


Abstract
Eighteen subclasses of S-adenosyl-l-methionine (AdoMet) radical proteins have been aligned in the first bioinformatics study of the AdoMet radical superfamily to utilize crystallographic information. The recently resolved X-ray structure of biotin synthase (BioB) was used to guide the multiple sequence alignment, and the recently resolved X-ray structure of coproporphyrinogen III oxidase (HemN) was used as the control. Despite the low 9% sequence identity between BioB and HemN, the multiple sequence alignment correctly predicted all but one of the core helices in HemN, and correctly predicted the residues in the enzyme active site. This alignment further suggests that the AdoMet radical proteins may have evolved from half-barrel structures (αβ)4 to three-quarter-barrel structures (αβ)6 to full-barrel structures (αβ)8. It predicts that anaerobic ribonucleotide reductase (RNR) activase, an ancient enzyme that, it has been suggested, serves as a link between the RNA and DNA worlds, will have a half-barrel structure, whereas the three-quarter barrel, exemplified by HemN, will be the most common architecture for AdoMet radical enzymes, and fewer members of the superfamily will join BioB in using a complete (αβ)8 TIM-barrel fold to perform radical chemistry. These differences in barrel architecture also explain how AdoMet radical enzymes can act on substrates that range in size from 10 atoms to 608 residue proteins.
INTRODUCTION
S-Adenosyl-l-methionine (AdoMet) radical proteins correspond to a newly identified superfamily with an estimated 600 unique sequences (1). They are involved in various biosynthetic pathways for vitamins, cofactors, antibiotics or DNA, and are called ‘Radical SAM’, ‘SAM radical’ or ‘AdoMet radical’ proteins based on their use of AdoMet as a substrate or cofactor (1,2). They all share a conserved consensus ‘CxxxCxxC’ motif demonstrated to be responsible for the binding of an Fe4S4 cluster (3–6), which is involved in the reductive cleavage of AdoMet to generate a 5′ deoxyadenosyl radical (5′dA·) (2,7). This radical species is then used to initiate radical-based chemistry on various substrates, according to the functional specificity of the enzyme. Among this superfamily, only a few members have been extensively characterized: biotin synthase (BioB), lipoate synthase (LipA), lysine-2,3-aminomutase (KamA), coproporphyrinogen III oxidase (HemN), pyruvate formate-lyase activating enzyme (PflA), class III RNR activating enzyme (NrdG) and spore photoproduct lyase (SplB) [reviewed in (2,8,9)].
The bioinformatics study on the AdoMet radical protein family by Sofia and co-workers (1) identified a common core of about 200 amino acids containing a few conserved patches of residues, but a full analysis was hindered by the lack of three-dimensional structure information. While sequence homology within a subclass (e.g. biotin synthase) is good, sequence homology between subclasses (e.g. biotin synthase and NrdG) is not as good (see Tables 1 and 2), making it difficult to align full-length sequences of one subclass to another without a three-dimensional structure framework.
Table 1. General information about sequences used in the Figure 2 alignment.
| Subclasses | BioB E.coli | Unk1 M.jannaschii | ThiH E.coli | Unk2 D.vulgaris | PylB M.mazei | CofG M.jannaschii | CofH M.jannaschii | LipA E.coli | MiaB E.coli | MoaA E.coli | NifB A.vinelandii | HemN E.coli | KamA C.subterminal | PflA E.coli | BssD T.aromatica | NrdG E.coli | NirJ P.aeruginosa | SplB B.subtilis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total number of residues | 346 | 375 | 376 | 322 | 349 | 361 | 359 | 321 | 474 | 329 | 502 | 457 | 426 | 245 | 331 | 154 | 387 | 342 |
| Number of residues used in alignment | 226 | 211 | 224 | 219 | 227 | 193 | 188 | 198 | 161 | 186 | 185 | 156 | 174 | 234 | 131 | 129 | 143 | |
| Sequence identity range within subclass | 59–99 | 54–69 | 36–95 | 40–53 | 44–86 | 38–68 | 50–63 | 47–97 | 75–99 | 58–98 | 55–87 | 48–99 | 48–74 | 47–97 | 62–80 | 61–99 | 99–43 | 74–90 |
| Number of sequences used in each subclass | 32 | 4 | 12 | 11 | 4 | 11 | 11 | 48 | 10 | 22 | 14 | 17 | 14 | 18 | 4 | 19 | 5 | 7 |
Table 2. Percentage sequence identity between the different sequences aligned in Figure 2.
| Subclasses | BioB E.coli | Unk1 M.jannaschii | ThiH E.coli | Unk2 D.vulgaris | PylB M.mazei | CofG M.jannaschii | CofH M.jannaschii | LipA E.coli | MiaB E.coli | MoaA E.coli | NifB A.vinelandii | HemN E.coli | KamA C.subterminal | PflA E.coli | BssD T.aromatica | NrdG E.coli | NirJ P.aeruginosa | SplB B.subtilis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BioB E.coli | — | 22 | 18 | 21 | 17 | 12 | 11 | 14 | 13 | 16 | 15 | 9 | 12 | 10 | 10 | 8 | 9 | 12 |
| Unk1 M.jannaschii | — | 20 | 18 | 21 | 22 | 20 | 12 | 12 | 14 | 9 | 9 | 17 | 11 | 8 | 8 | 11 | 13 | |
| ThiH E.coli | — | 15 | 18 | 14 | 10 | 11 | 8 | 20 | 11 | 10 | 10 | 11 | 8 | 11 | 15 | 9 | ||
| Unk2 D.vulgaris | — | 26 | 14 | 11 | 14 | 12 | 16 | 14 | 11 | 13 | 9 | 11 | 14 | 14 | 13 | |||
| PylB M.mazei | — | 17 | 11 | 12 | 12 | 16 | 9 | 15 | 10 | 10 | 8 | 11 | 17 | 10 | ||||
| CofG M.jannaschii | — | 20 | 15 | 10 | 11 | 6 | 9 | 6 | 7 | 5 | 5 | 9 | 9 | |||||
| CofH M.jannaschii | — | 10 | 8 | 12 | 9 | 11 | 10 | 10 | 7 | 11 | 9 | 13 | ||||||
| LipA E.coli | — | 12 | 14 | 21 | 11 | 12 | 9 | 10 | 10 | 9 | 8 | |||||||
| MiaB E.coli | — | 16 | 10 | 17 | 12 | 10 | 8 | 10 | 9 | 7 | ||||||||
| MoaA E.coli | — | 17 | 9 | 17 | 10 | 14 | 20 | 22 | 10 | |||||||||
| NifB A.vinelandii | — | 10 | 20 | 12 | 11 | 14 | 15 | 12 | ||||||||||
| HemN E.coli | — | 10 | 5 | 9 | 8 | 12 | 6 | |||||||||||
| KamA C.subterminal | — | 11 | 8 | 12 | 16 | 13 | ||||||||||||
| PflA E.coli | — | 22 | 16 | 14 | 11 | |||||||||||||
| BssD T.aromatica | — | 21 | 21 | 14 | ||||||||||||||
| NrdG E.coli | — | 16 | 17 | |||||||||||||||
| NirJ P.aeruginosa | — | 13 | ||||||||||||||||
| SplB B.subtilis | — |
Following the recent determination of the X-ray structure of biotin synthase from Escherichia coli (EcBioB) in our laboratory (10), we have extended the work of Sofia et al. and aligned full-length sequences of 18 different AdoMet radical protein subclasses. Interestingly, we found that the interactions with AdoMet observed in the EcBioB structure are conserved among the superfamily and we confirmed that all AdoMet radical proteins share the same structural core. This core or subdomain may be described as part of a TIM barrel, containing the elements required for radical generation. NrdG seems to correspond to the most compact version of an AdoMet radical protein, perhaps representing an ancestral form of this family. These proteins also contain a region that is highly divergent between each subclass, presumably dedicated to function or substrate specificity. We can predict those residues that line the active site pocket for the selected AdoMet radical proteins subclasses and can rationalize how the larger substrates reach the inside of the barrel. The recent determination of the X-ray structure of HemN from E.coli (EcHemN) (11), another member of the AdoMet radical proteins superfamily, confirmed both our structure-based sequence alignment and the predictions we deduced from it. In addition, our structure-based multiple sequence alignment gives insights into the origin and the evolution of AdoMet radical proteins as well as the TIM-barrel fold. Our results on AdoMet radical proteins support an assembly of (βα)8 fold from (βα)2n precursors (12), favoring the convergent evolution of a TIM barrel and suggesting high plasticity upon substrate specificity adaptation.
MATERIALS AND METHODS
Selection of subclasses and sequences
For our study, we selected 13 of the 31 subclasses of AdoMet radical enzymes identified by iterative profile methods by Sofia et al. (1). These 13 included all the well-characterized members of the superfamily (BioB, LipA, KamA, PflA, NrdG, HemN and SplB) and others that were selected to cover the sequence space as defined by the dendrogram in (1). Subclasses identified as ‘another subclass-like’ (e.g. HemN-like, NirJ-like) in (1) were excluded from this study. To these 13, we added two recently characterized AdoMet radical proteins involved in coenzyme F420 biosynthesis (CofH and CofG) (13), and three subclasses (Unk1, Unk2, PylB) whose similarity to BioB (∼17–22% identity) made them useful to bridge BioB sequences with more divergent sequences. Since one step in our alignment protocol involves manual intervention, the use of more than 18 subclasses was impractical.
For each AdoMet radical subclass, the sequence of the best-characterized protein was chosen as the reference sequence. Each set of AdoMet radical sequences was then amplified by a BLAST or PSI-BLAST search (14) either on the EXPASY server (15) or the National Center for Biotechnology Information websites, using the reference sequences as targets. A stringent E-value cut-off (between 1 × 10−100 and 1 × 10−60, depending on the degree of similarity between different subclasses) was used to avoid inclusion of protein sequences that belong to a ‘subclass-like’ group rather than the subclass itself. This selection was important to prevent a high background level during the first stages of multiple sequence alignments for each individual subclass. The number of sequences used for each subclass is shown in Table 1.
Identifying conserved patches for each subclass and between subclasses
For each subclass, a classical multiple sequence alignment was carried out using CLUSTAL (16) to identify core conserved regions. In the first stages of the analysis of these alignments, only the subclasses that contained more than 14 sequences were used, to increase the contrast between these conserved and non-conserved regions (see Tables 1 and 2). Each subclass exhibits multiple patches of three-plus conserved residues (10 patches in BioB sequences, MoaA 7, ThiH 9, NifB 8, HemN 9, LipA 9, KamA 5, PflA 7).
Next, conserved regions from each subclass were compared to find motifs shared among the 18 AdoMet radical families. In addition to the previously noted ‘CxxxCxxC’ motif, a series of highly conserved glycine and proline residues spaced throughout the AdoMet radical sequences were identified. In the EcBioB structure, these conserved glycine and proline residues are located at the beginning or end of most of the secondary structural elements (i.e. β-strands and α-helices) of the TIM barrel (Figure 1), where they are likely to play a role as secondary structure terminators (17). Glycine/proline residues seem to be particularly well conserved in AdoMet radical proteins (26 conserved glycine/proline residues in BioB sequences, 16 in MoaA, 24 in ThiH, 24 in NifB, 22 in HemN, 24 in LipA, 22 in KamA, 18 in PflA). This high conservation of glycine/proline residues appears to be a general feature of TIM-barrel proteins. For example, 34 sequences of triose phosphate isomerase show 19 conserved glycine/proline residues for ∼250 amino acids, and 34 sequences of 2-phosphoglycerate dehydratase show 16 for ∼350 amino acids. The spacing between these conserved glycine or proline residues in different AdoMet radical protein subclasses is also conserved, varying only about plus or minus one residue. In addition, these conserved glycines/prolines delimit the previously observed conserved patches, allowing us to define secondary structure element-containing sequence fragments to use in the multiple sequence alignments.
Figure 1.
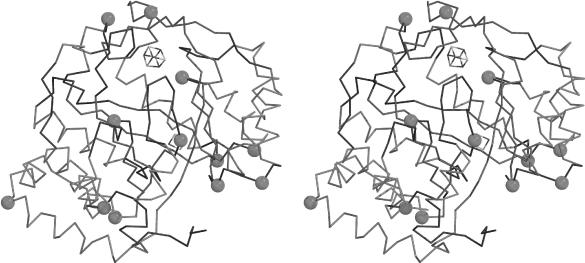
Cα trace of the EcBioB structure. Conserved glycine and/or proline residues within the BioB subclass are represented by a sphere at their Cα position. Strands are depicted in red, helices in blue and loops in black. The Fe4S4 binding loop is depicted in purple.
Creating the multiple sequence alignment
CLUSTAL (16) was used to align the sequence fragments. The alignments were checked manually and adjusted, based on the nature of the residues (i.e. hydrophilic/hydrophobic, large/small, positively/negatively charged), and based on the EcBioB structure and the principle of compensatory mutations. Sequence fragments were included in the alignment based on their length and their agreement with the amino acid pattern in the EcBioB structure and the other aligned sequences. During the first stages of the sequence alignment, only the most conserved sequence segments were included. For the most part, these sequence segments map onto the β-strands of the EcBioB structure in regions near the AdoMet binding site, and thus are likely to be responsible for AdoMet binding and/or radical generation. We subsequently used these conserved blocks as markers along the sequences to try to identify secondary structure elements containing weaker conservation in each subclass (corresponding mainly to helices). These weaker blocks were then incorporated step by step into the alignment, leading to the structure-based multiple sequences alignment presented in Figure 2.
Figure 2.

Multiple sequence alignment containing 18 different AdoMet radical subclasses. For clarity, only one member per subclass is presented here. Each individual selected sequence can be related to the other members of its subclass using standard sequence alignment programs. The alignment corresponds only to structurally homologous sequences in order to link AdoMet radical protein sequences to the EcBioB structure. The numbers in red boxes correspond to the number of omitted residues that were not found to be structurally homologous to the EcBioB structure. The light green boxes correspond to the highly conserved blocks between the different AdoMet radical protein subclasses. The black dashes indicate gaps at these positions. The colored boxes are derived from CLUSTAL (16) and correspond to conserved hydrophobic or aromatic residues (blue), conserved acidic residues (purple), conserved serines or threonines (green), conserved cysteines (pink), conserved lysines or arginines (light red), prolines (yellow) and glycines (orange). Only the parts of the sequences corresponding to the TIM-barrel-like domain are presented here. The secondary structure elements deduced from the EcBioB structure are shown at the top of the alignment, and the ones from EcHemN at the bottom. The parts of the EcHemN sequence represented in gray correspond to fragments that we either misassigned or did not assign. The sequences presented here correspond to E.coli biotin synthase (GI:16128743), the M.jannaschii protein of unknown function (sp|Q58195|), the putative E.coli thiamin synthase (sp|P30140|), the D.vulgaris protein of unknown function (contig 1529 obtained from The Institute for Genomic Research website at http://www.tigr.org), the M.mazei PylB protein (tr|Q8PWY2|), the M.jannaschii CofG and CofH subunits of the 7,8-didemethyl-8-hydroxy-5-deazariboflavin synthase (sp|Q57888| and sp|Q58826|), the E.coli lipoate synthase (sp|P25845|, the E. coli MiaB protein (GI:1786882), the E.coli MoaA protein (sp|P30745|), the A.vinelandii NifB protein (sp|P11067|), the E.coli HemN protein (sp|P32131|), the Clostridium subterminal lysine 2,3-aminomutase (GI:5410603), the E.coli pyruvate formate-lyase activase (sp|P09374|), the T.aromatica benzylsuccinate synthase activase (tr|O87941|), the E.coli class III RNR activase (GI:16132059), the Pseudomonas aeruginosa NirJ protein (tr|P95416|) and the Bacillus subtilis spore photoproduct lyase (GI:16078457).
X-ray structure of HemN as a control
Very recently, the structure of the oxygen-independent coproporphyrinogen III oxidase from E.coli (EcHemN) was solved (11). We used the available coordinates [Protein Data Bank (PDB) code 1OLT] to perform a structural comparison with the EcBioB structure. The structural superposition was performed using LSQMAN (18) and secondary structure elements assignments were deduced from the program DSSP (19) included in the program ESPript (20,21). EcHemN has one of the lowest sequence identities to EcBioB of the proteins used in this study (Table 2), making this structural comparison an important independent criterion to control our assignments and validate our approach.
RESULTS
AdoMet radical proteins have a conserved subdomain
The analysis of the multiple sequence alignments within each selected subclass of AdoMet radical proteins outlined blocks of amino acids with different degrees of conservation. In general, these blocks are not well conserved between subclasses. For example, the highly conserved ‘YNHNLDT’ motif in BioB sequences (residues 150–156 in EcBioB, see Figure 2) is structurally equivalent to the conserved ‘FNHNLEN’ motif in LipA (residues 191–197 in EcLipA), ‘LNTHFNH’ in KamA (residues 228–233 in CsKamA), ‘VMLDLKQ’ in PflA (residues 126–132 in EcPflA) and ‘LSMGVQD’ in HemN (residues 167–173 in EcHemN). This example illustrates why the comparison of sequences between the different subclasses is not easy without a structural scaffold to guide the alignments. With our structure-based approach, however, we have been able to predict a core fold for the AdoMet radical protein family based on (i) the presence of conserved patches of residues delimited by conserved glycines or prolines, (ii) a similar spacing between these patches, (iii) a similar amino acid pattern in the patches and (iv) three motifs, ‘CxxxCxΦC’ (where Φ is an aromatic residue), ‘GGE’ and ‘GxIxGxxE’ (see below and Figures 2 and 3). This core fold appears highly conserved in terms of length of helices and strands (Figure 2). Exceptions include HemN, LipA and MiaB which have a significantly longer helix α1 with a different amino acid pattern, and KamA and NrdG which do not seem to have the additional helix α4A (see Figure 2).
Figure 3.
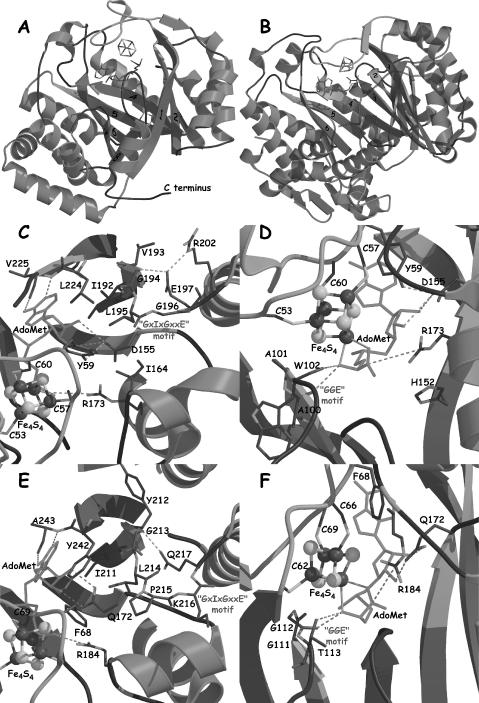
(A and B) Views of the EcBioB and EcHemN structures, respectively. Helices are depicted in blue, strands in red, and loops in black. The numbers indicate the strand number from the N-terminal extremity of the TIM barrel. The zones interacting with AdoMet are depicted in orange. The secondary structural elements that are different in the two structures are depicted in semi-transparent dark blue. (C and D) Views of protein:AdoMet (green) interactions in EcBioB. (E and F) Same views as (C and D) for EcHemN. The color code for secondary structures is the same as in (A).
The comparison of the available EcHemN structure (11) to EcBioB (10) reveals a similar fold (RMSD 2.14 Å for 98 Cα atoms). However, while EcBioB presents a complete (βα)8 TIM-barrel-like fold, EcHemN exhibits only a (βα)6 motif corresponding to three quarters of a barrel (11) (Figure 3B). The lack of closure of the barrel in the EcHemN structure causes the individual β-strands to be less inclined relative to the barrel axis, and the curvature of the β-sheet is not as tight. The observation that AdoMet radical enzymes can have (βα)6- and (βα)8-barrel folds has implications about TIM-barrel fold evolution, and about active site access for larger substrates in some AdoMet enzymes.
The comparison of the EcHemN structure to our secondary structure assignment for that particular protein shows that all but one of our predictions are correct (see Figure 2). With the exception of the assignment of helix α2, which does not share the same length or location as in the EcBioB structure, all the other predicted secondary structure elements correspond exactly to those observed in the X-ray structure (see Figure 2). We are able to predict some small differences between structures of EcBioB and EcHemN based on amino acid patterns. For example, the amino acid pattern differences suggested that the first helix of the (βα)6 subdomain in EcHemN would not be located at the same position as in EcBioB. Other small differences were harder to predict, such as the small deviation in the curvature of the beginning of strand β3 for EcHemN compared to EcBioB structure. The structure-based sequence alignment of EcBioB and EcHemN reveals only 9% identity, confirming their high divergence in comparison to the rest of the family (see Table 2). Thus, the agreement between our predictions for EcHemN and the X-ray structure validates the method. The similarities of the structures of EcHemN and EcBioB taken together with our multiple sequence alignment predict a part or whole TIM-barrel fold for all AdoMet radical proteins in this study.
Residues implicated in AdoMet binding and radical generation
The X-ray structure of EcBioB with AdoMet bound to the Fe4S4 cluster has allowed us to identify the residues involved in AdoMet binding and putatively in radical generation. The ‘CxxxCxΦC’ motif (C53 to C60 in EcBioB) is in a loop between strand β1 and helix α1 of the TIM barrel and is involved, as predicted, in the binding of the Fe4S4 cluster (3–6). AdoMet is the fourth ligand of the Fe4S4 cluster and binds the unique iron atom with its N and O atoms from the methionine moiety, again as predicted (22). The interactions between AdoMet and the protein can be divided into three parts: contacts to the methionyl moiety, the ribose and the adenine (Figure 3C and D). The amino N and the carboxylate of the methionyl moiety are positioned to the hydrogen bond with backbone O atoms of A100 and W102, and to form a salt bridge with the guanidinium group of R173, respectively. These interactions may be important for modulating the properties of the methionyl moiety of AdoMet to improve ligation to the fourth Fe atom of the Fe4S4 cluster. The AdoMet ribose is positioned such that hydrogen bonding is possible between the highly conserved D155 in EcBioB structure and O2′ and O3′ atoms of the ribose moiety. The AdoMet adenine interactions involve both hydrophobic stacking and hydrogen bonding. One side of the adenine portion stacks against Y59 (in the EcBioB structure) that belongs to the ‘CxxxCxΦC’ motif and I192, and hydrogen bonds involve the Watson–Crick site of the adenine moiety and the main chain N and O atoms of V225.
The structure-based multiple sequence alignment for the 18 families studied indicates that all the amino acids involved in interactions with AdoMet are part of conserved motifs at conserved positions, except for V225 (see Figure 2). A100, A101 and W102 in EcBioB are located at the C-terminal end of strand β2 (Figure 3D) and are structurally equivalent to a highly conserved ‘GGE’ motif in other AdoMet radical proteins. This glycine-rich motif seems to be important for the proper conformation of the loop following strand β2 to permit the hydrogen bonding with the N atom of the methionine moiety. Indeed, the EcHemN structure presents a similar interaction involving the carbonyl moiety G113 that belongs to the HemN-conserved ‘GGG’ motif (residues 111–113) at the end of strand β2 (11) (Figures 2 and 3F).
Residue D155 in the EcBioB structure is located at the C-terminal end of strand β4 and all AdoMet radical proteins except PflA and BssD (see below) present a highly conserved D, E, N or Q residue at that position (see Figure 2), which allows for hydrogen bonding with the ribose moiety. Interestingly, AdoMet-dependent methyltransferases lack a highly conserved AdoMet binding motif, but typically show the same hydrogen bond between the ribose hydroxyl groups and D or E (23). According to our sequence alignment (Figure 2), EcHemN should have a similar interaction, involving Q172, equivalent to D155 in EcBioB structure, and the hydroxyl groups O2′ and O3′ of the AdoMet ribose moiety. The comparison of EcBioB and EcHemN structures confirms this hydrogen bonding (Figure 3D and F). Furthermore, in both the EcBioB and EcHemN structures, the residue at this position presents unusual backbone torsion angles likely due to its functional role (10,11). Residue I192 in EcBioB belongs to another glycine-rich motif that we will refer to as the ‘GxIxGxxE’ motif. This motif is strictly conserved in the BioB subclass and highly conserved in the 18 AdoMet radical protein subclasses studied here. All sequences present a large hydrophobic residue at the position equivalent to I192 in EcBioB, suitable for stacking with the adenine moiety of AdoMet. The following highly conserved G (G194 in EcBioB) and conserved E or D residues (E197 in EcBioB) are located in the loop following strand β5. In the EcBioB structure, the side chain carboxylate group of E197 interacts with the main chain N atom of G194 and is likely to be important for the structure of this loop (Figure 3C), and for the maintenance of the AdoMet binding site. The HemN sequences present a similar ‘DxIxGxPxQ’ motif (residues 209–217 in EcHemN) with insertion of a strictly conserved proline residue between the strictly conserved G and Q (see Figure 2). Again, the EcHemN structure shows a conservation of the hydrophobic interaction between I211 and the adenine moiety of AdoMet, as well as conservation of the hydrogen bonding between the N atom of G213 and the side chain Oε1 atom of Q217. The presence of the strictly conserved P215 is likely to allow for an extra residue to fit in the loop while maintaining the loop's three-dimensional structure and interactions (Figure 3E).
Some interactions are conserved in the tertiary structure but not in the secondary or primary structures. According to our multiple sequence alignment, the arginine that forms a salt bridge with the carboxylate of the methionine moiety is only conserved in the primary structure in some AdoMet radical proteins such as BioB or ThiH. However, the presence of a positive charge counterpart facing the carboxylate moiety of AdoMet seems to be more conserved, as there are other conserved lysines or arginines in other subclasses of AdoMet radical proteins that could substitute for the arginine R173 in EcBioB. This assessment is again confirmed by the EcHemN structure, which presents a strictly conserved arginine residue (R184 in EcHemN) that interacts with the carboxylate moiety of AdoMet. Whereas these arginines do not occupy exactly the same position in EcBioB and EcHemN, neither in their sequences nor in their secondary structures, the guanidinium moieties sit at a very similar location, allowing for the conservation of the salt bridge.
No particular conserved residue or motif was found in HemN corresponding to V225 in the EcBioB structure, which is not surprising since backbone atoms of V225 are making the contacts. An alanine residue in an equivalent position at the end of a β-strand in the EcHemN structure is involved in similar contacts with the adenine moiety of AdoMet (Figure 3C and E). This multiple sequence alignment has been successful in predicting the residues involved in both AdoMet binding in HemN and in others that seem to be involved in maintaining the integrity of the structure. Such success with HemN validates this approach.
DISCUSSION
AdoMet radical proteins: a (βα)6 core
Although conserved regions involved in AdoMet binding are spread throughout the sequence, from the ‘CxxxCxΦC’ loop to strand β6, the multiple sequence alignment we obtained shows structural conservation only from strands β1 to β5, ending with the ‘GxIxGxxE’ motif (see Figure 2). Beyond that point, the sequences start to diverge between subclasses, and only some of them (Unk1, ThiH, Unk2, PylB and CofG) have sequences consistent with a hydrophobic strand β6, followed by a helical turn with the ‘GTPΦ’ motif (αGTPΦ in Figure 2) and by a mostly hydrophobic helix α6 that contains two conserved arginines or lysines (R245 and R251 in EcBioB). After strand β7, computer as well as manual alignments fail even for the most similar proteins. This observation of sequence divergence beyond strand β5 is in good agreement with the previous observation by Sofia and co-workers (1) that the conserved core of AdoMet radical proteins contains about 200 amino acids. Within each subclass, however, the story is different and sequence similarities can be observed in the C-terminal region. This leads us to conclude that AdoMet radical proteins have a common (αβ)6 structural core with a highly variable C-terminal region. The former contains the elements for the radical generation and is conserved among the AdoMet radical superfamily. The latter, which has no detectable homology between subclasses, is likely to be substrate specific and adopt a different fold as a function of substrate size and reaction type. This idea is consistent with the structure of EcHemN, which shares the first six strands of a barrel-like fold with BioB, but is missing the last two strands. With the exception of NrdG, which will be discussed in detail later, AdoMet radical enzymes appear to share a common structural subdomain equivalent to a three-quarters barrel.
Conservation near AdoMet binding site can be grouped by substrate type
X-ray structures of BioB and HemN show which residues in these enzymes contact the AdoMet and line the active site, and the multiple sequence alignment suggests which residues in other enzymes will play these roles. Interestingly, the residues contacting the AdoMet are not universally well conserved; instead, conservation is higher between AdoMet radical enzymes that use similar substrates. Such clustering of conservation suggests that residues near the AdoMet binding site, such as D155, N153 and N151 in EcBioB, or Q172, D209 and E145 in EcHemN (Figure 4A and B), are involved in more than AdoMet binding. An example of this clustering is found in BioB and LipA, enzymes that catalyze sulfur insertion reactions with the same stereochemistry on substrates with non-activated carbons (Figure 4D). BioB and LipA share a conserved asparagine patch, ‘YNHNLD’ in BioB and ‘FNHNLE’ in LipA (see Figure 2). A common role for these conserved asparagines in substrate binding is not obvious since N153 of this motif is involved in hydrogen bonding with the ureo moiety of dethiobiotin, and lipoic acid has neither the ureo moiety nor any hydrogen bond donor or acceptor at this location (Figure 4D). Instead, we propose that these residues may be involved in repositioning the 5′dA· species with respect to the highly similar substrates for H atom abstraction. These residues are not highly conserved among all AdoMet radical enzymes because the amount of conformational change of the 5′dA· should differ depending on substrate type and location and whether the AdoMet cleavage is reversible. Because BioB and LipA have similar substrates, we see higher conservation between these enzymes.
Figure 4.
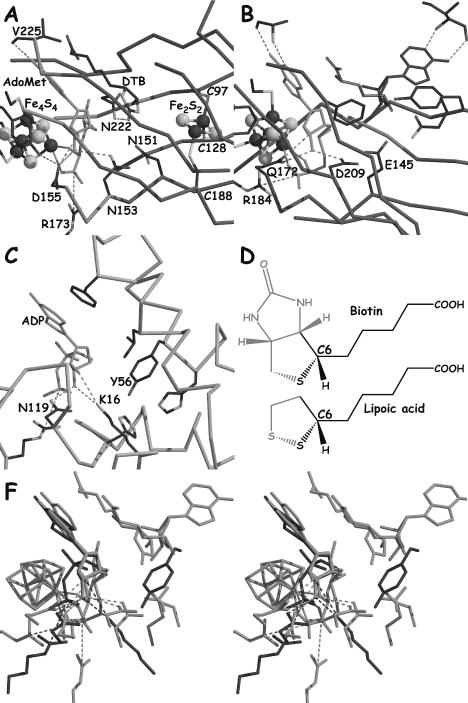
(A–C) A view of the active sites of EcBioB, EcHemN and DdNDPk, respectively, based on the superposition of their ribose moiety. AdoMet and ADP are depicted in green. (D) Comparison of the structures of biotin and lipoic acid. (E) Stereoview of the superposition of EcBioB, EcHemN and DdNDPk in green, orange and blue, respectively, in the same orientation as in (A–C).
Another example of conservation among groups of subclasses is found in full-length activase enzymes. Here, the sequence alignments suggest that a strictly conserved lysine, K131 and K191, in EcPflA and TaBssD, respectively, will occupy the same position as D155 in EcBioB and Q172 in EcHemN, and will interact with the ribose moiety of AdoMet (Figure 4A and B). The use of K to bind a ribose moiety has a precedent in NDP kinase from Dictyostelium discoideum (DdNDPk) (PDB code 1KDN) (24) (Figure 4C and F). The substitution of the typical D, N, E or Q in most AdoMet radical proteins by K in the full-length activases may be important for either AdoMet location or conformation, or to allow a direct H-atom abstraction from the target glycine.
How to accommodate such different substrates
There are fewer residues following strand β6 in proteins that interact with large substrates than for proteins that interact with small molecules substrates. For some of the proteins that bind large substrates, the binding of a protein or DNA substrate could serve to seal off the active site, and the proximity of the active site to AdoMet would still allow for direct hydrogen atom abstraction. In the EcBioB structure, the (αβ)6-barrel subdomain is followed by two short β-strands that complete the TIM-barrel structure. A long loop connecting strand β8 and helix α8 (in red Figure 5A) contributes to the closure of the barrel, and is expected to seal the barrel from solvent upon substrate binding in EcBioB (10). On the other hand, the three-quarter barrel in the EcHemN structure is complemented at its N-terminus by additional secondary structure elements which occupy a location similar to strands β7 and β8, and the N-terminal part of β1 in the EcBioB structure (Figure 5B and C). These different structural features in HemN lead to a large active site cavity, consistent with its larger heme substrate. Both HemN and BioB active sites are accessible from the same side of the barrel (Figure 5A and B).
Figure 5.
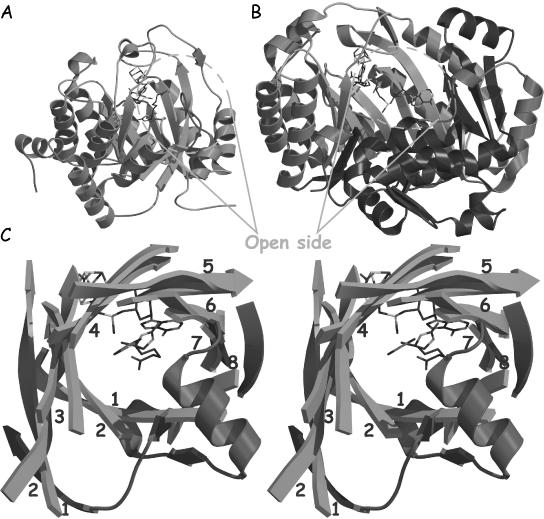
(A) A view of the EcBioB structure showing the more open side with shorter strands 7 and 8, and helices. The loop proposed to ‘close the door’ of the active site upon substrate binding is depicted in red. (B) A view of EcHemN in the same orientation as EcBioB in (A) showing the open side of the active site cavity. The C-terminal region is depicted in purple. (C) Stereoview of the superposition of the strands of the TIM barrel from EcBioB (green) and the equivalent the three-quarter barrel from EcHemN (light purple). The additional secondary structural elements that complement the three-quarter barrel structure in EcHemN are depicted in red.
Class III RNR activase NrdG is the simplest AdoMet radical protein
RNRs are thought to be ancient enzymes, potentially serving as a link between the RNA and the DNA worlds, and of the classes of RNRs, the anaerobic class III RNR is considered to be the most ancient (25). Our sequence alignments suggest that NrdG is structurally the simplest AdoMet radical protein, which leads to a further proposal that NrdG could resemble the ancestor of the AdoMet radical superfamily. Whereas all the other proteins in the AdoMet radical superfamily contain at least 300 residues, NrdG proteins contain only about 160 residues which appears too short to fit either a complete TIM barrel or a (βα)6 subdomain (see Figure 2). Proteins with (αβ)8 folds typically contain upwards of 228 residues (26). In addition to NrdG's short sequence length, sequence similarities between it and other AdoMet radical proteins ends at helix α4 (Figure 2). NrdG may contain a strand β5; however, this strand does not have the generic AdoMet-binding ‘GxIxGxxE’-like motif. Thus, NrdG appears to correspond to only one half of (βα)8 barrel, the half essential for radical generation.
If EcNrdG has a half-barrel structure, then there must be compensating changes to that structure to maintain the necessary interactions with the AdoMet that are provided by backbone atoms from strand β6 in EcBioB and EcHemN. According to our sequence alignment, NrdG does not have helix α4A. Thus, helix α4 may not be at the ‘outer’ side of the β-sheet, as in (αβ)8- or (αβ)6-barrel folds, but rather at the ‘inner’ side, as in flavodoxin-like folds, where it could contribute to the closure of the active site and provide interactions suitable for binding the adenine moiety of AdoMet. It should be noted that NrdG is the only AdoMet radical protein characterized so far without a conserved aromatic residue (Y59 in EcBioB) prior to the last cysteine in the ‘CxxxCxΦC’ motif (see Figure 2). Instead, a conserved aromatic residue is located just after the last cysteine. This alteration may be necessary to complement the difference in tertiary structure of NrdG.
According to sequence comparisons by Sofia et al. (1) and in this study (Table 2), BssD has the highest sequence similarity to NrdG. In terms of function, NrdG, BssD, and PflA all catalyze a glycyl radical formation on a target protein that has or is likely to have the same fold (27–30). However, BssD and PflA sequences are significantly longer than NrdG sequences (approximately 260 residues instead of approximately 160 for NrdG), and contain a conserved motif equivalent to the ‘GxIxGxxE’ motif at the end of strand β5, and a secondary structure element homologous to strand β6 (see Figures 2 and 3). Thus, PflA and BssD are likely to share the same subdomain as BioB and HemN, constituting three-quarters or a complete TIM barrel, and yet by sequence homology and function they are a link between the shorter sequences of NrdG proteins and the more typical length sequences of the majority of AdoMet radical enzymes.
Relationship between half-barrel domains and the flavodoxin fold
The idea of half-barrel (βα)4 proteins has received considerable attention following the X-ray structure determinations of two proteins involved in histidine biosynthesis, imidazoleglycerol phosphate synthase (HisF) and N′-[(5′-phosphoribosyl)formimino]-5-aminoimidazole-4-carboxamide-ribonucleotide isomerase (HisA) (31–33). For these proteins, amino acid sequences and X-ray structures show that the (βα)8 barrels are made up of two superimposable half-barrel subdomains (31). To test the idea that during evolution two (βα)4 half-barrel domains came together to form a functional (βα)8 barrel, Höcker et al. (32) prepared and characterized HisF-N (the N-terminal half of HisF) and HisF-C (the C-terminal half) separately and together. They found that alone HisF-N and HisF-C are inactive, but if co-expressed in vivo or refolded together in vitro, HisF-N and HisF-C assemble into a fully active complex, lending weight to the idea that (βα)8 barrels evolved in a simple gene duplication event from ancestral (βα)4 half-barrels (32). To identify any structures with (βα)4 folds currently deposited in the PDB, Höcker et al. searched the DALI server (http://www.ebi.ac.uk/dali/) using HisF-N and HisF-C as the search models (33). No (βα)4 folds were identified. Instead, besides HisF, HisA and the (αβ)8-barrel enzyme phosphoenolpyruvate mutase, the flavodoxin-like fold of methylmalonyl-CoA mutase (MCM) gave the best hit, yielding Z-scores of 6.3 and 6.4 for HisF-N and HisF-C, respectively (33). This structural homology was interpreted as evidence for a common evolutionary origin of flavodoxin-like fold and half-barrels folds (33). It is interesting to consider why no half-barrel folds are presently found in the PDB. Höcker and co-workers suggested that (αβ)4 half-barrels might have the tendency to aggregate and thus would have evolved an additional half-barrel to fix this problem [i.e. evolved into an (αβ)8 fold], or at least evolved another strand and helices to cover the exposed side of the β-sheet [i.e. evolved into a flavodoxin-like (α/β)5 fold] (33). If EcNrdG does have an (αβ)4 fold, that could explain why this protein is so unstable and prone to aggregation when purified alone (34). The stability of NrdG in vivo may be enhanced by dimerization or by a strong association with its substrate or by both methods. Indeed, NrdG proteins are known to dimerize (34) and the interaction between the class III RNR catalytic subunit and NrdG from E.coli is so strong that it is not possible to separate them by chromatography (34). The same behavior seems to occur for NrdG from bacteriophage T4 and Lactococcus lactis (35).
Evolution of AdoMet radical proteins
According to our structure-based multiple sequence alignment and the comparison between the X-ray structures of EcBioB and EcHemN, two different evolutionary pathways for AdoMet radical proteins can be proposed. The first one corresponds directly to the evolution to the (βα)8 fold from the (βα)4 ancestor by gene duplication and subsequent evolution of the C-terminal sequence to accommodate a wide range of substrates and reactivities, while conserving the radical generation function with the (βα)6 subdomain. This evolutionary pathway would be comparable to HisF and to the independent evolution of the C-terminal half-barrels of prokaryotic and eukaryotic phosphoinositide-specific phospholipases C (PI-PLCs). Indeed, both prokaryotic and eukaryotic PI-PLCs contain a distorted TIM-barrel-like fold. Whereas the first half-barrel is highly conserved and contains all the amino acids essential for function, the second half displays significant structural deviations (36). The second evolutionary pathway corresponds to the evolution from a (βα)4 motif corresponding to an NrdG-like structure, to a (βα)6 motif (HemN) and subsequently to a complete (βα)8 TIM-barrel fold (BioB) by successive addition of (βα)2 motifs. Several three-dimensional structures of different AdoMet radical proteins are required to discriminate between these two possibilities.
Summary
Using a structure-guided multiple sequence alignment approach, we have been able to align 18 subclasses of AdoMet radical proteins. We have found that this alignment correctly predicted all but one of the core helices in HemN, and correctly predicted the enzyme active site residues, despite the low 9% sequence identity between EcBioB and EcHemN. This alignment predicts that anaerobic RNR activase NrdG, an ancient enzyme proposed to serve as a link between the RNA and DNA worlds, will have a structure that most closely represents the progenitor of the AdoMet Radical superfamily; a half-barrel reminiscent of a flavodoxin-like fold. The three-quarter barrel, exemplified by HemN, will likely be the most common architecture for AdoMet radical enzymes, while fewer members will join BioB in using a complete TIM-barrel fold. These three putative architectures for AdoMet radical proteins, (αβ)4, (αβ)6 and (αβ)8, are consistent with the hypothesis that TIM barrels are built with (αβ)2 precursors, and that the TIM-barrel fold observed for BioB is not evolutionarily related to other TIM-barrel proteins but is rather the result of convergent evolution. These variations in barrel architecture also explain how AdoMet radical enzymes can act on substrates that range in size from 10 atoms to 608 residue proteins. DTB is small enough that a loop movement alone could provide access to the active site, whereas the use of a three-quarter barrel in HemN allows access to the active site for the larger substrate coproporphyrinogen. Finally, we have found that residues involved in AdoMet binding and radical generation will be contained in the first part of the barrel fold, the common core. Thus, AdoMet radical enzymes can be thought of as modular, containing a unit with the conserved AdoMet radical generating apparatus and another unit with the determinants for substrate binding and specificity.
Acknowledgments
ACKNOWLEDGEMENTS
This research was funded in part by grants from the NIH, Searle Scholars Program and Alfred P. Sloan Foundation.
REFERENCES
- 1.Sofia H.J., Chen,G., Hetzler,B.G., Reyes-Spindola,J.F. and Miller,N.E. (2001) Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res., 29, 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey P.A. and Booker,S.J. (2001) Radical mechanisms of S-adenosylmethionine-dependent enzymes. Adv. Protein Chem., 58, 1–45. [DOI] [PubMed] [Google Scholar]
- 3.Hewitson K.S., Baldwin,J.E., Shaw,N.M. and Roach,P.L. (2000) Mutagenesis of the proposed iron–sulfur cluster binding ligands in Escherichia coli biotin synthase. FEBS Lett., 466, 372–376. [DOI] [PubMed] [Google Scholar]
- 4.Hewitson K.S., Ollagnier-de Choudens,S., Sanakis,Y., Shaw,N.M., Baldwin,J.E., Munck,E., Roach,P.L. and Fontecave,M. (2002) The iron–sulfur center of biotin synthase: site-directed mutants. J. Biol. Inorg. Chem., 7, 83–93. [DOI] [PubMed] [Google Scholar]
- 5.Layer G., Verfurth,K., Mahlitz,E. and Jahn,D. (2002) Oxygen-independent coproporphyrinogen-III oxidase HemN from Escherichia coli. J. Biol. Chem., 277, 34136–34142. [DOI] [PubMed] [Google Scholar]
- 6.Tamarit J., Gerez,C., Meier,C., Mulliez,E., Trautwein,A. and Fontecave,M. (2000) The activating component of the anaerobic ribonucleotide reductase from Escherichia coli. An iron–sulfur center with only three cysteines. J. Biol. Chem., 275, 15669–15675. [DOI] [PubMed] [Google Scholar]
- 7.Fontecave M., Mulliez,E. and Ollagnier-de-Choudens,S. (2001) Adenosylmethionine as a source of 5′-deoxyadenosyl radicals. Curr. Opin. Chem. Biol., 5, 506–511. [DOI] [PubMed] [Google Scholar]
- 8.Cheek J. and Broderick,J.B. (2001) Adenosylmethionine-dependent iron–sulfur enzymes: versatile clusters in a radical new role. J. Biol. Inorg. Chem., 6, 209–226. [DOI] [PubMed] [Google Scholar]
- 9.Jarrett J.T. (2003) The generation of 5′-deoxyadenosyl radicals by adenosylmethionine-dependent radical enzymes. Curr. Opin. Chem. Biol., 7, 174–182. [DOI] [PubMed] [Google Scholar]
- 10.Berkovitch F., Nicolet,Y., Wan,J.T., Jarrett,J.T. and Drennan,C.L. (2004) Crystal structure of biotin synthase, an S-adenosylmethionine-dependent radical enzyme. Science, 303, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Layer G., Moser,J., Heinz,D.W., Jahn,D. and Schubert,W.D. (2003) Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of radical SAM enzymes. EMBO J., 22, 6214–6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlt J.A. and Raushel,F.M. (2003) Evolution of function in (beta/alpha)8-barrel enzymes. Curr. Opin. Chem. Biol., 7, 252–264. [DOI] [PubMed] [Google Scholar]
- 13.Graham D.E., Xu,H.M. and White,R.H. (2003) Identification of the 7,8-didemethyl-8-hydroxyz-5-deazariboflavin synthase required for coenzyme F420 biosythesis. Arch. Microbiol., 180, 455–464. [DOI] [PubMed] [Google Scholar]
- 14.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasteiger E., Gattiker,A., Hoogland,C., Ivanyi,I., Appel,R.D. and Bairoch,A. (2003) ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res., 31, 3784–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creighton T.E. (1984) Proteins, 1st edn. W.H. Freeman and Company, New York, NY. [Google Scholar]
- 18.Kleywegt G.J., Zou,J.Y., Kjeldgaard,M. and Jones,T.A. (2001) Around O. In Rossmann,M.G. and Arnold,E. (eds), International Tables for Crystallography, Volume F. Crystallography of Biological Macromolecules. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp. 353–367. [Google Scholar]
- 19.Kabsch W. and Sander,C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- 20.Gouet P., Courcelle,E., Stuart,D.I. and Metoz,F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- 21.Gouet P., Robert,X. and Courcelle,E. (2003) ESPript/ENDscript: extracting and rendering sequence and 3D information. Nucleic Acids Res., 31, 3320–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walsby C.J., Ortillo,D., Broderick,W.E., Broderick,J.B. and Hoffman,B.M. (2002) An anchoring role for FeS clusters: chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe–4S] cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc., 124, 11270–11271. [DOI] [PubMed] [Google Scholar]
- 23.Schubert H.L., Blumenthal,R.M. and Cheng,X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci., 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y.W., Morera,S., Janin,J. and Cherfils,J. (1997) AlF3 mimics the transition state of protein phosphorylation in the crystal structure of nucleoside diphosphate kinase and MgADP. Proc. Natl Acad. Sci. USA, 94, 3579–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reichard P. (2002) Ribonucleotide reductases: the evolution of allosteric regulation. Arch. Biochem. Biophys., 397, 149–155. [DOI] [PubMed] [Google Scholar]
- 26.Walden H., Bell,G.S., Russell,R.J., Siebers,B., Hensel,R., Taylor,G.L. and Taylor,G.L. (2001) Tiny TIM: a small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol., 306, 745–757. [DOI] [PubMed] [Google Scholar]
- 27.Leuthner B., Leutwein,C., Schulz,H., Horth,P., Haehnel,W., Schiltz,E., Schagger,H. and Heider,J. (1998) Biochemical and genetic characterization of benzylsuccinate synthase from Thauera aromatica: a new glycyl radical enzyme catalysing the first step in anaerobic toluene metabolism. Mol. Microbiol., 28, 615–628. [DOI] [PubMed] [Google Scholar]
- 28.Leppanen V.M., Merckel,M.C., Ollis,D.L., Wong,K.K., Kozarich,J.W. and Goldman,A. (1999) Pyruvate formate lyase is structurally homologous to type I ribonucleotide reductase. Structure, 7, 733–744. [DOI] [PubMed] [Google Scholar]
- 29.Becker A., Fritz-Wolf,K., Kabsch,W., Knappe,J., Schultz,S. and Volker Wagner,A.F. (1999) Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nature Struct. Biol., 6, 969–975. [DOI] [PubMed] [Google Scholar]
- 30.Logan D.T., Andersson,J., Sjöberg,B.-M. and Nordlund,P. (1999) A glycyl radical site in the crystal structure of a class III ribonucleotide reductase. Science, 283, 1499–1504. [DOI] [PubMed] [Google Scholar]
- 31.Lang D., Thoma,R., Henn-Sax,M., Sterner,R. and Wilmanns,M. (2000) Structural evidence for evolution of the beta/alpha barrel scaffold by gene duplication and fusion. Science, 289, 1546–1550. [DOI] [PubMed] [Google Scholar]
- 32.Hocker B., Beismann-Driemeyer,S., Hettwer,S., Lustig,A. and Sterner,R. (2001) Dissection of a (betaalpha)8-barrel enzyme into two folded halves. Nature Struct. Biol., 8, 32–36. [DOI] [PubMed] [Google Scholar]
- 33.Hocker B., Schmidt,S. and Sterner,R. (2002) A common evolutionary origin of two elementary enzyme folds. FEBS Lett., 510, 133–135. [DOI] [PubMed] [Google Scholar]
- 34.Fontecave M., Mulliez,E. and Logan,D.T. (2002) Deoxyribonucleotide synthesis in anaerobic microorganisms: the class III ribonucleotide reductase. Prog. Nucleic Acid Res. Mol. Biol., 72, 95–127. [DOI] [PubMed] [Google Scholar]
- 35.Torrents E., Eliasson,R., Wolpher,H., Graslund,A. and Reichard,P. (2001) The anaerobic ribonucleotide reductase from Lactococcus lactis. Interactions between the two proteins NrdD and NrdG. J. Biol. Chem., 276, 33488–33494. [DOI] [PubMed] [Google Scholar]
- 36.Heinz D.W., Essen,L.O. and Williams,R.L. (1998) Structural and mechanistic comparison of prokaryotic and eukaryotic phosphoinositide-specific phospholipases C. J. Mol. Biol., 275, 635–650. [DOI] [PubMed] [Google Scholar]