


Abstract
Hepatitis C virus (HCV) is the major etiological agent of non-A, non-B hepatitis. Current therapies are not effective in all patients and can result in the generation of resistant mutants, leading to a need for new therapeutic options. HCV has an RNA genome that contains a well-defined and highly conserved secondary structure within the 5′-untranslated region. This structure is known as the internal ribosomal entry site (IRES) and is necessary for translation and viral replication. Here, we test the hypothesis that antisense peptide nucleic acid (PNA) and locked nucleic acid (LNA) oligomers can bind key IRES sequences and block translation. We used lipid-mediated transfections to introduce PNAs and LNAs into cells. Our data suggest that PNAs and LNAs can invade critical sequences within the HCV IRES and inhibit translation. Seventeen base PNA or LNA oligomers targeting different regions of the HCV IRES demonstrated a sequence-specific dose–response inhibition of translation with EC50 values of 50–150 nM. Inhibition was also achieved by PNAs ranging in length from 15 to 21 bases. IRES-directed inhibition of gene expression widens the range of mechanisms for antisense inhibition by PNAs and LNAs and may provide further therapeutic lead compounds for the treatment of HCV.
INTRODUCTION
Currently, there are about 200 million people worldwide who are infected with the Hepatitis C virus (HCV). In the United States, 2.7 million people have chronic hepatitis C and the incidence of new symptomatic infections has been estimated to be 25 000 per year (1). There is no vaccine available and recombinant interferon-alpha (rIFNα) as a therapeutic treatment is effective in only a portion of the infected population (2).
The antiviral response of an HCV-infected host cell is to shut down global 7mG-cap-dependent translation (3). HCV evades this immune response by using an internal ribosomal entry site (IRES) within the 5′-untranslated region (5′-UTR). (4,5). IRESs form a well-defined and highly conserved RNA structure adjacent to AUG start sites and recruit host ribosomal subunits for cap-independent translation (6,7). HCV IRES binds eIF-3 and 40S ribosomal subunits in the absence of other cellular factors (8,9), and deletion and point mutations within the HCV IRES cause substantial decrease in viral translation (10). The importance of the IRES for the translation of HCV suggests that it would be an excellent target for oligonucleotide-based therapeutics (11–15). However, to be active, oligonucleotides must demonstrate the ability to invade structured RNA and block protein binding.
Several studies have investigated the use of antisense oligonucleotides targeted to the HCV IRES. Hanecak and co-workers (16,17) reported that oligonucleotides with phosphorothioate and 2′-modifications could inhibit HCV IRES-dependent translation. More recently, cationic phosphoramidate α-oligonucleotides were also shown to be effective (18). In cell-free studies, Toulme and co-workers (19) have demonstrated that antisense oligonucleotides compete for binding with the 40S ribosomal subunit, and Jang and co-workers (20) have used antisense oligonucleotides to help in identifying other proteins involved in IRES recognition (20). We hypothesized that oligomers capable of enhancing recognition of sequences embedded within nucleic acid structure might be superior agents for IRES recognition. Two such agents are peptide nucleic acids (PNAs) (21) and locked nucleic acids (LNAs) (22,23).
PNAs are DNA/RNA analogs with a neutral 2-aminoethylglycine backbone (21). PNAs are stable to digestion using nucleases and proteases (24), offer excellent discrimination for binding to match versus mismatch sequences (25), and provide a novel starting point for the design of biologically active agents. PNAs can be effective antisense agents inside cultured mammalian cells (26,27), but the ability of PNA to invade highly structured RNA sequences inside cells has not been well established.
LNA bases contain a bridging methylene carbon between the 2′ and 4′ positions of the ribose ring (22,23,28). This constraint preorganizes the oligonucleotide backbone and can increase Tm values by as much as 10°C per LNA substitution. LNA bases are introduced by standard DNA/RNA synthesis protocols, allowing the binding properties of chimeric oligonucleotides to be ‘fine-tuned’ for the recognition of specific targets. Chimeric LNAs have been demonstrated to be active antisense agents inside cultured mammalian cells (29–31) and can also block the association of HIV TAT protein with its RNA target TAR (32). Similar to PNAs, limited information is available on the ability of LNAs to target RNA secondary structure in cells.
Here, we show that the PNA and the chimeric LNA oligomers, which are complementary to HCV IRES can inhibit IRES-dependent gene expression inside cells.
MATERIALS AND METHODS
Materials
PNA monomers and other reagents necessary for PNA synthesis were obtained from Applied Biosystems (Foster City, CA). Amino acids were obtained from Advanced Chemtech (Louisville, KY) or Calbiochem-Novabiochem (La Jolla, CA). PNAs were made using an Expedite 8909 automated synthesizer (Applied Biosystems) (33). PNAs were analyzed and purified by reverse-phase high-performance liquid chromatography and mass spectral analysis by matrix-assisted laser desorption ionization time-of-flight (33). After purification, PNAs were freeze-dried and resuspended in water. LNAs were obtained from Proligo LLC (Boulder, CO). DNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Plasmid pRL-HL (34) was obtained from Dr Michael Gale (UT Southwestern Medical Center at Dallas, TX).
PNA:DNA hybridization
PNA:DNA duplex mixtures (50 μM) were annealed in the thin-walled PCR tubes in a thermocycler. Reductions in temperature occurred for 1 min with the hold times indicated: 95°C, 5 min; 85°C, 1 min; 75°C, 1 min; 65°C, 5 min; 55°C, 1 min; 45°C, 1 min; 35°C, 5 min; 25°C, 1 min; 15°C, 1 min; hold 15°C. After annealing, the PNA:DNA duplexes were maintained at −20°C until evaluation of Tm was performed. PNA:DNA melting temperature studies were performed by measuring the change in absorbance at 260 nM using a Cary 100Bio UV/Vis spectrophotometer (Varian Inc., Walnut Creek, CA) equipped with a 12-position sample holder and a Peltier temperature control accessory.
Lipid-mediated transfection of PNA:DNA duplexes and LNAs
Annealed PNA:DNA duplexes were prepared for transfection by equilibrating 12.8 μl of 50 μM PNA:DNA stock in 137 μl of Opti-MEM (Invitrogen). In a separate tube, 1.9 μl of LipofectAMINE (Invitrogen) was mixed with 148 μl of Opti-MEM and vigorously mixed for 15 s followed by an equilibration at room temperature for 5 min. The diluted PNA:DNA and LipofectAMINE solutions (150 μl each) were mixed together by agitating for 15 s. Lipid complexes were allowed to form by incubating the mixture at room temperature for 15 min in the dark. The solution containing the PNA:DNA:LipofectAMINE complex (300 μl) was diluted to 3.2 ml using Opti-MEM resulting in a final concentration of 200 nM. This 200 nM solution was serial diluted to the final working concentrations of 100 and 25 nM. LNA oligonucleotide transfections were performed in the same manner except that they were first heated to 80°C for 5 min before mixing with Opti-MEM.
CV-1 cells (CCL-70; American Type Culture Collection, Manassas, VA) were plated at 10 000 cells/well in 48-well plates using DMEM and glutamine supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 500 U/ml penicillin, 0.1 mg/ml streptomycin and 0.06 mg/ml tylosin reagent (Sigma). Cells were incubated at 37°C at 5% CO2 for a minimum of 8 h before transfection. The cells were then washed once with 200 μl of Opti-MEM, followed by 14–16 h transfection with the PNA:DNA:lipid complex (or LNA:lipid complex) or lipid only control.
Lipid-mediated transfection of reporter plasmid pRL-HL into CV-1 cells
Plasmid vector encoding luciferase was transfected 8–12 h after transfection of PNA or chimeric LNA. The bicistronic vector pRL-HL developed by Honda and co-workers (34) was prepared for transfection by equilibrating plasmid DNA (120 ng/well) in 19.5 μl/well of Opti-MEM. Likewise, 0.2 μl of LipofectAMINE (7 μg/ml) was mixed with 19.8 μl of Opti-MEM. The plasmid solution and the LipofectAMINE solution were mixed thoroughly and incubated for 20 min in the dark. The mixed solutions were diluted using Opti-MEM to a final concentration of 120 ng/well at 50 μl/well. Transfections were incubated for 8–10 h before the transfection medium was aspirated and replaced with fresh media.
The cells were harvested 36–48 h post-vector transfection and analyzed for firefly luciferase and renilla luciferase activities. The luciferase assays were performed at a time prior to confluency of the CV-1 cells in the 48-well plate. Therefore, cells should have been in the exponential phase of cell growth and not at the stationary plateau. The media was aspirated and the cells were lysed using 100 μl of 1× Passive Lysis Buffer (PLB; Promega) on ice for 20 min. Firefly luciferase assays were conducted in an opaque, flat-bottomed 96-well plate (Costar) with 20 μl of lysate, 100 μl of firefly assay buffer (21.5 mM MgCl2, 3.7 mM ATP in 0.1 M KH2PO4, pH 7.8) and 100 μl of 1 mM luciferin (Biosynth, Naperville, IL) dispensed via ML-3000 microplate luminescence system (Dynex technologies, Chantilly, VA). Data were recorded using enhanced flash parameters with BioLinx software v.2.22 and integrated from 0 to 5 s. Renilla luciferase assays were performed using 20 μl of lysate, 100 μl of renilla assay buffer (0.1 M KH2PO4, 0.5 M NaCl and 1 mM EDTA), and dispensed 100 μl of 0.18 μg/ml of coelenterazine (Biosynth). Data were integrated from 0 to 5 s. All experiments were performed at least three times and reported as the ratio of firefly luciferase to renilla luciferase, normalized to the lipid only control.
RESULTS AND DISCUSSION
Design of PNAs and LNAs to target sequences within HCV IRES
We obtained PNA and LNA oligomers (Figure 1 and Tables 1 and 2) to test the hypothesis that chemically modified oligomers could be effective inhibitors of IRES-mediated gene expression inside cells. The IRES sequence controlling expression of HCV was chosen as a target because it has been well characterized and inhibitory oligomers could become lead compounds for therapeutic development (15).
Figure 1.
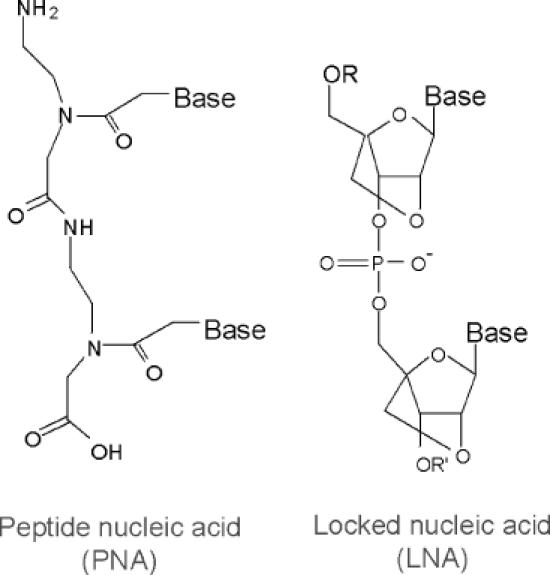
Chemical structures of PNA and LNA.
Table 1. PNA sequences.
| Name | Sequence | Mass expected/found | Tm (°C) | Target IRES sequence |
|---|---|---|---|---|
| P1 | GAGTGATCTATGGTGGA | 4868/4871 | 78 | 26–42 |
| P2 | ACGCCATGGCTAGACGC | 4743/4748 | 80 | 75–91 |
| P3 | GTTGATCCAAGAAAGGA | 4830/4827 | 74 | 191–207 |
| P3–19 | GTTGATCCAAGAAAGGACC | 5332/5335 | 83 | 191–209 |
| P3–21 | GTTGATCCAAGAAAGGACCCG | 5876/5879 | 88 | 191–211 |
| P4 | GCGGGGGCACGCCCAAA | 4793/4795 | 89 | 226–242 |
| P5 | TTTCGCGACCCAACACT | 4653/4664 | 81 | 260–276 |
| P5-scrambled | CATCATCGATCCTAGCC | 4653/4650 | 74 | Control |
| P5-sense | AGTGTTGGGTCGCGAAA | 4853/4850 | 80 | Control |
| P6–15 | ACGAGACCTCCCGGG | 4201/4202 | 83 | 315–329 |
| P6 | TACGAGACCTCCCGGGG | 4759/4764 | 85 | 314–330 |
| P6–19 | CTACGAGACCTCCCGGGGC | 5261/5261 | 88 | 313–331 |
| P6–21 | TCTACGAGACCTCCCGGGGCA | 5803/5800 | 90 | 312–332 |
| P7 | GTGCTCATGGTGCACGG | 4805/4810 | 83 | 333–349 |
| P9 | TGTCGTTCGCGGGCGCA | 4781/4780 | 90 | F-luc |
PNAs are listed from N- to C-termini. All PNAs contain a C-terminal lysine. Tm values are for hybridization to exactly complementary sequences. F-luc: a sequence within the coding region for firefly luciferase.
Table 2. Chimeric LNA sequences.
| Name | Sequence | Tm (°C) | Target IRES sequence |
|---|---|---|---|
| L3 | GTTGATCCAAGAAAGGA | 75 | 191–207 |
| L4 | GCGGGGGCACGCCCAAA | 92 | 226–242 |
| L6 | TACGAGACCTCCCGGGG | 92 | 314–330 |
| L7 | GTGCTCATGGTGCACGG | 83 | 333–349 |
| L8 | AGTGTTGGGTCGCGAAA | 83 | Control |
| L9 | GTCGTTCGCGGGCGC | 81 | F-luc |
Sequences are listed from 5′ to 3′. Boldfaced bases are LNA. Tm values are for hybridization to exactly complementary sequences. F-luc: a sequence within the coding region for firefly luciferase.
The binding of PNAs to RNA inside cells does not cause the degradation of RNA because PNA:RNA hybrids are not substrates for RNAse H (35). To allow direct comparison of the effects of LNAs and PNAs, the LNAs used in these studies (with the exception of L9) were mixtures of LNA and DNA bases, and were designed to have three or fewer consecutive DNA bases. Such designs have been demonstrated to have little or no ability to recruit RNAse H upon binding to RNA (36). The exception, L9, contained seven contiguous bases and would be expected to activate RNAse H (36). L9 was targeted to the coding region of reporter luciferase mRNA and had previously been shown to be a potent inhibitor of expression (29). It was included in these experiments as a positive control for antisense activity.
LNAs and PNAs were designed to be complementary to sites that had been identified to form critical interactions with proteins during translation (Figure 2). Melting temperature (Tm) analysis revealed that PNAs bound with similar stabilities to their target sequences (Table 1). PNA P2 targets a single-stranded apical domain previously determined to be important for IRES-mediated translation (37,38). PNA P3 and LNA L3 are complementary to a region known to bind p120, a subunit of eIF3 (39,40). PNA P4 and LNA L4 were designed to bind to the partially single-stranded region surrounding a four-way junction and disrupt the ion-dependent tertiary folding necessary to bind eIF3 (41).
Figure 2.
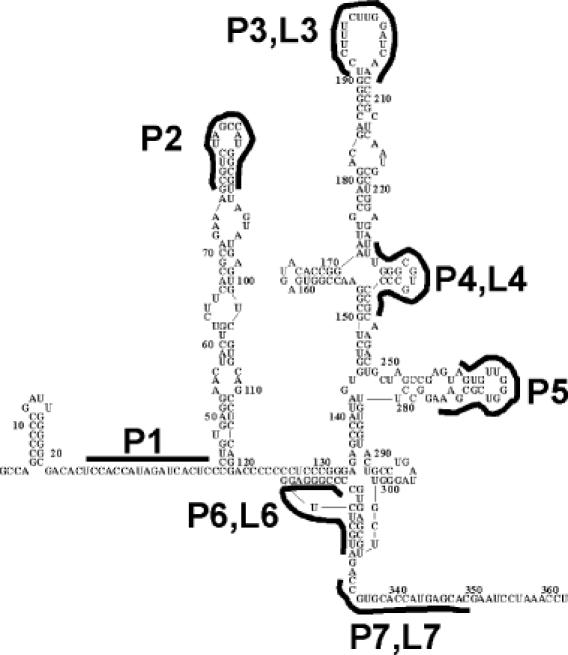
HCV 5′-UTR/IRES secondary structure and PNA or LNA target sites. Dark lines depict hybridization sites for PNAs P1–P7 and LNAs L3, L4, L6 and L7.
PNA P5 targets a site believed to come in contact with the 40S ribosomal subunit (42). PNA P6 and LNA L6 are specific for a conserved RNA pseudoknot (43). PNA P7 and LNA L7 target the AUG translation start site downstream from the IRES and is adjacent to the target site of a phosophorothioate oligonucleotide that is currently being tested in Phase II clinical trials for the treatment of HCV (44). PNA P1 is complementary to a sequence involved in long-range RNA–RNA interactions within the HCV IRES core (428–442 nt) not present in the bicistronic transcript (45). PNA P9 and LNA L9 target a sequence within the coding region of firefly luciferase.
Cellular delivery of PNA and LNA oligomers
PNAs have an uncharged backbone and cannot be delivered into cells by standard lipid-mediated transfection protocols. To facilitate the efficient uptake of PNAs by cultured cells, we anneal PNAs to DNA oligonucleotides and mix the PNA:DNA hybrid with cationic lipid (26,27). The lipid binds to the DNA and allows it to pass through the cell membrane. The hybridized PNA is then transported into the cell as cargo. LNA oligomers have a negatively charged phosphodiester backbone and were delivered into cells using standard lipid-mediated transfection protocols.
The carrier DNA oligonucleotides for transfection of PNAs were chosen to form PNA:DNA complexes in the measured melting temperatures of 65–75°C. Tm values >75°C may result in the PNA not being efficiently released from the DNA, whereas Tm values <65°C may indicate that the complex is too unstable. Typically, we test two or three DNA oligonucleotides per PNA and empirically determine the best combination for inhibiting gene expression. Consistent with previous results (46,47), fluoresence-assisted flow cytometry (FACS) indicated that fluorophore-labeled PNA and LNA oligomers were entering >90% of cells (data not shown).
Inhibition of IRES-dependent translation by PNAs
To investigate the recognition of HCV IRES by PNAs, we introduce several PNAs into CV-1 (monkey kidney) cells. We then introduce the bicistronic transcript expressing plasmid, pRL-HL (Figure 3A) (34). This plasmid encodes both renilla and firefly luciferase from a single transcript driven by a CMV promoter. Expression of renilla luciferase is driven by a cap-dependent mechanism and should not be affected by the addition of PNA. In contrast, the expression of firefly luciferase is under the control of the HCV IRES. Firefly luciferase activity should be inhibited if the PNAs bound to their IRES target sequences and if binding is adequate to disrupt protein association. There was no significant complementarity between the PNAs and the control renilla luciferase gene.
Figure 3.
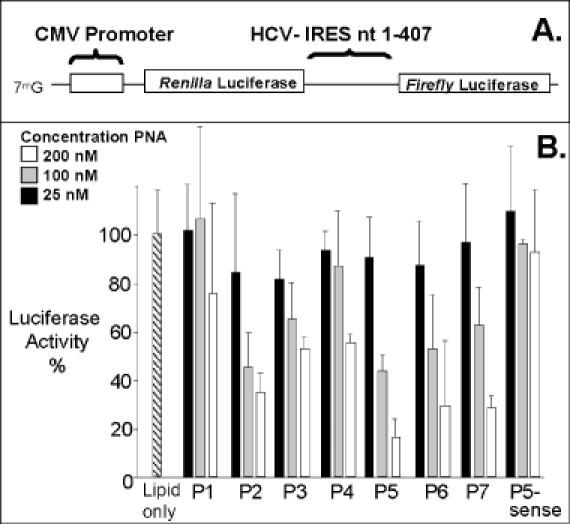
Effect of addition of PNAs on the inhibition of IRES-dependent translation of firefly luciferase. (A) Schematic representation of bicistronic report gene expressing both firefly and renilla luciferase (34). (B) Inhibition of luciferase expression by PNAs P1–P7. Firefly luciferase activity was normalized relative to renilla luciferase activity. Percentages are calculated relative to luciferase activity in the presence of added lipid only. Data are derived from at least three experiments.
Transfection procedures can reduce gene expression relative to untreated controls regardless of the complementarity of an oligomer for its target, because lipid–oligomer complexes can cause cell population growth rate to decrease. This is especially true at higher concentrations of lipid and oligomer. To control for this non-selective inhibition, firefly luciferase activity was normalized using the accompanying values for renilla luciferase activity. There was no correlation between cell growth rates and the observed decrease in IRES-mediated gene expression upon addition of PNAs.
We observed varying levels of inhibition by PNAs P2–P7 (Figure 3B). Inhibition was dose-dependent, with maximal effects observed upon transfection of 200 nM PNA. Addition of PNAs P2, P5, P6 and P7 yielded the highest levels of inhibition, whereas PNAs P3 and P4 were only marginally active. PNA P1, which was targeted to a sequence preceding the well-defined IRES structure, and PNA P5-sense were inactive. Combination of inhibitory PNAs did not significantly enhance the potency of inhibition (data not shown). Gene expression was not inhibited by the addition of lipid in the absence of PNA. These results suggest that the HCV IRES is susceptible to binding by PNAs at a wide range of target sequences.
Effect of PNA length of inhibition of gene expression
In a previous study, we had shown that antisense inhibition of expression of the human caveolin-1 required PNAs that should be at least 19 bases long (27). In our current study, however, we observed that 17 base PNAs were effective inhibitors of IRES-mediated expression (Figure 3B). To investigate this discrepancy, we synthesized PNAs of varied length that were analogous in sequence to PNAs P3 or P6. We observed that PNAs P6–15 or P6–17 appeared to be slightly more effective when compared with longer PNAs P6–19 or P6–21 for the inhibition of HCV IRES-mediated expression of luciferase (Figure 4). The small difference in efficiency between P6 and its longer analogs may be due to the fact that the extra PNA bases are not needed for the recognition but do increase the potential for non-target interactions that can interfere with the recognition of IRES mRNA. P6, P6–19 and P6–21 share a common carrier DNA, so differences in transfection efficiency probably do not account for any difference in efficacy. Seventeen base PNA P3 was approximately as potent an inhibitor as the longer analog PNAs P3-19 and P3-21 (Figure 4).
Figure 4.
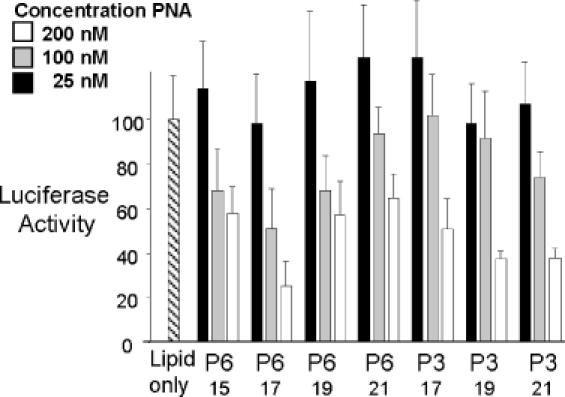
Effect of PNA length on the inhibition of IRES-dependent translation of firefly luciferase. PNA lengths are noted for derivatives of PNAs P6 and P3. Firefly luciferase activity was normalized relative to renilla luciferase activity. Percentages are calculated relative to luciferase activity in the presence of added lipid only. Data are derived from at least three experiments.
PNA length and mechanisms of antisense inhibition
Our observation that 15 or 17 base PNAs are inhibitors of expression stands in striking contrast to our results with PNAs targeted to mRNA encoding human caveolin-1, which showed little inhibition by 15 or 17 base PNAs (27). The PNAs in the caveolin study were targeted to sequences within the coding region of the mRNA and, to function, were required to block ribosome movement. The anti-HCV IRES PNAs, in contrast, probably function by preventing proteins from binding to the RNA. One explanation for the effectiveness of our anti-HCV IRES PNAs is that blocking protein binding may be less demanding than the need to block progress of a ribosome that has already begun translation.
This hypothesis is consistent with a previous study from our laboratory of antisense inhibition by 15 base PNAs (26). In this study, we had observed previously that 15 base PNAs directed to the extreme 5′ end of the transcript mRNA could efficiently block transcription, but that 15 PNAs targeted to downstream target sites were ineffective. From these experiments, we concluded that one active PNA functions by preventing the ribosome from initiating translation, and the binding of the inactive PNAs targeted to downstream sites was insufficient to prevent translation. Consistent with our previous findings, PNA P8, a 17 base PNA targeted to the luciferase-coding region, was inactive (data not shown).
Inhibition of IRES-dependent translation by LNAs
LNAs provide another option for using high affinity hybridization to improve nucleic acid recognition. The LNAs used in these studies were chimeric molecules containing mixtures of DNA and LNA bases. The chimeras were designed to be analogous to PNAs 2–5 (Table 2). No LNA containing more than three contiguous DNA bases and any LNA–RNA hybrids formed would not be predicted to be good substrates for RNAse H.
We transfected the LNAs into cells using cationic lipid and observed the effect of their addition on luciferase expression. Antisense chimeric LNAs L3 and L4 did not substantially inhibit luciferase expression (Figure 5). LNAs L6 and L7 were capable of inhibiting expression to some degree, but none performed as well as analogous PNAs P6 and P7 (Figure 5). Our observation that the inhibition by IRES-directed PNAs is more efficient than LNAs suggests that PNAs may be better able to invade the IRES, possibly due to their uncharged backbone linkages.
Figure 5.
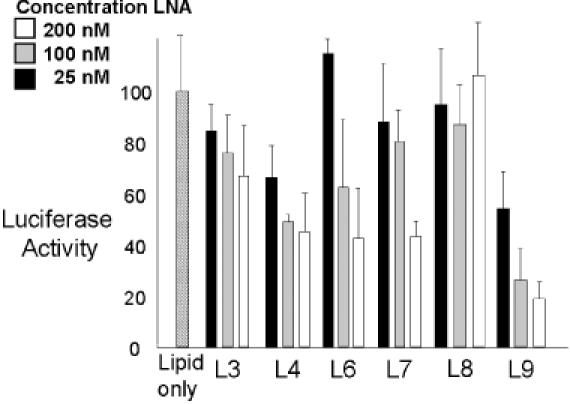
Effect of addition of LNAs on the inhibition of IRES-dependent translation of firefly luciferase. Firefly luciferase activity was normalized relative to renilla luciferase activity. Percentages are calculated relative to luciferase activity in the presence of added lipid only. Data are derived from at least three experiments.
An LNA that has the same sequence as the target sense strand, LNA L8, did not inhibit expression. But LNA L9, an LNA targeted to a downstream sequence within the coding region of luciferase mRNA, was able to inhibit expression.
Factors governing the potencies of anti-IRES PNAs and LNAs
In this study, we observe that some PNAs and LNAs function better than others. Moreover, as a group, the anti-IRES PNAs and LNAs described here are not as potent inhibitors as antisense PNAs and LNAs used in our previous studies of non-IRES targets (26,27,29). It is important to consider the origins of these differences.
One explanation is that transfection efficiencies may vary depending on the PNA or the LNA length or sequence. We do not believe this to be the case, because FACS analysis or fluorescence microscopy of fluorophore-labeled PNAs and LNAs routinely reveals >90% uptake by cells regardless of oligomer length or sequence [(46,47), data not shown, Kaihatsu, unpublished data]. A more likely explanation for the differences in efficiency is that the IRES structures differ in their stabilities and that the entry of PNAs and LNAs is easier at some IRES targets than others. The need to disrupt existing IRES structure would also explain why the levels of inhibition observed in this study are less than those observed in our previous studies of other mRNA targets (26,27,29).
IRES sequences as targets for PNAs and LNAs
IRES sequences are a distinct class of cellular targets for recognition by oligonucleotides and PNAs. Because IRES sequences often direct the synthesis of viral proteins, compounds that efficiently bind to IRES sequences provide lead compounds for therapeutic development. The challenge for the design of these compounds is that they must be able to disrupt RNA secondary structure and compete with proteins for binding at their target sites.
LNAs and PNAs share an ability to bind complementary sequences with high affinity, but also have many characteristics that differ. LNA monomers are more conformationally restricted, while PNAs are relatively flexible. LNA has negatively charged backbone linkages, whereas PNA linkages are uncharged. Finally, PNA is well known for its ability to invade duplex DNA. LNA has also been recently noted to bind duplex DNA (48), but the overall potential for LNA to bind structured nucleic acids is much less well characterized.
Our results demonstrate that PNAs and LNAs can inhibit IRES-mediated expression. Although we cannot exclude the possibility of PNAs and chimeric LNAs targeting pre-folded IRES RNA in our results, Toulme and co-workers (19) demonstrated similar results from in vitro assays of folded IRES RNA transcripts targeted with antisense 2′-O-methyloligoribonucleotides.
Achieving further improvement in the potency of IRES-targeted inhibition is a primary goal for future research. One strategy for improving the potency of inhibition by LNAs directed to IRES sequence is introduction of the ability to recruit RNAse H. We and others have noted that attachment of positive charges to PNAs can enhance strand invasion of duplex DNA (49,50), and similar modifications may also increase binding of RNA structures within HCV IRES. Finally, we and others have shown that the attachment of sugar moieties to PNAs allows liver cell-specific targeting through the asialoglycoprotein receptor, and such modifications may be useful for in vivo testing of anti-IRES PNAs (51,52).
Given the numerous options for improving the efficiency of IRES-mediated inhibition, our results should only be viewed as a starting point. It is likely that further improvements in the PNA and LNA chemistry and cellular delivery will lead to increasingly potent agents for IRES recognition.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Khalil Arar and Alex Amiet (Proligo LLC) for generously providing the LNA oligomers used in these studies and Dr Micheal Gale (UT Southwestern) for helpful discussions. This work was supported by grants from the National Institutes of Health (GM 60642) and the Robert A. Welch Foundation (I-1244).
REFERENCES
- 1.Armstrong G.L., Alter,M.J., McQuillan,G.M. and Margolis,H.S. (2000) The past incidence of hepatitis C virus infection: implications for the future burden of chronic liver disease in the United States. Hepatology, 31, 777–782. [DOI] [PubMed] [Google Scholar]
- 2.Ikeda K., Saitoh,S., Arase,Y., Chayama,K., Suzuki,Y., Kobayashi,M., Tsubota,A., Nakamura,I., Murashima,N., Kumada,H. and Kawanishi,M. (1999) Effect of interferon therapy on hepatocellular carcinogenesis in patients with chronic hepatitis type C: a long-term observation study of 1,643 patients using statistical bias correction with proportional hazard analysis. Hepatology, 29, 1124–1130. [DOI] [PubMed] [Google Scholar]
- 3.Thompson S.R. and Sarnow,P. (2000) Regulation of host cell translation by viruses and effects on cell function. Curr. Opin. Microbiol., 3, 366–370. [DOI] [PubMed] [Google Scholar]
- 4.Wang C., Sarnow,P. and Siddiqui,A. (1993) Translation of human hepatitis C virus RNA in cultured cells is mediated by an internal ribosome-binding mechanism. J. Virol., 67, 3338–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Honda M., Ping,L.H., Rihnbrand,R.C., Amphlett,E., Clarke,B., Rowlands,D. and Lemon,S.M. (1996) Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology, 222, 31–42. [DOI] [PubMed] [Google Scholar]
- 6.Sarnow P. (2003) Viral internal ribosome entry site elements: novel ribosome–RNA complexes and roles in viral pathogenesis. J. Virol., 77, 2801–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vagner S., Galy,B. and Pyronnet,S. (2001) Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Rep., 2, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spahn C.M., Kieft,J.S., Grassucci,R.A., Penczek,P.A., Zhou,K., Doudna,J.A. and Frank,J. (2001) Hepatitis C virus IRES RNA-induced changes in the conformation of the 40S ribosomal subunit. Science, 291, 1959–1962. [DOI] [PubMed] [Google Scholar]
- 9.Buratti E., Tsminetzky,S., Zotti,M. and Baralle,F.E. (1998) Functional analysis of the interaction between HCV 5′ UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res., 26, 3179–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukavsky P.J., Otto,G.A., Lancaster,A.M., Sarnow,P. and Puglisi,J.D. (2000) Structures of two RNA domains essential for hepatitis C virus internal ribosome entry site function. Nature Struct. Biol., 7, 1105–1110. [DOI] [PubMed] [Google Scholar]
- 11.McCaffrey A.P., Ohashi,K., Meuse,L., Shen,S., Lancaster,A.M., Lukavsky,P.J., Sarnow,P. and Kay,M.A. (2002) Determinants of hepatitis C translational initiation in vitro, in cultured cells and mice. Mol. Ther., 5, 676–684. [DOI] [PubMed] [Google Scholar]
- 12.Blight K.J., Kolyhalov,A.A., Reed,K.E., Agapov,E.V. and Rice,C.M. (1998) Molecular virology of hepatitis C virus: an update with respect to potential antiviral targets. Antivir. Ther., 3 (Suppl. 3), 71–81. [PubMed] [Google Scholar]
- 13.Gallego J. and Varani,G. (2002) The hepatitis C virus internal ribosome-entry site: a new target for antiviral research. Biochem. Soc. Trans., 30, 140–145. [DOI] [PubMed] [Google Scholar]
- 14.Klinck R., Westhof,E., Walker,S., Afshar,M., Collier,A. and Aboul-Ela,F. (2000) A potential RNA drug target in the hepatitis C virus internal ribosomal entry site. RNA, 6, 1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinand-Mari C., Lebleu,B. and Robbins,I. (2003) Oligonucleotide-based strategies to inhibit human hepatitis C virus. Oligonucleotides, 13, 539–548. [DOI] [PubMed] [Google Scholar]
- 16.Hanecak R., Brown-Driver,V., Fox,M.C., Azad,R.F., Furusako,S., Nozaki,C., Ford,C., Sasmor,H. and Anderson,K.P. (1996) Antisense oligonucleotide inhibition of hepatitis C virus gene expression in transformed hepatocytes. J. Virol., 70, 5203–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown-Driver V., Eto,T., Lesnik,E., Anderson,K.P. and Hanecak,R.C. (1999) Inhibition of translation of hepatitis C virus RNA by 2′-modified antisense oligonucleotides. Antisense Nucl. Acid Drug Dev., 9, 145–154. [DOI] [PubMed] [Google Scholar]
- 18.Michel T., Martinand-Mari,C., Debart,F., Lebleu,B., Robbins,I. and Vasseur,J.-J. (2003) Cationic phosphoramidate alpha-oligonucleotides efficiently target single-stranded DNA and RNA and inhibit hepatitis C virus IRES-mediated translation. Nucleic Acids Res., 31, 5282–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tallet-Lopez B., Aldaz-Carroll,L.,Chabas,S., Dausse,E., Staedel,C. and Toulme,J.-J. (2003) Antisense oligonucleotides targeted to the domain IIId of the hepatitis C virus IRES compete with 40S ribosomal subunit binding and prevent in vitro translation. Nucleic Acids Res., 31, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi K., Kim,J.H., Li,X., Paek,L.Y., Ha,S.H., Ryu,S.H., Wimmer,E. and Jang,S.K. (2004) Nucleic Acids Res., 32, 1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen P.G., Egholm,M., Berg,R.H. and Buchardt,O. (1991) Sequence-selective recognition of DNA by strand displacement with a thymine substituted polyamide. Science, 254, 1497–1500. [DOI] [PubMed] [Google Scholar]
- 22.Koshkin A.A. and Wengel,J. (1998) Synthesis of novel 2′,3′-linked bicyclic thymine ribonucleosides. J. Org. Chem., 63, 2778–2781. [DOI] [PubMed] [Google Scholar]
- 23.Obika S., Nanbu,D., Hari,Y., Andoh,J., Morio,K., Doi,T. and Imanishi,T. (1998) Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett., 39, 5401–5404. [Google Scholar]
- 24.Demidov V.V., Potaman,V.N., Frank-Kamenetskii,M.D., Egholm,M., Buchardt,O., Sonnichsen,S.H. and Nielsen,P.E. (1994) Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol., 48, 1310–1313. [DOI] [PubMed] [Google Scholar]
- 25.Egholm M., Buchardt,O., Christensen,L.R., Behrens,C., Freier,S.M., Driver,D.A., Berg,R.H., Kim,S.K., Norden,B. and Nielsen,P.E. (1993) PNA hybridizes to complementary oligonucleotides obeying Watson–Crick hydrogen bonding rules. Nature, 365, 566–568. [DOI] [PubMed] [Google Scholar]
- 26.Doyle D.F., Braasch,D.A., Simmons,C.G., Janowski,B.A. and Corey,D.R. (2001) Inhibition of gene expression inside cells by peptide nucleic acids: effect of mRNA target sequence, mismatched bases, and PNA length. Biochemistry, 40, 53–64. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y., Braasch,D.A., Nulf,C.J. and Corey,D.R. (2004) Efficient and isoform-selective inhibition of cellular gene expression by peptide nucleic acids. Biochemistry, 43, 1921–1927. [DOI] [PubMed] [Google Scholar]
- 28.Braasch D.A. and Corey,D.R. (2001) Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem. Biol., 8, 1–7. [DOI] [PubMed] [Google Scholar]
- 29.Braasch D.B., Liu,Y. and Corey,D.R. (2002) Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. Nucleic Acids Res., 30, 5160–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grunweller A., Wyszko,E., Bieber,B., Jahnel,R., Erdmann,V.A. and Kurreck,J. (2003) Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res., 31, 3185–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frieden M., Christensen,S.M., Mikkelsen,N.D., Rosenbohm,C., Thrue,C.A., Westergaard,M., Hansen,H.F., Orum,H. and Koch,T. (2003) Expanding the design horizon of antisense oligonucleotides with alpha-L-LNA. Nucleic Acids Res., 31, 6365–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arzumanov A., Walsh,A.P., Rajwanshi,V.K., Kumar,R., Wengel,J. and Gait,M.J. (2001) Inhibition of HIV-1 Tat-dependent trans activation by steric block chimeric 2′-O-methyl/LNA oligoribonucleotides. Biochemistry, 40, 14645–14654. [DOI] [PubMed] [Google Scholar]
- 33.Mayfield L.D. and Corey,D.R. (1999) Automated synthesis of peptide nucleic acids and peptide nucleic acid-peptide conjugates. Anal. Biochem., 268, 401–404. [DOI] [PubMed] [Google Scholar]
- 34.Honda M., Kaneko,S., Matsushita,E., Kobayashi,K., Abell,G.A. and Lemon,S.M. (2000) Cell cycle regulation of hepatitis C virus internal ribosomal entry site-directed translation. Gastroenterology, 118, 152–162. [DOI] [PubMed] [Google Scholar]
- 35.Knudsen H. and Nielsen,P.E. (1996) Antisense properties of duplex- and triplex-forming PNAs. Nucleic Acids Res., 24, 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurreck J., Wyszko,E., Gillen,C. and Erdmann,V.A. (2002). Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res., 30, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Honda M., Ping,L.H., Rijnbrand,R.C., Amphlett,E., Clarke,B., Rowlands,D. and Lemon,S.M. (1996) Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology, 222, 31–42. [DOI] [PubMed] [Google Scholar]
- 38.Honda M., Rijnbrand,B., Abell,G., Kim,D. and Lemon,S.M. (1999) Natural variation in translational activities of the 5′ nontranslated RNAs of hepatitis C virus genotypes 1a and 1b: evidence for a long-range RNA–RNA interaction outside of the internal ribosomal entry site. J. Virol., 73, 4941–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Buratti E., Tisminetzky,S., Zotti,M. and Baralle,F.E. (1998) Functional analysis of the interaction between HCV 5′UTR and putative subunits of eukaryotic translation initiation factor eIF3. Nucleic Acids Res., 26, 3179–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collier A.J., Gallego,J., Klinck,R., Cole,P.T., Harris,S.J., Harrison,G.P., Aboul-Ela,F., Varani,G. and Walker,S. (2002) A conserved RNA structure within the HCV IRES eIF3-binding site. Nature Struct. Biol., 9, 375–380. [DOI] [PubMed] [Google Scholar]
- 41.Kieft J.S., Zhou,K., Jubin,R., Murray,M.G., Lau,J.Y. and Doudna,J.A. (1999) The hepatitis C virus internal ribosome entry site adopts an ion-dependent tertiary fold. J. Mol. Biol., 292, 513–529. [DOI] [PubMed] [Google Scholar]
- 42.Kolupaeva V.G., Pestova,T.V. and Hellen,C.U. (2000) An enzymatic footprinting analysis of the interaction of 40S ribosomal subunits with the internal ribosomal entry site of hepatitis C virus. J. Virol., 74, 6242–6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C., Le,S.Y., Ali,N. and Siddiqui,A. (1995) An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region. RNA, 1, 526–537. [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H., Hanecak,R., Brown-Driver,V., Azad,R., Conklin,B., Fox,M.C. and Anderson,K.P. (1999) Antisense oligonucleotide inhibition of hepatitis C virus (HCV) gene expression in livers of mice infected with an HCV-vaccinia virus recombinant. Antimicrob. Agents Chemother., 43, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y.K., Lee,S.H., Kim,C.S., Seol,S.K. and Jang,S.K. (2003) Long-range RNA–RNA interaction between the 5′ nontranslated region and the core-coding sequences of hepatitis C virus modulates the IRES-dependent translation. RNA, 9, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamilton S.E., Simmons,C.G., Kathriya,I. and Corey,D.R. (1999) Cellular delivery of peptide nucleic acids and inhibition of human telomerase. Chem. Biol., 6, 343–351. [DOI] [PubMed] [Google Scholar]
- 47.Elayadi A.N., Braasch,D.A. and Corey,D.R. (2002) Implications of high affinity hybridization by locked nucleic acids for inhibition of human telomerase. Biochemistry, 41, 9973–9981. [DOI] [PubMed] [Google Scholar]
- 48.Hertoghs K.M., Ellis,J.H. and Catchpole,I.R. (2003) Use of locked nucleic acid oligonucleotides to add functionality. Nucleic Acids Res., 31, 5817–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaihatsu K., Shah,R.H., Zhao,X. and Corey,D.R. (2003) Extending recognition by peptide nucleic acids (PNAs): binding to duplex DNA and inhibition of transcription by tail-clamp PNA-peptide conjugates. Biochemistry, 42, 13996–14003. [DOI] [PubMed] [Google Scholar]
- 50.Bentin T. and Nielsen,P.E. (1996) Enhanced peptide nucleic acid binding to supercoiled DNA: possible implications for DNA “breathing” dynamics. Biochemistry, 35, 8863–8869. [DOI] [PubMed] [Google Scholar]
- 51.Zhang X., Simmons,C.G. and Corey,D.R. (2001) Synthesis and intracellular delivery of lactose-labeled PNAs. Bioorg. Med. Chem. Lett., 11, 1269–1272. [DOI] [PubMed] [Google Scholar]
- 52.Hamzavi R., Dolle,F., Tavitian,B., Dahl,O. and Nielsen,P.E. (2003) Modulation of the pharmacokinetic properties of PNA: preparation of galactosyl, mannosyl, fucosyl, N-acetylgalactosaminyl, and N-acetylglucosaminyl derivatives of aminoethylglycine peptide nucleic acid monomers and their incorporation into PNA oligomers. Bioconj. Chem., 14, 941–954. [DOI] [PubMed] [Google Scholar]