

Abstract
Streptococcus mutans, a major pathogen of dental caries, may promote systemic infections after accessing the bloodstream from oral niches. In this study, we investigate pathways of complement immunity against S. mutans and show that the orphan regulator CovR (CovRSm) modulates susceptibility to complement opsonization and survival in blood. S. mutans blood isolates showed reduced susceptibility to C3b deposition compared to oral isolates. Reduced expression of covRSm in blood strains was associated with increased transcription of CovRSm-repressed genes required for S. mutans interactions with glucans (gbpC, gbpB, and epsC), sucrose-derived exopolysaccharides (EPS). Consistently, blood strains showed an increased capacity to bind glucan in vitro. Deletion of covRSm in strain UA159 (UAcov) impaired C3b deposition and binding to serum IgG and C-reactive protein (CRP) as well as phagocytosis through C3b/iC3b receptors and killing by neutrophils. Opposite effects were observed in mutants of gbpC, epsC, or gtfBCD (required for glucan synthesis). C3b deposition on UA159 was abolished in C1q-depleted serum, implying that the classical pathway is essential for complement activation on S. mutans. Growth in sucrose-containing medium impaired the binding of C3b and IgG to UA159, UAcov, and blood isolates but had absent or reduced effects on C3b deposition in gtfBCD, gbpC, and epsC mutants. UAcov further showed increased ex vivo survival in human blood in an EPS-dependent way. Consistently, reduced survival was observed for the gbpC and epsC mutants. Finally, UAcov showed an increased ability to cause bacteremia in a rat model. These results reveal that CovRSm modulates systemic virulence by regulating functions affecting S. mutans susceptibility to complement opsonization.
INTRODUCTION
Streptococcus mutans is a common species of the oral cavity of humans involved in the pathogenesis of dental caries, which can promote infective endocarditis and other systemic infections after gaining access to the bloodstream (1–4). However, factors involved in S. mutans survival in the bloodstream are unknown but likely include mechanisms to evade host immunity. S. mutans expresses the orphan response regulator CovR (CovRSm) (also known as GcrR) (5–8), which is an orthologue of the CovR protein of the two-component regulatory system (TCS) CovRS (also known as CsrRS) of the pathogenic species Streptococcus pyogenes (group A Streptococcus [GAS]) and Streptococcus agalactiae (group B Streptococcus [GBS]). In GAS, S. pyogenes CovR (CovRSpy) typically functions as a repressor of a panel of virulence genes involved in the evasion of host immunity and tissue invasiveness (9). In S. mutans, CovRSm represses virulence factors involved in the establishment of S. mutans in dental biofilms (7, 8, 10, 11), but its role in systemic virulence is unknown. Genes directly repressed by CovRSm include gtfB and gtfC, which encode glucosyltransferases B and C, respectively, required for the extracellular synthesis of glucans from sucrose (7), major structural exopolysaccharides (EPS) of cariogenic biofilms (1, 2). CovRSm also inhibits the expression of several genes involved in cell wall biogenesis and surface interactions with EPS, including GbpB (glucan-binding protein B), GbpC (glucan-binding protein C), EpsC (enzyme for exopolysaccharide synthesis [UDP-N-acetylglucosamine 2-epimerase]), LysM (lysine motif protein), and WapE (cell wall protein E) (8, 10–12).
An isogenic mutant of covRSm obtained from UA159 (serotype c) shows impaired susceptibility to phagocytosis by human polymorphonuclear leukocytes (PMNs) in a blood-dependent manner (13). Among the four known S. mutans serotypes (serotypes c, e, f, and k), serotype c is the most prominent serotype in the oral cavity (∼70 to 80% of strains) and is frequently associated with systemic infections, being detected in 30.3 and 65.5% of S. mutans-positive specimens of cardiac valves and atheromatous plaques, respectively, from patients subjected to cardiac surgeries (14, 15). Serotype c strain MT8148 survives during 1 to 2 days in the bloodstream of rats (16), further suggesting mechanisms of evasion of blood immunity. In this study, we investigated the roles of CovRSm in the susceptibility of S. mutans strains to complement immunity mediated by C3b, a major opsonin present in blood and other host fluids (17, 18). Profiles of C3b deposition on strains isolated from blood of patients with bacteremia and/or infective endocarditis and on strains from the oral cavity were compared to assess diversity in susceptibility to complement immunity. The low susceptibility to C3b deposition observed for blood isolates was then compared to transcript levels of covRSm and of CovRSm-repressed genes. Effects of covRSm deletion in strain UA159 (serotype c) on the binding of C3b, IgG antibodies, and C-reactive protein (CRP) and on phagocytosis mediated by C3b/iC3b or IgG receptors and killing by human neutrophils (PMNs) were determined. Mechanisms of CovRSm regulation of S. mutans susceptibility to complement immunity were then investigated by assessing the effects of the deletion of CovRSm-regulated genes (gtfB, gtfC, gbpC, epsC, lysM, and wapE) on C3b- and antibody-mediated immunity in the presence or absence of sucrose-derived EPS. Finally, strains were compared regarding ex vivo survival in human blood and in a rat model of bacteremia and infective endocarditis.
MATERIALS AND METHODS
Studied strains and culture conditions.
Strains used in this study are described in Table 1. Strains were grown (37°C with 10% CO2) from frozen stocks in brain heart infusion (BHI) agar (Difco). BHI agar or chemically defined medium (CDM) (10) with or without sucrose (0.01 and 0.1%) was used in the experiments. Erythromycin (10 μg/ml), spectinomycin (200 μg/ml), or kanamycin (500 μg/ml) (Merck Labs, Germany) was added to media for cultivation of deletion and complemented mutants.
TABLE 1.
Strains used in this study
| Strain | Relevant characteristic(s) | Source or reference |
|---|---|---|
| UA159 | Oral isolate, caries-affected child; Erms Specs Kans | ATCC |
| UAcovR | ΔcovR::Ermr | 13 |
| UAwapE | ΔwapE::Ermr | 11 |
| UAlysM | ΔlysM::Ermr | 11 |
| UAepsC | ΔepsC::Ermr | 11 |
| UAgbpC | ΔgbpC::Ermr | This study |
| UAcovR+ | ΔcovR::Ermr pDL278::SMU.1924; Specr | 13 |
| UAwapE+ | ΔwapE::Ermr pDL278::SMU.1091; Specr | 11 |
| UAlysM+ | ΔlysM::Ermr pDL278::SMU.2147c; Specr | 11 |
| UAepsC+ | ΔepsC::Ermr pDL278::SMU.1437c; Specr | 11 |
| UAgbpC+ | ΔgbpC::Ermr; pDL278::SMU.1396; Specr | This study |
| MT8148 | Oral isolate, healthy Japanese child | 19 |
| C1 | ΔgbpC::Kanr; mutant of MT8148 | 19 |
| S5 | ΔgtfBC::Ermr; double mutant of MT8148 | 20 |
| BC7s | ΔgtfD::Ermr ΔgtfBC::Kanr; triple mutant of MT8148 | 20 |
| 2ST1 | Oral isolate, caries-affected child | 21 |
| 2VS1 | Oral isolate, caries-affected child | 21 |
| 3SN1 | Oral isolate, caries-free child | 21 |
| 4SM1 | Oral isolate, caries-free child | 21 |
| 4VF1 | Oral isolate, caries-affected child | 21 |
| 5SM3 | Oral isolate, caries-free child | 21 |
| 8ID3 | Oral isolate, caries-free child | 21 |
| 11A1 | Oral isolate, caries-free child | 21 |
| 11SSST2 | Oral isolate, caries-free child | 21 |
| 11VS1 | Oral isolate, caries-free child | 21 |
| 15JP3 | Oral isolate, caries-free child | 21 |
| 15VF2 | Oral isolate, caries-affected child | 21 |
| SA12 | Blood, infective endocarditis | 22 |
| SA13 | Blood, bacteremia | 22 |
| SA14 | Blood, infective endocarditis | 22 |
| SA15 | Blood, bacteremia | 22 |
| SA16 | Blood, infective endocarditis | 22 |
| SA17 | Blood, bacteremia | 22 |
| SA18 | Blood, infective endocarditis | 22 |
| D39 | Streptococcus pneumoniae serotype 2 (NCTC 7466) | NCTC |
| TIGR4 | Streptococcus pneumoniae serotype 4 (ATCC BAA-334) | ATCC |
Construction of gbpC deletion and complemented mutants.
The nonpolar gbpC deletion mutant was obtained from strain UA159 (UAgbpC) by double-crossover recombination with a null allele (of 2,315 bp) constructed by PCR ligation (23). In the recombinant allele, an internal sequence of 1,455 bp of the encoding region of gbpC was replaced by an erythromycin resistance cassette (Ermr) obtained from plasmid pVA838. The complemented gbpC mutant (UAgbpC+) was obtained by transforming UAgbpC with plasmid pDL278 containing an intact copy of gbpC and the spectinomycin resistance gene. Primers used for the construction of mutants are shown in Table 2.
TABLE 2.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′–3′)a | Product size; positions or relevant characteristicb |
|---|---|---|
| ermE1-AscI | TTGGCGCGCCTGGCGGAAACGTAAAAGAAG | 998 bp; amplicon containing the Ermr gene from pVA838 |
| ermE2-XhoI | TTCTCGAGGGCTCCTTGGAAGCTGTCAGT | |
| gbpCP1 | CCCTCAACACACTCTGCTAA | 473 bp; 323 bp upstream to 150 bp downstream of the gbpC ORF |
| gbpCP2-AscI | TTGGCGCGCCCGGTTCTGATGCTTGTGTAT | |
| gbpCP3-XhoI | TTCTCGAGGGAGAAATGCGTGTTAGAGA | 387 bp; 1,605 bp upstream to 240 bp downstream of the encoding region of gbpC |
| gbpCP4 | CTTACCCATCACAAAAACCA | |
| C1-SacI | GGGAGCTCCCCTCAACACACTCTGCTAA | 2,139 bp; amplicon containing the encoding region of gbpC for mutant complementation |
| C2-SphI | GGGCATGCAACAAGAACTGCTGCTCAAG |
Underlined sequences indicate restriction enzyme linkers.
ORF, open reading frame.
RNA isolation, reverse transcription, and qPCR.
RNA was purified from strains at the mid-log phase of growth (A550 of 0.3) by using an RNeasy kit (Qiagen, Germany) and treated with Turbo DNase (Ambion, USA), as described previously (11). The cDNA was obtained from 1 μg of RNA by using random primers (24) and SuperScript III (Life Technologies, USA), according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed with a StepOne real-time PCR system (Life Technologies) with cDNA (10 ng), 10 μM each primer, and 1× Power SYBR green PCR master mix (Lifetech) in a total volume of 10 μl. The cycling conditions were 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, the optimal temperature for primer annealing (Table 2) for 15 s, and 72°C for 30 s. Tenfold serial dilutions of genomic DNA (300 ng to 0.003 ng) were used to generate standard curves for the absolute quantification of RNA expression levels. Melting curves were obtained for each primer set. Results were normalized against S. mutans 16S rRNA gene expression values (24). Assays were performed in duplicate with at least two independent RNA samples.
S. mutans interaction with EPS.
Cell aggregation mediated by sucrose-derived EPS was assessed as described previously (25). Briefly, strains were grown in BHI medium (37°C with 10% CO2 for 18 h), and an equal number of cells was transferred to fresh BHI medium supplemented with 0.1% sucrose and incubated for 24 h (37°C with 10% CO2). Cell aggregation was then visually inspected.
Surface-associated EPS was analyzed by scanning electron microscopy (SEM) in strains grown in BHI medium or CDM with or without 0.1% sucrose. Briefly, cultures grown during 18 h in BHI medium or CDM were 100-fold diluted with fresh medium containing or not containing 0.1% sucrose and incubated to reach an A550 of 0.3. Cells from volumes of 500 μl were then harvested by centrifugation, washed with phosphate-buffered saline (PBS), and processed for SEM analysis, as previously described (12). Samples were analyzed with a scanning electron microscope (JSM 5600LV; JEOL, Japan).
Volunteers, sera, and blood samples.
Blood samples from six healthy subjects (three males and three females; mean age, 30 years [range, 25 to 45 years]) were collected by venipuncture in heparin vacuum tubes (BD Vacutainer), according to standard protocols previously approved by the Ethical Committee of the Piracicaba Dental School, State University of Campinas (protocol number 031/2012). Serum samples were stored in aliquots at −70°C until use. Levels of C3 in serum samples were determined as described below and were within normal levels in all volunteers (mean, 1.91 mg/ml; standard deviation [SD], 0.68 mg/ml; range, 1.18 to 2.88 mg/ml) (26). Mean levels of IgG and IgM in the same samples were also determined and were, respectively, 11.68 (±1.82) mg/ml and 1.35 (±0.64) mg/ml. Serum samples from one volunteer, which were representative of C3, IgG, and IgM levels, were used as controls. Commercial human serum depleted of C1q was obtained from Calbiochem (MA, USA). The Calbiochem C1q-depleted serum is free of EDTA and retains alternative pathway activity (27). As a control for integrity, C1q-depleted serum was supplemented with purified human C1q (Calbiochem) to a physiological concentration range (75 μg/ml in 100% serum). Heat-inactivated sera (56°C for 20 min) were also used as negative controls in preliminary experiments and showed minimal effects on comparative analyses of C3b deposition between strains.
Determination of total levels of C3, IgG, and IgM antibodies in serum.
The serum concentrations of C3, IgG, and IgM antibodies were determined by enzyme-linked immunosorbent assays (ELISAs) using commercial systems for the quantification of human complement C3 (Molecular Innovations, MI, USA), human IgG, and human IgM (Bethyl Laboratories, Inc., TX, USA), respectively. Briefly, 100 μl of serum samples diluted in dilution buffer (1:100,000, 1:500,000, and 1:10,000, respectively) was added to 96-well plates coated with anti-C3, anti-IgG, or anti-IgM and then incubated for 30 min at room temperature (RT). After a series of three washes with wash buffer, 100-μl aliquots of antibodies specific to C3, human IgG, or human IgM were added per well, and plates were incubated (RT) for 1 h. After a new series of washes, 100 μl per well of secondary horseradish peroxidase (HRP)-conjugated antibodies (1:50,000) was added, and incubation continued for 30 min. After a new series of washes, 100 μl per well of a chromogenic HRP substrate (3,3′,5,5′-tetramethylbenzidine) was added, and plates were incubated for 30 min. Reactions were stopped by the addition of 1 N H2SO4 to the mixture. Absorbances (A450) were measured in a microtiter plate reader (Versa Max) and converted to micrograms per milliliter using standard curves for C3 (0.02 to 10 ng/ml), IgG (0.69 to 167 ng/ml), or IgM (1.03 to 250 ng/ml) antibodies.
C3b deposition on S. mutans.
Deposition of C3b on the surface of serum-treated S. mutans strains was determined as described previously (27, 28), with some modifications. Briefly, ∼107 CFU of strains at the mid-log phase of growth (A550 of 0.3) were harvested by centrifugation (10,000 × g at 4°C), washed two times with PBS (pH 7.4), and suspended in 20 μl of 20% serum (diluted in PBS). Samples were then incubated (37°C for 30 min) and washed twice with PBS–0.05% Tween (PBST). Cells were then incubated on ice (40 min) with fluorescein isothiocyanate (FITC)-conjugated polyclonal goat anti-human C3 IgG antibody (ICN, CA, USA) (1:300 in PBST). After two washes with PBST, bacterial cells were fixed in 3% paraformaldehyde in PBS and analyzed on a FACSCalibur flow cytometer (BD Biosciences) using forward- and side-scatter parameters to gate at least 25,000 bacteria. Results were expressed as the geometric mean fluorescence intensity (MFI) of C3b-positive cells or as the mean fluorescence index (FI) (percentage of positive cells multiplied by the MFI) (29, 30). Control samples included bacteria treated only with PBS instead of serum.
PMN isolation, opsonophagocytosis, and killing assays.
Human PMNs were isolated from fresh heparinized blood samples from one reference volunteer by centrifugation over a double gradient composed of Histopaque-1119 and Histopaque-1083 (Sigma-Aldrich), as previously described, with modifications (31). Red blood cells were eliminated by hypotonic lysis. Isolated PMNs were suspended in RPMI 1640 medium (Gibco, Life Technologies, NY, USA) supplemented with inactivated 10% fetal bovine serum. Cell viability (>98%) was monitored by trypan blue exclusion. Cell purity (>95%) was monitored by May-Grunwald-Giemsa staining.
For opsonophagocytosis assays, bacteria were previously labeled with FITC as described previously (32), with some modifications. Briefly, 500 μl of bacterial strains (A550 of 0.3) was washed two times with PBS, suspended in 600 μl of carbonate buffer (0.15 M Na2CO3, 0.9% NaCl [pH 9]) with 1.7 mg/ml of FITC (Sigma), and incubated for 1 h (with shaking at RT in the dark). Cells were then harvested and washed three times with PBST, and aliquots were stored overnight in 10% glycerol at −70°C. Bacterial labeling was analyzed with a fluorescence microscope (Leica) and by flow cytometry (FACSCalibur; BD).
For C3b deposition, aliquots containing 107 CFU of FITC-labeled bacteria were incubated with 20% serum and added to wells of 96-well plates containing 2 × 105 PMNs in 50 μl of RPMI medium to a multiplicity of infection (MOI) of 200 bacteria per PMN. After incubation (37°C and 10% CO2 with gentle shaking) for 5 or 30 min, reaction mixtures were fixed by the addition of 100 μl of 3% of paraformaldehyde. PMNs were then analyzed by using a FACSCalibur instrument (BD Biosciences), and the frequency of phagocytosis was expressed as the number of PMN cells with intracellular bacteria, within a total of 10,000 PMNs analyzed (33). The MOI was determined in preliminary experiments testing MOIs of 20 to 200 bacteria per PMN, after 5 to 60 min of incubation. Data from flow cytometry analysis was compared to data from light microscopy analysis of samples stained by using May-Grunwald-Giemsa stain, as previously described (13). These comparisons confirmed that most of the PMN-associated bacteria were internalized.
To assess phagocytosis by PMNs through C3b/iC3b receptors, similar assays were performed with PMNs previously incubated (37°C for 30 min) with mouse anti-CD35 monoclonal antibodies (MAbs) (BioLegend, CA, USA) or anti-CD11b/CD18 MAbs to block CR1 or CR3 receptors (34, 35), respectively. As a reference, PMNs incubated with anti-CD32 (FcγRII) (eBioscience, CA, USA) or anti-C16 (FcγRIII) (BioLegend, CA, USA) MAbs were also tested.
Opsonophagocytic killing was assessed as previously described (36, 37), with modifications. Briefly, preopsonized bacteria (20% human serum for 30 min at 37°C) were added to samples of human PMNs in RPMI with 10% human serum at an MOI of 200:1. After incubation (37°C for 10 and 30 min) with shaking, reactions were stopped at 4°C. PMNs were harvested by centrifugation (500 × g for 8 min at 4°C), washed twice with PBS (pH 7.2), and lysed with 2% saponin (12 min at 37°C), and viable counts of intracellular bacteria were determined by plating serial dilutions onto BHI agar. Bacterial counts were also determined in control wells with identically treated samples without PMNs. Viable bacteria were counted in culture supernatants of PMN samples to monitor the number of extracellular bacteria. Percent intracellular survival was calculated as follows: (CFU ml−1 test well)/(CFU ml−1 control well) × 100.
Determination of binding of serum IgG, IgM, and CRP to S. mutans.
Binding of serum IgG, IgM, or CRP to S. mutans was determined as previously described (27, 38), with some modifications. Briefly, bacterial strains were harvested from 500 μl of cultures of UA159 (A550 of 0.3), washed twice with PBS (pH 7.0), incubated with 20% serum, and washed with PBST. To assess surface levels of IgG, IgM, or CRP, cells were then incubated with polyclonal goat IgG anti-human IgG Fc conjugated with FITC (Novus Biological, USA) (1:900), polyclonal mouse IgG anti-human IgM Fc conjugated with allophycocyanin (APC) (1:1,000) (BioLegend, USA), or goat IgG anti-human CRP conjugated with FITC (GeneTex, USA) (1:100), respectively. Streptococcus pneumoniae strains D39 and TIGR4 were used as controls for CRP binding, because the S. pneumoniae cell wall contains known CRP ligands (phosphorylcholine [PCho]) (39).
After 40 min of incubation on ice, bacterial cells were washed twice with 300 μl of PBST, harvested (16,000 × g for 2 min), and suspended in 300 μl of 3% paraformaldehyde. Flow cytometry analyses were performed as described above, using forward- and side-scatter parameters to gate at least 25,000 bacteria. Bacterial samples treated with PBS instead of serum were used as negative controls.
Ex vivo survival of S. mutans strains in human blood.
Bacteria from cultures grown in BHI medium (A550 of 0.3) were harvested (11,000 × g for 2 min), washed twice in PBS, and resuspended in 1 ml of fresh whole human blood. Samples were then incubated (37°C and 5% CO2 with gentle agitation), and aliquots were collected at different time points (5, 30, 60, 120, and 240 min), serially diluted, and plated onto BHI agar for determination of bacterial counts. Aliquots collected just after bacterial suspension in blood were used for initial measurements of CFU per milliliter (time zero). Changes in numbers of viable bacteria were then calculated in relation to counts at time zero to reduce the influence of variations in blood-mediated aggregation between strains on the numbers of CFU per milliliter. Three independent experiments were performed in duplicate with blood samples from one volunteer with reference levels of C3, IgG, and IgM (2.8, 14.6, and 1.5 μg/ml of serum, respectively).
Survival of S. mutans strains in a rat model of bacteremia and infective endocarditis.
Protocols for the animal experiments were approved by the institutional animal care and use committee of the Osaka University Graduate School of Dentistry (approval number 24-019). All rats were treated humanely in accordance with the National Institute of Health and AERI-BBRI Animal Care and Use Committee guidelines. The rat infective endocarditis model was used according to methods described previously, with some modifications (40). In brief, 21 Sprague-Dawley male rats (400 to 500 g each) were anesthetized with a mixture of xylazine and midazolam (0.1 ml–100 g). A sterile polyethylene catheter with a guide wire was surgically placed across the aortic valve of each animal via the right carotid artery, and the tip was positioned and placed at the aortic valve in the left ventricle. A bacterial suspension (109 cells per body, from cultures in BHI medium) in PBS was intravenously administered through the jugular vein. Bacterial clearance was examined by measuring the numbers of bacteria in blood samples from the jugular veins, which were taken at 1, 3, 6, and 24 h and 7 days after the initial infection. The blood samples were placed onto Mitis-Salivarius agar (Difco Laboratories, Detroit, MI, USA) plates containing bacitracin (0.2 U/ml; Sigma Chemical Co., St. Louis, MO, USA) and 15% (wt/vol) sucrose (MSB agar) and incubated at 37°C for 48 h. Seven days after bacterial infection, the rats were sacrificed by an overdose of anesthesia, and the aortic valves were extirpated, transversely sectioned, and subjected to Gram staining and to bacterial recovery for microbial counting.
Statistical analyses.
Flow cytometry data (percentages of positive bacteria or PMNs and MFI or FI values) were analyzed by comparing means of data from at least three independent experiments using a nonparametric Kruskal-Wallis test with Dunn's post hoc test. Comparisons of the mean MFI or FI values for surface-bound C3b between oral and blood isolates were performed by using a Mann-Whitney U test. Spearman's rank correlation was applied to analyze associations between MFI values of surface C3b and those of surface IgG. Ex vivo survival in blood was compared between strains by testing differences in relative bacterial counts (log CFU per milliliter) at each time point of incubation in relation to initial counts in blood suspensions. Bacterial counts in the rat bloodstream were also compared between strains. Relative or absolute bacterial counts were compared between strains at each time point by using a Kruskal-Wallis test with Dunn's post hoc test, using correction for repeated measures (30).
RESULTS
S. mutans strains isolated from blood show reduced susceptibility to C3b deposition compared to oral isolates.
Although complement immunity is recognized as an important blood defense against streptococcal species (18, 28, 41), the profiles of S. mutans susceptibility to complement-mediated opsonization were unknown. Thus, we compared patterns of C3b deposition on strains isolated from the bloodstream of patients with bacteremia associated or not with infective endocarditis (n = 7) and isolates of the oral cavity (n = 13), including reference strain UA159. As shown in Fig. 1, blood isolates showed reduced levels of C3b deposition compared to oral isolates. Two blood serotype c strains (SA13 and SA18) showed the lowest MFI values for C3b (mean MFI values, 148.9 ± 65.8 and 215.7 ± 112.0, respectively) and low FI values (mean FI values, 2,069.0 ± 802.8 and 3,019.9 ± 1,056.4, respectively). Mean MFI and FI values of strain UA159 were 623.2 (±100.3) and 17,678.4 (±2,908.7), respectively.
FIG 1.
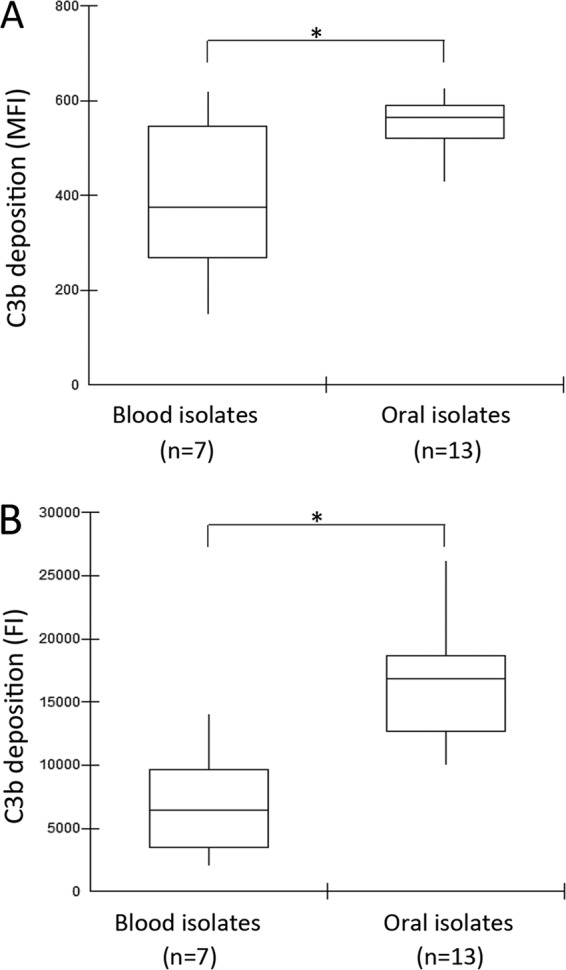
Box plot comparisons of C3b depositions between S. mutans strains isolated from blood samples and those of strains isolated from the oral cavity. C3b bound to serum-treated strains was probed with anti-C3 antibody conjugated with FITC for flow cytometry analysis. (A) Levels of C3b bound to S. mutans strains are expressed as geometric mean fluorescence intensity (MFI) values. (B) Fluorescence index (FI) values were obtained by multiplying the percentage of C3b-positive cells by MFI values for C3b. MFI and FI data are means of results from three independent experiments. Asterisks indicate significant differences between groups (P < 0.05 as determined by a Mann-Whitney U test).
S. mutans blood strains show reduced activity of covRSm and increased transcription of CovRSm target genes required for surface interaction with EPS.
To investigate the role of CovRSm in strain susceptibilities to C3b deposition, transcript levels of covRSm and downstream genes of four blood strains showing the lowest levels of C3b deposition (SA13, SA15, SA16, and SA18) were compared with those of four oral isolates with the highest levels of binding to C3b (2VS1, 11A1, 11SSST2, and 8ID3) and reference strain UA159. The CovRSm-repressed genes selected were those affecting S. mutans cell surface properties, including lysM, wapE, epsC, gbpB, gbpC, gtfB, gtfC, and gtfD (glucosyltransferase D-encoding gene) (7, 11, 42). As shown in Fig. 2, blood strains showed lower levels of covRSm transcripts than did the oral isolates. Consistently, blood isolates showed increased transcription of epsC, gbpC, and gbpB, which are genes involved in S. mutans surface interactions with EPS (11, 12, 43). No significant differences in transcript levels of lysM, wapE, gtfB, gtfC, or gtfD were detected (Fig. 2). Levels of C3b deposition in the analyzed strains negatively correlated with transcript levels of epsC (Spearman correlation [r], −0.45; P < 0.05), gbpB (r, −0.21; P < 0.05), and gbpC (r, −0.35; P < 0.05). Thus, diversity in the transcriptional activities of covRSm and of CovRSm-repressed genes is associated with differences in levels of C3b deposition in S. mutans strains.
FIG 2.

Reverse transcription-qPCR comparisons of transcript levels of covRSm and CovRSm-regulated genes (lysM, wapE, epsC, gbpC, gbpB, gtfB, and gtfC) in blood (n = 4) and oral (n = 5) strains of S. mutans. The gtfD gene, which is not regulated by CovRSm, was tested as a control. Asterisks indicate significant differences in mean levels of transcripts between groups (P < 0.05 as determined by analysis of variance with Tukey's post hoc test).
S. mutans blood strains show an increased capacity to bind EPS produced in the presence of sucrose, similarly to the covRSm isogenic mutant.
Because gbpB, gbpC, and epsC encode proteins for S. mutans binding to sucrose-derived EPS (glucan) (11, 12, 43), we compared the capacities of aggregation in the presence of sucrose of blood and oral isolates. Isogenic mutants of covRSm and of CovRSm-repressed genes (gbpC, epsC, gtfB, gtfC, lysM, and wapE) were also tested, except for gbpB, which is essential for S. mutans viability (12). As shown in Fig. 3A, blood isolates showed a higher capacity to aggregate in BHI medium containing 0.1% sucrose than did oral isolates. Blood strains SA13 and SA18 showed aggregation phenotypes similar to that of the UAcov strain (Fig. 3B). As anticipated, the gbpC isogenic mutant did not aggregate, while only weak aggregation was detected in the epsC mutant (Fig. 3C). In addition, because gtfB, gtfC, and gtfD are required for the synthesis of glucan from sucrose, mutants of these genes obtained from strain MT8148 did not aggregate (Fig. 3C). The aggregation phenotypes of the gbpC mutants obtained from MT8148 (Fig. 3C) and UA159 (Fig. 3B) were similar. SEM analysis supported data from a previous report (11) on the increased interaction of UAcov with sucrose-derived EPS in biofilms and confirmed the strain capacities to bind sucrose-derived EPS under the growth conditions applied in the C3b binding assays (data not shown).
FIG 3.
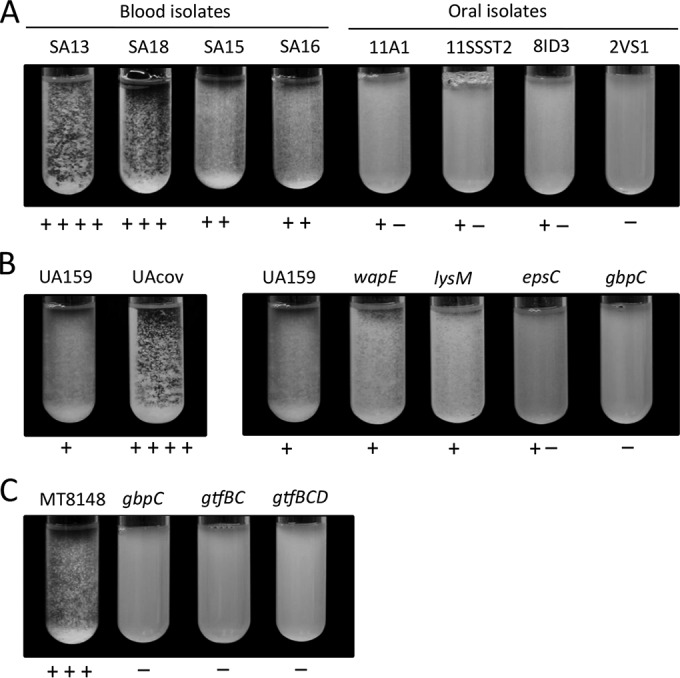
Comparisons of S. mutans capacities to aggregate in the presence of sucrose-derived EPS. Strains were incubated in BHI medium supplemented with 0.1% sucrose during 24 h for visual analysis of clump formation. Intensities of cell aggregation were determined by a blind examiner and are indicated below the respective images. (A) Comparisons between blood and oral strains. (B) Comparisons between parent strain UA159 and the respective knockout mutants. (C) Comparisons of parent strain MT8148 with the respective knockout mutants.
Inactivation of covRSm impairs deposition of C3b, phagocytosis mediated by C3b/iC3b receptors, and opsonophagocytic killing by human PMNs.
UAcov shows low susceptibility to phagocytosis by human PMNs in a serum-dependent way (13), suggesting that CovRSm regulates surface components affecting serum opsonization. As shown in Fig. 4A, deposition of C3b was impaired in UAcov. This phenotype was completely restored in the complemented UAcov+ mutant. Significant reductions in the frequencies of phagocytosis of UAcov in the presence of 20% serum were also observed in comparison to parental strain UA159 or UAcov+ (Fig. 4B). Importantly, blocking of CR1 and/or CR3 receptors of PMNs with anti-CD35 (CR1) or anti-CD11b/CD18 (CR3) antibodies reduced differences in the frequencies of phagocytosis between UA159 and UAcov (Fig. 4B). Additionally, the simultaneous blockage of CR1 and CR3 receptors abolished differences in the frequencies of phagocytosis between these strains (Fig. 4B), reflecting the multiple and cooperative functions of CR1 and CR3 in bacterial phagocytosis mediated by C3b/iC3b (44). Treatment of PMNs with anti-CD32 (FcγRII) or anti-CD16 (FcγRIII) antibodies also reduced the phagocytosis of UA159, although blockage of both Fcγ receptors did not eliminate differences in the frequencies of phagocytosis between UA159 and UAcov (Fig. 4B).
FIG 4.

Comparisons of C3b deposition, opsonophagocytosis, and killing by PMNs between covR mutant (UAcov), parent (UA159), and complemented (+) strains. (A) Intensities of C3b deposition (MFI) in strains treated with a reference serum or pools of sera obtained from six volunteers were determined by flow cytometry. (B) Percentages of PMNs with associated bacteria were assessed after 5 min of exposure of PMNs to FITC-labeled bacteria in the presence of 20% serum. Untreated PMNs and PMNs treated with MAbs to block CR1 (CD35), CR3 (CD11b/CD18), FcγRII (CD32), and/or FcγRIII (CD16) receptors were tested. (C) Percentages of intracellular survival in PMNs after 10 min of incubation with preopsonized bacteria were calculated in relation to bacterial counts of no-PMN control samples. Columns represent means of data from three independent experiments. Bars indicate standard deviations. Asterisks indicate significant differences in relation to UA159 under the same conditions (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
To examine if the reduced phagocytosis of UAcov was associated with reduced killing by PMNs, strains were compared in opsonophagocytic killing assays. As shown in Fig. 4C, UAcov showed increased survival to PMN during 10 min of incubation. Similar results were obtained after exposure of PMNs to the tested strains during 30 min (data not shown). Viable bacteria in PMN culture supernatants were monitored, confirming the reduced phagocytosis of UAcov compared to UA159; mean counts of UAcov bacteria in culture fluids were significantly higher than UA159 counts (P < 0.05). These data establish the strong influence of C3b/iC3b deposition on S. mutans phagocytosis and imply that reduced deposition of C3b/iC3b on UAcov not only impairs phagocytosis mediated by C3b/iC3b receptors but also is associated with reduced killing by PMNs.
C3b deposition on S. mutans is strongly dependent on C1q of the classical pathway of complement activation.
We hypothesized that EPS bound to the S. mutans surface could compromise antibody recognition of immunogenic surface proteins, thus affecting the classical pathway of complement activation. In this pathway, the proteolytic cascade initiates with C1q binding to different host components bound to the bacterial surface, most prominently IgG or IgM antibodies but also acute-phase proteins of innate immunity, e.g., CRP (17, 18). Therefore, we analyzed the effect of covRSm inactivation on levels of antibodies bound to S. mutans (from pools of sera from six volunteers) and investigated the effect of the classical pathway on binding of C3b to S. mutans. Because the classical pathway can also be activated by CRP bound to the surface of streptococcal species containing CRP ligands (27, 45), we additionally assessed the binding of this acute-phase protein to S. mutans. As shown in Fig. 5A, significant reductions in the binding of IgG antibodies were observed for UAcov compared to the parent strain. In addition, although levels of CRP bound to S. mutans UA159 were low compared to those for the S. pneumoniae control strains, UAcov showed reduced binding to CRP compared to UA159 (Fig. 5B). Importantly, levels of surface-bound IgG and CRP were restored in complemented mutant strain UAcov+ (Fig. 5A and B). No significant changes in the binding of IgM antibodies to S. mutans were observed (data not shown).
FIG 5.

Binding of serum IgG, CRP, and C3b to S. mutans strains in the presence of serum. (A and B) Strains were treated with 20% human serum, and levels of surface IgG (A) and CRP (B) were determined by flow cytometry (MFI). S. pneumoniae strains D39 and TIGR4 were used as controls for CRP binding. (C) Levels of C3b binding were measured after treatment of bacteria with reference serum, serum depleted of C1q (C1q−), or serum depleted of C1q and supplemented with purified C1q (C1q+). Columns represent means of results from three independent experiments; bars represent standard deviations. Strains were compared by using a Kruskal-Wallis test with Dunn's post hoc test. Asterisks indicate significant differences in relation to UA159 under the same conditions (P < 0.05).
C3b deposition was minimal when strains were treated with C1q-depleted serum compared to normal serum. Supplementation of C1q-depleted serum with purified C1q (to physiological levels) restored C3b deposition (Fig. 5C). These data indicate that most of the C3b bound to the S. mutans surface resulted from the activation of the C1 component of the classical pathway. Thus, covRSm inactivation impairs S. mutans surface binding of serum IgG antibodies and CRP, serum components that trigger the classical pathway of complement activation, a major pathway involved in C3b deposition on S. mutans.
C3b deposition on S. mutans is affected by interaction with sucrose-derived EPS.
S. mutans accesses the bloodstream from oral niches, where it is exposed to dietary sucrose to synthesize EPS, including glucan. covRSm inactivation in serotype c strains upregulates not only genes for the synthesis of sucrose-derived glucan (gtfB, gtfC, and gtfD) but also genes encoding glucan-binding proteins (gbpB and gbpC) and EpsC (epsC), which are involved in surface binding to these polymers (7, 8, 10, 11, 43). Therefore, to address whether sucrose-derived EPS on the S. mutans surface influences C3b deposition, we compared levels of C3b binding of the parent strain to those of isogenic covRSm, gtfBCD, gbpC, and epsC mutants previously grown in medium with different concentrations of sucrose, which were then harvested, washed, and exposed to serum. As shown in Fig. 6A, the parent and covRSm strains grown in the presence of sucrose showed reduced levels of C3b compared to those of the same strain grown in medium without sucrose. C3b deposition was more intensely reduced in UAcov in the presence of sucrose. When grown in CDM supplemented with 0.1% sucrose, mean levels of C3b (MFI) in UAcov were 4-fold lower than those in the parent strain UA159 (82.3 versus 326.4). In sucrose-free CDM, levels of C3b in UAcov were only 1.9-fold lower than C3b levels in the parent strain UA159 (507.4 versus 991.5). In BHI medium, UAcov showed a 4-fold reduction in C3b deposition, but in BHI medium with 0.1% sucrose, UAcov showed a 9.4-fold reduction in C3b deposition compared to that of the parent strain (33.4 versus 315.4). Although the level of C3b binding was higher in strains grown in sucrose-free CDM than in strains grown in BHI medium (Fig. 6A), the addition of 0.01% sucrose to CDM was sufficient to eliminate these differences. This result suggests that there may be trace amounts of sucrose in complex BHI medium, although we can not rule out that unknown BHI medium components adsorbed to the S. mutans surface could also influence C3b deposition.
FIG 6.

Effects of previous growth in medium supplemented with sucrose on strain susceptibilities to C3b deposition. Strains grown in BHI medium or CDM supplemented or not with 0.01 or 0.1% sucrose were harvested, washed with PBS, and treated with human serum for C3b deposition. Columns represent mean MFI values for C3b determined by flow cytometry in three independent experiments. Bars represent standard deviations. Asterisks indicate significant differences between groups (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
Because sucrose-derived glucans are synthesized by GtfB, GtfC, and GtfD, we further confirmed if deletion of multiple gtf genes (double gtfBC and triple gtfBCD mutants) would eliminate the effect of previous exposure to sucrose on S. mutans susceptibility to C3b deposition. GtfB synthesizes insoluble glucan (rich in α1-3 linkages), while GtfC synthesizes a mixture of insoluble and soluble (rich in α1-6 linkages) glucans, and GtfD synthesizes only soluble glucan (1). As expected, significant increases in C3b deposition were observed for the gtfBC and gtfBCD mutants compared to parent strain MT8148 under all culture conditions (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test). Supplementation of growth medium with sucrose did not significantly affect C3b deposition on these mutants (Fig. 6B). Therefore, sucrose-derived EPS impacts C3b deposition on the S. mutans surface. Of note, analyses of the effects of gtfBCD on C3b opsonization were performed with mutants previously obtained from strain MT8148, because gbpC inactivation affected C3b deposition on MT8148 in a fashion similar to that observed for UA159 (see below).
Because levels of sucrose in the bloodstream seem to be minimal (46), we assessed whether the capacity of S. mutans to bind sucrose-derived EPS influences C3b deposition. Thus, levels of C3b in the gbpC mutant (UAgbpC) were compared to those in parent strain UA159 previously grown in the presence or absence of sucrose. As shown in Fig. 6C, deletion of gbpC promoted a significant increase in C3b deposition compared to the parent strain, especially when strains were recovered from sucrose-containing medium: BHI medium containing 0.1% sucrose (mean 4.6-fold increase in the C3b MFI) and CDM containing 0.1% sucrose (mean 3.2-fold increase in the C3b MFI) (P < 0.05 as determined by a Kruskal-Wallis test with a post hoc test). These results are compatible with the inability of UAgbpC to bind EPS produced from sucrose (Fig. 3B and SEM analysis data not shown). Similar results were observed for the gbpC mutant obtained from strain MT8148 (data not shown). The influence of sucrose-derived products on C3b deposition was further analyzed in the epsC mutant. Previous growth of the epsC mutant in the presence of sucrose reduced C3b deposition (Fig. 6C), which is compatible with the finding that UAepsC retained some capacity to bind EPS (Fig. 3B). Thus, the expression of proteins that bind sucrose-derived EPS significantly affects S. mutans susceptibility to C3b deposition. Of note, wild-type strain MT8148 showed lower levels of binding to C3b than did the UA159 strain (Fig. 6A and B), consistent with its higher capacity to interact with sucrose-derived EPS than UA159 (Fig. 3C).
Finally, to confirm that the reduced susceptibility of blood isolates to C3b deposition was promoted by sucrose-derived EPS, we compared levels of C3b deposition on blood strains SA13 and SA18 grown in the four growth media. As a reference, oral strain 2VS1 (with reduced binding to sucrose-derived EPS) (Fig. 3A) was also tested. As expected, levels of C3b deposition on SA13 and SA18 were increased when the strains were grown in sucrose-free CDM (Fig. 6D). Levels of C3b deposition on 2VS1 were significantly higher than those observed for the blood strains under all growth conditions (P < 0.05 as determined by a Kruskal-Wallis test), but the addition of sucrose to media did not significantly affect C3b binding to this strain (Fig. 6D). Thus, the low susceptibility of blood strains to C3b deposition is influenced by sucrose-derived EPS in a fashion similar to that observed for the covR mutant.
Influence of sucrose-derived EPS on binding of serum IgG to the S. mutans surface and on frequencies of opsonophagocytosis and killing by human PMNs.
Because complement activation on S. mutans was found to be strongly dependent on the classical pathway, we investigated whether changes in C3b deposition in S. mutans promoted by previous growth in the presence of sucrose could be associated with reduced binding to serum IgG. As shown in Fig. 7A, levels of IgG bound to UA159 and UAcov were impaired when these strains were grown in medium with added sucrose. In addition, exposure to sucrose significantly reduced phagocytosis and killing by PMNs, especially in UAcov (Fig. 7B and C). Consistent with data from flow cytometry analyses of phagocytosis (Fig. 7B), mean counts of extracellular UAcov bacteria in the supernatants of PMNs analyzed in killing assays were 1.6- and 6.3-fold higher than those of extracellular UA159 bacteria when strains were respectively grown in CDM and CDM with 0.1% sucrose (data not shown). In addition, similarly to UAcov, the growth of blood strains SA13 and SA18 in medium supplemented with sucrose impaired the binding of serum IgG (Fig. 7D), reduced the frequency of phagocytosis (Fig. 7E), and increased resistance to killing by PMNs (Fig. 7F). In contrast, medium supplementation with sucrose promoted limited effects on IgG binding and phagocytosis in 2VS1 (Fig. 7D and E). These results support the role of surface-associated EPS in the evasion of opsonophagocytosis and killing by PMNs.
FIG 7.

Effects of previous growth of S. mutans strains in medium supplemented with sucrose on binding to serum IgG, susceptibility to phagocytosis by PMNs, and killing by PMNs. (A and D) Strains grown in BHI medium or CDM supplemented or not with 0.01 or 0.1% sucrose were harvested, washed with PBS, and treated with human serum for IgG binding. Levels of IgG binding were determined by flow cytometry and are expressed as MFI values. (B and E) FITC-labeled strains grown in different media were incubated with PMNs isolated from human peripheral blood in the presence of 20% serum during 5 min. (C and F) Strains grown in CDM supplemented or not with 0.1% sucrose were preopsonized and incubated with PMNs (10 min). Percentages of intracellular survival were calculated in relation to viable counts determined for the no-PMN control samples. Columns represent means of data from three independent experiments. Bars indicate standard deviations. Asterisks indicate significant differences in comparison to the parent strain (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
Binding of serum IgG to the S. mutans surface and rates of serum-mediated phagocytosis were also assessed in mutants of CovRSm-repressed genes involved in the synthesis of and/or interaction with EPS (Fig. 8). Significant increases in IgG binding to the S. mutans surface were promoted by the inactivation of epsC, gbpC (Fig. 8A), and gtfBCD (Fig. 8C). The inactivation of lysM and wapE had modest effects on IgG binding (Fig. 8A), compatible with the limited effects of these genes on binding to sucrose-derived EPS (Fig. 3B). Increases in phagocytosis were also observed for the epsC, gbpC, and gtfBCD mutants (Fig. 8B and D). As observed for C3b deposition, complementation of the gbpC and epsC mutants restored levels of IgG binding and phagocytosis (Fig. 8A and B). These findings strengthen the influence of S. mutans interactions with sucrose-derived EPS on strain susceptibilities to opsonic phagocytosis by human PMNs.
FIG 8.

Comparisons of binding to serum IgG (A and C) and of phagocytosis by human PMNs (B and D) between mutants of genes regulated by CovRSm. Mutant or complemented strains were compared with the respective parent strains (UA159 or MT8148). Columns represent means of data from three independent experiments; bars indicate standard deviations. Asterisks indicate significant differences in relation to the parent strain (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
Inactivation of covRSm and of the CovRSm-repressed genes gbpC and epsC affects survival of S. mutans in human blood and systemic virulence.
Because complement immunity is an important mechanism of blood clearance of streptococcal pathogens (28, 30, 47), we investigated the effects of the inactivation of covRSm and downstream genes on the ex vivo survival of S. mutans in human blood. The UAcov mutant showed an increased capacity to survive in human blood, which was completely restored in the UAcov+ complemented mutant (Fig. 9A). Consistent with the role of CovRSm as a direct repressor of gbpC and epsC, the UAgbpC and UAepsC mutants showed reduced survival in blood compared to the parent strain and the respective complemented mutants (Fig. 9B and C). To further confirm the effects of EPS on the increased ex vivo survival of UAcov in blood, assays were performed with strains grown in sucrose-free CDM and in CDM with 0.1% sucrose. As shown in Fig. 9D, differences in survival in blood between UAcov and UA159 were eliminated when strains were grown in CDM, whereas CDM supplementation with sucrose increased differences between UAcov and parent strains (Fig. 9E). Thus, sucrose-derived EPS are involved in the increased survival of UAcov in human blood.
FIG 9.

Ex vivo viability in human blood. Numbers of viable bacteria (log CFU per milliliter) were expressed in relation to initial counts in blood suspensions (time zero). Strains were grown in BHI medium (A to C), CDM (D), or CDM supplemented with 0.1% sucrose (E). Data represent means of results from triplicates of one representative experiment. Bars indicate standard deviations. Differences in relation to the parent strain at each time point were tested by a Kruskal-Wallis test with Dunn's post hoc test (*, P < 0.05).
Comparisons of bacterial counts in the bloodstream of rats confirmed the findings of ex vivo survival in human blood for UAcov. As shown in Table 3, the UAcov mutant survives for longer periods and at higher counts in the rat bloodstream than the parent or complemented strains. Two rats infected with UAcov died during the experiment, while no deaths occurred in the UA159-infected group. Higher S. mutans counts were found in valves of UAcov-infected rats (mean, 20,090 ± 44,467 CFU/ml; median, 40 CFU/ml) than in valves of UA159-infected animals (mean, 1,352 ± 2,723 CFU/ml; median, 0 CFU/ml), although differences between strains did not reach statistical significance (P > 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
TABLE 3.
Bacterial counts in blood of rats (n = 7)
| Infecting strain | Mean CFU/ml of blood ± SD (no. of rats with bacteria recovered)a |
||||
|---|---|---|---|---|---|
| 1 h | 3 h | 6 h | 24 h | 7 days | |
| UA159 | 1,627 ± 1,029 (7) | 247 ± 135 (7) | 19 ± 19 (4) | 0 (0) | 0 (0) |
| UAcov | 1,767 ± 1,053 (7) | 194 ± 147 (7) | 119 ± 98* (6) | 0 (0) | 16 ± 36* (5) |
| UAcov+ | 2,501 ± 2,309 (7) | 104 ± 64* (7) | 16 ± 15 (4) | 3 ± 5 (2) | 0 (0) |
Asterisks indicate significant differences in relation to the parent strain at the same time period (P < 0.05 as determined by a Kruskal-Wallis test with Dunn's post hoc test).
DISCUSSION
The complement system plays multiple roles in the elimination of microorganisms, both as part of the innate immune system and by augmenting antibody-mediated immunity (17, 18). The reduced susceptibilities to C3b deposition found in S. mutans blood strains (Fig. 1) indicate that evasion of complement immunity is important for the systemic virulence of S. mutans. In GAS, natural mutations in covRSpy were detected in strains involved in human infections, and inactivation of the CovRSSpy TCS enhanced strain virulence in murine models (48–50). Virulence genes repressed by CovRSpy include genes involved in complement evasion (e.g., has operon for hyaluronic acid capsule synthesis) (50–52), which are not present in S. mutans genomes (53, 54). Reduced transcript levels of covRSm in blood isolates associated with increased transcription of CovRSm-repressed genes (gbpC, gbpB, and epsC) suggest that the diversity in covRSm activities influences the capacities of S. mutans strains to survive in the bloodstream.
In GAS and GBS, the CovRSSpy/S. agalactiae CovRS (CovRSSag) regulons show strain specificity (55, 56). The CovRSm regulon was assessed in serotype c strain UA159 (8, 11), but its diversity remains to be investigated in strains associated with systemic infections. S. mutans serotype c was detected with higher frequencies in S. mutans-positive specimens of heart valves and atheromatous plaques from patients subjected to cardiovascular surgeries (30.3 and 65.5% of specimens, respectively) than serotype k (detected in 9.1 and 25% of these specimens, respectively) (14), which was previously implicated in systemic infections (57). Interestingly, 77% of serotype k-positive specimens were also positive for serotype c (41), suggesting synergy of S. mutans serotypes for systemic virulence. The systemic virulence of serotype k is associated with the expression of the collagen-binding proteins Cnm and Cbm, which are involved in the capacity of S. mutans to invade endothelial cells in vitro (40, 58–60) and to form vegetations on injured heart valves in a rat model of infective endocarditis (40). However, serotype c strains rarely harbor these genes (22), and there is no report that CovRSm regulates cnm or cbm.
A major function of complement immunity against Gram-positive bacteria is to covalently bind C3b/iC3b opsonins on the bacterial surface through the activity of C3 convertases on C3 (17, 18). C3 convertases result from proteolytic cascades initiated by different mechanisms, known as the classical, mannan-binding lectin, and alternative pathways (17). Functions of each pathway in complement immunity against streptococci seem to be species specific (27, 28, 30, 45) and are usually circumvented by multiple evasion mechanisms (41, 61, 62). Here, we show that the classical pathway plays a major role in complement deposition on S. mutans (Fig. 5C), which is consistent with the reduced binding of IgG antibodies to UAcov (Fig. 5A). Because C1q is activated through its binding to IgG on the bacterial surface, assessing individual roles of complement and IgG in S. mutans opsonization is difficult. In addition, although S. mutans seems to not have prototypical CRP ligands (63), covRSm inactivation also reduced binding to CRP (Fig. 5B). CRP levels are increased in the bloodstream of subjects with biofilm-dependent oral diseases, e.g., gingivitis and periodontitis (64); thus, the role of acute-phase proteins in S. mutans blood clearance needs to be investigated.
The major known role of CovRSm in S. mutans biology is to regulate the expression of secreted enzymes for the synthesis of EPS from sucrose and cell surface components involved in interactions with EPS (5, 6, 10, 11). Some of these genes, e.g., gtfBC, ftf, and wapE, are controlled by a complex regulatory circuit (11, 65–67), which might explain the lack of associations between gtfBC transcription and profiles of covRSm expression among the tested strains (Fig. 2). Because the secreted GtfBC enzymes are stable in saliva and bind to several oral bacteria (2), strains with increased binding to EPS could benefit from Gtf-producing members of the same ecological niche. Thus, an increased capacity to bind EPS would be more significant for systemic virulence than the ability to produce Gtfs itself. In serotype c strain V403, the deletion of multiple genes required for the synthesis of EPS from sucrose (gtfB, gtfC, and ftf) increased S. mutans phagocytosis by human granulocytes and reduced virulence in an animal model of infectious endocarditis (68). Consistently, our gtfBC and gtfBCD mutants showed high susceptibility to C3b opsonization even when grown in the presence of sucrose (Fig. 6). However, the production of sucrose-derived EPS impaired C3b deposition only in strains that were able to bind these polymers; the gbpC mutant was susceptible to C3b deposition even when grown in the presence of sucrose (Fig. 6). Thus, the expression of gbpC, and perhaps other glucan-binding proteins upregulated in blood isolates, e.g., GbpB (Fig. 2), might be critical for EPS-mediated complement evasion.
The increased binding of the gbpC mutant to serum IgG (Fig. 6) further indicates that surface EPS may impair antibody-mediated activation of complement in a way analogous to that of capsules of S. pneumoniae (45, 47, 69). Besides GbpC, EpsC showed a prominent influence on complement opsonization (Fig. 6 and 8). In Gram-positive bacteria, EpsC is required for the production of UDP-ManNAc, an intermediate for the synthesis of EPS which is also required for the attachment of teichoic acids to the cell wall (70–73). The epsC mutant retained some degree of binding to sucrose-derived EPS (Fig. 3B), which might explain, at least in part, the remaining influence of sucrose on the binding of C3b to this mutant (Fig. 6). Alternatively, EpsC could also affect sucrose-independent mechanisms of S. mutans evasion of complement immunity. Functional analyses of EpsC might shed new light on its roles in complement susceptibility.
Although the effects of the covR deletion on C3b opsonization were more clearly observed when strains were grown in sucrose-containing media, reductions in levels of C3b binding to UAcov were still observed in sucrose-free CDM (Fig. 6), which suggests that CovRSm regulates additional functions of complement evasion. The lower levels of C3b binding to blood strains grown in sucrose-free CDM (especially in SA13) (Fig. 6) account for the hypothesis that S. mutans strains apply multiple mechanisms of complement evasion. In GAS strains, CovRSpy plays multiple roles in complement evasion besides regulating capsule production (50, 52, 74). Studies are under way to identify additional factors affecting S. mutans susceptibility to complement immunity.
The increased persistence of UAcov in human blood mediated by sucrose-derived EPS (Fig. 9) and its ability to cause bacteremia in rats (Table 3) further strengthen the role of CovRSm in systemic virulence. Reduced C3b/IgG opsonization of UAcov is, at least in part, explained by the upregulation of epsC and gbpC, because the inactivation of these genes reduced survival in human blood (Fig. 9A and B). Different from this study, no reduction in survival in the bloodstream of rats was observed for a gbpC mutant (C1) obtained from MT8148 compared to a streptomycin-resistant MT8148 variant (MT8148R) (75). Although we found that C1 has an increased susceptibility to C3b deposition compared to the MT8148 parent strain (data not shown), C3b deposition in the MT8148R variant is unknown. As shown in this study, differences in growth media can affect S. mutans susceptibility to complement opsonization. Furthermore, bacterial aggregation mediated by blood components could affect bacterial counts in blood suspensions. UAcov shows increased aggregation in blood compared to UA159 (data not shown), which could explain the initial reductions in UAcov counts in the ex vivo assays, although bacterial loads were normalized by initial blood counts (Fig. 9). Increased aggregation of UAcov could occur in the rat bloodstream; thus, survival rates of UAcov (Table 3) might be underestimated.
There may also be differences between human blood and rat blood in complement activation on S. mutans. S. mutans is an exclusive species of humans; thus, levels and epitope specificities of IgG antibodies to S. mutans may differ in human and rat sera. There are further differences in the production and structure of CRP between rats and humans (76). In addition, CR1, shown to be important for S. mutans opsonophagocytosis, is also involved in blood clearance by human erythrocytes through immune adherence (77). Because rodent erythrocytes do not express CR1 (77), a more complete analysis of the influence of C3b deposition on blood clearance of S. mutans would require animal models designed to assess CR1-mediated immune adherence (78). Apart from the limitations of our model, significant increases in viable counts of UAcov bacteria in the rat bloodstream compared to those of UA159 bacteria were detected (Table 3). At 6 h postinfection, the counts of UAcov mutant bacteria were 6.3-fold higher (detected in 85.7% of the animals) than those of the parent strain (detected in 57.1% of animals). Although UAcov counts in heart valves were higher than UA159 counts, these differences did not reach significance. Because only the numbers of viable bacteria were assessed, we cannot exclude the possibility that increased differences in tissue infection between strains might have been observed if total levels of bacteria in heart valve specimens were measured by using culture-independent methods. In addition, survival of UAcov in the rat bloodstream would likely increase if strains were previously grown in medium with 0.1% sucrose added. Therefore, studies are required to improve in vivo models for assessing the influence of complement evasion on the systemic virulence of S. mutans.
In summary, this study provides evidence that systemic virulence of S. mutans strains involves reduced susceptibility to complement-mediated opsonization. Roles of CovRSm in resistance to complement immunity involves regulation of the capacity of S. mutans to interact with EPS, which in turn affects complement activation. Two CovRSm-repressed genes, gbpC and epsC, were identified as playing important roles in resistance to complement immunity and survival in blood, as revealed by transcriptional profiles of these genes in isolates from systemic infections and by molecular analyses of isogenic mutants.
ACKNOWLEDGMENTS
We thank Satu Alaluusua for providing the blood isolates analyzed in this study. We thank Daniel J. Smith for critical reading of the manuscript.
This study was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) (grant numbers 2012/50966-6 and 2015/12940-3). L.A.A. was supported by FAPESP (fellowship numbers 2012/04222-5 and 2015/07237-1). E.N.H.-C. was supported by FAPESP (fellowship number 2009/50547-0) and CAPES-PNPD (2013). This work was also supported by KAKENHI grant number 15K11363 from the Japan Society for the Promotion of Science.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Banas JA, Vickerman MM. 2003. Glucan-binding proteins of the oral streptococci. Crit Rev Oral Biol Med 14:89–99. doi: 10.1177/154411130301400203. [DOI] [PubMed] [Google Scholar]
- 2.Bowen WH, Koo H. 2011. Biology of Streptococcus mutans-derived glucosyltransferases: role in extracellular matrix formation of cariogenic biofilms. Caries Res 45:69–86. doi: 10.1159/000324598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakano K, Ooshima T. 2009. Serotype classification of Streptococcus mutans and its detection outside the oral cavity. Future Microbiol 4:891–902. doi: 10.2217/fmb.09.64. [DOI] [PubMed] [Google Scholar]
- 4.Nakano K, Nemoto H, Nomura R, Inaba H, Yoshioka H, Taniguchi K, Amano A, Ooshima T. 2009. Detection of oral bacteria in cardiovascular specimens. Oral Microbiol Immunol 24:64–68. doi: 10.1111/j.1399-302X.2008.00479.x. [DOI] [PubMed] [Google Scholar]
- 5.Sato Y, Yamamoto Y, Kizaki H. 2000. Construction of region-specific partial duplication mutants (merodiploid mutants) to identify the regulatory gene for the glucan-binding protein C gene in vivo in Streptococcus mutans. FEMS Microbiol Lett 186:187–191. doi: 10.1111/j.1574-6968.2000.tb09102.x. [DOI] [PubMed] [Google Scholar]
- 6.Idone V, Brendtro S, Gillespie R, Kocaj S, Peterson E, Rendi M, Warren W, Michalek S, Krastel K, Cvitkovitch D, Spatafora G. 2003. Effect of an orphan response regulator on Streptococcus mutans sucrose-dependent adherence and cariogenesis. Infect Immun 71:4351–4360. doi: 10.1128/IAI.71.8.4351-4360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biswas S, Biswas I. 2006. Regulation of the glucosyltransferase (gtfBC) operon by CovR in Streptococcus mutans. J Bacteriol 188:988–998. doi: 10.1128/JB.188.3.988-998.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dmitriev A, Mohapatra SS, Chong P, Neely M, Biswas S, Biswas I. 2011. CovR-controlled global regulation of gene expression in Streptococcus mutans. PLoS One 6:e20127. doi: 10.1371/journal.pone.0020127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Churchward G. 2007. The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol 64:34–41. doi: 10.1111/j.1365-2958.2007.05649.x. [DOI] [PubMed] [Google Scholar]
- 10.Biswas I, Drake L, Biswas S. 2007. Regulation of gbpC expression in Streptococcus mutans. J Bacteriol 189:6521–6531. doi: 10.1128/JB.00825-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stipp RN, Boisvert H, Smith DJ, Hofling JF, Duncan MJ, Mattos-Graner RO. 2013. CovR and VicRK regulate cell surface biogenesis genes required for biofilm formation in Streptococcus mutans. PLoS One 8:e58271. doi: 10.1371/journal.pone.0058271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duque C, Stipp RN, Wang B, Smith DJ, Hofling JF, Kuramitsu HK, Duncan MJ, Mattos-Graner RO. 2011. Downregulation of GbpB, a component of the VicRK regulon, affects biofilm formation and cell surface characteristics of Streptococcus mutans. Infect Immun 79:786–796. doi: 10.1128/IAI.00725-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Negrini TC, Duque C, Vizoto NL, Stipp RN, Mariano FS, Hofling JF, Graner E, Mattos-Graner RO. 2012. Influence of VicRK and CovR on the interactions of Streptococcus mutans with phagocytes. Oral Dis 18:485–493. doi: 10.1111/j.1601-0825.2011.01896.x. [DOI] [PubMed] [Google Scholar]
- 14.Nakano K, Nemoto H, Nomura R, Homma H, Yoshioka H, Shudo Y, Hata H, Toda K, Taniguchi K, Amano A, Ooshima T. 2007. Serotype distribution of Streptococcus mutans a pathogen of dental caries in cardiovascular specimens from Japanese patients. J Med Microbiol 56:551–556. doi: 10.1099/jmm.0.47051-0. [DOI] [PubMed] [Google Scholar]
- 15.Nakano K, Nomura R, Matsumoto M, Ooshima T. 2010. Roles of oral bacteria in cardiovascular diseases—from molecular mechanisms to clinical cases: cell-surface structures of novel serotype k Streptococcus mutans strains and their correlation to virulence. J Pharmacol Sci 113:120–125. doi: 10.1254/jphs.09R24FM. [DOI] [PubMed] [Google Scholar]
- 16.Nakano K, Tsuji M, Nishimura K, Nomura R, Ooshima T. 2006. Contribution of cell surface protein antigen PAc of Streptococcus mutans to bacteremia. Microbes Infect 8:114–121. doi: 10.1016/j.micinf.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Dunkelberger JR, Song WC. 2010. Complement and its role in innate and adaptive immune responses. Cell Res 20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 18.Walport MJ. 2001. Complement. First of two parts. N Engl J Med 344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 19.Nakano K, Matsumura M, Kawaguchi M, Fujiwara T, Sobue S, Nakagawa I, Hamada S, Ooshima T. 2002. Attenuation of glucan-binding protein C reduces the cariogenicity of Streptococcus mutans: analysis of strains isolated from human blood. J Dent Res 81:376–379. doi: 10.1177/154405910208100604. [DOI] [PubMed] [Google Scholar]
- 20.Ooshima T, Matsumura M, Hoshino T, Kawabata S, Sobue S, Fujiwara T. 2001. Contributions of three glycosyltransferases to sucrose-dependent adherence of Streptococcus mutans. J Dent Res 80:1672–1677. doi: 10.1177/00220345010800071401. [DOI] [PubMed] [Google Scholar]
- 21.Mattos-Graner RO, Smith DJ, King WF, Mayer MP. 2000. Water-insoluble glucan synthesis by mutans streptococcal strains correlates with caries incidence in 12- to 30-month-old children. J Dent Res 79:1371–1377. doi: 10.1177/00220345000790060401. [DOI] [PubMed] [Google Scholar]
- 22.Nakano K, Lapirattanakul J, Nomura R, Nemoto H, Alaluusua S, Gronroos L, Vaara M, Hamada S, Ooshima T, Nakagawa I. 2007. Streptococcus mutans clonal variation revealed by multilocus sequence typing. J Clin Microbiol 45:2616–2625. doi: 10.1128/JCM.02343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lau PC, Sung CK, Lee JH, Morrison DA, Cvitkovitch DG. 2002. PCR ligation mutagenesis in transformable streptococci: application and efficiency. J Microbiol Methods 49:193–205. doi: 10.1016/S0167-7012(01)00369-4. [DOI] [PubMed] [Google Scholar]
- 24.Stipp RN, Goncalves RB, Hofling JF, Smith DJ, Mattos-Graner RO. 2008. Transcriptional analysis of gtfB, gtfC, and gbpB and their putative response regulators in several isolates of Streptococcus mutans. Oral Microbiol Immunol 23:466–473. doi: 10.1111/j.1399-302X.2008.00451.x. [DOI] [PubMed] [Google Scholar]
- 25.Palmer SR, Miller JH, Abranches J, Zeng L, Lefebure T, Richards VP, Lemos JA, Stanhope MJ, Burne RA. 2013. Phenotypic heterogeneity of genomically-diverse isolates of Streptococcus mutans. PLoS One 8:e61358. doi: 10.1371/journal.pone.0061358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O. 1998. Reference distributions for immunoglobulins A, G, and M: a comparison of a large cohort to the world's literature. J Clin Lab Anal 12:371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuste J, Sen A, Truedsson L, Jonsson G, Tay LS, Hyams C, Baxendale HE, Goldblatt F, Botto M, Brown JS. 2008. Impaired opsonization with C3b and phagocytosis of Streptococcus pneumoniae in sera from subjects with defects in the classical complement pathway. Infect Immun 76:3761–3770. doi: 10.1128/IAI.00291-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown JS, Hussell T, Gilliland SM, Holden DW, Paton JC, Ehrenstein MR, Walport MJ, Botto M. 2002. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc Natl Acad Sci U S A 99:16969–16974. doi: 10.1073/pnas.012669199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Exley RM, Shaw J, Mowe E, Sun YH, West NP, Williamson M, Botto M, Smith H, Tang CM. 2005. Available carbon source influences the resistance of Neisseria meningitidis against complement. J Exp Med 201:1637–1645. doi: 10.1084/jem.20041548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuste J, Ali S, Sriskandan S, Hyams C, Botto M, Brown JS. 2006. Roles of the alternative complement pathway and C1q during innate immunity to Streptococcus pyogenes. J Immunol 176:6112–6120. doi: 10.4049/jimmunol.176.10.6112. [DOI] [PubMed] [Google Scholar]
- 31.Baudhuin J, Migraine J, Faivre V, Loumagne L, Lukaszewicz AC, Payen D, Favier B. 2013. Exocytosis acts as a modulator of the ILT4-mediated inhibition of neutrophil functions. Proc Natl Acad Sci U S A 110:17957–17962. doi: 10.1073/pnas.1221535110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Falk P, Roth KA, Boren T, Westblom TU, Gordon JI, Normark S. 1993. An in vitro adherence assay reveals that Helicobacter pylori exhibits cell lineage-specific tropism in the human gastric epithelium. Proc Natl Acad Sci U S A 90:2035–2039. doi: 10.1073/pnas.90.5.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clatworthy MR, Smith KG. 2004. FcgammaRIIb balances efficient pathogen clearance and the cytokine-mediated consequences of sepsis. J Exp Med 199:717–723. doi: 10.1084/jem.20032197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weineisen M, Sjobring U, Fallman M, Andersson T. 2004. Streptococcal M5 protein prevents neutrophil phagocytosis by interfering with CD11b/CD18 receptor-mediated association and signaling. J Immunol 172:3798–3807. doi: 10.4049/jimmunol.172.6.3798. [DOI] [PubMed] [Google Scholar]
- 35.Schwartz JT, Barker JH, Long ME, Kaufman J, McCracken J, Allen LA. 2012. Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via complement receptors 1 and 3 in nonimmune serum. J Immunol 189:3064–3077. doi: 10.4049/jimmunol.1200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis KM, Akinbi HT, Standish AJ, Weiser JN. 2008. Resistance to mucosal lysozyme compensates for the fitness deficit of peptidoglycan modifications by Streptococcus pneumoniae. PLoS Pathog 4:e1000241. doi: 10.1371/journal.ppat.1000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuste J, Sen A, Truedsson L, Jonsson G, Hyams C, Cohen JM, Camberlein E, Sriskandan S, Brown JS. 2010. Impaired opsonization with complement and phagocytosis of Streptococcus pyogenes in sera from subjects with inherited C2 deficiency. Microbes Infect 12:626–634. doi: 10.1016/j.micinf.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Domenech M, Ramos-Sevillano E, Garcia E, Moscoso M, Yuste J. 2013. Biofilm formation avoids complement immunity and phagocytosis of Streptococcus pneumoniae. Infect Immun 81:2606–2615. doi: 10.1128/IAI.00491-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hakenbeck R, Madhour A, Denapaite D, Bruckner R. 2009. Versatility of choline metabolism and choline-binding proteins in Streptococcus pneumoniae and commensal streptococci. FEMS Microbiol Rev 33:572–586. doi: 10.1111/j.1574-6976.2009.00172.x. [DOI] [PubMed] [Google Scholar]
- 40.Nomura R, Otsugu M, Naka S, Teramoto N, Kojima A, Muranaka Y, Matsumoto-Nakano M, Ooshima T, Nakano K. 2014. Contribution of the interaction of Streptococcus mutans serotype k strains with fibrinogen to the pathogenicity of infective endocarditis. Infect Immun 82:5223–5234. doi: 10.1128/IAI.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lambris JD, Ricklin D, Geisbrecht BV. 2008. Complement evasion by human pathogens. Nat Rev Microbiol 6:132–142. doi: 10.1038/nrmicro1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biswas I, Drake L, Erkina D, Biswas S. 2008. Involvement of sensor kinases in the stress tolerance response of Streptococcus mutans. J Bacteriol 190:68–77. doi: 10.1128/JB.00990-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato Y, Senpuku H, Okamoto K, Hanada N, Kizaki H. 2002. Streptococcus mutans binding to solid phase dextran mediated by the glucan-binding protein C. Oral Microbiol Immunol 17:252–256. doi: 10.1034/j.1399-302X.2002.170408.x. [DOI] [PubMed] [Google Scholar]
- 44.Underhill DM, Ozinsky A. 2002. Phagocytosis of microbes: complexity in action. Annu Rev Immunol 20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 45.Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. 2010. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun 78:704–715. doi: 10.1128/IAI.00881-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sutherland LR, Verhoef M, Wallace JL, Van Rosendaal G, Crutcher R, Meddings JB. 1994. A simple, non-invasive marker of gastric damage: sucrose permeability. Lancet 343:998–1000. doi: 10.1016/S0140-6736(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 47.Hyams C, Trzcinski K, Camberlein E, Weinberger DM, Chimalapati S, Noursadeghi M, Lipsitch M, Brown JS. 2013. Streptococcus pneumoniae capsular serotype invasiveness correlates with the degree of factor H binding and opsonization with C3b/iC3b. Infect Immun 81:354–363. doi: 10.1128/IAI.00862-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Engleberg NC, Heath A, Miller A, Rivera C, DiRita VJ. 2001. Spontaneous mutations in the CsrRS two-component regulatory system of Streptococcus pyogenes result in enhanced virulence in a murine model of skin and soft tissue infection. J Infect Dis 183:1043–1054. doi: 10.1086/319291. [DOI] [PubMed] [Google Scholar]
- 49.Levin JC, Wessels MR. 1998. Identification of csrR/csrS, a genetic locus that regulates hyaluronic acid capsule synthesis in group A Streptococcus. Mol Microbiol 30:209–219. [DOI] [PubMed] [Google Scholar]
- 50.Graham MR, Virtaneva K, Porcella SF, Barry WT, Gowen BB, Johnson CR, Wright FA, Musser JM. 2005. Group A Streptococcus transcriptome dynamics during growth in human blood reveals bacterial adaptive and survival strategies. Am J Pathol 166:455–465. doi: 10.1016/S0002-9440(10)62268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heath A, DiRita VJ, Barg NL, Engleberg NC. 1999. A two-component regulatory system, CsrR-CsrS, represses expression of three Streptococcus pyogenes virulence factors, hyaluronic acid capsule, streptolysin S, and pyrogenic exotoxin B. Infect Immun 67:5298–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agrahari G, Liang Z, Glinton K, Lee SW, Ploplis VA, Castellino FJ. 2016. Streptococcus pyogenes employs strain-dependent mechanisms of C3b inactivation to inhibit phagocytosis and killing of bacteria. J Biol Chem 291:9181–9189. doi: 10.1074/jbc.M115.704221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ajdic D, McShan WM, McLaughlin RE, Savic G, Chang J, Carson MB, Primeaux C, Tian R, Kenton S, Jia H, Lin S, Qian Y, Li S, Zhu H, Najar F, Lai H, White J, Roe BA, Ferretti JJ. 2002. Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc Natl Acad Sci U S A 99:14434–14439. doi: 10.1073/pnas.172501299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cornejo OE, Lefebure T, Bitar PD, Lang P, Richards VP, Eilertson K, Do T, Beighton D, Zeng L, Ahn SJ, Burne RA, Siepel A, Bustamante CD, Stanhope MJ. 2013. Evolutionary and population genomics of the cavity causing bacteria Streptococcus mutans. Mol Biol Evol 30:881–893. doi: 10.1093/molbev/mss278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sumby P, Whitney AR, Graviss EA, Deleo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang SM, Ishmael N, Dunning HJ, Puliti M, Tissi L, Kumar N, Cieslewicz MJ, Tettelin H, Wessels MR. 2008. Variation in the group B Streptococcus CsrRS regulon and effects on pathogenicity. J Bacteriol 190:1956–1965. doi: 10.1128/JB.01677-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakano K, Nomura R, Nakagawa I, Hamada S, Ooshima T. 2004. Demonstration of Streptococcus mutans with a cell wall polysaccharide specific to a new serotype, k, in the human oral cavity. J Clin Microbiol 42:198–202. doi: 10.1128/JCM.42.1.198-202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abranches J, Miller JH, Martinez AR, Simpson-Haidaris PJ, Burne RA, Lemos JA. 2011. The collagen-binding protein Cnm is required for Streptococcus mutans adherence to and intracellular invasion of human coronary artery endothelial cells. Infect Immun 79:2277–2284. doi: 10.1128/IAI.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nomura R, Nakano K, Taniguchi N, Lapirattanakul J, Nemoto H, Gronroos L, Alaluusua S, Ooshima T. 2009. Molecular and clinical analyses of the gene encoding the collagen-binding adhesin of Streptococcus mutans. J Med Microbiol 58:469–475. doi: 10.1099/jmm.0.007559-0. [DOI] [PubMed] [Google Scholar]
- 60.Nomura R, Nakano K, Naka S, Nemoto H, Masuda K, Lapirattanakul J, Alaluusua S, Matsumoto M, Kawabata S, Ooshima T. 2012. Identification and characterization of a collagen-binding protein, Cbm, in Streptococcus mutans. Mol Oral Microbiol 27:308–323. doi: 10.1111/j.2041-1014.2012.00649.x. [DOI] [PubMed] [Google Scholar]
- 61.Blom AM, Hallstrom T, Riesbeck K. 2009. Complement evasion strategies of pathogens—acquisition of inhibitors and beyond. Mol Immunol 46:2808–2817. doi: 10.1016/j.molimm.2009.04.025. [DOI] [PubMed] [Google Scholar]
- 62.Zipfel PF, Hallstrom T, Riesbeck K. 2013. Human complement control and complement evasion by pathogenic microbes—tipping the balance. Mol Immunol 56:152–160. doi: 10.1016/j.molimm.2013.05.222. [DOI] [PubMed] [Google Scholar]
- 63.Gillespie SH, McWhinney PH, Patel S, Raynes JG, McAdam KP, Whiley RA, Hardie JM. 1993. Species of alpha-hemolytic streptococci possessing a C-polysaccharide phosphorylcholine-containing antigen. Infect Immun 61:3076–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu T, Trevisan M, Genco RJ, Falkner KL, Dorn JP, Sempos CT. 2000. Examination of the relation between periodontal health status and cardiovascular risk factors: serum total and high density lipoprotein cholesterol, C-reactive protein, and plasma fibrinogen. Am J Epidemiol 151:273–282. doi: 10.1093/oxfordjournals.aje.a010203. [DOI] [PubMed] [Google Scholar]
- 65.Senadheera MD, Guggenheim B, Spatafora GA, Huang YC, Choi J, Hung DC, Treglown JS, Goodman SD, Ellen RP, Cvitkovitch DG. 2005. A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB, and ftf expression, biofilm formation, and genetic competence development. J Bacteriol 187:4064–4076. doi: 10.1128/JB.187.12.4064-4076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Browngardt CM, Wen ZT, Burne RA. 2004. RegM is required for optimal fructosyltransferase and glucosyltransferase gene expression in Streptococcus mutans. FEMS Microbiol Lett 240:75–79. doi: 10.1016/j.femsle.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida A, Ansai T, Takehara T, Kuramitsu HK. 2005. LuxS-based signaling affects Streptococcus mutans biofilm formation. Appl Environ Microbiol 71:2372–2380. doi: 10.1128/AEM.71.5.2372-2380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Munro CL, Macrina FL. 1993. Sucrose-derived exopolysaccharides of Streptococcus mutans V403 contribute to infectivity in endocarditis. Mol Microbiol 8:133–142. [DOI] [PubMed] [Google Scholar]
- 69.Yother J. 2011. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol 65:563–581. doi: 10.1146/annurev.micro.62.081307.162944. [DOI] [PubMed] [Google Scholar]
- 70.Campbell RE, Mosimann SC, Tanner ME, Strynadka NC. 2000. The structure of UDP-N-acetylglucosamine 2-epimerase reveals homology to phosphoglycosyl transferases. Biochemistry 39:14993–15001. doi: 10.1021/bi001627x. [DOI] [PubMed] [Google Scholar]
- 71.Kiser KB, Bhasin N, Deng L, Lee JC. 1999. Staphylococcus aureus cap5P encodes a UDP-N-acetylglucosamine 2-epimerase with functional redundancy. J Bacteriol 181:4818–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McLoon AL, Guttenplan SB, Kearns DB, Kolter R, Losick R. 2011. Tracing the domestication of a biofilm-forming bacterium. J Bacteriol 193:2027–2034. doi: 10.1128/JB.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Velloso LM, Bhaskaran SS, Schuch R, Fischetti VA, Stebbins CE. 2008. A structural basis for the allosteric regulation of non-hydrolysing UDP-GlcNAc 2-epimerases. EMBO Rep 9:199–205. doi: 10.1038/sj.embor.7401154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Agrahari G, Liang Z, Mayfield JA, Balsara RD, Ploplis VA, Castellino FJ. 2013. Complement-mediated opsonization of invasive group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J Biol Chem 288:27494–27504. doi: 10.1074/jbc.M113.494864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nomura R, Nakano K, Ooshima T. 2004. Contribution of glucan-binding protein C of Streptococcus mutans to bacteremia occurrence. Arch Oral Biol 49:783–788. doi: 10.1016/j.archoralbio.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 76.de Beer FC, Baltz ML, Munn EA, Feinstein A, Taylor J, Bruton C, Clamp JR, Pepys MB. 1982. Isolation and characterization of C-reactive protein and serum amyloid P component in the rat. Immunology 45:55–70. [PMC free article] [PubMed] [Google Scholar]
- 77.Jacobson AC, Weis JH. 2008. Comparative functional evolution of human and mouse CR1 and CR2. J Immunol 181:2953–2959. doi: 10.4049/jimmunol.181.5.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li J, Glover DT, Szalai AJ, Hollingshead SK, Briles DE. 2007. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect Immun 75:5877–5885. doi: 10.1128/IAI.00839-07. [DOI] [PMC free article] [PubMed] [Google Scholar]