

Abstract
Ehrlichia chaffeensis is an obligately intracellular bacterium that reprograms the mononuclear phagocyte through diverse effector-host interactions to modulate numerous host cell processes, including transcription. In a previous study, we reported that E. chaffeensis TRP32, a type 1 secreted effector, interacts with multiple host nucleus-associated proteins and also autoactivates reporter gene expression in yeast. In this study, we demonstrate that TRP32 is a nucleomodulin that binds host DNA and alters host gene transcription. TRP32 enters the host cell nucleus via a noncanonical translocation mechanism that involves phosphorylation of Y179 located in a C-terminal trityrosine motif. Both genistein and mutation of Y179 inhibited TRP32 nuclear entry. An electromobility shift assay (EMSA) demonstrated TRP32 host DNA binding via its tandem repeat domain. TRP32 DNA-binding and motif preference were further confirmed by supershift assays, as well as competition and mutant probe analyses. Using chromatin immunoprecipitation with next-generation sequencing (ChIP-seq), we determined that TRP32 binds a G-rich motif primarily within ±500 bp of the gene transcription start site. An ontology analysis identified genes involved in processes such as immune cell differentiation, chromatin remodeling, and RNA transcription and processing as primary TRP32 targets. TRP32-bound genes (n = 1,223) were distributed on all chromosomes and included several global regulators of proliferation and inflammation such as those encoding FOS, JUN, AKT3, and NRAS and noncoding RNA genes microRNA 21 (miRNA 21) and miRNA 142. TRP32 target genes were differentially regulated during infection, the majority of which were repressed, and direct repression/activation of these genes by TRP32 was confirmed in vitro with a cellular luciferase reporter assay.
INTRODUCTION
Ehrlichia chaffeensis is a Gram-negative, obligately intracellular bacterium and the etiologic agent of human monocytotropic ehrlichiosis (HME), an emerging life-threatening tick-borne zoonosis. In humans, E. chaffeensis infects mononuclear phagocytes, causing an acute infection that manifests as an undifferentiated febrile illness. The presence of severe symptoms and the absence of a high bacterial load suggest that the disease has an immunopathological basis, caused in part by alterations in infected mononuclear phagocyte function (1).
During E. chaffeensis infection, there are significant changes in host gene expression associated with direct/indirect host-pathogen interactions that serve to promote bacterial survival and replication within the host cell. The genes most affected are those of the early innate and cell-mediated immune response, cell cycle, cell differentiation, apoptosis programming, membrane trafficking, and intracellular signaling (2, 3). The mechanisms whereby E. chaffeensis directs these changes in gene transcription are not fully understood. However, manipulation of host transcription appears to be an important mechanism in the overall molecular strategy of ehrlichial subversion of the host cell.
Nucleomodulins are an emerging class of bacterial effectors that function by entering and reprogramming the host cell nucleus. They have been described in a variety of organisms and typically function by mimicking some aspect of host biology (4–6). Nucleomodulins may function by directly modulating host cell gene transcription, as is observed with the transcription activator-like (TAL) effectors of the plant pathogens Xanthomonas and Ralstonia (6). The TAL effectors directly interact with host DNA using a novel tandem repeat DNA-binding domain to activate expression of genes that facilitate infection (7).
Nucleomodulins were recently identified in tick-borne, obligately intracellular bacteria from the family Anaplasmataceae. Anaplasma phagocytophilum AnkA interacts with AT-rich sequences found in regulatory regions within the promoters of host genes and leads to the downregulation of genes involved in host defense via an epigenetic mechanism (8, 9). E. chaffeensis Ank200 was also found to bind to repetitive AT-rich sequences called Alu elements located within the promoters and intergenic regions of genes involved in transcriptional regulation, ATPase activity, and apoptosis, and Ank200 targets were differentially regulated during infection (10). Most recently, we reported that the E. chaffeensis tandem repeat effector TRP120 has a tandem repeat DNA-binding domain similar to that described in the TAL effectors of Xanthomonas. During infection, TRP120 enters the host nucleus and binds to a GC-rich motif within the regulatory regions of specific host genes. Targets include genes involved with transcriptional regulation, signal transduction, and apoptosis. The TRP120 target genes examined were upregulated during infection or when TRP120 was directly introduced into the cell (11). Although the mechanisms by which TRP120 regulates host gene expression are not fully understood, TRP120 interactions with host epigenetic regulators may play a role (12). Despite lacking classical nuclear localization signals (NLSs), multiple E. chaffeensis TRPs have been detected in the host cell nucleus during infection; however, their functions within the nucleus are not well defined (11, 12, 15, 16).
E. chaffeensis TRPs are type I secretion system substrates that have defined interactions with a wide variety of host cell proteins, including many that are associated with the nucleus or host cell transcription (12, 15, 16). TRP32-interacting host proteins include several nuclear proteins and transcription factors, including DAZAP2, a Wnt target gene activator, the hematopoietically expressed homeobox (HHEX), a transcription factor required for hematopoietic cell differentiation, the elongation factor 1-alpha-1 (EF1A1), a transcription factor in T cells, and p53 inducible protein 11 (TP53I11), which binds DNA nonspecifically and induces apoptosis (16–19). Interestingly, TRP32 constructs containing only the tandem repeat domain were independently capable of activating gene transcription in a eukaryotic system (16).
The mechanisms of host transcriptional modulation by pathogens are diverse and complex. In this study, we have determined that E. chaffeensis TRP32 is a nucleomodulin that accesses the nucleus through a tyrosine phosphorylation-dependent manner and binds host DNA through a tandem repeat domain to modulate host gene transcription.
MATERIALS AND METHODS
Cell culture and infection.
Ehrlichia chaffeensis (strain Arkansas) was propagated in a human monocytic cell line (THP-1). THP-1 cells were maintained in RPMI 1640 with HEPES (25 mM; Gibco) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 5 mM l-glutamine, 1% sodium pyruvate, and 12.5 ml 10% glucose at 37°C in a 5% CO2 atmosphere. HeLa cells (human cervical epithelial) for transfection were grown in modified Eagle medium (MEM; Gibco) supplemented with 10% FBS (HyClone, Logan, UT) and maintained in a 5% CO2 atmosphere.
Expression and purification of recombinant TRP32.
Full-length and truncated TRP32 gene fragments were PCR amplified from E. chaffeensis genomic material and cloned into pGEX-6p1 vectors (GE). The constructs were transformed into Escherichia coli BL21 (Genlantis) for protein expression. Glutathione S-transferase (GST)-tagged TRP32 recombinant proteins were purified using glutathione Sepharose 4B (GE) according to the manufacturer's instructions.
Nuclear localization.
TRP32 localization during infection was examined by immunofluorescent and laser confocal microscopy. E. chaffeensis-infected and uninfected THP-1 cells were cytocentrifuged onto glass slides at 24, 48, and 72 h postinfection (hpi) and fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature. Cells were then permeabilized using 1% Triton X-100 with 5% bovine serum albumin (BSA) in PBS for 1 h. After permeabilization, cells were incubated with rabbit anti-Dsb (1:1,000) and mouse anti-TRP32 (1:1,000) antibodies (20) for 30 min and then washed and labeled with Alexa Fluor 488-IgG(H+L)-conjugated goat anti-mouse and Alexa Fluor 594-IgG(H+L)-conjugated goat anti-rabbit (1:100; Molecular Probes) secondary antibodies for 30 min before mounting with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen). Samples were examined using a Zeiss LSM 510 Meta laser scanning confocal microscope configured with an Axiovert 200M inverted microscope using a c-Apochromat 63×/1.2 numerical aperture water immersion lens. UV argon, visible argon ion, and green helium neon lasers were used and emissions read using band pass filters of 385 to 470 nm (for DAPI), 505 to 530 nm (for Alexa Fluor 488 conjugate), and 560 to 615 nm (for Alexa Fluor 594 conjugate), respectively. Images were analyzed with LSM 510 software, and z-stacks were constructed by imaging optical slices at 1-μm intervals. FIJI was used for subsequent image processing, and only linear adjustments (i.e., brightness, contrast) were made (21).
The full-length, N-terminal and tandem repeat (amino acids [aa] 1 to 138), and C-terminal (amino acids 138 to 198) constructs were created as previously described (16). The C-terminal truncations were PCR amplified from the full-length plasmid. Primer sequences are shown in Table S1 in the supplemental material. TRP32 truncation constructs were cloned into the vector pAC-GFP-C1 (Clontech). These vectors were amplified in E. coli TOP10 and purified using the PerfectPrep EndoFree Plasmid Maxi kit (5Prime). Purified plasmids were transfected into HeLa cells using Lipofectamine 2000 (Thermo Fisher) according to the manufacturer's instructions. Cells were acetone fixed and mounted with ProLong Gold with DAPI (Invitrogen). Localization of ectopically expressed constructs was examined at 24 h posttransfection (hpt) using fluorescence microscopy and by immunoblotting of nuclear/cytosolic separations.
ChIP-seq.
E. chaffeensis-infected THP-1 cells harvested at 3 days postinfection (dpi) were used for chromatin immunoprecipitation with next-generation sequencing (ChIP-seq) using EZ Magna ChIP (EMD Millipore). Briefly, cells were cross-linked using a final concentration of 1% formaldehyde for 10 min. Cells were then pelleted and lysed, and the lysate was sonicated on ice using Sonics Vibra Cell. Sonication (12 cycles, 30 s at 5-W output, 30-s rest) was used to generate chromatin fragments less than 1 kb in length. TRP32 was immunoprecipitated using rabbit anti-TRP32, and preimmune serum was used as a control. Cross-links were reversed, and nucleic acid was purified according to the manufacturer's protocol. Library preparation was performed, involving PCR amplification with adapter ligation by the UTMB Next Generation Sequencing Core, and samples were tracked using index tags incorporated into the adapters. Library quality was evaluated, and quantification of library DNA templates was performed. Samples were sequenced using an Illumina HiSeq 1500 to generate paired-end 50-bp sequence reads. Sequence reads were analyzed by base calling and sequence quality-filtering scripts using Illumina Pipeline software. Sequences were aligned to the human genome (NCBI build 37) using bowtie2, allowing a maximum of two mismatches to the reference genome, and a binary alignment/map (BAM) file was generated. Model-based analysis of ChIP-seq (MACs) was used for peak calling, and peaks were visualized using the Integrated Genomics Viewer (IGV) by the Broad Institute.
Gene annotation and ontology.
A browser extensible data (BED) file consisting of highly significant peaks (P < 10−20) was submitted to Genomic Regions Enrichment of Annotations Tool (GREAT v3.0) (22). These peaks were associated with nearby genes using the Basal Plus Extension rule, which assigns genes a proximal regulatory region consisting of −5 kb to +1 kb from the transcription start site and a distal regulatory region extending up to 1,000 kb in either direction but ending at any other gene's proximal regulatory region. Additionally, a set of curated regulatory domains from the literature was utilized. After the ChIP-seq regions were assigned to nearby genes, the annotations from those genes were used to calculate statistically enriched categories using hypergeometric and binomial tests. In order to calculate binding site percentages, the Nearest Gene rule was used to associate peaks to genes. This rule associates peaks to genes similarly to the Basal Plus Extension rule but allows only one gene to be assigned to any one genomic region. This analysis did not alter the top 10 significantly enriched gene ontologies (GOs) in any category. In order to determine enrichment at the promoter region, experimental data were compared to 10 randomly generated data sets and a one-sample t test was used to determine significance.
TRP32 binding sites.
The DNA-binding motif was identified using the Multiple EM Motif Elicitation (MEME) ChIP software suite (23). The regions ±250 bp from the TRP32 binding peak were submitted to MEME in fasta format. Motifs that were statistically enriched in these regions were returned in the form of probability matrices ranked by E value and similarity. Motifs were tested by electrophoretic mobility shift assay (EMSA). The MEME program SPAMO (spaced motif analysis) was used to identify binding sites for other transcription factors that were significantly enriched at fixed up- or downstream intervals from the TRP32 motif. The program FIMO (Finding Individual Motif Occurrences), also part of the MEME suite, was used to confirm the presence of the motif in TRP32 bound regions. The 500-bp fasta sequences of highly significant TRP32 bound regions was queried using the six-nucleotide motif, which was entered as GG[AT]GGC. Both strands of the TRP32 bound regions were queried, and a cutoff P value of ≤0.01 was used.
EMSA.
Whole genomic DNA was isolated from healthy THP-1 cells and sheared using the protocol and enzyme from the ChIP-IT Express Enzymatic kit (Active Motif) without cross-linking. The resulting sheared genomic material was then purified using the QIAquick PCR Purification kit (Qiagen) and biotin labeled using the LabelIT nucleic acid labeling kit (Mirus). EMSAs were performed using the Lightshift chemiluminescent EMSA kit (Thermo Fisher). Briefly, 5 ng of labeled whole genomic DNA was incubated with 2 μg of purified protein in a 10 mM Tris buffer with 50 mM KCl, 1 mM dithiothreitol (DTT), 5 mM MgCl2, 2.5% glycerol, 0.05% NP-40, and 50 ng/μl poly(dI · dC). Samples were incubated at 4°C for 1 h before separation on a 6% DNA retardation gel (Thermo Fisher) at 100 V for 90 min. Reaction mixtures were then transferred to Biodyne B nylon membranes (Thermo Fisher) at 20 V for 1 h. Transferred DNA was cross-linked to the membrane by placing the membrane face down on a transilluminator for 10 min or in a CL-1000 UV cross-linker (UVP) for 5 min. Reaction products were imaged using streptavidin-horseradish peroxidase (HRP) and film. Supershift assays were performed using rabbit anti-TRP32 antibody. Antibody at 1:200, 1:50, and 1:20 dilutions was added to the EMSA reaction mixture after 30 min of incubation, and the mixture was allowed to incubate for 30 min. For EMSAs utilizing a probe, 400 pM labeled DNA was incubated with 1 μg of purified protein. Competition assays were performed using a 1,000-fold molar increase of unlabeled nucleic acid. Protein and unlabeled DNA were incubated for 30 min at 4°C before adding labeled DNA and incubating for the remaining 30 min.
Oligonucleotide probes (IDT) modeled after TRP32-bound sequences were resuspended in annealing buffer (10 mM Tris [pH 7.5 to 8.0], 50 mM NaCl, 1 mM EDTA), and complementary strands were annealed by heating to 95°C in a heat block for 5 min followed by slow cooling to room temperature. Before use, probes were diluted to the above-mentioned concentrations in annealing buffer. Probe sequences are listed in Table S2 in the supplemental material.
qPCR.
Expression of TRP32 host gene targets was measured by reverse transcription-quantitative PCR (qRT-PCR). The genes that corresponded to the top 100 most significantly enriched peaks were chosen for initial analysis; however, 25 were excluded because they were histone-coding genes, which are difficult to differentiate by qPCR due to high sequence identity. Primers were designed for the remaining 75 genes using NCBI Primer Blast. The primers were designed to detect mRNA sequences and were all designed to span exon-exon junctions to prevent amplification of corresponding genomic sequences (see Table S3 in the supplemental material). All primers were tested by melt-curve and gel analysis for specificity, and primers that amplified multiple products or amplified a product that was not the predicted size were excluded.
RNA was extracted from healthy THP-1 cells and from cells at 24, 48, and 72 hpi using the RNeasy minikit (Qiagen). cDNA was synthesized from 1 μg of RNA using the qScript cDNA synthesis kit (Quanta Biosciences). qPCR was performed with 5 ng of cDNA in triplicate using iQ SYBR green Supermix (Bio-Rad) according to the manufacturer's instructions. Fold changes were calculated using the ΔΔCT method (where CT is threshold cycle) using the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene as the reference gene (24).
Luciferase gene expression assay.
Promoter regions containing the TRP32 binding sites as determined by GREAT were cloned into pGL4.10 (Promega), a promoterless luciferase vector. Each promoter construct (200 ng) was transfected into HeLa cells using Lipofectamine 2000 (Thermo Fisher) along with various concentrations of GFP-TRP32 or an empty green fluorescent protein (GFP)-expressing vector. Equal amounts of total DNA were transfected into each cell. After 24 h, cells were harvested and the Dual Glo Luciferase reagent (Promega) was added to the cells according to the manufacturer's protocol. Relative light output was measured using a Veritas microplate luminometer. Relative expression levels were calculated for each gene compared to the control, and the P value was calculated using the Student t test.
RESULTS
TRP32 is translocated into the host nucleus in a tyrosine phosphorylation-dependent manner.
Although lacking a canonical nuclear localization signal, TRP32 is transported into the host nucleus in a temporally regulated manner. At early points in infection (24 hpi), TRP32 remains around the morulae. However, at 48 hpi TRP32 was observed in the perinuclear region, and by 72 hpi, increasing amounts of TRP32 were detected in the perinuclear region and in the nucleus (Fig. 1A). The nuclear localization of TRP32 was confirmed using orthogonal projections of a z-stack, which showed TRP32 diffusely throughout the nucleus and in nuclear puncta (Fig. 1B). In order to elucidate the mechanism for TRP32 nuclear translocation, GFP-tagged truncation constructs were created and transfected into HeLa cells and their localization was observed. We determined that the C terminus (60 aa) alone was sufficient for nuclear localization. Additional C-terminal domain truncation constructs were generated to explore the role of two trityrosine motifs in nuclear translocation. These constructs were ectopically expressed, and localization was observed by fluorescence microscopy. We identified two trityrosine motifs that were in many of the constructs translocated to the nucleus (Fig. 2). The central tyrosine of each of these motifs was predicted to be phosphorylated, and when we performed immunoprecipitation of infected THP-1 cells using an anti-phosphotyrosine antibody, we found that TRP32 was enriched in the experimental IP compared to the control (Fig. 3A). This led to the hypothesis that tyrosine phosphorylation might be involved in TRP32 nuclear localization. Hence, subcellular localization of wild-type (WT) TRP32 was examined in HeLa cells treated with vehicle alone (control) or with the tyrosine kinase inhibitor genistein. In cells treated with genistein, TRP32 was observed primarily in the perinuclear region, while TRP32 in control cells exhibited the characteristic nuclear localization (Fig. 3B). In order to examine the role of specific tyrosine residues, single and double phenylalanine mutants of the tyrosine residues (Y168 and Y179) were generated, and the localization of these ectopically expressed constructs was observed. Although both single mutants exhibited reduced nuclear localization, the Y179F mutant showed a greater decrease in nuclear localization than did the Y168F mutant (Fig. 3C). These data support the conclusion that phosphorylation of Y179 facilitates nuclear localization and that TRP32 is trafficked to the host cell nucleus in a tyrosine phosphorylation-dependent manner. A similar mechanism has been described with regard to STAT1 nuclear access, which homodimerizes in a phosphorylation-dependent manner to create a dimer-dependent NLS (25). Moreover, phosphorylation of these residues may be required for interaction with another protein that has an NLS.
FIG 1.

TRP32 localizes to the nucleus of E. chaffeensis-infected THP-1 cells. (A) E. chaffeensis-infected and uninfected THP-1 cells were fixed and probed with rabbit anti-TRP32 (green), anti-DSB (red, morula), and DAPI (blue, DNA) and then visualized using confocal microscopy. Early (24 hpi) TRP32 is associated primarily with the morulae. At 48 hpi, TRP32 localized with morulae and in the perinuclear region. At 72 hpi, TRP32 localized with the morulae but was also observed at the perinuclear region and in the nucleus of the host cell. TRP32 was not observed in uninfected cells. Bar, 10 μm. (B) Orthogonal projections of optical slices from a z-stack of an E. chaffeensis-infected THP-1 cell at 72 hpi showed both diffuse and punctate TRP32 within the host nucleus. Top panels show a y-z projection with left panels showing an x-z projection. The positions of the x and y axes within the projections denote the z depth of the slice shown in the center.
FIG 2.

The C-terminal trityrosine motif is important for E. chaffeensis TRP32 nuclear localization. HeLa cells were transfected with GFP-tagged TRP32 constructs. After 24 h of expression, cells were fixed and visualized using fluorescence microscopy. (A) Both full-length and C-terminal TRP32 constructs (green) localized to the nucleus (blue), but the N-terminal plus tandem repeat construct did not. Bar, 10 μm. (B) Schematic of TRP32 C-terminal truncation constructs. (C) TRP32 C-terminal truncations C2 and C3, which both contain trityrosine repeats, were located primarily in the nucleus, while other C-terminal constructs lacking the trityrosine repeat showed diffuse nuclear and cytosolic localization. Bar, 10 μm.
FIG 3.

Phosphorylation of E. chaffeensis TRP32 at tyrosine 179 is required for nuclear localization. (A) Tyrosine phosphorylation of TRP32 was detected during infection. E. chaffeensis-infected THP-1 cells were harvested at 72 hpi and immunoprecipitated using an antiphosphotyrosine antibody (PY99; Santa Cruz) or an IgG control. The blot was probed using rabbit anti-TRP32. (B) Tyrosine kinase inhibitor genistein inhibits TRP32 (green) localization to the nucleus (blue). HeLa cells were transfected with GFP-tagged TRP32, and 24 hpt cells were treated with 10 μM genistein (bottom) or vehicle (dimethyl sulfoxide [DMSO], top) for 15 min and then fixed and visualized using fluorescence microscopy. Genistein-treated cells show decreased nuclear localization of TRP32 compared to the control. Bar, 10 μm. (C) TRP32 tyrosine 179 (Y179) is required for nuclear localization. Wild-type TRP32 and tyrosine mutant TRP32 constructs were transfected into HeLa cells, and fluorescence microscopy was performed 24 hpt. TRP32 localized primarily to the cell nucleus, but Y179F mutants (3rd down) exhibited predominately cytosolic localization.
TRP32 contains two copies of a nine-amino-acid transactivation domain.
Previously, we showed that full-length TRP32 and its internal tandem repeat domain are able to activate transcription of a reporter gene in a eukaryotic system. Upon further investigation, we identified two 9-aa, trans-activating domains (TADs; aa 52 to 60 and 82 to 90) within the tandem repeats (Fig. 4). This 9-aa TAD is a fuzzy motif consisting of two hydrophobic clusters separated by a hydrophilic cluster and is found in several yeast and mammalian transcription factors, including VP16, p53, HSF1, nuclear factor interleukin-6 (NF-IL-6), NF-κB, and NFAT1 (26).
FIG 4.

E. chaffeensis TRP32 putative 9-amino-acid transactivation domain. (A) TRP32 amino acid sequence with predicted transactivating domains (TAD) highlighted in red. (B) Sequence alignment of TRP32 predicted TAD with previously described eukaryotic transactivation domains from myeloid/lymphoid or mixed-lineage leukemia protein (MLL), transcription factor 3 (E2A), and galactose-induced regulatory protein (Gal4). The putative TRP32 TADs are highly similar to previously described eukaryotic transactivating domains with a leucine residue (L, red) followed by an amphipathic motif. Hydrophobic residues (ζ) are shown in green and hydrophilic residues (Φ) in blue.
TRP32 binds to host genomic DNA via its tandem repeat DNA-binding domain.
Our previous studies of TRP120 combined with TRP32 activation of reporter gene transcription in a yeast two-hybrid (Y2H) assay and nuclear localization led to the hypothesis that TRP32 might be binding host genomic DNA via its TR domain to modulate host cell transcription. We investigated TRP32 DNA binding using EMSAs with recombinant full-length TRP32 and truncation constructs. We determined that the full-length TRP32, as well as the TR domain alone, bound host genomic DNA (gDNA) (Fig. 5). Consistent with E. chaffeensis TRP120, which also binds DNA via its TR domain, no homology to known eukaryotic DNA-binding domains was identified by in silico modeling. Although the TRP DNA-binding domains are not predicted to be structurally similar to any described DNA-binding domain, the presence of internal tandem repeats with high identity is similar to what is observed with the Xanthomonas transcription activator-like (TAL) effectors, which also bind DNA via highly similar internal repeats. This feature suggests that Ehrlichia TRP effector DNA-binding domains may interact with DNA similarly. X-ray crystallography studies are needed to determine whether the TRP32 tandem repeat DNA-binding domain is structurally similar to the Xanthomonas tandem repeat DNA-binding domain.
FIG 5.
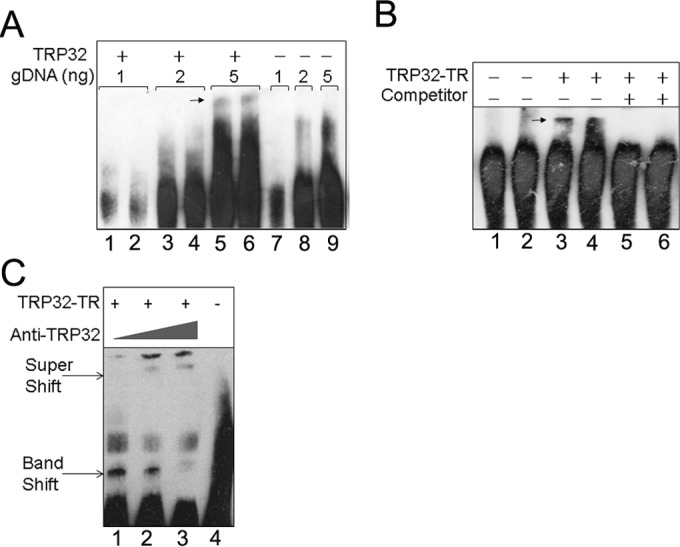
E. chaffeensis TRP32 binds host gDNA via its tandem repeat domain. (A) TRP32 binds to host genomic DNA. Various amounts of purified host gDNA were incubated with recombinant, full-length TRP32 and then separated by EMSA. Band shifts are indicated by an arrow (lanes 5 and 6). A band shift was not observed in the corresponding lane to which TRP32 was not added (lane 9). (B) The tandem repeat domain of TRP32 is the DNA-binding domain. EMSA was performed using a TRP32 tandem repeat construct (TRP32-TR) and 5 ng of host gDNA. The TRP32 band shift is indicated by an arrow (lanes 3 and 4). Binding was abolished when excess unlabeled competitor was added (lanes 5 and 6). (C) Supershift assays in which TRP32-TR was incubated with host DNA and increasing concentrations of anti-TRP32 antibody were performed. Band shifts and super shifts are labeled with arrows.
TRP32 binds within the promoter region of host genes involved in a variety of cellular processes.
ChIP-seq was used to identify TRP32 genomic targets. We found 1,587 genomic regions significantly enriched (P < 10−4) compared to the negative control. Of these, 240 regions were highly significant, with a P value of <10−20. The enriched regions mapped to all chromosomes (Fig. 6A and B), and all of the highly significant regions mapped to at least one human gene. These highly significant peaks were significantly enriched within the promoter region (±5 kb from the transcription start site) compared to random control (P < 0.0001), and the majority, 60.8% (146/240), were within ±1 kb. This location is consistent with transcription factor binding (27). The remaining highly significant peaks were found downstream from the transcription start site (TSS) (22.9%) or further upstream (16.3%) (Fig. 6C and D). When spaced motif analysis (SPAMO) was performed on all significant, TRP32-bound sequences, binding sites for other transcription factors were significantly enriched at fixed up- or downstream intervals, including those for HHEX (17 bp, P = 2.98e−52) and TCF7L2 (44 bp, P = 2.13e−23) (28). When significant genomic regions were probed for their association with various biological processes using gene ontology analysis, we observed the enrichment of genes associated with cell differentiation, chromatin remodeling, RNA transcription and processing, and regulation of translation. The TRP32 targets were also enriched for genes specific for cellular compartments, most notably the nucleosome and ribosome (Fig. 7). Several host cellular pathways were also significantly enriched among TRP32 targets, including DNA replication and immune cell activation pathways. There was also enrichment of genes associated with apoptotic pathways; however, these did not reach the threshold of significance (false discovery rate [FDR] P value = 0.09) (Table 1). Additionally, it was noted that although the majority of TRP32 target genes were protein coding, some were also RNA genes. Among TRP32 noncoding RNA targets were several microRNAs (miRNAs), including miRNA 21 and miRNA 142 (Fig. 8).
FIG 6.

E. chaffeensis TRP32 binds host genes within promoter regions. (A) TRP32 chromatin immunoprecipitation resulted in significant enrichment of 1,587 regions (P < 10−4) compared to serum control, and 240 peaks were highly significant (P < 10−20). (B) Number of TRP32 enriched regions/megabase of DNA for each chromosome. (C) TRP32 highly significant peaks are enriched within the promoter regions of host genes. A histogram showing the association of highly significant peaks with host genes demonstrates that TRP32 preferentially associates with host genes within ±5 kb from the transcription start site (TSS). Some peaks corresponded to more than one host gene and were counted multiple times in this histogram. (D) The majority of highly significant peaks clustered within ±500 bp from the TSS.
FIG 7.

E. chaffeensis TRP32 targets include genes in several categories relevant to infection. (A) Terms from gene ontology (GO) of biological processes that were significantly enriched by both binomial region and hypergeometric fold enrichment are presented by hypergeometric fold enrichment. (B) Significantly enriched terms of cellular compartments presented by hypergeometric fold enrichment. (C) Significantly enriched gene ontologies of molecular processes presented by hypergeometric fold enrichment.
TABLE 1.
TRP32 binds genes in several cellular pathways
| Pathway | Fold enrichment | FDR Q-value | Gene (distance in bp from TSS) |
|---|---|---|---|
| DNA replication | 20.52 | 3.34 × 10−9 | H3F3B (+3,557), H3F3B (−38), HIST1H3B (−22), HIST1H3I (−985), HIST1H3C (+146), HIST1H3J (−2,477), HIST1H3F (+687), HIST1H3A (+324), HIST1H3G (+241), XRN2 (+36,927), HIST1H3H (−2,348) |
| T cell activation | 5.09 | 3.41 × 10−2 | AKT3 (+119,847), B2M (−49), CALM2 (+246), FOS (+4,069), HLA-DOA (+37,266), JUN (+94), NRAS (+249) |
| B cell activation | 5.84 | 2.79 × 10−2 | BTK (−4,889), CALM2 (+248), FOS (+4,089), JUN (+94), NRAS (+249), PTPN6 (+2,363) |
| Apoptosis signaling | 3.83 | 9.14 × 10−2 | JUN (+94), ATF4 (+2,578), TNF (+101), JDP2 (−149,291), FOS (+4,089), TMBIM8 (−205), AKT3 (+119,847) |
FIG 8.

E. chaffeensis TRP32 binds to noncoding RNA genes in the host cell. (A) TRP32 targets include RNA-coding genes. When all significant TRP32 bound regions of chromatin were examined, 24% mapped to RNA-coding genes. When the RNA-coding genes were examined, the majority were snoRNAs (33%) followed by miRNAs (29%), antisense RNAs (17%), and noncoding RNAs (17%). A small percentage corresponded to pseudogenes (4%). (B) A table of the characterized noncoding RNAs bound by TRP32 shows that many are involved in cell fate determination: NR_027349 (51, 52); NR_029493 (53–55); NR_029683 (56–59); NR_002819 (60, 61); NR_029779 (62–64); NR_030230 (65).
TRP32 DNA motif.
In order to define the DNA motif bound by TRP32, the nucleotide sequences of the ChIP peaks were entered into the pattern-mining program MEME-ChIP. MEME-ChIP uses multiple algorithms to search for sequences that are enriched in the data set. It returns any statistically significant results in the form of position weight matrices (PWM) and can then compare the resultant sequences with known transcription factor motifs. Using this program, several significant motifs were returned. The three most highly significant motifs were selected for analysis. Notably, all three motifs were highly similar, with two containing centrally located GGAGGC and GGTGGC sequences that may be variants of the same motif. The other motif also contained a similar sequence (CCAGGC), but it was not centrally located. Real sequences representing the three most highly significant motifs were used to construct biotin-labeled probes to test TRP32 binding affinity by EMSA. Using this system, we found that the two probes containing the centrally located GG[A/T]GGC sequence were bound with high affinity. The probe that contained the peripherally located CCAGGC was bound with much lower affinity. The specificity of these interactions was confirmed using unlabeled probes as specific competitors (Fig. 9). The results from the EMSA suggested that a GGTGGC-like sequence approximated the consensus motif for TRP32. Additionally, when the 240 highly significant regions were examined for the presence of this sequence using the program FIMO (Finding Individual Motif Occurrences) with a threshold for P of <0.01, they were all found to contain this motif (24). In order to confirm that this was indeed the TRP32 binding motif, wild-type constructs containing this motif and mutant constructs with an alanine substitution in the predicted motif or the 10 nucleotides located at the 5′ or 3′ end were synthesized. These constructs were examined by EMSA and compared to the WT sequence. We found that the Δ1 mutant, which had a disrupted GGTGGC sequence, showed decreased binding by TRP32 and that the Δ2 mutant, which entirely lacked the GGTGGC sequence, showed an even greater decrease in binding, as expected. However, surprisingly, the Δ3 mutant also showed decreased binding by TRP32. When this probe sequence was examined further, an inverted repeat of the predicted motif was identified, which was disrupted in that mutant (Fig. 9D).
FIG 9.

E. chaffeensis TRP32 binds a G-rich motif. (A) TRP32 was predicted to bind to multiple, highly similar, G-rich motifs. Probability matrices of the top three predicted TRP32 DNA motifs from MEME-ChIP are shown. (B) The predicted motifs from panel A were tested by EMSA. TRP32-TR was incubated with biotin-labeled probes in the presence and absence of unlabeled specific competitor. Relative densities are indicated. Motif 3 was identified as the putative DNA motif due to its strong binding, which was significantly decreased (P = 0.002) in the presence of competitor. (C) The TRP32-TR construct was incubated with the wild-type motif 3 (WT) and with three mutant probes. TRP32 bound the wild-type probe with greater affinity than the mutants. All of the mutants showed decreased binding. Relative densities are indicated. (D) Sequences of the wild-type (WT) and mutant probes are shown with the putative motif boxed in blue and the inverted repeat boxed in green. Mutated sequences are bolded.
TRP32 modulates expression of target genes during infection and in a luciferase reporter assay.
To examine TRP32's effect on transcription of identified target genes, transcriptional activity was measured by qPCR in healthy and infected cells at 24, 48, and 72 hpi, and relative fold change of transcription activity in infected compared to uninfected controls was calculated. Of the 57 TRP32 target genes that were measured, 46 (80%) were differentially expressed during infection, with the majority (60%) being highly downregulated; however, some genes were highly upregulated (Fig. 10A).
FIG 10.

E. chaffeensis TRP32 has a direct effect on target gene expression. (A) TRP32 targets are differentially regulated during infection. The most highly enriched genes from the ChIP-seq were assayed by qPCR at 24, 48, and 72 hpi. Data are presented as fold changes from noninfected. Colors represent the fold changes from −10 to +10. Data are representative of two (24 and 48 hpi) or three (72 hpi) experiments. (B) TRP32 modulates gene expression in a luciferase reporter assay. Several genes were chosen, and the promoter regions, including the TRP32 binding site, were cloned into a luciferase reporter. Promoter constructs were transfected into HeLa cells along with various concentrations of a TRP32-expressing or empty GFP vector. Luciferase expression was measured after 24 h. Luciferase expression is reported as fold change from control. Data are representative of 3 to 5 experiments. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
In order to determine the direct role of TRP32 in regulating gene expression, promoters from identified target genes were tested for transcriptional activity using luciferase transcriptional reporter assays. These constructs were transfected into HeLa cells along with various concentrations of TRP32-GFP and an empty GFP control plasmid, and expression of luciferase was measured at 24 h posttransfection. We found that TRP32 expression resulted in differential luciferase expression from the target gene promoters, consistent with gene expression observed during infection. This differential regulation occurred in a dose-dependent manner, and both activation and repression of luciferase gene expression due to TRP32 were seen in this system. When the TRP32 binding site was deleted in the JUN promoter (ΔJUN), no differential regulation was seen (Fig. 10B).
DISCUSSION
All obligately intracellular bacteria, including Ehrlichia, must subvert innate host defenses and exploit normal host processes in order to survive. One of the strategies that E. chaffeensis uses is to target conserved host pathways that act as nexuses of cell fate through moonlighting effectors, which have multiple roles/functions during infection (29, 30). Many of these nexuses involve regulating the function of a transcription factor. Recently, bacterial nucleomodulins that directly alter host gene transcription have been described. This enables intracellular pathogens with small genomes and a limited effector repertoire to efficiently reprogram the host cell (31).
Despite the mounting evidence that E. chaffeensis TRPs are a novel family of transcription factor-like effectors that access the host nucleus, a conundrum has been that most lack a classical nuclear localization signal. Our preliminary studies have determined that TRPs enter the host nucleus by diverse means; however, a common theme of “piggy-backing” into the nucleus via protein-protein interactions seems to exist. We found that TRP32 residue Y179 is required for TRP32 nuclear localization via a regulated tyrosine phosphorylation-dependent mechanism. In fact, TRP32 localization at 48 hpi is consistent with the perinuclear localization of TRP32 in genistein-treated cells and in Y179 mutants. This is suggestive that phosphorylation of TRP32 is required for interaction with an NLS-containing partner, and indeed, the relocalization seen after genistein treatment is consistent with that of a phosphoregulated nucleocytoplasmic shuttling protein such as ERK1/2 or SMADs (32–34). Additionally, the PYYY motif seen in TRP32 can be involved in interactions with ubiquitin ligases such as the Cbl family, which binds to phosphorylated tyrosines or the NEDD family ubiquitin ligases, which are themselves regulated by tyrosine phosphorylation (35–37). It is possible that ubiquitination may be the ultimate signal for TRP32 nuclear localization. The temporal differences seen in TRP32 localization support the idea of a regulated means of nuclear translocation, which is quite unlike TRP120 nuclear localization, which begins early during infection (3 hpi) and continues to accumulate in the nucleus as the infection progresses (11). The exact mechanisms for TRP32 nuclear trafficking, i.e., phosphorylation alone or phosphorylation-triggered ubiquitination and the interacting partners, that potentiate these modifications remain to be determined.
TRP32 bound 1,587 genomic regions significantly, which is similar to the numbers of peaks identified for mammalian transcription factors in other reported ChIP-seq experiments (38). This value is approximately 1 order of magnitude less than the number of regions bound by TRP120; however, the two studies yielded similar numbers of highly significant peaks (P ≤ 10−20; TRP120, 582; TRP32, 240). When only the highly significant peaks were examined, these peaks mapped to human genes, and 70% were located within the promoter region. Although transcriptional regulatory regions may reside quite distally from the TSS, most well-studied transcription factors bind their target genes within the proximal promoter and binding in this region has been linked to the regulatory function of transcription factors (39, 40). Interestingly, some of the genomic regions contained known promoter elements. For example, TRP32 binds several histone H4 genes within a described suppressor element (41). Intriguingly, histone H4 gene transcription was downregulated during infection (data not shown). When expression of TRP32 targets was examined, we found that 80% were differentially regulated during infection. This is an expected result, as transcription factors typically bind many nonfunctional sites, with reports of as many as ∼50 to 99.9% of transcription factor-bound sites not having a clear functional role (42, 43). Most gene targets showed differences in expression primarily at 72 hpi, although some showed changes at 48 hpi as well. This is consistent with TRP32 localization data, which show that TRP32 surrounds and begins to enter the nucleus at 48 hpi and accumulates in the nucleus at 72 hpi.
TRP32 is able to directly modulate transcription of genes during a luciferase gene expression assay. In this assay, targets are both up- and downregulated, which is similar to what is seen during infection and suggests that TRP32 may act as both a transcription repressor and an activator. This differential expression occurs in a dose-dependent manner and is ablated when the TRP32 binding site is deleted. All together, these data strongly suggest that TRP32 is directly regulating gene expression of its targets. This is different from what was previously seen with TRP120, which primarily activated gene expression of its targets and for which a direct mechanism of action was not confirmed (11). For a few genes, the regulation seen during the luciferase assay did not correspond to the changes in gene expression seen during infection. The most notable example of this was JUN, which was highly upregulated during infection but downregulated by TRP32 in the luciferase assay. This is not unexpected and could be the result of multiple factors. Some differences might be due to cell type; however, other infection-related processes are likely involved. Cell stress, the host innate immune response, and effector-mediated processes not only modulate gene transcription but can also affect posttranscriptional processing of mRNAs. JUN mRNA is highly posttranscriptionally regulated (44).
The ability to act as a transcriptional repressor or activator is a common feature of many eukaryotic transcription factors. Dual-function transcription factors can be regulated by several factors, including variations in the motif they bind, posttranslational modifications of the transcription factor, interaction with different transcription coregulators, or the general transcriptional milieu (i.e., cell cycle, cell type, cell activation status) (45–47). Posttranslational modification of TRP32 may play a role in regulating TRP32 transcription factor function. TRP32 is predicted to be ubiquitinated at lysine residues adjacent to the putative 9-aa TADs. The 9-aa TAD is reported to be the minimal required element for interaction with several transcriptional cofactors, including CBP, p300, and MED15 (48), and ubiquitination at these residues could theoretically alter TRP32's ability to interact with these transcriptional coactivators. Interaction with other transcription factors and transcription initiation complex components can also regulate transcription factor function. TRP32 is known to interact with several host transcription regulators, including HHEX and DAZAP2, and SPAMO determined that these interactions likely occur at the promoter and may thus be important for TRP32-mediated transcriptional regulation. Hence, their differential activation and recruitment may affect TRP32 function (16, 49).
When TRP32 target genes are examined for cellular process ontologies, several categories with relevance to infection were statistically enriched, including hematopoietic cell differentiation and proliferation, chromatin remodeling, transcription, RNA processing, and regulation of translation. Additionally, several global regulators of host cell proliferation and inflammation were among TRP32 gene targets, including FOS, JUN, AKT3, tumor necrosis factor (TNF), and NRAS. TRP32 manipulation of host differentiation and proliferation may be a factor in the leukopenia that is a common clinical feature in HME. A previous study examining the host transcriptome during infection showed that genes regulating cell cycle, differentiation, and apoptosis and the innate immune response, among others, were differentially expressed during infection (2). Although in that study only early time points (up to 24 hpi) during infection were examined, the results included many gene categories targeted by multiple ehrlichial nucleomodulins, including TRP32. Overall, although distinct differences in TRP target genes are noted, there is some overlap in functional categories of genes targeted by E. chaffeensis nucleomodulins. Previously, TRP120 was shown to interact with genes involved in regulating transcription, posttranslational modifications, and apoptosis (11). Ank200 targets also include genes involved in transcriptional regulation and apoptosis as well as genes coding for structural proteins associated with the nucleus and membrane-bound organelles (10). Moreover, there is also overlap in specific gene targets. Both TRP32 and Ank200 target histone-coding genes, for example, and all three nucleomodulins target the inflammatory mediator TNF-α. This poses the question of whether these nucleomodulins may be functioning cooperatively at the promoter or if they might be regulating gene expression separately in a coordinated manner. When TRP32 and TRP120 common target genes were examined, we found that both bound within the proximal promoter only ∼3% of the time and less than 1% of the time were they within 150 bp. This suggests that they do not typically function as part of the same regulatory complex. Temporal differences in nuclear localization also suggest that TRP32 and TRP120 may function independently, although the possibility of cooperative function cannot be eliminated.
When TRP32 target sequences were examined, several highly similar G-rich motifs were statistically overrepresented. When the most statistically enriched motifs were examined by EMSA, TRP32 was shown to bind them with differing affinity and specificity. These differences in binding affinity seem to relate to the position of the GG[A/T]GGC-like sequence within the probe and the presence or absence of an imperfect repeat, with the probe that contained a peripherally located motif and a truncated repeat showing decreased binding affinity compared to probes with a centrally located [A/T]GGC sequence. Additionally, mutational analysis of the probe suggests that TRP32 may bind as a dimer. Although binding to a single GGTGGC was observed, it was considerably weaker than when two motifs were present in the form of imperfect inverted repeats, a finding consistent with other dimeric transcription factors, such as EBF (50). Inverted repeats can also indicate that the DNA is self-annealing to form secondary structures that are recognized by the DNA-binding protein. However, the binding pattern seen in our experiments is consistent with transcription factor dimers binding to DNA, since when either repeat is deleted, the DNA-binding affinity decreases; however, it is not abolished as would be expected if the interaction were dependent on DNA secondary structure.
In this work, we present the first report of an ehrlichial effector directly regulating host gene expression. TRP32 was found to bind and regulate to specific host genes both during infection and in a cell-based luciferase reporter assay. Additionally, we show that TRP32 enters the host nucleus in a tyrosine phosphorylation-dependent manner primarily after 48 hpi. This is different from what has been reported for previously studied TRPs, which enter the host cell nucleus very early in infection. Finally, we show that TRP32 interaction with host genes occurs via binding to a repeated GG[A/T]GGC motif found in the promoter region of specific host genes. Further studies will define the role of TRP32 in influencing host cell differentiation and function and will identify how the various ehrlichial nucleomodulins interact to regulate the host and promote ehrlichial survival.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to all of the lab members both current and past and to Yuriy Fofanov for their insight and advice and for many useful discussions. We also thank Thomas Wood and Steven Widen from the UTMB Next Generation Sequencing Core Facility for their time and expertise.
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases grants AI105536 and AI106859 and by funding from the Clayton Foundation for Research (to Jere W. McBride). Tierra Farris was supported by the NIAID T32 AI 007526-15 and by a McLaughlin predoctoral fellowship. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00657-16.
REFERENCES
- 1.Dunphy PS, Luo T, McBride JW. 2013. Ehrlichia moonlighting effectors and interkingdom interactions with the mononuclear phagocyte. Microbes Infect 15:1005–1016. doi: 10.1016/j.micinf.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang JZ, Sinha M, Luxon BA, Yu XJ. 2004. Survival strategy of obligately intracellular Ehrlichia chaffeensis: novel modulation of immune response and host cell cycles. Infect Immun 72:498–507. doi: 10.1128/IAI.72.1.498-507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride JW, Walker DH. 2011. Molecular and cellular pathobiology of Ehrlichia infection: targets for new therapeutics and immunomodulation strategies. Expert Rev Mol Med 13:e3. doi: 10.1017/S1462399410001730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silmon de Monerri NC, Kim K. 2014. Pathogens hijack the epigenome: a new twist on host-pathogen interactions. Am J Pathol 184:897–911. doi: 10.1016/j.ajpath.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bierne H, Hamon M, Cossart P. 2012. Epigenetics and bacterial infections. Cold Spring Harb Perspect Med 2(12):a010272. doi: 10.1101/cshperspect.a010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bierne H, Cossart P. 2012. When bacteria target the nucleus: the emerging family of nucleomodulins. Cell Microbiol 14:622–633. doi: 10.1111/j.1462-5822.2012.01758.x. [DOI] [PubMed] [Google Scholar]
- 7.Saijo Y, Schulze-Lefert P. 2008. Manipulation of the eukaryotic transcriptional machinery by bacterial pathogens. Cell Host Microbe 4:96–99. doi: 10.1016/j.chom.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Garcia JC, Rennoll-Bankert KE, Pelly S, Milstone AM, Dumler JS. 2009. Silencing of host cell CYBB gene expression by the nuclear effector AnkA of the intracellular pathogen Anaplasma phagocytophilum. Infect Immun 77:2385–2391. doi: 10.1128/IAI.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Garcia JC, Barat NC, Trembley SJ, Dumler JS. 2009. Epigenetic silencing of host cell defense genes enhances intracellular survival of the rickettsial pathogen Anaplasma phagocytophilum. PLoS Pathog 5:e1000488. doi: 10.1371/journal.ppat.1000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu B, Nethery KA, Kuriakose JA, Wakeel A, Zhang X, McBride JW. 2009. Nuclear translocated Ehrlichia chaffeensis ankyrin protein interacts with a specific adenine-rich motif of host promoter and intronic Alu elements. Infect Immun 77:4243–4255. doi: 10.1128/IAI.00376-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu B, Kuriakose JA, Luo T, Ballesteros E, Gupta S, Fofanov Y, McBride JW. 2011. Ehrlichia chaffeensis TRP120 binds a G+C-rich motif in host cell DNA and exhibits eukaryotic transcriptional activator function. Infect Immun 79:4370–4381. doi: 10.1128/IAI.05422-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo T, Kuriakose JA, Zhu B, Wakeel A, McBride JW. 2011. Ehrlichia chaffeensis TRP120 interacts with a diverse array of eukaryotic proteins involved in transcription, signaling, and cytoskeleton organization. Infect Immun 79:4382–4391. doi: 10.1128/IAI.05608-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reference deleted.
- 14.Reference deleted.
- 15.Wakeel A, Kuriakose JA, McBride JW. 2009. An Ehrlichia chaffeensis tandem repeat protein interacts with multiple host targets involved in cell signaling, transcriptional regulation, and vesicle trafficking. Infect Immun 77:1734–1745. doi: 10.1128/IAI.00027-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo T, McBride JW. 2012. Ehrlichia chaffeensis TRP32 interacts with host cell targets that influence intracellular survival. Infect Immun 80:2297–2306. doi: 10.1128/IAI.00154-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maruyama T, Nara K, Yoshikawa H, Suzuki N. 2007. Txk, a member of the non-receptor tyrosine kinase of the Tec family, forms a complex with poly(ADP-ribose) polymerase 1 and elongation factor 1alpha and regulates interferon-gamma gene transcription in Th1 cells. Clin Exp Immunol 147:164–175. doi: 10.1111/j.1365-2249.2006.03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong X-F, Li H, Cao E-H. 2007. PIG11 protein binds to DNA in sequence-independent manner in vitro. Biochem Biophys Res Commun 358:29–34. doi: 10.1016/j.bbrc.2007.04.048. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Liu XM, Wang XJ, Zhang Y, Liang XQ, Cao EH. 2009. PIG11 is involved in hepatocellular carcinogenesis and its over-expression promotes Hepg2 cell apoptosis. Pathol Oncol Res 15:411–416. doi: 10.1007/s12253-008-9138-5. [DOI] [PubMed] [Google Scholar]
- 20.Luo T, Zhang X, Wakeel A, Popov VL, McBride JW. 2008. A variable-length PCR target protein of Ehrlichia chaffeensis contains major species-specific antibody epitopes in acidic serine-rich tandem repeats. Infect Immun 76:1572–1580. doi: 10.1128/IAI.01466-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 25.Fagerlund R, Melén K, Kinnunen L, Julkunen I. 2002. Arginine/lysine-rich nuclear localization signals mediate interactions between dimeric STATs and importin α5. J Biol Chem 277:30072–30078. doi: 10.1074/jbc.M202943200. [DOI] [PubMed] [Google Scholar]
- 26.Piskacek S, Gregor M, Nemethova M, Grabner M, Kovarik P, Piskacek M. 2007. Nine-amino-acid transactivation domain: establishment and prediction utilities. Genomics 89:756–768. doi: 10.1016/j.ygeno.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 27.Koudritsky M, Domany E. 2008. Positional distribution of human transcription factor binding sites. Nucleic Acids Res 36:6795–6805. doi: 10.1093/nar/gkn752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitington T, Frith MC, Johnson J, Bailey TL. 2011. Inferring transcription factor complexes from ChIP-seq data. Nucleic Acids Res 39:e98. doi: 10.1093/nar/gkr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo T, Dunphy PS, Lina TT, McBride JW. 2015. Ehrlichia chaffeensis exploits canonical and noncanonical host Wnt signaling pathways to stimulate phagocytosis and promote intracellular survival. Infect Immun 84:686–700. doi: 10.1128/IAI.01289-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lina TT, Dunphy PS, Luo T, McBride JW. 2016. Ehrlichia chaffeensis TRP120 activates canonical Notch signaling to downregulate TLR2/4 expression and promote intracellular survival. mBio 7:e00672-16. doi: 10.1128/mBio.00672-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran Van Nhieu G, Arbibe L. 2009. Genetic reprogramming of host cells by bacterial pathogens. F1000 Biol Rep 1:80. doi: 10.3410/B1-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Köster M, Frahm T, Hauser H. 2005. Nucleocytoplasmic shuttling revealed by FRAP and FLIP technologies. Curr Opin Biotechnol 16:28–34. doi: 10.1016/j.copbio.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 33.Nicolás FJ, De Bosscher K, Schmierer B, Hill CS. 2004. Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci 117:4113–4125. doi: 10.1242/jcs.01289. [DOI] [PubMed] [Google Scholar]
- 34.Birbach A, Gold P, Binder BR, Hofer E, de Martin R, Schmid JA. 2002. Signaling molecules of the NF-κB pathway shuttle constitutively between cytoplasm and nucleus. J Biol Chem 277:10842–10851. doi: 10.1074/jbc.M112475200. [DOI] [PubMed] [Google Scholar]
- 35.Leykauf K, Salek M, Bomke J, Frech M, Lehmann W-D, Dürst M, Alonso A. 2006. Ubiquitin protein ligase Nedd4 binds to connexin43 by a phosphorylation-modulated process. J Cell Sci 119:3634–3642. doi: 10.1242/jcs.03149. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen LK, Kolch W, Kholodenko BN. 2013. When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal 11:52–52. doi: 10.1186/1478-811X-11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao N, Dodge I, Band H. 2002. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol 71:753–763. [PubMed] [Google Scholar]
- 38.Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. 2012. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, Slightham C, Jiang L, Hyman AA, Kim SK, Waterston RH, Gerstein M, Snyder M, Reinke V. 2011. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21:245–254. doi: 10.1101/gr.114587.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sikora-Wohlfeld W, Ackermann M, Christodoulou EG, Singaravelu K, Beyer A. 2013. Assessing computational methods for transcription factor target gene identification based on ChIP-seq data. PLoS Comput Biol 9:e1003342. doi: 10.1371/journal.pcbi.1003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Last TJ, Birnbaum M, van Wijnen Stein AJGS, Stein JL. 1998. Repressor elements in the coding region of the human histone H4 gene interact with the transcription factor CDP/cut. Gene 221:267–277. doi: 10.1016/S0378-1119(98)00415-6. [DOI] [PubMed] [Google Scholar]
- 42.Cusanovich DA, Pavlovic B, Pritchard JK, Gilad Y. 2014. The functional consequences of variation in transcription factor binding. PLoS Genet 10:e1004226. doi: 10.1371/journal.pgen.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spivakov M. 2014. Spurious transcription factor binding: non-functional or genetically redundant? Bioessays 36:798–806. doi: 10.1002/bies.201400036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sherman ML, Stone RM, Datta R, Bernstein SH, Kufe DW. 1990. Transcriptional and post-transcriptional regulation of c-jun expression during monocytic differentiation of human myeloid leukemic cells. J Biol Chem 265:3320–3323. [PubMed] [Google Scholar]
- 45.Gordon S, Akopyan G, Garban H, Bonavida B. 2006. Transcription factor YY1: structure, function, and therapeutic implications in cancer biology. Oncogene 25:1125–1142. doi: 10.1038/sj.onc.1209080. [DOI] [PubMed] [Google Scholar]
- 46.Truscott M, Raynal L, Premdas P, Goulet B, Leduy L, Bérubé G, Nepveu A. 2003. CDP/Cux stimulates transcription from the DNA polymerase α gene promoter. Mol Cell Biol 23:3013–3028. doi: 10.1128/MCB.23.8.3013-3028.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauer DC, Buske FA, Bailey TL. 2010. Dual-functioning transcription factors in the developmental gene network of Drosophila melanogaster. BMC Bioinformatics 11:366–366. doi: 10.1186/1471-2105-11-366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piskacek M, Vasku A, Hajek R, Knight A. 2015. Shared structural features of the 9aaTAD family in complex with CBP. Mol Biosyst 11:844–851. doi: 10.1039/C4MB00672K. [DOI] [PubMed] [Google Scholar]
- 49.Stampfel G, Kazmar T, Frank O, Wienerroither S, Reiter F, Stark A. 2015. Transcriptional regulators form diverse groups with context-dependent regulatory functions. Nature 528:147–151. doi: 10.1038/nature15545. [DOI] [PubMed] [Google Scholar]
- 50.Hagman J, Gutch MJ, Lin H, Grosschedl R. 1995. EBF contains a novel zinc coordination motif and multiple dimerization and transcriptional activation domains. EMBO J 14:2907–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mendell JT. 2008. miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mogilyansky E, Rigoutsos I. 2013. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ 20:1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krichevsky AM, Gabriely G. 2009. miR-21: a small multi-faceted RNA. J Cell Mol Med 13:39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang CH, Yue J, Fan M, Pfeffer LM. 2010. IFN induces miR-21 through a signal transducer and activator of transcription 3-dependent pathway as a suppressive negative feedback on IFN-induced apoptosis. Cancer Res 70:8108–8116. doi: 10.1158/0008-5472.CAN-10-2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buscaglia LEB, Li Y. 2011. Apoptosis and the target genes of microRNA-21. Chin J Cancer 30:371–380. doi: 10.5732/cjc.011.10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lagrange B, Martin RZ, Droin N, Aucagne R, Paggetti J, Largeot A, Itzykson R, Solary E, Delva L, Bastie J-N. 2013. A role for miR-142-3p in colony-stimulating factor 1-induced monocyte differentiation into macrophages. Biochim Biophys Acta 1833:1936–1946. doi: 10.1016/j.bbamcr.2013.04.007. [DOI] [PubMed] [Google Scholar]
- 57.Isobe T, Hisamori S, Hogan DJ, Zabala M, Hendrickson DG, Dalerba P, Cai S, Scheeren F, Kuo AH, Sikandar SS, Lam JS, Qian D, Dirbas FM, Somlo G, Lao K, Brown PO, Clarke MF, Shimono Y. 2014. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 3:e01977. doi: 10.7554/eLife.01977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naqvi AR, Fordham JB, Nares S. 2015. miR-24, miR-30b and miR-142-3p regulate phagocytosis in myeloid inflammatory cells. J Immunol 194:1916–1927. doi: 10.4049/jimmunol.1401893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fordham JB, Naqvi AR, Nares S. 2015. Regulation of miR-24, miR-30b, and miR-142-3p during macrophage and dendritic cell differentiation potentiates innate immunity. J Leukoc Biol 98:195–207. doi: 10.1189/jlb.1A1014-519RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tripathi V, Shen Z, Chakraborty A, Giri S, Freier SM, Wu X, Zhang Y, Gorospe M, Prasanth SG, Lal A, Prasanth KV. 2013. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet 9:e1003368. doi: 10.1371/journal.pgen.1003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang F, Yi F, Han X, Du Q, Liang Z. 2013. MALAT-1 interacts with hnRNP C in cell cycle regulation. FEBS Lett 587:3175–3181. doi: 10.1016/j.febslet.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 62.Wendlandt EB, Graff JW, Gioannini TL, McCaffrey AP, Wilson ME. 2012. The role of MicroRNAs miR-200b and miR-200c in TLR4 signaling and NF-κB activation. Innate Immun 18:846–855. doi: 10.1177/1753425912443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kopp F, Wagner E, Roidl A. 2014. The proto-oncogene KRAS is targeted by miR-200c. Oncotarget 5:185–195. doi: 10.18632/oncotarget.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chuang T-D, Khorram O. 2014. miR-200c regulates IL8 expression by targeting IKBKB: a potential mediator of inflammation in leiomyoma pathogenesis. PLoS One 9:e95370. doi: 10.1371/journal.pone.0095370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamamoto Y, Yoshioka Y, Minoura K, Takahashi R-U, Takeshita F, Taya T, Horii R, Fukuoka Y, Kato T, Kosaka N, Ochiya T. 2011. An integrative genomic analysis revealed the relevance of microRNA and gene expression for drug-resistance in human breast cancer cells. Mol Cancer 10:1. doi: 10.1186/1476-4598-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.