

Abstract
Centella asiatica (雷公根 léi gōng gēn) is a traditional medicinal herb with high antioxidant activity, which decreases amyloid-β (Aβ) deposition in the brain. At the same time, aggregated Aβ-induced oxidative stress is the trigger in the pathogenesis of Alzheimer's disease (AD). Here, we investigated the ability of C. asiatica ethanol extract (CAE) to protect PC12 and IMR32 cells from Aβ1–40-induced production of reactive oxygen species (ROS) and concomitant neurotoxicity. Aggregated Aβ1–40 treatment resulted in reduced cell viability, which can be reversed by cotreatment with 25, 50, and 100 μg/mL CAE. Moreover, CAE eliminated the Aβ1–40-mediated increase in ROS production. Thus, CAE-mediated protection against aggregated Aβ1–40-induced neurotoxicity is attributable to modulation of the antioxidative defense system in cells, including the activities of superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, and levels of glutathione and glutathione disulfide by CAE. This emphasizes the potential therapeutic and preventive value of CAE in the treatment of AD.
Keywords: Alzheimer's disease, Amyloid β, Centella asiatica, Antioxidative defence system, Oxidative stress, Reactive oxygen species
Graphical abstract
1. Introduction
Amyloid β (Aβ) peptides have been proposed as the pathognomonic indicators of the pathogenesis of Alzheimer's disease (AD), the most prevalent neurodegenerative disease worldwide, which is characterized by impaired memory and loss of neurons in the central nervous system.1, 2 Aβ peptide, a byproduct of the degradation process of amyloid precursor protein (APP), consists of two major peptides of varying lengths: Aβ1–40 and Aβ1–42. Plasma concentrations of Aβ1–40 and Aβ1–42 increase with age and are elevated in individuals with mutations that cause early-onset AD. Both peptides are important components of plaques in AD and have been proposed to induce neuronal death and neurotoxicity in both in vivo and in vitro studies.3, 4, 5 A primary Aβ1–40 mechanism of action is the induction of excess reactive oxygen species (ROS), including superoxide, hydrogen peroxide (H2O2), and singlet oxygen. This increase in intracellular ROS causes oxidative stress. Moreover, a combination of Aβ-mediated ROS induction and excessive Ca2+ influx has been reported to lead to neuronal loss and cellular apoptosis.6 Given the importance of ROS-related mechanisms in AD, several studies have used antioxidants, such as vitamin E7 and Ginkgo biloba (銀杏 yín xìng) extract,8 or have enhanced the activities of enzymes in the antioxidative defence system, such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPx), and glutathione reductase (GR)9 to protect neuronal cells from the Aβ-induced ROS. These studies have shown that use of antioxidants and activation of the antioxidative defence system could suppress the neurotoxicity of Aβ in in vivo and in vitro models. Therefore, agents capable of attenuating oxidative stress may contribute to a superior therapeutic strategy for the treatment of Aβ-induced neurotoxicity and may lead to improved neurological outcomes in AD.
Centella asiatica (雷公根 léi gōng gēn), a member of the family Apiaceae (Umbelliferae), has been used as a traditional medicinal herb in Asia for over 2000 years. A number of medicinal functions and biological activities have been found in C. asiatica, both in the whole plant and its extract, including the ethanolic and aqueous extracts. The major active components of C. asiatica ethanol extract (CAE) are the triterpenoids, including asiatic acid and asiaticoside.10 CAE is considered to possess excellent antioxidant capabilities for scavenging 2,2-diphenyl-1-picrylhydrazyl (DPPH), reducing Fe3+,11 and activating the antioxidative defence system in the brain.12 Feeding aged rats CAE for 60 consecutive days delayed the aging process by improving oxidative status and reducing lipid peroxidation in the rat brain.12 Additionally, CAE has been used in the treatment of neurodegenerative diseases, such as AD. CAE could enhance the capability of rats in performing several memory tasks, including the Morris water maze and the passive avoidance test.13 The use of CAE in an AD transgenic mouse model was reported to reduce the deposition of Aβ in the hippocampus and improve the behavioral symptoms of mice.14, 15 However, the mechanism underlying the inhibition of Aβ deposition or prevention of Aβ neurotoxicity remains unclear.
The rat pheochromocytoma (PC12) cells are commonly used in the neuronal cell study. It is well known that exogenous stimuli such as Nerve Growth Factor (NGF) induce neurite outgrowth. Many studies looking to elucidate mechanisms involved in neuronal gene expression have been conducted in PC12 cells as these cells take on a cholinergic phenotype when differentiated with NGF. However, the IMR32 cell line has been identified for studying tau regulation as these cells have been shown to develop fibrillar structures that react to imunoprobes for paired helical filaments, the main constituents of neurofibrillary tangles. In theory, IMR32 cells being of human neuronal origin may be a more appropriate cell line to study APP-processing in relation to Alzheimer's disease than the rat phaeochromocytoma PC12 cell line. Therefore, these detected differences warrant further investigation. To understand how CAE modulates Aβ-mediated neurotoxicity, we investigated whether the addition of CAE to differentiated PC12 and IMR32 cells expressing aggregated Aβ1–40 could affect Aβ1–40-induced cell death and excessive ROS generation. We also measured cellular levels of a variety of antioxidative enzymes and oxidative molecules, including SOD, catalase, GPx, GR, glutathione (GSH), and glutathione disulphide (GSSG), to further define the CAE-mediated enhancement of the antioxidative defence system in AD.
2. Materials and methods
2.1. Preparation of aggregated Aβ peptide
The Aβ1–40 aggregation method was modified from our previous study.16 One milligram of Aβ1–40 peptide was dissolved in phosphate-buffered saline (PBS, pH 7.4) at a concentration of 500 μM and incubated at 37 °C for 24 h. After incubation, the peptide was stored at −20 °C as the stock solution. To prepare the aggregated Aβ1–40 peptide solution, the stock solution was diluted to 230 μM by PBS and incubated at 37 °C for 7 days. In all experiments, the aggregated Aβ1–40 peptide solution was diluted to the indicated experimental concentrations by cell culture medium. Confirmation of the aggregated Aβ peptide was measured by the thioflavin T (ThT) Aβ1–40 aggregation kit (Ana Spec Inc., Fremont, CA, USA). The aggregation of Aβ1–40 in the presence and absence of 50 μM morin and tannic acid, known inhibitors of fibril formation. Reactions were assembled at a final volume of 100 μL at room temperature in a black 96-well plate according to the assay protocol.
2.2. Preparation of C. asiatica ethanolic (雷公根 léi gōng gēn) extract (CAE)
The whole dried C. asiatica plant provided by Wei Chuan Corporation (Taipei, Taiwan) was ground into powder by a homogenizer (Model PRO 200; PRO Scientific Inc., Oxford, CT, USA). Ten grams of C. asiatica powder was soaked in 100 mL of 20% ethanol and incubated at 37 °C for 1 day. The ethanolic extract was filtered and freeze-dried and the final CAE powder was stored in the dark at −20 °C until use.
2.3. HPLC analysis of C. asiatica extracts active compounds
C. asiatica (1 g) was extracted with 10 mL of 20 % ethanol at 37 °C for 24 h. The extract was filtered with a 0.45-μm filter and analyzed by HPLC. HPLC analyses were performed on a LC-2000 series apparatus (Jasco) with a PU-2089 plus pump and a MD-2010 plus diode array detector, equipped with a LUNA C18 column (250 × 4.6 mm inner diameter; 5-μm particle size; Phenomenex, Torrance, CA, USA). The wavelength of diode array detector was set at 206 nm. The analytical method was based on previous study.17
2.4. PC12 and IMR32 cell culture and treatment
Rat PC12 pheochromocytoma cells and human IMR32 neuroblastoma cells were purchased from Bioresource Collection and Research Center (BCRC, Hsinchu, Taiwan). PC12 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium with 10% horse serum (HS), 5% fetal bovine serum (FBS), and 2 mM l-glutamine at 37 °C in a 5% CO2 humidified environment. IMR32 cells were cultured in minimum essential media (MEM) medium with 10% FBS at 37 °C in a 5% CO2 humidified environment. Poly lysine was prepared by adding 50 mL sterile tissue culture grade water to 5 mg poly lysine. Coated cell culture surface with 1 mL/25 cm² culture surface. After 15 min removed solution by aspiration and thoroughly rinsed surface with sterile tissue culture grade water. Finally, the cell culture was sterilized under the hood and UV light for 10–15 min. PC12 cells were seeded on poly-l-lysine-coated plates and cultured for 24 h, followed by culturing in the medium containing 100 ng/mL nerve growth factor for 6 days. Differentiated PC12 cells were treated with or without aggregated Aβ1–40 peptide in the presence of CAE (0, 25, 50, or 100 μg/mL).
2.5. 1,3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell viability assay
Differentiated PC12 and IMR32 cells seeded in 24-well plates (5 × 104 cell/well) were incubated with culture medium containing CAE (0, 25, 50 or 100 μg/mL) for 24 or 48 h and then the cells were incubated with 1,3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (0.5 mg/mL) at 37 °C in the dark for 2 h. The optical density (OD) of each well was determined by the microplate reader (F3; Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 570 nm after dissolving with dimethyl sulphoxide.
2.6. 5,5ʹ-dithiobis-2-nitrobenzoic acid (DTNB), 2,7-dichlorofluorescin diacetate (DCFH-DA) fluorescent assay
DCFH-DA was used as a ROS probe to monitor the intracellular accumulation of ROS. To measure ROS production, differentiated PC12 cells seeded in 12-well plates (1 × 105 cell/well) were treated with aggregated Aβ1–40 and CAE (0, 25, 50 or 100 μg/mL) for 6 and 24 h. After treatment, the cells were collected, washed twice with phosphate buffered saline (PBS), and stained with DCFH-DA for 30 min, then the cells were washed and analyzed by flow cytometry (FACSCanto II; BD Biosciences, Franklin Lakes, NJ, USA).
2.7. Analysis of SOD, catalase, GR and GPx activity
The activity of SOD, catalase, GR and GPx were determined in cell samples (by triplicate each one) using commercial kits (Cayman Co., Ann Arbor, MI, USA) following the manufacturer's protocol for each kit. The data were expressed as relative percentages of the control group which was not treated with CAE and Aβ1–40.
2.8. Measurement of intracellular GSSG and GSH concentrations
To measure the concentration of GSH, 10 μL of cell lysate was mixed with 95 μL reaction solution containing 2 U/mL GR, 200 μM nicotinamide adenine dinucleotide phosphate (NADPH), and 2 mM ethylenediaminetetraacetic acid (EDTA). After adding 100 μL DTNB (10 mM dissolved in 50 mM phosphate buffer, pH 7.2) to the mixture, the OD value at 405 nm was measured at 1-min intervals for 5 min. The GSH concentration was calculated by a GSH standard curve. To measure the concentration of GSSG, 70 μL cell lysate was mixed with 4 μL 1-methyl-2-vinylpyridinium trifluoromethanesulphonate (M2VP; 10 mM) and incubated for 1 h to remove GSH in the lysate. The lysate was then treated as described above for the measurement of GSH, and the GSSG concentration was calculated by a GSSG standard curve.
2.9. Statistical analysis
All data were expressed as mean ± standard deviation (SD). Statistical analysis was calculated with one-way analysis of variance (ANOVA), followed by Duncan's test, which was provided by SPSS13.0 statistical software (SPSS Institute, Inc., Chicago, IL, USA). The P value of two groups less than 0.05 was considered as significantly different between the values compared.
3. Results
3.1. Effect of CAE on Aβ1–40-induced neurotoxicity in differentiated PC12 and IMR32 cells
We first treated differentiated PC12 and IMR32 cells with CAE alone to examine their toxicity. Cell viabilities of differentiated PC12 and IMR32 cells were maintained above 85.5% and 133.3%, respectively, upon treatment with varying concentrations of CAE for 24 and 48 h. Results indicate that CAE was not significantly cytotoxic (P > 0.05 versus control for both) at concentrations of less than 100 μg/mL (Table 1). Therefore, we selected a CAE concentration in the range of 25–100 μg/mL for subsequent experiments.
Table 1.
The effect of CAE treatment on the viability of differentiated PC12 and IMR32 cells.
| Group | CAE concentration (μg/mL) |
||||
|---|---|---|---|---|---|
| 0 |
25 |
50 |
100 |
||
| Cell viability (%) | |||||
| PC 12 | 24 h | 100.3 ± 6.2 | 119.3 ± 4.9* | 90.5 ± 3.5** | 85.5 ± 0.5** |
| IMR32 | 102.2 ± 1.6 | 109.2 ± 2.4* | 112.4 ± 2.5* | 121.2 ± 2.6* | |
| PC 12 | 48 h | 100.2 ± 5.9 | 101.4 ± 4.5 | 96.9 ± 0.4** | 96.9 ± 6.2** |
| IMR32 | 103.3 ± 3.1 | 109.1 ± 2.2 | 120.8 ± 4.1* | 133.3 ± 3.1* | |
*P < 0.05 compared to control; increase in cell viability. **P < 0.05 compared to control, decrease in cell viability.
CAE: Centella asiatica 20% ethanol extract.
Treatment of differentiated PC12 cells with varying concentrations of aggregated Aβ1–40 (2–12 μM) significantly reduced the cell viability of differentiated PC12 cells; after 24 h, from 79.5% at 2 μM to 62.2% at 12 μM (P < 0.05, Fig. 1A). Treatment of IMR32 cells with varying concentrations of aggregated Aβ1–40 (0.4–2.8 μM) significantly reduced the cell viability of IMR32 cells; after 24 h, from 96.5% at 0.4 μM to 60.3% at 2.8 μM (P < 0.05). When the concentration of aggregated Aβ1–40 exceeded 2.0 μM, cell viability was reduced to less than 56.5% after 48 h (Fig. 1B). These results indicate that IMR32 cells were more sensitive than PC12 cells to Aβ1–40-induced cell toxicity.
Fig. 1.

CAE attenuates Aβ1–40-induced neurotoxicity. Differentiated PC12 (A) and IMR32 (B) cells were treated with varying concentrations of aggregated Aβ1–40, and cell viability was measured at 24 and 48 h after treatment. To determine the effect of CAE on Aβ1–40 neurotoxicity, differentiated PC12 (C) and IMR32 (D) cells were treated with aggregated Aβ1–40 and CAE at indicated concentrations for 24 and 48 h, and cell viability was measured. In all experiments, cells left untreated served as controls. Values are expressed as mean ± SD (n = 6). *Significantly different (P < 0.05) vs. the control group. #Significantly different (P < 0.05) vs. the Aβ1–40 treatment alone.
To monitor the effect of CAE on Aβ1–40-induced neurotoxicity in differentiated PC12 cells, cells were treated with 8 μM aggregated Aβ1–40 in the presence of varying concentrations of CAE. We observed that without CAE cell viability of differentiated PC12 cells was significantly reduced to 67.8% and 60.4% at 24 and 48 h after Aβ1–40 treatment, respectively (P < 0.05 versus control, Fig. 1C). We also observed that the neurotoxicity of aggregated Aβ1–40 was significantly diminished by treatment with CAE in the concentration range of 25–100 μg/mL at 24 h (P < 0.05 versus Aβ1–40 treatment alone, Fig. 1C), indicating that CAE could protect differentiated PC12 cells from aggregated Aβ1–40-induced neurotoxicity. Interestingly, this protective effect became less significant when cells were treated for 48 h, except for the 100 μg/mL CAE treatment (Fig. 1C). Viability of IMR32 cells was significantly reduced to 66.9%, 77.0%, and 70.2% at 24 h; 68.6%, 79.0%, and 68.9% at 48 h, respectively after treating with 2.0 μM aggregated Aβ1–40 (P < 0.05 versus control, Fig. 1D). Furthermore, the addition of CAE in the concentration range of 25–100 μg/mL significantly protected IMR32 cells from the neurotoxicity induced by aggregated Aβ1–40 at 24 and 48 h (P < 0.05 versus Aβ1–40 treatment alone, Fig. 1D).
3.2. Chromatogram of C. asiatica (雷公根 léi gōng gēn) extracts in high performance liquid chromatography (HPLC)
We calculated the content in CA extracts with HPLC. After comparing the chromatogram of CA extracts with four standard solutions, including madecoside (MS), asiaticoside (AS), madecassic acid (MA) and asiatic acid (AA), we confirmed that MS and AS were present, but not MA and AA. After quantitative analysis, MS content accounted for 1.20% of the CA extracts, while AS content accounted for only 0.17%. Because the most effective dose in CA extracts was 50 μg/mL, we used this concentration to calculate the most effective concentration of MS and AS. Using this value, the concentration of MS was found to be 6.0 μg/mL, calculated as 50 μg/mL × 1.20%, while the concentration of AS was 0.085 μg/mL, calculated as 50 μg/mL × 0.17% (Fig. 2A and B).
Fig. 2.

Chromatogram of Centella asiatica (雷公根 léi gōng gēn) extracts in HPLC. (A) Chromatogram of standards with HPLC method. (B) Chromatogram of Centella asiatica extracts with the HPLC method. Standard solutions including madecoside (MS), asiaticoside (AS), madecassic acid (MA) and asiatic acid (AA).
3.3. Effect of CAE on Aβ1–40-stimulated ROS production
Accumulation of ROS in cells is an important index for monitoring the neurotoxicity caused by aggregated Aβ1–40. Aggregated Aβ1–40 treatment for 6 and 24 h significantly increased ROS levels in differentiated PC12 cells to approximately 130% that of the control (P < 0.05 versus control; Fig. 3). Treatment of PC12 cells with 25, 50, and 100 μg/mL CAE and Aβ1–40 for 6 h reduced ROS to normal levels (P < 0.05 versus Aβ1–40 treatment alone); however, this effect was no longer significantly present after 24 h (Fig. 3). These results suggested that CAE may only suppress the induction of ROS at an early stage after aggregated Aβ1–40 treatment.
Fig. 3.

Effect of CAE on Aβ1–40-induced ROS production. The ROS levels in differentiated PC12 cells were measured after cotreatment with CAE and aggregated Aβ1–40. Cells left untreated served as the control. Relative fluorescence intensity = (fluorescence intensity of treatment/fluorescence intensity of control) × 100. Values are expressed as mean ± SD (n = 6). *P < 0.05 compared to control, #P < 0.05 compared to Aβ1–40 treatment alone.
3.4. Effect of CAE and Aβ1–40 treatment on SOD and catalase activities
A primary ROS in the human body is superoxide, which can be generated by the effect of aggregated Aβ1–40 on mitochondria.18 Our data showed that after 24 h of aggregated Aβ1–40 treatment, the SOD activity of differentiated PC12 and IMR32 cells was significantly inhibited to approximately 90% and 60% of control levels, respectively (P < 0.05 versus control; Fig. 4A and B). However, CAE clearly reversed the inhibition of SOD activity (P < 0.05 versus Aβ1–40 treatment alone) caused by aggregated Aβ1–40. Additionally, CAE at concentrations of 25, 50, and 100 μg/mL mediated the increase of SOD activity to normal levels (Fig. 4A and B). Interestingly, SOD activities observed in the presence of different concentrations of CAE did not demonstrate any clear dose-dependence in PC12 cells, but rather demonstrated peak activation at the lowest dose (25 μg/mL). We suggest that these results are attributable to complex components of CAE, some of which are activating, while others are inhibitory; at the lowest dose, the sum of all CAE-based effects may be activating in nature (a hypothesis that requires further examination).
Fig. 4.

Effect of CAE on SOD and catalase activities in Aβ1–40-treated cells. SOD activity in differentiated PC12 (A) and IMR32 (B) cells was measured at 24 h after treatment with aggregated Aβ1–40 and CAE at indicated concentrations. Catalase activity in differentiated PC12 (C) and IMR32 (D) cells was measured at 24 h after treatment with aggregated Aβ1–40 and CAE. In all experiments, cells left untreated served as controls. SOD and catalase activities were presented as a percentage of the control. Values are expressed as mean ± SD (n = 6). *P < 0.05 compared to control, #P < 0.05 compared to Aβ1–40 treatment alone.
Hydrogen peroxide, a primary ROS generated upon buildup of aggregated Aβ1–40 in cells, is converted to H2O and oxygen by the action of catalase. As shown in Fig. 4C, when the differentiated PC12 cells were treated with 8 μM aggregated Aβ1–40 for 24 h, catalase activity was significantly reduced to approximately 85% that of control (P < 0.05 versus control). However, when the cells were cotreated with Aβ1–40 and CAE at concentrations of 25, 50, and 100 μg/mL, the reduction in catalase activity observed in cells treated with Aβ1–40 alone was markedly reversed (P < 0.05 versus Aβ1–40 treatment alone) to 116.3%, 123.6%, and 129.6% of the control, respectively (Fig. 4C).
When IMR32 cells were treated with 2.0 μM aggregated Aβ1–40 for 24 h, catalase activity was significantly reduced to approximately 66.3% that of control (P < 0.05 versus control, Fig. 4D). However, when the cells were cotreated with CAE at concentrations of 25, 50, and 100 μg/mL, the reduction in catalase activity observed in cells treated with Aβ1–40 alone was markedly reversed (P < 0.05 versus Aβ1–40 treatment alone) to 70.2%, 78.3%, and 82.2% of the control, respectively (Fig. 4D). These findings suggest that CAE has the ability to modulate catalase activity in the cell to reduce ROS generation caused by Aβ1–40 accumulation.
3.5. Effect of CAE and Aβ1–40 treatment on the glutathione system
GR and GPx are antioxidant enzymes that mediate the breakdown of H2O2 into non-toxic products. GPx catalyzes the transfer of electrons from GSH to GSSG, accompanied by the conversion of H2O2 into water. By contrast, GR catalyzes the digestion of GSSG into GSH, thereby providing sufficient reactive molecules for GPx activity. Our results indicated that the activities of GR and GPx after treatment with aggregated Aβ1–40 for 24 h did not differ significantly from those of the control (P < 0.05 versus control). However, after treatment with a combination of Aβ1–40 and varying concentrations of CAE (25, 50, and 100 μg/mL), the activities of GR and GPx in differentiated PC12 cells were significantly enhanced to approximately GR 110% and GPx 112% that of the Aβ treatment (P < 0.05 versus Aβ treatment alone; Fig. 5A and C), while activities of GR and GPx in IMR32 cells were significantly enhanced to approximately GR 113% and GPx 117% that of the Aβ treatment (P < 0.05 versus Aβ treatment alone; Fig. 5B and D). This finding suggests that CAE could stimulate the activities of GR and GPx in differentiated PC12 and IMR32 cells to modulate oxidative stress in these cells, irrespective of whether the activities of GR and GPx were affected by aggregated Aβ1–40. In addition, we measured the GSH/GSSG ratio, a strong indicator of oxidative stress in the cells. The ratio of GSH/GSSG in the cells was significantly reduced after treatment with Aβ1–40, from approximately 31.5 to 18.3, and from approximately 34.5 to 19.3 in differentiated PC12 and IMR32 cells, respectively (Fig. 5E). However, cotreatment with CAE resulted in a significant increase in the ratio of GSH/GSSG, from approximately 19.9 to 23.1 and from approximately 21.3 to 30.2 in PC12 and IMR32 cells, respectively (P < 0.05 versus Aβ1–40 treatment alone). These results suggested that differentiated PC12 and IMR32 cells treated with Aβ1–40 were existing under conditions of high oxidative stress, and treatment of CAE can reduces Aβ1–40-induced toxicity in neuronal cells.
Fig. 5.

Effect of CAE on the glutathione system in Aβ1–40-treated cells. Differentiated PC12 (A, C) and IMR32 cells (B, D) were treated with aggregated Aβ1–40 and CAE at indicated concentrations for 24 h, and GR (A, B) and GPx (C, D) activities were measured. GR and GPx activities are displayed as a percentage of the control. (E) The GSH/GSSG ratio in differentiated PC12 (■) and IMR32 (□) cells were measured at 24 h after cotreatment with CAE and aggregated Aβ1–40. In all experiments, cells left untreated served as controls. Values are expressed as mean ± SD (n = 6). *P < 0.05 compared to control, #P < 0.05 compared to Aβ1–40 treatment alone.
4. Discussion
The Aβ peptide Aβ1–40, and Aβ1–42 to an even greater extent, is a critical factor in triggering AD. The primary mechanisms of Aβ peptide-mediated action in the development of AD involve reactions with metals, such as Cu+ or Fe2+,19 or binding to receptors, such as the N-methyl-d-aspartate (NMDA) receptor. Additionally, after Aβ enters the cell, it interferes with the electron transport chain in mitochondria, leading to mitochondrial dysfunction and the generation of excessive superoxide levels.20, 21 In all these respects, both Aβ types could be classified as ROS inducers.
Plasma concentrations of both Aβ1–40 and Aβ1–42 increase with age in individuals over the age of 65 years,22 in individuals who carry mutations that cause early-onset familial AD,23 and in patients with Down's syndrome, who are at heightened risk of developing AD.24 Additionally, plasma Aβ levels are elevated in first degree relatives of people with AD, who are also at an increased risk of developing the disease.25 The plasma concentrations of Aβ1–40 and Aβ1–42 were associated with risk and subtypes of dementia, a prospective population-based cohort study of men and women aged 55 y and older.24 High plasma concentrations of Aβ1–40 were associated with an increased risk of dementia, particularly in individuals who have concomitantly low concentrations of Aβ1–42; these individuals had an over 10-fold increased risk of dementia compared with individuals with low concentrations of both Aβ1–40 and Aβ1–42.26
In this study, we selected Aβ1–40 as an inducer of neurotoxicity in neuronal cells to investigate the relationship between CAE and AD. We had established this cell-based model in a previous study.16 Our data showed that the Aβ1–40 peptide treatment resulted in a marked elevation of ROS levels in differentiated PC12 cells, and caused markedly elevated rates of cell death (Fig. 1, Fig. 3), suggesting that the model of Aβ1–40 treatment in differentiated PC12 cells could faithfully replicate ROS-related, Aβ1–40-induced neurotoxicity, and could function as a suitable platform for examining the role of CAE in Aβ1–40-induced neurotoxicity. Aβ1–40 triggered the reduction of SOD and catalase activities (Fig. 4A–D), as well as a decrease in the concentration of GSH (Fig. 5E). These results are consistent with the findings of previous studies showing that aggregated Aβ1–40 could damage the antioxidative defence system in cells, particularly in neurons.27 These findings also confirm that our selection of Aβ1–40 for investigating the antioxidative defence system was appropriate.
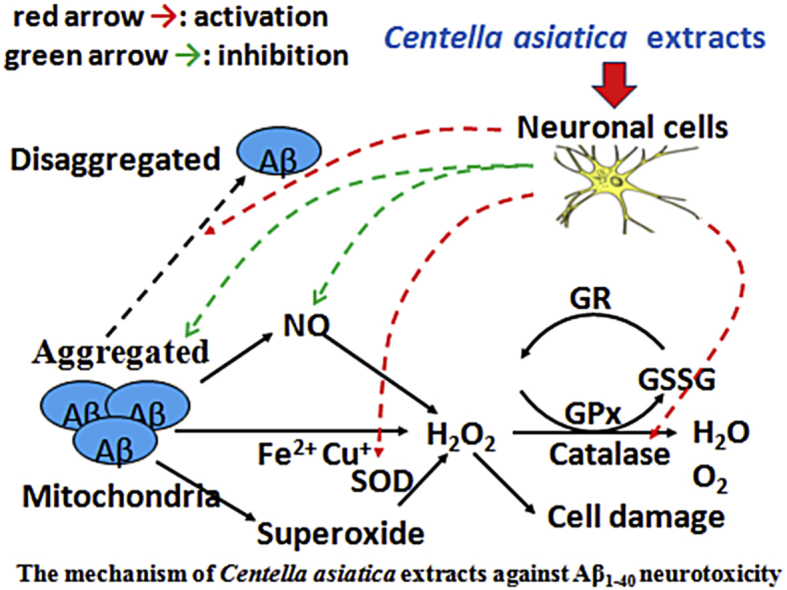
In the current study, we showed that treatment with CAE greatly facilitated the recovery of cells from Aβ1–40-induced neurotoxicity (Fig. 1C and D). Mechanism by which CAE inhibits Aβ1–40-induced neurotoxicity at least partially involves activation of the antioxidative enzymes (Fig. 4, Fig. 5, Fig. 6). CAE significantly reduced the ROS level upon cotreatment with Aβ1–40 at 6 h. This result suggests that CAE not only modulates the antioxidative defence system, but also protects the cell from Aβ1–40-induced neurotoxicity by direct elimination of imbalanced ROS production at an early stage (Fig. 3). Previous studies have shown that C. asiatica (雷公根 léi gōng gēn) extracts, including both aqueous and ethanolic extracts, possess DPPH radical-scavenging capability and high reducing potential.11 Thus, we propose that CAE can protect neuronal cells against Aβ1–40-induced neurotoxicity via multiple mechanisms, which together may account for the observation that CAE improves behavioral symptoms in the mouse AD model.15
Fig. 6.

The mechanism of Centella asiatica extracts against Aβ1–40 neurotoxicity.
Several strategies have been proposed to treat AD; for example, the use of pharmaceuticals that target the acetylcholine system28; compounds that clear the deposition of Aβ or interrupt the generation of Aβ29, 30; and antioxidants that reduce ROS levels or elevate the antioxidative defence system to protect neurons from Aβ-induced toxicity.8, 9 We believe that a common thread among all these strategies for the prevention and cure of AD is the primary importance ascribed to neuronal protection, particularly by enhancing the antioxidative defence system. Because most other strategies primarily focus on more advanced stages of AD and on managing symptoms of AD, their therapeutic efficacy cannot assist patients with AD to fully return to a normal life. Only by elevating the capacity of the antioxidative defence system can the goal of preventing AD or other ROS-related diseases be achieved. We observed a strong correlation between CAE and activation of the antioxidative defence system. However, CAE may prevent AD development, not only by simply activating the antioxidative defence system, but also by possessing the capability to directly eliminate ROS or to interfere with Aβ accumulation, as suggested by the reduction of the Aβ1–42 and Aβ1–40 deposition in transgenic mice administered CAE.14 Therefore, CAE possesses great potential for being developed into a functional food consumed to suppress or prevent AD, although this requires additional in vivo studies.
5. Conclusion
Our findings suggest that CAE can suppress Aβ-induced neurotoxicity by enhancing the antioxidative defence system in differentiated PC12 and IMR32 cells and provides a plausible basis for the development of therapeutic treatment or prophylaxis for AD.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by the National Science Council (NSC 98-2622-B-002-005-CC2) and assistance in the operation of some equipment was provided by the TechComm of National Taiwan University.
Footnotes
Peer review under responsibility of The Center for Food and Biomolecules, National Taiwan University.
References
- 1.Selkoe D.J. Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J Alzheimers Dis. 2001;3:75–80. doi: 10.3233/jad-2001-3111. [DOI] [PubMed] [Google Scholar]
- 2.Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 3.Behl C., Davis J.B., Lesley R.S., Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 4.Mattson M.P. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saido T.C., Iwata N. Metabolism of amyloid beta peptide and pathogenesis of Alzheimer's disease. Towards presymptomatic diagnosis, prevention and therapy. Neurosci Res. 2006;54:235–253. doi: 10.1016/j.neures.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 6.Demuro A., Parker I., Stutzmann G. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463–12468. doi: 10.1074/jbc.R109.080895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conti M., Couturier M., Lemonnier A., Lemonnier F. Effects of alpha-tocopherol on antioxidant enzyme activity in human fibroblast cultures. Int J Vitam Nutr Res. 1993;63:71–76. [PubMed] [Google Scholar]
- 8.Smith J.V., Luo Y. Elevation of oxidative free radicals in Alzheimer's disease models can be attenuated by Ginkgo biloba extract EGb 761. J Alzheimers Dis. 2003;5:287–300. doi: 10.3233/jad-2003-5404. [DOI] [PubMed] [Google Scholar]
- 9.Jeong J.C., Yoon C.H., Lee W.H. Effects of Bambusae concretio Salicea (Chunchukhwang) on amyloid beta-induced cell toxicity and antioxidative enzymes in cultured rat neuronal astrocytes. J Ethnopharmacol. 2005;98:259–266. doi: 10.1016/j.jep.2004.12.034. [DOI] [PubMed] [Google Scholar]
- 10.James J.T., Dubery I.A. Pentacyclic triterpenoids from the medicinal herb, Centella asiatica (L.) Urban. Molecules. 2009;14:3922–3941. doi: 10.3390/molecules14103922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mukherjee S., Dugad S., Bhandare R. Evaluation of comparative free-radical quenching potential of Brahmi (Bacopa monnieri) and Mandookparni (Centella asiatica) Int J Ayurveda Res. 2011;32:258–264. doi: 10.4103/0974-8520.92549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Subathra M., Shila S., Devi M.A., Panneerselvam C. Emerging role of Centella asiatica in improving age-related neurological antioxidant status. Exp Gerontol. 2005;40:707–715. doi: 10.1016/j.exger.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 13.Veerendra-Kumar M.H., Gupta Y.K. Effect of different extracts of Centella asiatica on cognition and markers of oxidative stress in rats. J Ethnopharmacol. 2002;79:253–260. doi: 10.1016/s0378-8741(01)00394-4. [DOI] [PubMed] [Google Scholar]
- 14.Dhanasekaran M., Holcomb L.A., Hitt A.R. Centella asiatica extract selectively decreases amyloid beta levels in hippocampus of Alzheimer's disease animal model. Phytother Res. 2009;23:14–19. doi: 10.1002/ptr.2405. [DOI] [PubMed] [Google Scholar]
- 15.Soumyanath A., Zhong Y.P., Henson E. Centella asiatica extract improves behavioral deficits in a mouse model of Alzheimer's disease: investigation of a possible mechanism of action. Int J Alzheimers Dis. 2012;2012 doi: 10.1155/2012/381974. 381974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C.L., Wang J.J., Pan T.M. Red mold rice extract represses amyloid beta peptide-induced neurotoxicity via potent synergism of anti-inflammatory and antioxidative effect. Appl Microbiol Biotechnol. 2008;79:829–841. doi: 10.1007/s00253-008-1480-8. [DOI] [PubMed] [Google Scholar]
- 17.Rafamantnana M.H., Rozet E., Raoelison G.E. An improved HPLC-UV method for the simultaneous quantification of triterpenic glycosides and aglycones in leaves of Centella asiatica (L.) Urb (APIACEAE) J Chromatogr B Anal Technol Biomed Life Sci. 2009;887:2396–2402. doi: 10.1016/j.jchromb.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 18.Anantharaman M., Tangpong J., Keller J.N. β-Amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLh/NLh X PS-1P264L/P264L double knock-in mouse model of Alzheimer's disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clementi M.E., Marini S., Coletta M., Orsini F., Giardina B., Misiti F. Abeta (31-35) and Abeta (25-35) fragments of amyloid beta-protein induce cellular death through apoptotic signals: role of the redox state of methionine-35. FEBS Lett. 2005;579:2913–2918. doi: 10.1016/j.febslet.2005.04.041. [DOI] [PubMed] [Google Scholar]
- 20.Aliev G., Seyidova D., Lamb B.T. Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer's disease-like pathology in transgenic mice. Neurol Res. 2003;25:665–674. doi: 10.1179/016164103101201977. [DOI] [PubMed] [Google Scholar]
- 21.Manczak M., Anekonda T.S., Henson E., Park B.S., Quinn J., Reddy P.H. Mitochondria are a direct site of A beta accumulation in Alzheimer's disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 22.van Dijk E.J., Prins N.D., Vermeer S.E. Plasma amyloid beta, apolipoprotein E, lacunar infarcts, and white matter lesions. Ann Neurol. 2004;55:570–575. doi: 10.1002/ana.20050. [DOI] [PubMed] [Google Scholar]
- 23.Scheuner D., Eckman C., Jensen M. Secreted amyloid betaprotein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 24.Schupf N., Patel B., Silverman W. Elevated plasma amyloid beta-peptide 1-42 and onset of dementia in adults with down syndrome. Neurosci Lett. 2001;301:199–203. doi: 10.1016/s0304-3940(01)01657-3. [DOI] [PubMed] [Google Scholar]
- 25.Younkin S.G. The role of A beta 42 in Alzheimer's disease. J Physiol Paris. 1998;92:289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- 26.van Oijen M., Hofman A., Soares H.D., Koudstaal P.J., Breteler M.M. Plasma Aβ1–40 and Aβ1–42 and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006;5:655–660. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 27.Gunasingh M.J., Philip J.E., Ashok B.S. Melatonin prevents amyloid protofibrillar induced oxidative imbalance and biogenic amine catabolism. Life Sci. 2008;83:96–102. doi: 10.1016/j.lfs.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 28.Wilcock G., Howe I., Coles H. A long-term comparison of galantamine and donepezil in the treatment of Alzheimer's disease. Drugs Aging. 2003;20:777–789. doi: 10.2165/00002512-200320100-00006. [DOI] [PubMed] [Google Scholar]
- 29.Donahue J.E., Flaherty S.L., Johanson C.E. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 30.van Marum R.J. Current and future therapy in Alzheimer's disease. Fundam Clin Pharmacol. 2008;22:265–274. doi: 10.1111/j.1472-8206.2008.00578.x. [DOI] [PubMed] [Google Scholar]