow rate of 200 nL/min. The spray voltage was set to 2.2 kV, and the temperature of the heated capillary was set to 280 °C. Full MS scans were acquired from m/z 300 to 2000 at 120k resolution, and MS2 scans following collision-induced fragmentation were collected in the ion trap for the most intense ions in the Top-Speed mode within a 3 s cycle using Fusion instrument software (v1.1, Thermo Fisher Scientific). The raw spectra were searched against the human protein database (UniProt, Oct. 2014) using SEQUEST (Proteome Discoverer 1.4, Thermo Fisher Scientific) with full MS peptide tolerance of 20 ppm and MS2 peptide fragment tolerance of 0.5 Da, and filtered using ProteoIQ (v2.7, Premier Biosoft) at the protein level to generate a 1% false-discovery rate for protein assignments. Proteins detected at 1% false-discovery rate in the negative controls (no recombinant enzyme added, data not shown) were excluded from the final lists of proteins identified in respective conditions. UniProt was used to define cellular localization. Quantification was performed by normalizing the spectral counts generated by ProteoIQ (v2.7, Premier Biosoft).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institute of General Medical Sciences (P01GM107012) from the US National Institutes of Health. We thank Dr. Heather Flanagan-Steet for assistance with the confocal imaging.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b04049.
Figures S1 and S2, Tables S1–S8, chemical synthesis, and NMR spectra (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Sletten EM, Bertozzi CR. Angew. Chem., Int. Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grammel M, Hang HC. Nat. Chem. Biol. 2013;9:475–484. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Saxon E, Bertozzi CR. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]; (b) Schilling CI, Jung N, Biskup M, Schepers U, Brase S. Chem. Soc. Rev. 2011;40:4840–4871. doi: 10.1039/c0cs00123f. [DOI] [PubMed] [Google Scholar]
- (3).(a) Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]; (b) Soriano Del Amo D, Wang W, Jiang H, Besanceney C, Yan AC, Levy M, Liu Y, Marlow FL, Wu P. J. Am. Chem. Soc. 2010;132:16893–16899. doi: 10.1021/ja106553e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kennedy DC, McKay CS, Legault MC, Danielson DC, Blake JA, Pegoraro AF, Stolow A, Mester Z, Pezacki JP. J. Am. Chem. Soc. 2011;133:17993–18001. doi: 10.1021/ja2083027. [DOI] [PubMed] [Google Scholar]
- (4).(a) Jewett JC, Bertozzi CR. Chem. Soc. Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Debets MF, van Berkel SS, Dommerholt J, Dirks AT, Rutjes FP, van Delft FL. Acc. Chem. Res. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- (5).(a) Whiteheart SW, Hart GW. Anal. Biochem. 1987;163:123–135. doi: 10.1016/0003-2697(87)90102-3. [DOI] [PubMed] [Google Scholar]; (b) Gross HJ, Brossmer R. Eur. J. Biochem. 1988;177:583–589. doi: 10.1111/j.1432-1033.1988.tb14410.x. [DOI] [PubMed] [Google Scholar]; (c) Gross HJ, Brossmer R. Glycoconjugate J. 1995;12:739–746. doi: 10.1007/BF00731233. [DOI] [PubMed] [Google Scholar]; (d) Blixt O, Allin K, Bohorov O, Liu X, Andersson-Sand H, Hoffmann J, Razi N. Glycoconjugate J. 2008;25:59–68. doi: 10.1007/s10719-007-9062-z. [DOI] [PubMed] [Google Scholar]; (e) Zheng T, Jiang H, Gros M, del Amo DS, Sundaram S, Lauvau G, Marlow F, Liu Y, Stanley P, Wu P. Angew. Chem., Int. Ed. 2011;50:4113–4118. doi: 10.1002/anie.201100265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chaubard JL, Krishnamurthy C, Yi W, Smith DF, Hsieh-Wilson LC. J. Am. Chem. Soc. 2012;134:4489–4492. doi: 10.1021/ja211312u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Mbua NE, Li X, Flanagan-Steet HR, Meng L, Aoki K, Moremen KW, Wolfert MA, Steet R, Boons GJ. Angew. Chem., Int. Ed. 2013;52:13012–13015. doi: 10.1002/anie.201307095. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Mercer N, Ramakrishnan B, Boeggeman E, Verdi L, Qasba PK. Bioconjugate Chem. 2013;24:144–152. doi: 10.1021/bc300542z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Li Q, Li Z, Duan X, Yi W. J. Am. Chem. Soc. 2014;136:12536–12539. doi: 10.1021/ja5054225. [DOI] [PubMed] [Google Scholar]; (j) Rouhanifard SH, Lopez-Aguilar A, Wu P. ChemBioChem. 2014;15:2667–2673. doi: 10.1002/cbic.201402433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Yu SH, Zhao P, Sun T, Gao Z, Moremen KW, Boons GJ, Wells L, Steet R. J. Biol. Chem. 2016;291:3982–3989. doi: 10.1074/jbc.M115.700369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Kajihara Y, Kamitani T, Sato R, Kamei N, Miyazaki T, Okamoto R, Sakakibara T, Tsuji T, Yamamoto T. Carbohydr. Res. 2007;342:1680–1688. doi: 10.1016/j.carres.2007.05.029. [DOI] [PubMed] [Google Scholar]; (b) Yu CC, Withers SG. Adv. Synth. Catal. 2015;357:1633–1654. [Google Scholar]
- (8).Yao JZ, Uttamapinant C, Poloukhtine A, Baskin JM, Codelli JA, Sletten EM, Bertozzi CR, Popik VV, Ting AY. J. Am. Chem. Soc. 2012;134:3720–3728. doi: 10.1021/ja208090p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, Ballabio A. EMBO J. 2010;29:3607–3620. doi: 10.1038/emboj.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; (b) Cabeza C, Figueroa A, Lazo OM, Galleguillos C, Pissani C, Klein A, Gonzalez-Billault C, Inestrosa NC, Alvarez AR, Zanlungo S, Bronfman FC. Mol. Neurodegener. 2012;7:11. doi: 10.1186/1750-1326-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mbua NE, Flanagan-Steet H, Johnson S, Wolfert MA, Boons GJ, Steet R. Proc. Natl. Acad. Sci. U. S. A. 2013;110:10207–10212. doi: 10.1073/pnas.1221105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yu H, Yu H, Karpel R, Chen X. Bioorg. Med. Chem. 2004;12:6427–6435. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- (11).Friscourt F, Ledin PA, Mbua NE, Flanagan-Steet HR, Wolfert MA, Steet R, Boons GJ. J. Am. Chem. Soc. 2012;134:5381–5389. doi: 10.1021/ja3002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McCombs JE, Zou C, Parker RB, Cairo CW, Kohler JJ. ACS Chem. Biol. 2016;11:185–192. doi: 10.1021/acschembio.5b00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ghosh P, Dahms NM, Kornfeld S. Nat. Rev. Mol. Cell Biol. 2003;4:202–212. doi: 10.1038/nrm1050. [DOI] [PubMed] [Google Scholar]
- (14).Elia G. Proteomics. 2008;8:4012–4024. doi: 10.1002/pmic.200800097. [DOI] [PubMed] [Google Scholar]
- (15).Ramya TN, Weerapana E, Cravatt BF, Paulson JC. Glycobiology. 2013;23:211–221. doi: 10.1093/glycob/cws144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Arnott D, Kishiyama A, Luis EA, Ludlum SG, Marsters JC, Jr., Stults JT. Mol. Cell. Proteomics. 2002;1:148–156. doi: 10.1074/mcp.m100027-mcp200. [DOI] [PubMed] [Google Scholar]; (b) Elortza F, Nuhse TS, Foster LJ, Stensballe A, Peck SC, Jensen ON. Mol. Cell. Proteomics. 2003;2:1261–1270. doi: 10.1074/mcp.M300079-MCP200. [DOI] [PubMed] [Google Scholar]; (c) Wu CC, MacCoss MJ, Howell KE, Yates JR., 3rd Nat. Biotechnol. 2003;21:532–538. doi: 10.1038/nbt819. [DOI] [PubMed] [Google Scholar]; (d) Watarai H, Hinohara A, Nagafune J, Nakayama T, Taniguchi M, Yamaguchi Y. Proteomics. 2005;5:4001–4011. doi: 10.1002/pmic.200401258. [DOI] [PubMed] [Google Scholar]; (e) Kaji H, Yamauchi Y, Takahashi N, Isobe T. Nat. Protoc. 2006;1:3019–3027. doi: 10.1038/nprot.2006.444. [DOI] [PubMed] [Google Scholar]; (f) Lewandrowski U, Moebius J, Walter U, Sickmann A. Mol. Cell. Proteomics. 2006;5:226–233. doi: 10.1074/mcp.M500324-MCP200. [DOI] [PubMed] [Google Scholar]
- (17).Wollscheid B, Bausch-Fluck D, Henderson C, O’Brien R, Bibel M, Schiess R, Aebersold R, Watts JD. Nat. Biotechnol. 2009;27:378–386. doi: 10.1038/nbt.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moremen KW, Tiemeyer M, Nairn AV. Nat. Rev. Mol. Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Pipalia NH, Hao M, Mukherjee S, Maxfield FR. Traffic. 2007;8:130–141. doi: 10.1111/j.1600-0854.2006.00513.x. [DOI] [PubMed] [Google Scholar]; (b) Fernandez-Monreal M, Brown TC, Royo M, Esteban JA. J. Neurosci. 2012;32:13200–13205. doi: 10.1523/JNEUROSCI.0061-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Barb AW, Meng L, Gao Z, Johnson RW, Moremen KW, Prestegard JH. Biochemistry. 2012;51:4618–4626. doi: 10.1021/bi300319q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.