

Abstract
We report herein full details of the preparation of 4′-thiouridine, -cytidine, -adenosine and -guanosine phosphoramidites based on our synthetic protocol via the Pummerer reaction. Fully modified 4′-thioRNAs containing four kinds of 4′-thioribonucleoside units were prepared according to the standard RNA synthesis. The Tm values and thermodynamic parameters of a series of duplexes were determined by UV melting and differential scanning calorimetry (DSC) measurements. The resulting overall order of thermal stabilities for the duplexes was 4′-thioRNA:4′-thioRNA >> 4′-thioRNA:RNA > RNA:RNA > RNA:DNA > 4′-thioRNA:DNA. In addition, it was shown that the dominant factor in the stability of the duplexes consisting of 4′-thioRNA was enthalpic in character. The CD spectra of not only 4′-thioRNA:RNA and 4′-thioRNA:4′-thioRNA but also 4′-thioRNA:DNA were all similar to those of duplexes in the A-conformation. The stability of 4′-thioRNA in human serum was 600 times greater than that of natural RNA. Neither the RNA:RNA nor the 4′-thioRNA:4′-thioRNA duplexes were digested under the same conditions. The first example of a post-modification of an RNA aptamer by 4′-thioribonucleoside units was demonstrated. Full modification of the aptamer thioRNA3 resulted in complete loss of binding activity. In contrast, modifications at positions other than the binding site were tolerated without loss of binding activity. The post-modified RNA aptamer thioRNA5 was thermally stabilized and resistant toward nuclease digestion. The results presented in this paper will, it is hoped, contribute to the development of 4′-thioRNA as a new generation of artificial RNA.
INTRODUCTION
It is generally acknowledged that RNA is more than merely a messenger between DNA and protein, and that it possesses a variety of biological functions through RNA–DNA, RNA–RNA and RNA–protein interactions. In the last decade, the development of functional RNA molecules has received much attention because of the potential of these molecules in antisense (1), ribozyme (2) and RNA aptamer (3) technology. The epoch-making scientific discovery of RNA interference, especially small interfering RNA (siRNA) (4), has also opened the way for RNA research. However, since RNA is a transient molecule and readily hydrolyzed by nucleases, interest in developing an artificial RNA molecule which exhibits improved stability toward enzyme digestion and high hybridization ability for therapeutic application of RNA molecules has recently increased. Numerous efforts have been devoted to the study of the structural modification of RNA, especially in antisense methodology. To date, a series of 2′-O-alkylated RNAs appears highly promising as artificial RNA molecules (5). As an alternative, the 2′,4′-bridged nucleic acid [BNA or locked nucleic acid (LNA)] also has been developed and used for biological applications (6–8). Although both of the modified RNAs exhibit high nuclease resistance and hybridization properties, the 2′-OH group, a distinctive characteristic of RNA, is absent in these molecules. As mentioned above, since RNAs, including siRNA, have currently gained new research interest, a new class of artificial RNA would be desirable.
Accordingly, 4′-thioRNA, which consists of 4′-thioribonucleoside units, would be an ideal artificial RNA molecule. Imbach et al. have already reported the synthesis of 4′-thio RNAs consisting of 4′-thiouridine (9), and 4′-thiouridine, -cytidine and -adenosine (10). Investigation of their properties revealed that the 4′-thioRNA forms a thermally more stable duplex with its complementary natural RNA than the natural RNA duplex, and the 4′-thioRNA hybridizes more tightly to natural RNA than to natural DNA. Moreover, the 4′-thioRNA showed high resistance not only toward exonucleases but also toward endonucleases. However, despite these favorable properties of 4′-thioRNA, no further investigation of this molecule has been reported thus far, probably due to the difficulty in efficiently synthesizing the 4′-thioribonucleoside units. In view of these preliminary results and having recently completed an efficient stereoselective synthesis of 4′-thioribonucleosides (11), we were prompted to re-evaluate the properties of 4′-thioRNA with an aim toward developing a new generation of artificial RNA.
In this paper, we report the practical synthesis of the four 4′-thioribonucleoside phosphoramidite units and the synthesis of 4′-thioRNA, including all 4′-thioribonucleoside units. The resulting fully modified 15mer RNA (4′-thioRNA) forms a 4′-thioRNA:RNA duplex with a higher thermal stability relative to the corresponding RNA:RNA duplex, as is previously reported (10). In addition, it was shown that a 4′-thioRNA:4′-thioRNA duplex exhibited significantly increased thermal stability relative to the RNA:RNA duplex. The 4′-thioRNA was 600 times more stable than the natural RNA in human serum. Its application to a post-modification of the RNA aptamer toward NF-κB was also demonstrated.
MATERIALS AND METHODS
General methods
Physical data were measured as follows: 1H, 13C and 31P NMR spectra were recorded at 270 and 100 MHz instruments, respectively, in CDCl3 or DMSO-d6 as the solvent with tetramethylsilane as an internal standard. Chemical shifts are reported in parts per million (δ), and signals are expressed as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) or br (broad). Mass spectra were measured on a JEOL JMS-D300 spectrometer. TLC was done on Merck Kieselgel F254 precoated plates. The silica gel used for column chromatography was Merck silica gel 5715.
Synthesis of 4′-thioribonucleoside phosphoroamidite units
1-[2-O-(tert-Butyldimethylsilyl)-3,5-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-4-thio-β-d-ribofuranosyl]uracil (7)
Compound 1 (11) (1.0 g, 1.50 mmol) was dissolved in methylamine in MeOH (40%, 40 ml), and the mixture was kept for 5 h at room temperature. The solvent was removed in vacuo to give crude 4. The residue was coevaporated with toluene (twice). The resulting 4 was dissolved in dry CH2Cl2 (25 ml), and 2,6-lutidine (2.8 ml, 24.0 mmol) and TBSOTf (2.8 ml, 12.0 mmol) were added. The mixture was stirred for 23 h at room temperature. The reaction was quenched by addition of ice. The reaction mixture was partitioned between AcOEt and 1 M HCl. The separated organic layer was washed with saturated aqueous NaHCO3 (three times), followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (4:1), to give 7 (835 mg, 90% as a white foam): FAB-LRMS m/z 617 (M+); FAB-HRMS calcd for C27H53N2O6SSi3 (MH+) 617.2933, found 617.2910; 1H NMR (CDCl3) δ: 8.78 (br s, 1H), 8.45 (d, 1H, J = 8.2 Hz), 5.64 (d, 1H, J = 8.2 Hz), 5.52 (s, 1H), 4.11–3.98 (m, 4H), 3.62 (dd, 1H, J = 2.4, 9.8 Hz), 1.12–0.86 (m, 37H), 0.20 and 0.10 (each s, each 3H); 13C NMR (CDCl3) δ: 163.3, 150.7, 142.0, 101.3, 79.7, 72.0, 66.4, 58.3, 49.6, 26.0, 18.1, 17.7, 17.5, 17.4, 17.3, 17.2, 17.0, 16.9, 16.8, 13.7, 13.3, 13.1, 12.6, −5.5.
1-[2-O-(tert-Butyldimethylsilyl)-4-thio-β-d-ribofuranosyl]uracil (11)
To a solution of 7 (510 mg, 0.82 mmol) in THF (10 ml) was added AcOH (94 μl, 1.64 mmol) and TBAF (1 M in THF, 1.64 ml, 1.64 mmol) at 0°C. After being stirred for 30 min at the same temperature, the reaction mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with water, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (1:2 to 1:3), to give 11 (190 mg, 61% as a white solid): FAB-LRMS m/z 375 (M+); FAB-HRMS calcd for C15H27N2O5SSi (MH+) 375.1410, found 375.1390; 1H NMR (DMSO-d6) δ: 11.32 (br s, 1H), 8.08 (d, 1H, J = 8.0 Hz), 5.89 (d, 1H, J = 6.6 Hz), 5.68 (d, 1H, J = 8.0 Hz), 5.21 (t, 1H, J = 5.2 Hz), 5.18 (d, 1H, J = 4.1 Hz), 4.22 (m, 1H), 3.96 (m, 1H), 3.63 (m, 1H), 3.24 (m, 1H), 0.81 (s, 9H), 0.08 and 0.03 (each s, each 3H); 13C NMR (DMSO-d6) δ: 162.7, 150.8, 141.4, 101.9, 78.4, 72.7, 62.6, 62.5, 52.9, 25.5, 17.7, −4.6, −5.0.
1-[2-O-(tert-Butyldimethylsilyl)-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl]uracil (14) (9)
To a solution of 11 (538 mg, 1.44 mmol) in dry pyridine (20 ml) was added DMTrCl (728 mg, 2.15 mmol), and the reaction mixture was stirred for 1 h at room temperature. The reaction was quenched by addition of ice. The reaction mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (3:2), to give 14 (807 mg, 83% as a yellow foam): 1H NMR (CDCl3) δ: 8.13 (s, 1H), 8.05 (d, 1H, J = 8.3 Hz), 7.44 (m, 2H), 7.34–7.21 (m, 7H), 6.85 (m, 4H), 5.97 (d, 1H, J = 4.3 Hz), 5.45 (dd, 1H, J = 2.4, 8.3 Hz), 4.28 (m, 1H), 4.13 (m, 1H), 3.80 (s, 6H), 3.53 (m, 2H), 3.42 (m, 2H), 2.34 (d, 1H, J = 6.2 Hz), 0.91 (s, 9H), 0.15 and 0.11 (each s, each 3H).
1-{2-O-(tert-Butyldimethylsilyl)-3-O-[(N,N-diisopropylamino) methoxyphosphino]-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl}uracil (17) (9)
To a solution of 14 (500 mg, 0.74 mmol) in dry CH2Cl2 (5 ml) was added N,N-diisopropylethylamine (516 μl, 2.96 mmol), DMAP (18 mg, 0.15 mmol) and N,N-diisopropylmethylphosphonamidic chloride (359 μl, 1.85 mmol), and the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was diluted with CHCl3, and washed with H2O (twice) and saturated brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (3:1), to give 17 (510 mg, 82% as a white foam): 31P NMR (CDCl3) δ: 150.5 and 150.2.
1-[2-O-(tert-Butyldimethylsilyl)-5-O-(4,4′-dimethoxytrityl)-3-O-succinyl-4-thio-β-d-ribofuranosyl]uracil (20)
To a solution of 14 (150 mg, 0.22 mmol) in dry acetonitrile (3 ml) was added succinic anhydride (88 mg, 0.88 mmol), triethylamine (123 μl, 0.88 mmol), and DMAP (27 mg, 0.22 mmol). The reaction mixture was stirred for 28 h at room temperature and partitioned between AcOEt and H2O. The separated organic layer was washed with H2O, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with AcOEt, to give 20 (98 mg, 57% as a white foam): 1H NMR (CDCl3 + D2O) δ: 8.33 (d, 1H, J = 8.6 Hz), 7.44 (m, 2H), 7.34−7.24 (m, 7H), 6.84 (m, 4H), 5.72 (d, 1H, J = 2.6 Hz), 5.38 (dd, 1H, J = 1.3, 8.6 Hz), 5.17 (dd, 1H, J = 2.6, 7.9 Hz), 4.33 (t, 1H, J = 2.6 Hz), 3.80 (s, 6H), 3.73 (dd, 1H, J = 3.3, 7.9 Hz), 3.47 (dd, 1H, J = 3.3, 9.9 Hz), 3.37 (dd, 1H, J = 3.3, 9.9 Hz), 2.55 (m, 4H),0.90 (s, 9H), 0.17 and 0.11 (each s, each 3H).
N4-Benzoyl-1-[2-O-(tert-butyldimethylsilyl)-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl]cytosine (15) (10)
To a solution of 12 (12) (538 mg, 1.44 mmol) in dry pyridine (20 ml) was added DMTrCl (728 mg, 2.15 mmol), and the reaction mixture was stirred for 1 h at room temperature. The reaction was quenched by addition of ice. The reaction mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with saturated aqueous NaHCO3, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (3:2), to give 15 (807 mg, 83% as a yellow foam): 1H NMR (CDCl3) δ: 8.68 (m, 2H), 7.98 (m, 2H), 7.63 − 7.28 (m, 13H), 6.89 (m, 4H), 6.01 (d, 1H, J = 2.9 Hz), 4.28 (t, 1H, J = 2.9 Hz), 4.13 (m, 1H), 3.84 (s, 6H), 3.55 (m, 3H), 2.18 (d, 1H, J = 8.8 Hz), 0.93 (s, 9H), 0.29 and 0.16 (each s, each 3H).
N4-Benzoyl-1-{2-O-(tert-butyldimethylsilyl)-3-O-[(N,N-diisopropylamino)methoxyphosphino]-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl}cytosine (18) (10)
In the similar manner as described for 17, 15 (450 mg, 0.58 mmol) in dry CH2Cl2 (15 ml) was treated with N,N-diisopropylethylamine (402 μl, 2.31 mmol), DMAP (14 mg, 0.12 mmol), and N, N-diisopropylmethylphosphonamidic chloride (280 μl, 1.44 mmol) to give 18 (429 mg, 79% as a yellow foam): 31P NMR (CDCl3) δ: 151.0 and 149.3.
N4-Benzoyl-1-[2-O-(tert-butyldimethylsilyl)-5-O-(4,4′-dimethoxytrityl)-3-O-succinyl-4-thio-β-d-ribofuranosyl]cytosine (21)
In the similar manner as described for 20, 15 (263 mg, 0.34 mmol) in dry acetonitrile (5 ml) was treated with succinic anhydride (136 mg, 1.36 mmol), triethylamine (190 μl, 1.36 mmol) and DMAP (42 mg, 0.34 mmol), to give 21 (290 mg, 97% as a white foam): 1H NMR (CDCl3 + D2O) δ: 8.78 (d, 1H, J = 7.6 Hz), 7.94 (m, 2H), 7.55 – 7.22 (m, 13H), 6.84 (m, 4H), 5.83 (d, 1H, J = 1.8 Hz), 5.18 (dd, 1H, J = 2.9, 8.8 Hz), 4.41 (m, 1H), 3.79 (s, 6H), 3.75 (m, 1H), 3.45 (m, 1H), 3.37 (m, 1H), 2.56 (m, 4H), 0.89 (s, 9H), 0.21 and 0.07 (each s, each 3H).
9-[3,5-O-(1,1,3,3-Tetraisopropyldisiloxane-1,3-diyl)-4-thio-β-d-ribofuranosyl]adenine (6)
Compound 3 (11) (2.12 g, 2.99 mmol) was dissolved in ethanolic ammonia (saturated at 0°C, 100 ml), and the mixture was heated at 80°C for 6 h in a steel container. The solvent was removed in vacuo. The residue was dissolved in methylamine in MeOH (40%, 50 ml), and the mixture was kept for 2 h at room temperature. The solvent was removed in vacuo, and the residue was purified by a silica gel column, eluted with MeOH in CHCl3 (2%), to give 6 (1.47 g, 94% as a white solid): FAB-LRMS m/z 526 (M+); FAB-HRMS calcd for C22H40N5O4SSi2 (MH+) 526.2340, found 526.3233; 1H NMR (CDCl3) δ: 8.37 (s, 1H), 8.28 (s, 1H), 5.92 (s, 1H), 5.66 (br s, 2H), 4.77 (dd, 1H, J = 4.0, 9.2 Hz), 4.42 (br d, 1H, J = 4.0 Hz), 4.14 (m, 2H), 3.76 (ddd, 1H, J = 2.6, 3.3, 9.2 Hz), 3.39 (br s, 1H), 1.15−0.92 (m, 28H); 13C NMR (CDCl3) δ: 155.5, 153.0, 149.5, 139.8, 120.2, 78.9, 73.3, 61.8, 58.8, 50.2, 17.5, 17.4, 17.2, 17.1, 17.0, 13.3, 13.2, 13.0, 12.5.
9-[2-O-(tert-Butyldimethylsilyl)-3,5-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-4-thio-β-d-ribofuranosyl]adenine (9)
In the similar manner as described for 7, 6 (1.46 g, 2.78 mmol) in dry CH2Cl2 (50 ml) was treated with 2,6-lutidine (2.7 ml, 22.8 mmol) and TBSOTf (2.6 ml, 11.4 mmol) to give 9 (1.56 g, 88% as a white solid): FAB-LRMS m/z 640 (M+); FAB-HRMS calcd for C28H54N5O4SSi3 (MH+) 640.3204, found 640.3233; 1H NMR (CDCl3) δ: 8.46 (s, 1H), 8.35 (s, 1H), 5.66 (s, 1H), 5.51 (br s, 2H), 4.35 (m, 2H), 4.17 (dd, 1H, J = 2.6, 12.5 Hz), 4.06 (d, 1H, J = 12.5 Hz), 3.75 (m, 1H), 1.16 − 0.98 (m, 28H), 0.83 (s, 9H), 0.30 and 0.14 (each s, each 3H); 13C NMR (CDCl3) δ: 155.0, 152.8, 149.9, 139.9, 120.2, 79.2, 72.2, 62.5, 57.8, 49.1, 25.7, 18.1, 17.5, 17.4, 17.3, 17.2, 17.1, 17.0, 16.9, 16.8, 13.3, 13.2, 13.1, 12.6, −4.6, −4.7.
N6-Benzoyl-9-[2-O-(tert-butyldimethylsilyl)-3,5-O-(1,1,3,3-tetraisopropyldisiloxane-1,3-diyl)-4-thio-β-d-ribofuranosyl] adenine (10)
To a solution of 9 (1.56 g, 2.44 mmol) in dry pyridine (50 ml) was added BzCl (0.85 ml, 7.32 mmol), and the reaction mixture was stirred for 5 h at room temperature. The reaction was quenched by addition of aqueous ammonia. The mixture was partitioned between AcOEt and H2O. The separated organic layer was washed with water, followed by brine. The organic layer was dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with hexane/AcOEt (3:1), to give 10 (1.49 g, 82% as a white foam): FAB-LRMS m/z 744 (M+); FAB-HRMS calcd for C35H58N5O5SSi3 (MH+) 744.3461, found 744.3483; 1H NMR (CDCl3) δ: 9.02 (br s, 1H), 8.83 (s, 1H), 8.66 (s, 1H), 8.03 (m 2H), 7.56 (m, 3H), 5.74 (s, 1H), 4.35 (m, 2H), 4.17 (dd, 1H, J = 2.6, 13.2 Hz), 4.07 (d, 1H, J = 13.2 Hz), 3.78 (m, 1H), 1.16–0.99 (m, 28H), 0.83 (s, 9H), 0.31 and 0.15 (each s, each 3H); 13C NMR (CDCl3) δ: 164.3, 152.5, 151.4, 149.2, 142.3, 133.6, 132.6, 128.7, 127.6, 123.4, 79.1, 72.2, 62.7, 57.7, 49.3, 25.7, 18.1, 17.5, 17.4, 17.1, 17.0, 16.9, 16.8, 13.3, 13.2, 13.1, 12.6, −4.6, −4.7.
N6-Benzoyl-9-[2-O-(tert-butyldimethylsilyl)-4-thio-β-d-ribofuranosyl]adenine (13)
In the similar manner as described for 11, 10 (755 mg, 1.01 mmol) in THF (30 ml) was treated with AcOH (116 μl, 2.0 mmol) and TBAF (1 N in THF, 2.0 ml, 2.0 mmol) to give 13 (380 mg, 75% as a white solid): FAB-LRMS m/z 502 (M+); FAB-HRMS calcd for C23H32N5O4SSi (MH+) 502.1944, found 502.1919; 1H NMR (DMSO-d6 + D2O) δ: 8.84 (s, 1H), 8.74 (s, 1H), 8.02 (m, 2H), 7.63 (m, 1H), 7.53 (m, 2H), 5.99 (d, 1H, J = 5.9 Hz), 4.73 (dd, 1H, J = 3.5, 5.9 Hz), 4.20 (m, 1H), 3.85 (dd, 1H, J = 5.6, 11.5 Hz), 3.71 (dd, 1H, J = 5.6, 11.5 Hz), 3.38 (m, 1H), 0.73 (s, 9H), −0.03 and −0.20 (each s, each 3H); 13C NMR (DMSO-d6) 165.4, 154.6, 152.2, 151.4, 150.2, 143.4, 133.2, 132.3, 128.3, 125.7, 78.8, 72.4, 62.6, 61.7, 53.2, 25.4, 17.6, −4.8, −5.2.
N6-Benzoyl-9-[2-O-(tert-butyldimethylsilyl)-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl]adenine (16) (10)
In the similar manner as described for 14, 13 (370 mg, 0.74 mmol) in dry pyridine (20 ml) was treated with DMTrCl (376 mg, 1.11 mmol) to give 16 (526 mg, 88% as a yellow foam): 1H NMR (CDCl3) δ: 9.00 (br s, 1H), 8.74 (s, 1H), 8.31 (s, 1H), 8.02 (m 2H), 7.61–7.23 (m, 12H), 6.84 (m, 4H), 5.99 (d, 1H, J = 4.6 Hz), 4.72 (dd, 1H, J = 4.6, 8.6 Hz), 4.20 (dd, 1H, J = 5.3, 8.6 Hz), 3.79 (m, 6H), 3.67 (m, 1H), 3.59 (dd, 1H, J = 5.9, 9.9 Hz), 3.49 (dd, 1H, J = 5.9, 9.9 Hz), 2.42 (d, 1H, J = 5.3 Hz), 0.82 (s, 9H), −0.02 and −0.12 (each s, each 3H).
N6-Benzoyl-9-{2-O-(tert-butyldimethylsilyl)-3-O-[(N,N-diisopropylamino)methoxyphosphino]-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl}adenine (19) (10)
In the similar manner as described for 17, 16 (520 mg, 0.65 mmol) in dry CH2Cl2 (15 ml) was treated with N,N-diisopropylethylamine (453 μl, 2.60 mmol), DMAP (16 mg, 0.13 mmol), and N,N-diisopropylmethylphosphonamidic chloride (316 μl, 1.63 mmol) to give 19 (471 mg, 75% as a white foam): 31P NMR (CDCl3) δ: 151.1 and 150.8.
N2-(N′,N′-Dimethylformamidine)-9-(4-thio-β-d-ribofuranosyl)guanine (23)
To a suspension of 22 (11) (598 mg, 2.0 mmol) in dry DMF (10 ml) was added N,N-dimethylformamide dimethyl acetal (400 μl, 3.0 mmol), and the reaction mixture was stirred for 23 h at room temperature. The solvent was removed in vacuo, and the residue was purified by a silica gel column, eluted with MeOH in CHCl3 (5–35%), to give 23 (650 mg, 91% as a yellow foam): FAB-LRMS m/z 355 (M+); FAB-HRMS calcd for C13H19N6O4S (MH+) 355.1188, found 355.1187; 1H NMR (DMSO-d6 + D2O) δ: 8.56 (s, 1H), 8.10 (s, 1H), 5.73 (d, 1H, J = 6.6 Hz), 4.56 (m, 1H), 4.17 (m, 1H), 3.75 (dd, 1H, J = 6.6, 11.2 Hz), 3.62 (dd, 1H, J = 5.9, 11.2 Hz), 3.28 (m, 1H), 3.16 and 3.02 (each s, each 3H); 13C NMR (DMSO-d6) 157.8, 157.4, 156.9, 137.3, 119.5, 77.1, 73.1, 63.2, 60.9, 53.3, 40.7, 34.7.
N2-(N′,N′-Dimethylformamidine)-9-[5-O-(4,4′-dimethoxytrityl)- 4-thio-β-d-ribofuranosyl]guanine (24)
To a solution of 23 (640 mg, 1.8 mmol) in dry pyridine (7 ml) was added DMTrCl (794 mg, 2.3 mmol), and the reaction mixture was stirred for 3.5 h at room temperature. The reaction was quenched by addition of ice. The reaction mixture was diluted with H2O, and extracted with CHCl3 (three times). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with MeOH in CHCl3 (0–8%), to give 24 (875 mg, 74% as a yellow foam): FAB-LRMS m/z 657 (M+); FAB-HRMS calcd for C34H37N6O6S (MH+) 657.2496, found 657.2483; 1H NMR (CDCl3 + D2O) δ: 8.40 (s, 1H), 7.66 (s, 1H), 7.45–7.16 (m, 9H), 6.79 (m, 4H), 5.97 (d, 1H, J = 5.6 Hz), 4.57 (m, 1H), 4.38 (m, 1H), 3.75 (s, 6H), 3.69 (m, 1H), 3.48 (m, 1H), 3.40 (m, 1H), 2.94 and 2.84 (each s, each 3H); 13C NMR (CDCl3) 158.4, 158.3, 156.5, 150.9, 144.5, 137.1, 135.7, 130.0, 128.1, 127.8, 126.8, 119.1, 113.1, 86.5, 79.0, 75.0, 65.6, 61.5, 55.2, 50.9, 41.4, 35.1.
N2-(N′,N′-Dimethylformamidine)-9-[2-O-(tert-butyldimethylsilyl)- 5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl]guanine (26)
To a solution of 24 (868 mg, 1.32 mmol) in dry pyridine (12 ml) was added imidazole (215 mg, 3.17 mmol) and TBSCl (240 mg, 1.59 mmol), and the reaction mixture was stirred at room temperature. After 13 h, additional imidazole (108 mg, 1.58 mmol) and TBSCl (120 mg, 0.8 mmol) were added, and the reaction mixture was stirred for an additional 1 h at the same temperature. The reaction was quenched by addition of ice. The reaction mixture was diluted with H2O, and extracted with CHCl3 (three times). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. The residue was purified by a silica gel column, eluted with MeOH in CHCl3 (0–4%), to give the fast eluting fractions containing 26 (277 mg, 27% as a white foam). The slow eluting fractions were collected and concentrated to give material containing mainly 25 (624 mg, 61%). To a solution of 25 in MeOH (12 ml) was added Et3N (240 μl), and the reaction mixture was stirred at room temperature overnight for equilibration. The solvent was removed in vacuo, and the residue was purified by a silica gel column to give additional 26 (276 mg, total 54%): FAB-LRMS m/z 771 (M+); FAB-HRMS calcd for C40H51N6O6SSi (MH+) 771.3361, found 771.3359; 1H NMR (CDCl3) δ: 8.73 (br s, 1H), 8.57 (s, 1H), 7.88 (s, 1H), 7.47–7.21 (m, 9H), 6.83 (m, 4H), 5.85 (d, 1H, J = 4.6 Hz), 4.37 (dd, 1H, J = 5.3, 8.6 Hz), 4.14 (dd, 1H, J = 4.6, 8.6 Hz), 3.78 (s, 6H), 3.60 (m, 1H), 3.44 (m, 2H), 3.10 and 3.08 (each s, each 3H), 2.47 (d, 1H, J = 5.3 Hz), 0.83 (s, 9H), −0.02 and −0.03 (each s, each 3H); 13C NMR (CDCl3) 158.5, 157.8, 156.6, 150.5, 144.3, 136.8, 135.5, 130.0, 128.0, 127.8, 126.9, 120.4, 113.2, 86.8, 80.8, 75.6, 65.0, 61.7, 55.2, 49.9, 41.2, 35.2, 25.6, 17.9, −4.9, −5.0.
N2-(N′,N′-Dimethylformamidine)-9-{2-O-(tert-butyldimethylsilyl)-3-O-[(N,N-diisopropylamino)methoxyphosphino]-5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl}guanine (27)
In the similar manner as described for 17, 26 (353 mg, 0.46 mmol) in dry CH2Cl2 (10 ml) was treated with N,N-diisopropylethylamine (320 μl, 1.84 mmol), DMAP (11 mg, 0.09 mmol), and N,N-diisopropylmethylphosphonamidic chloride (223 μl, 1.15 mmol) to give 27 (335 mg, 78% as a white foam): FAB-LRMS m/z 932 (M+); FAB-HRMS calcd for C47H67N7O7PSSi (MH+) 932.4330, found 932.4327; 31P NMR (CDCl3) δ: 150.5.
Synthesis of the controlled pore glass support
To a solution of 20 (95 mg, 0.12 mmol) in dry DMF (3 ml) was added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI•HCl, 23 mg, 0.12 mmol) and LCA-CPG (342 mg, 0.03 mmol, 89.2 μmol/g), and the mixture was kept for 16 h at room temperature. The solid support was filtered and washed with pyridine. The remaining amino groups were capped by treatment with 0.1 M DMAP and 10% Ac2O in pyridine. The resulting solid support was filtered and washed with MeOH and acetone, and dried under reduced pressure to give 4′-thiouridine unit loaded CPG support. The loading amount of 4′-thiouridine unit was estimated by DMTr cation assay to be 18.1 μmol/g. In the similar manner as described above, 21 (100 mg, 0.12 mmol) in dry DMF (2 ml) was treated with EDCI•HCl (23 mg, 0.12 mmol) and LCA-CPG (342 mg, 0.03 mmol, 89.2 μmol/g) at 50°C for 25 h to give 4′-thiocytidine unit loaded CPG support (24.5 μmol/g).
Synthesis of thioRNA1–5 (9,13)
Support bound thioRNA1–5 were synthesized on an Applied Biosystem 3400 DNA synthesizer using 4′-thioribonucleoside phosphoramidite units 17–19 and 27, and commercially available ribonucleoside phosphoramidite units at a 1.0 μmol scale following the standard procedure described for oligoribonucleotides (13). Each of phosphoramidite units was used at a concentration of 0.1 M in dry acetonitrile and the coupling time was extended to 10 min for each step. After completion of the synthesis, the CPG support was treated with a mixture of thiophenol, triethylamine and dioxane (1:2:2, 0.5 ml) for 1 h at room temperature, and washed with MeOH. The support was then treated with concentrated NH4OH at 55°C for 16 h, and the support was filtered off. The filtrate was concentrated and the residue was treated with TBAF (1 M in THF, 1.5 ml). After being stirred for 24 h at room temperature, the reaction was quenched with a 0.1 N TEAA buffer (pH 7.0, 5 ml), and the oligoribonucleotide protected by a DMTr group at the 5′ end was chromatographed on a C-18 silica gel column with a linear gradient of acetonitrile (from 5 to 40%) in 0.1 N TEAA buffer (pH 7.0). The fractions were combined and concentrated. The residue was treated with aqueous HCl (pH 2.0) for 15 min at room temperature, and then the solution was neutralized with diluted NH4OH. The solution was concentrated and the residue was purified on reversed-phase high-performance liquid chromatography (HPLC), using a J'sphere ODS-M80 column (4.6 × 150 mm, YMC) with a linear gradient of acetonitrile (from 5 to 25%) in 0.1 N TEAA buffer (pH 7.0). The structure of each thioRNA was confirmed by matrix-assisted laser desorption/ionization time-of-flight mass (MALDI-TOF/MS) spectrometry on a Voyager-DE pro: thioRNA1, calculated mass: C142H177N55O88P14S15 4976.9 (M–H), observed mass: 4978.2; thioRNA2, calculated mass: C143H177N57O88P14S15 5016.9 (M–H), observed mass: 5018.2; thioRNA3, calculated mass: C294H363N113O187P30S31 10394.9 (M–H), observed mass: 10392.3; thioRNA4, calculated mass: C294H363N113-O210P30S8 10025.4 (M–H), observed mass: 10029.6; thioRNA5, calculated mass: C294H363N113O205P30S13 10105.7 (M–H), observed mass: 10098.1.
Determination of thermodynamic parameters
UV melting experiment
Thermally induced transitions were monitored at 260 nm on a Beckman DU 650 spectrophotometer. Samples were prepared as follows.
Duplex formation: a solution containing an appropriate oligonucleotide and a complementary sequence (3 μM each) in a buffer of 10 mM sodium cacodylate (pH 7.0) containing 100 mM NaCl was heated at 90°C for 5 min, then cooled gradually to room temperature and used for the thermal denaturation study. The sample temperature was increased at a rate of 0.5°C/min.
Thermal stability of the RNA aptamer: the solution containing the RNA aptamer (5 μM) in a buffer of 10 mM sodium cacodylate (pH 7.0) containing 10 mM NaCl was prepared and the measurement was conducted in the same manner as described above.
Differential scanning calorimetry (DSC) measurements
DSC measurements were performed on a VP-DSC Microcalorimeter. The solution containing an appropriate oligonucleotide and a complementary sequence (25 μM each) in a buffer of 10 mM sodium cacodylate (pH 7.0) containing 10 mM NaCl was prepared and scanned from 1 to 110°C at a scan rate 0.5K/min. The apparent molar heat capacity versus temperature profiles were obtained by subtracting buffer versus buffer curves from the sample versus buffer curves. The data were normalized with regard to the concentration and sample volume. The excess heat capacity function, ΔCP, was obtained after baseline subtraction, assuming that the baseline is given by the linear temperature dependence of the native state heat capacity. The process enthalpies, ΔH°, were obtained by integrating the area under the heat capacity versus temperature curves. Tm is the temperature corresponding to the maximum of each DSC peak. The process entropies, ΔS°, were determined by integrating the curve obtained and dividing the heat capacity curve by the absolute temperature, i.e. ΔS° = ∫(ΔCP/T)ΔT. The free energies, ΔG°(37°C), were determined at T = 310.15K by ΔG°(37°C) = ΔH° − TΔS°.
CD measurements
CD spectra were obtained at 25°C on a Jasco J720. The solution containing samples in a buffer of 10 mM sodium cacodylate (pH 7.0) containing 100 mM NaCl was prepared, and the sample spectra were subtracted from the buffer spectrum. The molar ellipticity was calculated from the equation [θ] = θ/cl, where θ is the relative intensity, c the sample concentration and l the cell path length in centimeters.
Assays for human serum stability
Each RNA sample labeled with 32P at the 5′ -end (5 pmol) was mixed with the corresponding unlabeled RNA (100 pmol). The RNA sample was incubated in phosphate-buffered saline (PBS) (40 μl) containing 50% human serum at 37°C. At appropriate periods, 3 μl aliquots of the reaction mixture were added to 16 μl of loading buffer (1× TBE, 7 M urea, 0.1% bromophenol blue, 0.1% xylene cyanol). The mixtures were then analyzed by electrophoresis on 20% polyacrylamide gel containing 7 M urea. Radioactive densities of the gel were visualized by a Bio-imaging analyzer (Bas 2500, Fuji Co., Ltd).
Competition assay (14)
The ability of various competitor RNAs to block protein binding to κB sites was measured in competition experiments which included radiolabeled DNA duplex (10 nM) and the indicated competitor RNA. Binding reactions of 20 μl contained 1 μg of poly(dI–dC), 10 mM HEPES, pH 7.5, 0.1 M NaCl, 1 mM DTT and 10% glycerol. Protein [NF-κB (p50), human, recombinant, (Promega), 0.1 gel shift unit] was then added, followed by incubation for 30 min at 25°C and electrophoresis through 10% native polyacrylamide gels in 0.5× TBE buffer. Radioactive densities of the gel were visualized by a Bio-imaging analyzer.
RESULTS AND DISCUSSION
Synthesis
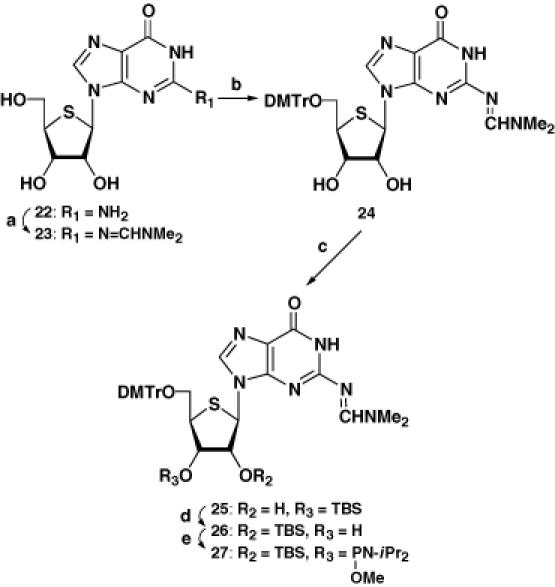
The synthesis of the phosphoramidite units for RNA is usually more challenging than that for DNA because of the discrimination of the 2′- and 3′-OH groups, when the synthesis starts from the free ribonucleoside. Our synthetic protocol avoids this problem (Scheme 1), except in the case of the 4′-thioguanosine phosphoramidite 27. Thus, the 3,5-dimethoxybenzoyl (DMBz) group of 1, a substrate obtained by the Pummerer reaction (11), was removed by treatment with methylamine to give 4. After protection of the resulting free 2′-OH group with a tert-butyldimethylsilyl (TBS) group, the 1,1,3,3-tetraisopropyldisiloxane-1,3-diyl (TIPDS) group was selectively removed by treatment of 7 with tetrabutylammonium fluoride (TBAF) in the presence of acetic acid at 0°C to give 11. Protection of the resulting 5′-OH group with a dimethoxytrityl (DMTr) group, followed by reaction with methyl N,N-diisopropylchlorophosphoramidite (15) in the presence of Hünig's base gave the 4′-thiouridine phosphoramidite 17 (9). The corresponding 4′-thiocytidine and -adenosine phosphoramidites 18 and 19 were also prepared in a similar manner (10). For the synthesis of 4′-thioguanosine phosphoramidite 27, the synthetic route shown in Scheme 2 proved to be more practical. The 2-amino group of 4′-thioguanosine (22) was protected with an N,N-dimethylformamidine group to give 23. After protection of the 5′-OH group with a DMTr group, N2-(N′,N′-dimethylformamidine)-9-[5-O-(4,4′-dimethoxytrityl)-4-thio-β-d-ribofuranosyl]guanine (24) was treated with TBSCl in pyridine in the presence of imidazole to give a mixture of the 3′-O-TBS derivative 25 (major) and the desired 2′-O-TBS derivative 26 (minor). When the separated 25 was treated with a catalytic amount of triethylamine in MeOH, migration of the TBS group to the 2′-OH group was observed and the desired 26 was finally obtained in 54% yield. Compound 26 was then converted to the corresponding phosphoramidite 27. We also prepared the phosphoramidite units possessing a 2-cyanoethyl group which is common for DNA synthesis. However, purification by silica gel chromatography was somewhat difficult and the coupling yields for the 4′-thioRNA synthesis were lower (data not shown). In order to attach the 4′-thiouridine or the 4′-thiocytidine unit to LCA-CPG, 14 and 15 were treated with succinic anhydride in the presence of triethylamine and 4-(dimethylamino)pyridine (DMAP) in acetonitrile to give compounds 20 and 21, respectively (Scheme 1). Compound 20 was then loaded on to LCA-CPG by treatment with EDCI•HCl in DMF at room temperature to give the CPG support derivatized with 4′-thiouridine. The loading amount of the 4′-thiouridine unit was estimated by DMTr cation assay to be 18.1 μmol/g. In the case of the CPG support derivatized with 4′-thiocytidine, the loading amount was quite low when the reaction was carried out at room temperature. Therefore, the reaction for the loading to CPG was carried out at 50°C to give the support derivatized with 4′-thiocytidine (24.5 μmol/g).
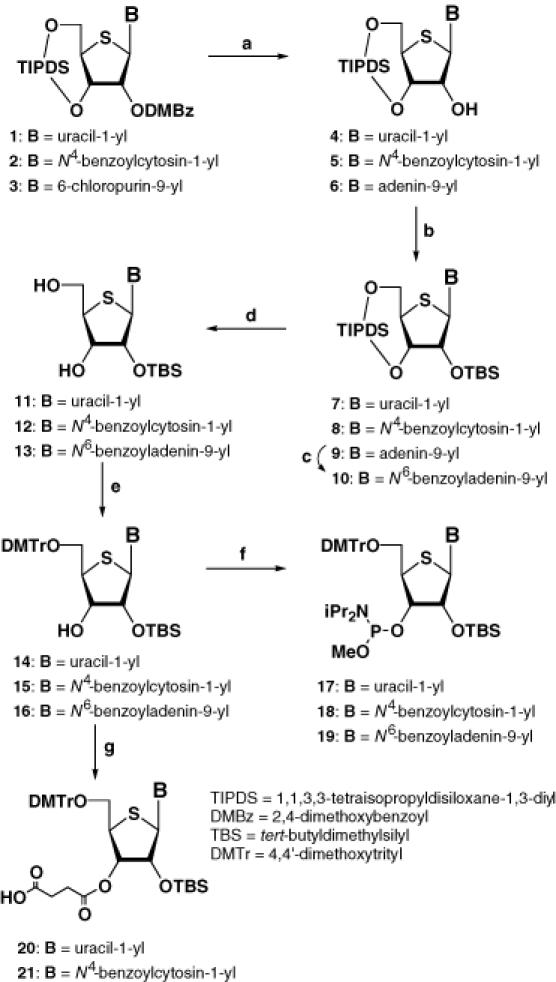
Scheme 1. (a) MeNH2, MeOH, for 4; (12) for 5; NH3/EtOH, 80°C then MeNH2, MeOH, for 6; (b) TBSOTf, 2,6-lutidine, CH2Cl2; (c) BzCl, pyridine then NH4OH; (d) TBAF, AcOH, THF; (e) DMTrCl, pyridine; (f) N,N-diisopropylmethylphosphonamidic chloride, iPr2NEt, DMAP, CH2Cl2; (g) succinic anhydride, Et3N, DMAP, CH3CN.
Scheme 2. (a) N,N-dimethylformamide dimethyl acetal, DMF; (b) DMTrCl, pyridine; (c) TBSCl, imidazole, pyridine; (d) Et3N, MeOH; (e) N,N-diisopropylmethylphosphonamidic chloride, iPr2NEt, DMAP, CH2Cl2.
A series of fully modified 4′-thioRNAs (thioRNA1–3; Figures 1 and 4) and RNAs partially modified, containing 4′-thioribonucleoside units (thioRNA4 and thioRNA5; Figure 4) was synthesized in a DNA synthesizer using the methyl-protected phosphoramidites 17 to 19 and 27, and/or commercially available cyanoethyl-protected phosphoramidites of the natural nucleosides. A solution of 0.1 M of each of the phosphoramidites in acetonitrile was used and the coupling time was set to 10 min. The fully protected RNAs were deprotected and purified, and the structures of the resulting RNAs were confirmed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (see the Materials and Methods).
Figure 1.

Sequences of RNA, 4′-thioRNA and DNA.
Figure 4.
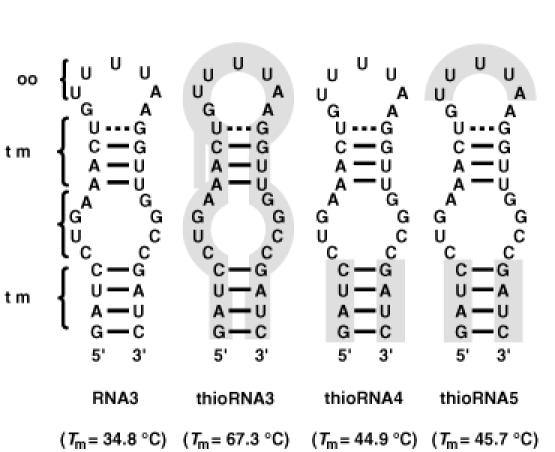
Secondary structures of unmodified and modified RNA aptamers for NF-κB. The shaded regions were substituted by 4′-thioribonucleoside units. The numbers in parentheses showed Tm values obtained by UV melting experiments.
Duplex formation and stability
First, thermal stabilities of the complementary duplexes by UV melting experiments were evaluated using thioRNA1, thioRNA2 and the corresponding RNA and DNA sequences (Table 1). The measurements were carried out in a buffer of 10 mM sodium cacodylate (pH 7.0) containing 100 mM NaCl. The duplexes consisting of natural RNA and DNA, namely RNA1:RNA2 and RNA1:DNA1, had Tm values of 66.2 ± 0.2 and 51.6 ± 0.1°C, respectively. When RNA1 was changed to thioRNA1, the corresponding Tm values were changed depending on the complementary sequences. Thus, the duplex of thioRNA1:RNA2 showed a higher Tm value than RNA1:RNA2 (74.5 ± 0.3°C versus 66.2 ± 0.2°C), while the duplex of thioRNA1:DNA1 had a lower Tm value than RNA1:DNA1 (45.5 ± 0.9°C versus 51.6 ± 0.1°C). These results of the hybridization properties of 4′-thioRNA were similar to those reported by Imbach et al. (10), except for a moderate enhancement (∼0.5°C per base pair) of the thermal stability of RNA1:RNA2 versus thioRNA1:RNA2.
Table 1. Thermodynamic parameters for duplex formation determined from UV melting and DSC measurementsa,b.
| Duplex | UV melting | DSC | |||
|---|---|---|---|---|---|
| Tm (°C) | Tm (°C) | ΔH° (kcal mol−1) | ΔS° (cal mol−1 K−1) | ΔG° (37°C) (kcal mol−1) | |
| RNA1:RNA2 | 66.2 ± 0.2 | 53.9 ± 1.3 | −85.2 ± 3.1 | −263.2 ± 9.7 | −3.58 ± 0.37 |
| thioRNA1:RNA2 | 74.5 ± 0.3 | 66.9 ± 0.1 | −100.8 ± 0.9 | −299.4 ± 2.8 | −7.90 ± 0.21 |
| thioRNA1:thioRNA2 | >99 | 91.0 ± 0.6 | −126.9 ± 1.0 | −350.8 ± 3.2 | −18.1 ± 0.1 |
| RNA1:DNA1 | 51.6 ± 0.1 | 43.9 ± 0.2 | −87.8 ± 2.2 | −279.5 ± 9.8 | −0.98 ± 0.99 |
| thioRNA1:DNA1 | 45.5 ± 0.9 | 35.9 ± 0.1 | −61.7 ± 2.2 | −200.8 ± 14.3 | 3.31 ± 0.37 |
aErrors reflect standard deviation from three independent experiments.
bExperimental conditions are described in Materials and Methods.
A drastic enhancement of thermal stability was observed in a duplex of thioRNA1:thioRNA2 (>99°C), and no obvious single transition corresponding to a helix-to-coil transition was observed under the conditions. Consequently, the concentration of NaCl was reduced to 10 mM instead of 100 mM in the buffer, and the measurements were carried out by UV melting (the data and UV melting curves are presented in the Supplementary Material), and differential scanning calorimetry (DSC) to evaluate the thermodynamic parameters along with the Tm value. In the study using DSC, the Tm value was defined according to the temperature corresponding to the maximum of each DSC peak. The resulting Tm values showed the same tendency as obtained by the UV melting experiments. In addition, the Tm of thioRNA1:thioRNA2 was estimated to be 91.0 ± 0.6°C in the buffer containing 10 mM NaCl. Since the ΔTm of RNA1:RNA2 was 37.1°C, the thioRNA1:thioRNA2 was stabilized ∼2.5°C per base pair. The overall order of thermal stabilities for the five duplexes was thioRNA1: thioRNA2 >> thioRNA1:RNA2 > RNA1:RNA2 > RNA1: DNA1 > thioRNA1:DNA1. 2′-O-alkylated RNA and LNA have been developed as artificial RNAs, as described in the Introduction. Although both RNA molecules show high hybridization properties to complementary RNAs, duplex formation with complementary DNAs is also thermally stabilized (16). However 4′-thioRNA prefers RNA as a complementary partner much more than DNA in duplex formation which is a characteristic of 4′-thioRNA. For the duplex formation of modified RNA:modified RNA, the conformationally restricted LNA:LNA duplex would be the most thermally stable duplex thus far (17). Despite the more flexible sugar conformation in 4′-thioRNA, the drastic enhancement of thermal stability of a 4′-thioRNA:4′-thioRNA duplex also should be noted.
The thermodynamic parameters obtained by DSC measurements are also shown in the last three columns of Table 1. In the case of duplex formation with RNA (RNA1:RNA2 versus thioRNA1:RNA2), the effect of the substitution of thioRNA1 increased the stability of the duplex by giving rise to a favorable contribution to the enthalpy of formation while effecting the entropy in an unfavorable manner. The net result of the substitution to thioRNA1 was to increase the stability of the duplex, and thus, this increasing in stability is the result of the more favorable contribution to enthalpy. The same tendency was also observed in the duplex formation of thioRNA1:thioRNA2. However, in the case of duplex formation with DNA (RNA1:DNA1 versus thioRNA1:DNA1), the same substitution gave rise to an unfavorable contribution to the enthalpy of formation, while effecting the entropy in a favorable manner. The net result of the substitution was a decrease in the stability of the duplex. This reduction in stability is the result of the more unfavorable contribution to enthalpy. Although the enthalpic and entropic contributions were opposite between RNA and DNA, in total, the factor dominating the stability of the duplex consisting of 4′-thioRNA was enthalpic in character.
To investigate the structural aspects of the duplexes consisting of 4′-thioRNA, CD spectra were measured together with those of the corresponding RNA:RNA and RNA:DNA duplexes (Figure 2). The CD spectrum of RNA1:RNA2 had a strong positive band near 260 nm and a negative band near 210 nm, which are characteristic of A-conformation (18). The spectrum of RNA1:DNA1 was slightly red-shifted and had a decreased positive band near 260 nm. This spectral feature is a key factor in describing the A-like character of the conformation. The spectrum of thioRNA1:RNA2 showed an equal intensity in the positive band, while a reduced negative band near 210 nm compared with that of RNA1:RNA2. When both strands were replaced by 4′-thioRNAs, i.e. the thioRNA1:thioRNA2 duplex, the intensity of the positive and negative bands were both reduced (Figure 2A). For the duplex of thioRNA1:DNA1, the spectrum differed from that of RNA1:DNA1, but was close to that of RNA1:RNA2 (Figure 2B). In short, all of the duplexes consisting of 4′-thioRNA had roughly similar positive and negative bands >200 nm as expected for A-conformation. In the CD spectra of nucleic acid analogs of the same sequences, the spectra at longer wavelengths will be affected by the number of bases per helical turn and the inclination of the bases with respect to the helical axis (19), while those at shorter wavelengths will be affected by conformational changes of the phosphate–sugar backbone (20). Consequently, it may be thought that not only duplexes of thioRNA1:RNA2 and thioRNA1:thioRNA2, but also the thioRNA1:DNA1 duplex adopt the A-conformation for their mode of base stacking. In contrast, the structures of the phosphate–sugar backbone will be different for each of the duplexes arising from the substitution of a furanose ring oxygen by a sulfur atom (probably affecting the minor groove of the duplex) (21). Similar spectral changes of CD were observed in duplexes of RNA:RNA, LNA:RNA, LNA:LNA and LNA:DNA (17).
Figure 2.
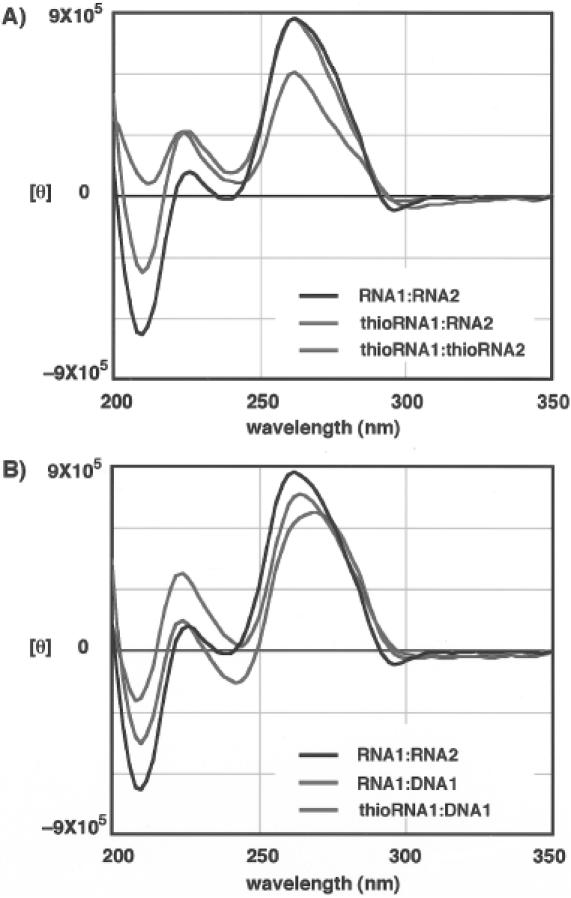
CD spectra of unmodified and modified duplexes.
As mentioned above, the thermal stability of thioRNA1: RNA2 was much higher than that of thioRNA1:DNA1. Since the strength of the CD bands in each duplex was almost the same, it is difficult to explain the difference in thermal stability based on the CD spectra. In the thermodynamic parameters, the factor dominating the thermal stability of the duplex consisting of 4′-thioRNA was enthalpic in character. When hybridizing with a complementary RNA, we observed a favorable enthalpic contribution, which was further enhanced in the 4′-thioRNA:4′-thioRNA duplex. Such enthalpically favored duplex formation has been reported in oligonucleotides containing N3′ → P5′ phosphoramidates (22), and was explained by extensive hydration of the duplex (23). One possible explanation is that the conformational change of the phosphate–sugar backbone and/or the 2′-OH groups of 4′-thioRNA may be favorable for hydration when hybridizing with RNA molecules, whereas favorable hydration may be disturbed when hybridizing with DNA, despite the conformational similarity of the two duplexes (presumed from the CD spectra). Further investigation, such as NMR and X-ray structural studies, is needed before a detailed discussion of the conformational aspects can be presented.
Stability of 4′-thioRNA in human serum
We next investigated the stability of single-stranded and double-stranded 4′-thioRNA compared with natural RNA. The RNAs were labeled at the 5′-end with 32P and incubated in PBS containing 50% human serum. The reactions were then analyzed by PAGE under denaturing conditions. Figure 3 shows the results of single-stranded RNAs. As can be seen in the results of RNA1 (left), natural RNA was rapidly hydrolyzed, while thioRNA1 was highly resistant against hydrolysis (right). The half-lives (t1/2s) were estimated based on the ratio of full-length RNA at each time, and the t1/2s of RNA1 and thioRNA1 were calculated as <10 s and 107 ± 27 min, respectively. To our surprise, the double-stranded RNAs, namely RNA1:RNA2 and thioRNA1:thioRNA2, were not hydrolyzed at all under the same conditions for 24 h (data not shown).
Figure 3.
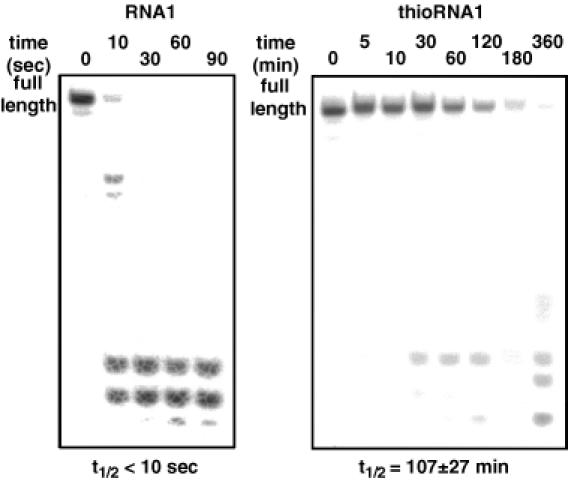
Stabilities of single-stranded RNA1 and thioRNA1 in human serum. PAGE of 5′-32P-labeled RNA1 and thioRNA1 incubated in PBS containing 50% human serum at 37°C.
Imbach et al. (9,10) have already reported the high stability of 4′-thioRNA consisting of the uridine unit (6mer) and uridine, cytidine and adenosine units (12mer) in cell culture medium containing 10% heat inactivated fetal calf serum. Our results supported the stability of single-stranded 4′-thioRNA even in human serum. One difference between Imbach's results and ours was the mode of digestion of the full-length RNAs. In their results, one nucleotide sequence shorter at the 3′-end was observed as a first metabolite by detection of reversed-phase HPLC. This mode of digestion was explained by the predominant contribution of a 3′-exonuclease activity in serum (24). In contrast, no shorter sequence like the one above was observed in either RNA1 or thioRNA1 by PAGE analysis, and thus, a smaller contribution of 3′-exonuclease activity is evident in our results. Thus far, numerous modified DNA and RNA analogs have been prepared, and their stability in serum has been tested. In the case of DNA analogs, such as those possessing phosphorothioate linkages (PS-DNA), the predominant activity of a 3′-exonuclease for digestion was observed (24). However, Shimayama et al. (25) reported a predominant contribution of endoribonucleases such as RNase A for the digestion of RNA analogs in serum. Thus it may be difficult to determine which is the main nuclease activity for nucleic acid digestion in serum. Nevertheless, our results excluding 3′-exonuclease activity, support the latter case. Although the mode of digestion of single-stranded 4′-thioRNA was different from that reported by Imbach et al., thioRNA1 showed high nuclease resistance and the stability was estimated to be 600 times greater than that of RNA1.
For the stability of double-stranded RNA, neither the natural RNA nor the 4′-thioRNA duplex was digested in human serum, which was an unexpected result. Quite recently, Braasch et al. (26) reported the stability of natural RNA and RNA analogs possessing phosphorothioate linkages (PS-RNA). In their report, either single-stranded natural RNA or PS-RNA was digested within an hour in a medium containing 5% fetal bovine serum, while the corresponding double-stranded RNAs were both stable over 24 h under the same conditions. Our results agree with theirs for duplex stability. In addition, their results showing that single-stranded PS-RNA is not stable in serum differ from those with PS-DNA, well-known nuclease resistant antisense molecule. These results suggest that 4′-thioRNA is much more stable than PS-RNA, and that the contribution of nucleases for the digestion of nucleic acid analogs may be different for DNA versus RNA analogs, as discussed above.
Post-modification of RNA aptamer toward NF-κB
As described above, 4′-thioRNA formed thermally stable duplexes with not only complementary RNA, but also 4′-thioRNA. In addition, 4′-thioRNA showed a high stability in human serum. In contrast, the tertiary structure of duplexes consisting of 4′-thioRNA is still unclear though an A-conformation was suggested from the CD spectra. Since our goal is the development of 4′-thioRNA as a new generation of artificial RNA, we examined the post-modification of a known RNA aptamer by 4′-thioribonucleoside units to demonstrate its utility. Thus far, numerous RNA aptamers specific for a variety of small and large molecules have been isolated by SELEX (27–29). From among them we chose the RNA aptamer toward the transcription factor NF-κB p50 homodimer (p502) isolated by Lebruska and Maher (14) for post-modification by 4′-thioribonucleoside units for the following reasons: (i) the length of the RNA aptamer is rather short (31 nt in length); (ii) the secondary structure of the aptamer is well elucidated; and (iii) the modification of the aptamer to DNA and 2′-O-methyl analogs resulted in loss of binding activity to NF-κB, a result which is explained by the necessity of the 2′-OH group(s).
We first prepared the fully modified RNA aptamer (Figure 4), thioRNA3 (shaded region represents substitution by 4′-thioribonucleoside units), and its binding affinity to NF-κB was examined in a competition assay, which included radiolabeled κB DNA duplex [the sequences of the κB DNA duplex; see (14)]. Although thioRNA3 showed much higher thermal stability for its overall structure relative to RNA3, the RNA aptamer for NF-κB isolated by Lebruska and Maher (67.3°C versus 34.8°C), this modified RNA regretably showed no binding affinity to NF-κB (data not shown). During the course of this study, Huang et al. (30) reported the X-ray crystal structure of the NF-κB (p50)/RNA aptamer complex, in which the sequence of the RNA aptamer is not the same as RNA3, but quite similar. In the resulting crystal structure, it was revealed that the regions of stem 1 and the bulge (Figure 4) were critical positions to bind NF-κB. Based on this report, we next prepared thioRNA4 (modified in the stem 2 region) and thioRNA5 (modified in the stem 2 and loop regions), in which the critical positions for the binding were not modified. The results of gel mobility shift assays monitoring the ability of unlabeled RNA3, thioRNA4 and thioRNA5 to compete with the radiolabeled κB DNA duplex are shown in Figure 5A. Competition with the κB DNA duplex was observed not only in RNA3, but also in thioRNA4 and thioRNA5, depending on the concentrations of the RNAs. To evaluate the 50% inhibitory concentration (IC50) values of each RNA competitor, the concentrations of the competitors tested were divided more finely from 0.001 to 1 μM (Figure 5B). The calculated IC50 values of thioRNA4 and thioRNA5 were 11.3 ± 3.2 and 6.3 ± 4.2 nM, respectively, and these values were the same as that of RNA3 (5.7 ± 4.0 nM). The thermal stabilities of thioRNA4 and thioRNA5 were obtained as 44.9 and 45.7°C, respectively.
Figure 5.
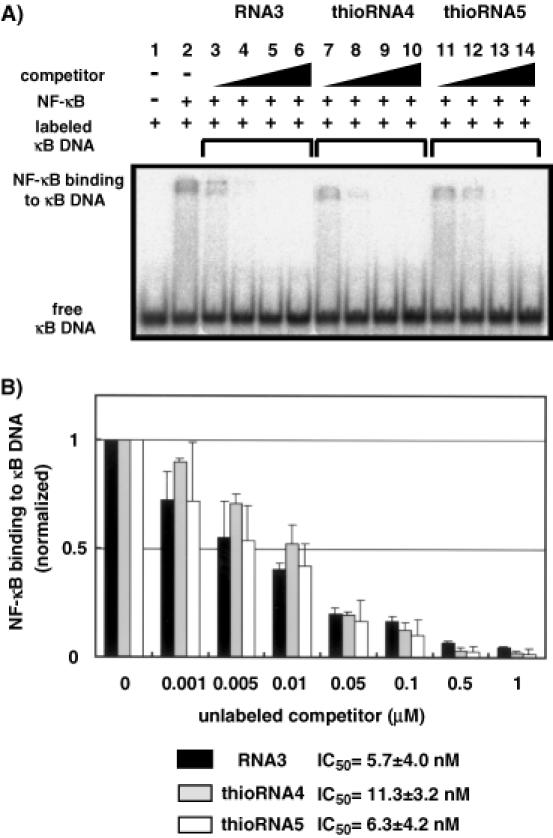
Unmodified and modified RNA aptamers compete with κB DNA duplex. (A) Labeled κB DNA duplex was incubated in the absence of NF-κB (lane 1), in the presence of NF-κB (lane 2), or together with various unlabeled competitors (lanes 3–6 for RNA3, lanes 7–10 for thioRNA4, lanes 11–14 for thioRNA5). Triangles indicate increasing unlabeled competitor concentrations (0.01, 0.1, 1 and 10 μM). (B) Quantitation of competitive inhibition of κB DNA duplex binding. Error bars reflect standard deviation from three independent experiments. Binding data are presented as values relative to the level of labeled κB DNA duplex binding in the absence of competitor.
In the isolated RNA aptamer obtained by SELEX, not only the aptamer sequence, but also the overall structure of the aptamer is critical for its binding to the target molecule. As described above, modification of RNA3 to thioRNA3 resulted in complete loss of binding activity, which was the same for the modification of RNA3 to DNA and 2′-O-methyl analogs. These results indicate that the overall structure is more important than the 2′-OH group(s) of the RNA aptamer for binding. Although the CD spectra indicated similar helical structures of the duplexes consisting of 4′-thioRNA, the conformational change of thioRNA3 would probably be small but could not be tolerated without loss of the binding activity in this case. In contrast, modifications at positions other than the binding site were tolerated without loss of the binding activity. Modifications in the stem 2 and loop regions did not affect the structure of the binding site, and both thioRNA4 and thioRNA5 showed the same high affinity to NF-κB as did RNA3. In addition, both of the modified RNAs were thermally stabilized. The resulting Tm value of thioRNA4 was 10.1°C higher than that of RNA3, which is in agreement with the results obtained in experiments on duplex stability (stabilized ∼2.5°C per base pair). The Tm value of thioRNA5 was slightly higher than that of thioRNA4, indicating that the introduction of 4′-thioribonucleoside units on a loop would be thermally stabilized. The stability of thioRNA5 in human serum was also examined and thioRNA5 was found to be three times more resistant relative to RNA3 (data not shown). Since thioRNA5 has a single-stranded natural RNA sequence on the bulge region, nuclease digestion was observed at this position. Although the resistance of thioRNA5 toward nuclease digestion may be insufficient in the present form, our results indicated that post-modification of the RNA aptamer by 4′-thioribonucleoside units: (i) is useful for stabilizing the overall structure of the RNA aptamer without loss of binding activity, except for excessive modification in the binding site and (ii) improves nuclease resistance.
CONCLUSION
In this paper, we have presented full details of the preparation of 4′-thiouridine, -cytidine, -adenosine and -guanosine phosphoramidites. Using these phosphoramidite units, 4′-thioRNAs were prepared based on the standard RNA synthesis. The resulting 4′-thioRNA formed a thermally stable duplex with the complementary RNA, while duplex formation with the complementary DNA proved unfavorable. These results indicate that 4′-thioRNA prefers RNA to DNA for duplex formation, which agrees with the previous results (10). We also demonstrated that the duplex consisting of 4′-thioRNAs was dramatically thermally stabilized by 2.5°C per base pair. The overall order of thermal stabilities for the duplexes examined was: 4′-thioRNA:4′-thioRNA >> 4′-thioRNA:RNA > RNA:RNA > RNA:DNA > 4′-thioRNA:DNA. Further investigations using DSC showed the factor dominating the thermal stability of the duplex consists of 4′-thioRNA was enthalpic in character. The CD spectra of the duplexes of not only 4′-thioRNA:RNA and 4′-thioRNA:4′-thioRNA but also of 4′-thioRNA:DNA were all similar to those in the A-conformation. The stability of 4′-thioRNA in human serum was 600 times greater than that of natural RNA, and the mode of digestion of 4′-thioRNA indicated the predominant contribution of endoribonuclease activities. With these favorable properties of 4′-thioRNA in hand, we examined the post-modification of an RNA aptamer toward NF-κB by 4′-thioribonucleoside units. Full modification of the aptamer, thioRNA3, resulted in complete loss of binding activity, presumably due to conformational changes. In contrast, modifications at positions other than the binding site were tolerated without loss of the binding activity. The post-modified RNA aptamer thioRNA5 was thermally stabilized and resistant toward nuclease digestion. Although our demonstration is just one example, the results presented in this paper should encourage the development of 4′-thioRNA as a new generation of artificial RNA. Since our synthetic protocol affords a practical synthesis of 4′-thioRNA, further applications are under way.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Ms Y. Misawa (Hokkaido University) for technical assistance and Ms S. Oka (Center for Instrumental Analysis, Hokkaido University) for technical assistance with MS. This work was supported in part by Grant-in-Aid for Scientific Research from the Japan Society for Promotion of Science (No. 15025203 and 15510169 to N.M. and No. 15209003 to A.M.). This paper constitutes part 228 of Nucleosides and Nucleotides [for part 227 in this series, see (31)].
REFERENCES
- 1.Kurreck J. (2003) Antisense technologies: improvement through novel chemical modifications. Eur. J. Biochem., 270, 1628–1644. [DOI] [PubMed] [Google Scholar]
- 2.Burlina F., Favre,A. and Fourrey,J.-L. (1997) Chemical engineering of RNase resistant and catalytically active hammerhead ribozymes. Bioorg. Med. Chem., 5, 1999–2010. [DOI] [PubMed] [Google Scholar]
- 3.Jayasene S.D. (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostic. Clin. Chem., 45, 1628–1650. [PubMed] [Google Scholar]
- 4.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 5.Manoharan M. (1999) 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochem. Biophys. Acta, 1489, 117–130. [DOI] [PubMed] [Google Scholar]
- 6.Koshkin A.A., Singh,S.K., Nielsen,P., Pajwanshi,V.K., Kumar,R., Meldgaard,M., Olsen,C.E. and Wengel,J. (1998) LNA (locked nucleic acids): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron, 54, 3607–3630. [Google Scholar]
- 7.Imanishi T. and Obika,S. (2002) BNAs: novel nucleic acid analogs with a bridged sugar moiety. Chem. Commun., 1653–1659. [DOI] [PubMed] [Google Scholar]
- 8.Wahlestedt C., Salmi,P., Good,L., Kela,J., Johnsson,T., Hokfelt,T., Broberger,C., Porreca,F., Lai,J., Ren,K. et al. (2000) Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl Acad. Sci. USA, 97, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellon L., Barascut,J.-L., Maury,G., Divita,G., Goody,R. and Imbach, J.-L. (1993) 4′-Thio-oligo-β-d-ribonucleotides: synthesis of β-4′-thio-oligouridylates nuclease resistance base pairing properties and interaction with HIV-1 reverse transcriptase. Nucleic Acids Res., 21, 1587–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leydier C., Bellon,L., Barascut,J.-L., Morvan,F., Rayner,B. and Imbach,J.-L. (1995) 4′-Thio-RNA: synthesis of mixed base 4′-thio-oligoribonucleotides, nuclease resistance, and base pairing properties with complementary single and double strand. Antisense Res. Dev., 5, 167–174. [DOI] [PubMed] [Google Scholar]
- 11.Naka T., Minakawa,N., Abe,H., Kaga,D. and Matsuda,A. (2000) The stereoselective synthesis of 4′-β-thioribonucleosides via the Pummerer reaction. J. Am. Chem. Soc., 122, 7233–7243. [Google Scholar]
- 12.Minakawa N., Kaga,D., Kato,Y., Endo,K., Tanaka,M., Sasaki,T. and Matsuda,A. (2002) Synthesis and structural elucidation of 1-(3-C-ethynyl-4-thio-β-d-ribofuranosyl)cytosine (4′-thioECyd). J. Chem. Soc., Perkin Trans., 1, 2182–2189. [Google Scholar]
- 13.Gait M.J. (1984) Oligonucleotides Synthesis: A Practical Approach. IRL Press, Oxford, UK. [Google Scholar]
- 14.Lebruska L.L. and Maher,L.J. (1999) Selection and characterization of an RNA decoy for transcription factor NF-κB. Biochemistry, 38, 3168–3174. [DOI] [PubMed] [Google Scholar]
- 15.Usman N., Ogilvie,K.K., Jiang,M.-Y. and Cedergren,R.J. (1987) Automated chemical synthesis of long oligoribonucleotides using 2′-O-silylated ribonucleosides 3′-O-phosphoramidites on a controlled-pore glass support: synthesis of a 43-nucleotide sequence similar to the 3′-half molecule of an Escherichia coli formylmethionine tRNA. J. Am. Chem. Soc., 109, 7845–7854. [Google Scholar]
- 16.Wengel J., Petersen,M., Nielsen,K.E., Jensen,G.A., Hakansson,A.E., Kumar,R., Sorensen,M.D., Rajwanshi,V.K., Bryld,T. and Jacobsen,J.P. (2001) LNA (locked nucleic acid) and the diastereoisomeric α-L-LNA: conformational tuning and high-affinity recognition of DNA/RNA targets. Nucleosides Nucleotides Nucleic Acids, 20, 389–396. [DOI] [PubMed] [Google Scholar]
- 17.Koshkin A.A., Nielsen,P., Meldgaard,M., Pajwanshi,V.K., Singh,S.K. and Wengel,J. (1998) LNA (locked nucleic acid): an RNA mimic forming exceedingly stable LNA:LNA duplexes. J. Am. Chem. Soc., 120, 13252–13253. [Google Scholar]
- 18.Gray D.M., Hung,S. and Johnson,K.H. (1995) Absorption and circular dichroism spectroscopy of nucleic acid duplexes and triplexes. Methods Enzymol., 246, 19–34. [DOI] [PubMed] [Google Scholar]
- 19.Johnson C.W. (1996) In Fasman,G.D. (ed.), Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum Press, NY, pp. 433–468. [Google Scholar]
- 20.Gray D.M., Ratliff,R.L. and Vaughan,M.R. (1992) Circular-dichroism spectroscopy of DNA. Methods Enzymol., 211, 389–406. [DOI] [PubMed] [Google Scholar]
- 21.Egli M. (1996) Structural aspects of nucleic acid analogs and antisense oligonucleotides. Angew. Chem. Int. Ed. Engl., 35, 1894–1909. [Google Scholar]
- 22.Gryaznov S.M., Lloyd,D.H., Chen,J., Schultz,R.G., DeDionisio,L.A., Ratmeyer,L. and Wilson,W.D. (1995) Oligonucleotide N3′→P5′ phosphoramidates. Proc. Natl Acad. Sci. USA, 92, 5798–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Egli M. (1998) Conformational preorganization, hydration, and nucleic acid duplex stability. Antisense Nucleic Acid Drug Dev., 8, 123–128. [DOI] [PubMed] [Google Scholar]
- 24.Hoke G.D., Draper,K., Freier,S.M., Gonzalez,C., Driver,V.B., Zounes,M.C. and Ecker,D.J. (1991) Effects of phosphorothioate capping on antisense oligonucleotide stability, hybridization and antiviral efficacy versus herpes simplex virus infection. Nucleic Acids Res., 19, 5743–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimayama T., Nishikawa,F., Nishikawa,S. and Taira,K. (1993) Nuclease-resistant chimeric ribozymes containing deoxyribonucleotides and phosphorothioate linkages. Nucleic Acids Res., 21, 2605–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braasch D.A., Jensen,S., Liu,Y., Kaur,K., Arar,K., White,M.A. and Corey,D.R. (2003) RNA interference in mammalian cells by chemically-modified RNA. Biochemistry, 42, 7967–7975. [DOI] [PubMed] [Google Scholar]
- 27.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T7 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 28.Famulok M. and Szostak,J.W. (1992) In vitro selection of specific ligand-binding nucleic acids. Angew. Chem. Int. Ed. Engl., 31, 979–988. [Google Scholar]
- 29.Hermann T. and Patel,D.J. (2000) Adaptive recognition by nucleic acid aptamers. Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 30.Huang D., Vu,D., Cassiday,L.A., Zimmerman,J.M., Maher,L.J. and Ghosh,G. (2003) Crystal structure of NF-κB (p50)2 complexed to a high-affinity RNA aptamer. Proc. Natl Acad. Sci. USA, 100, 9268–9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yamamoto Y., Shuto,S., Tamura,Y., Kodama,T., Hoshika,S., Ichikawa,S., Ohtsuka,E., Komatsu,Y. and Matsuda,A. (2004) Oligonucleotides having a loop consisting of 3′-deoxy-4′-C-(2-hydroxyethyl)thymidines form stable hairpins, Biochemistry, 43, 8690–8699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.