

Abstract
BACKGROUND & AIMS
Core 1- and 3-derived mucin-type O-linked oligosaccharides (O-glycans) are major components of the colonic mucus layer. Defective forms of colonic O-glycans, such as Tn antigen, are frequently observed in patients with ulcerative colitis and colorectal cancer, but it is not clear if they contribute to pathogenesis. We investigated whether and how impaired O-glycosylation contributes to development of colitis-associated colorectal cancer using mice lacking intestinal core 1- and 3-derived O-glycans.
METHODS
We generated mice that lack the core 1- and 3-derived intestinal O-glycans (DKO mice) and analyzed them, along with mice that lack the intestinal epithelial core 1 O-glycans (IEC C1galt1−/− mice) or mice that lack core 3 O-glycans (C3Gnt−/− mice). Intestinal tissues were collected at different time points and analyzed for levels of mucin and Tn antigen, development of colitis, and tumor formation using imaging, quantitative PCR, immunoblot, and ELISA techniques. We also used cellular and genetic approaches, as well as intestinal microbiota depletion, to identify inflammatory mediators and pathways that contribute to disease in DKO and wild-type littermates (controls).
RESULTS
Intestinal tissues from DKO mice contained higher levels of Tn antigen and had more severe spontaneous chronic colitis than tissues from IEC C1galt1−/− mice, whereas spontaneous colitis was absent in C3GnT−/− and control mice. IEC C1galt1−/− mice and DKO mice developed spontaneous colorectal tumors, although the onset of tumors in the DKO mice was earlier (age 8–9 months) than that in IEC C1galt1−/− mice (around age 12 months). Antibiotic depletion of the microbiota did not cause loss of Tn antigen but did reduce the development of colitis and cancer formation in DKO mice. Colon tissues from DKO mice, but not control mice, contained active forms of caspase-1 and increased caspase-11, which were reduced after antibiotic administration. Supernatants from colon tissues of DKO mice contained increased levels of interleukin-1β and interleukin-18, compared to those from control mice. Disruption of the caspase 1 and caspase 11 genes in DKO mice (DKO/Casp1/11−/− mice) decreased development of colitis, characterized by reduced colonic thickening, hyperplasia, and inflammatory infiltrate, compared with DKO mice.
CONCLUSIONS
Impaired expression of O-glycans causes colonic mucus barrier breach and subsequent microbiota-mediated activation of caspase 1-dependent inflammasomes in colonic epithelial cells of mice. These processes could contribute to colitis-associated colon cancer in humans.
Keywords: mucin-type O-glycans, mucus barrier, ulcerative colitis, mouse model
Introduction
Colorectal cancer may be initiated by mutations in key proto-oncogenes or tumor suppressors that occur somatically or are inherited,1 or induced by unresolved chronic intestinal inflammation such as inflammatory bowel disease (IBD).2 The pathogenesis of inflammation-associated cancer is less well understood relative to sporadic colorectal cancer, although components of the innate immune system and microbiota are regarded as pivotal players.3, 4
The colonic mucus layer constitutes an innate defense barrier for homeostasis of the host with the microbiota.5, 6 The mucin MUC2 forms the major structural basis of the mucus layer in humans and mice.6 MUC2 is primarily composed of O-glycans, which have two main subtypes known as core 1– and core 3–derived O-glycans.7, 8 The primary O-glycan structure linked to Ser or Thr is referred to as Tn antigen (GalNAcα-O-Ser/Thr).9 Tn antigen is normally extended to form core 1–derived glycans by the core 1 β1,3-galactosyltransferase (C1GALT1), which is expressed in most tissues,9 or core 3–derived O-glycans by core 3 β1,3-N-acetylglucosaminyltransferase (C3GnT), which is expressed mainly in the intestine.7
Both ulcerative colitis (UC) and colorectal cancer tissues show defective mucin O-glycosylation, including exposure of Tn antigen.10, 11 Tn antigen is considered a tumor-associated carbohydrate antigen,12 although its role in tumor development in vivo remains elusive. We previously demonstrated that mice lacking core 3–derived O-glycans are susceptible to chemically induced colitis and colon cancer,7 and that mice lacking intestinal core 1–derived O-glycans (IEC C1galt1−/−) develop spontaneous colitis.8 The primary defect in both strains, especially IEC C1galt1−/− mice, was impaired mucus barrier function, enabling rapid breach of the mucus layer by bacteria, which initiates colitis in a myeloid cell–dependent manner.8 However, Toll-like receptors (TLRs), which are important for microbial recognition, are not essential for bacterial-driven disease in IEC C1galt1−/− mice because loss of TLR4, or the TLR family adaptor Myd88, does not protect these mice from developing colitis.8
Recently, much attention has focused on deciphering the role of mucosal inflammasomes in IBD and cancer.13, 14,3, 15 Caspases 1 and 11 are key components of canonical and non-canonical inflammasomes, respectively, which are multiprotein complexes whose formation is dependent on diverse stimuli of microbial, environmental, or endogenous origin, and usually require additional components including members of the Nod-like receptor (NLR) family and adaptor proteins.16 Both types of inflammasomes function to activate caspase 1 or 11 leading to proteolytic processing of proIL-1β and proIL-18 that are then secreted and promote inflammation.16, 17 Caspase 1– and 11–dependent inflammasomes modulate inflammation and/or carcinogenesis in chemical models of colitis,18, 19 but their role in spontaneous colitis and cancer is unclear.
Here we show that mice lacking both core 3 and intestinal core 1 O-glycans spontaneously develop colitis and colorectal cancer. Our results show that defective O-glycosylation causes impaired mucin expression and defective mucus barrier that leads to microbiota-driven chronic colitis and colitis-associated tumorigenesis independent of Tn antigen expression. Furthermore, we found that both colitis and colonic tumorigenesis are mediated by epithelial-derived mucosal inflammasomes.
Materials and Methods
Generation of Mice
Mice lacking core 3-derived O-glycans (C3GnT−/−), or intestinal core 1-derived O-glycans (C1galt1f/f;VillinCre or IEC C1galt1−/−) were generated as described.7, 8 Mice lacking both types of O-glycans (DKO) were generated through crossing IEC C1galt1−/− with C3GnT−/− mice. DKO mice were crossed with mice lacking both caspase 1 and 11 (Casp1/11−/−) to generate DKO mice lacking major canonical and non-canonical inflammasomes (DKOΔinfl). Mixed sexes were used throughout the studies as phenotypes did not exhibit gender dependency. Broad-spectrum antibiotics to deplete the microbiota were administered as described.8
Tissue Preparation and Staining
Fresh tissues were harvested from mice, fixed and processed (paraffin-embedding, cryopreservation) for histology and immunostaining as described.8 Immunohistochemical and/or epifluorescent staining was performed as described for all antibodies.8
Bacterial Analysis
Bacterial analysis in antibiotic or non-treated mice was carried out by FISH using the universal EUB338 probe, and by qPCR of 16S rRNA gene from gDNA extracted from fecal materials.6
Protein Extraction and Western Blotting
Cell or tissue lysates were quantified, separated by SDS PAGE, transferred to PVDF membrane and probed with given antibodies. Bands were detected by standard chemiluminescence and imaged for densitometry using Image J.
RNA Extraction and Quantitative RT-PCR
Total RNA was extracted from tissues, and cDNA was synthesized for qPCR assays. Supplementary Table S1 lists primer sequences and reaction conditions for all genes analyzed.
Organ Culture and ELISA
Colon tissue pieces were incubated in media for 24 hrs, and supernatants of tissues were analyzed by ELISA.
BM Chimera Studies
BM cells were collected from WT, DKO, Casp1/11−/−, DKOΔinfl mice and equal numbers were injected into the appropriate pre-irradiated groups. Mice were euthanized 9 weeks later for analysis.
Statistics
All data represent the mean ± SEM unless otherwise indicated. An unpaired two-tailed or Student’s t-test or Mann-Whitney U-test was used to assess the statistical significance of differences between two groups. One-way ANOVA followed by the Bonferroni post-test was used to analyze the significance of differences between three or more groups. Fisher’s exact test was used to determine the significance of differences in percent incidence among groups. A P value less than 0.05 was considered to be significant. Data were plotted and analyzed using Microsoft Excel and GraphPad Prism V5.0.
Results
Mice lacking both core 1- and core 3-derived O-glycans exhibit spontaneous colitis-associated colon cancer
Our published data show that mice with loss of core 1-derived O-glycans (IEC C1galt1−/−) exhibit spontaneous colitis.7, 8 Recently, we have found that IEC C1galt1−/− mice exhibited colon tumors at an older age relative to their WT littermates (Figure S1A and B). Histology and IHC confirmed that these tumors were invasive adenocarcinomas (Figure S1B and C). Consistent with our previous data, the tumor tissues exhibited dramatically reduced acidic glycans vs. WT littermates as revealed by Alcian blue (AB) staining, and expressed abundant Tn antigen (Figure S1 D & E). The colon tumors occurred in ~90% of mice between 18 and 24 months, with an average of 3 tumors/colon and variable in size (Figure S1F). These data indicate that unresolved chronic colitis in IEC C1galt1−/− mice is associated with spontaneous tumorigenesis.
Core 3-derived O-glycans, whose formation is controlled by the glycosyltransferase C3GnT, are important in the colon.7, 8 To determine whether additional loss of core 3-derived O-glycans impacted spontaneous tumorigenesis in IEC-C1galt1−/− mice, we used our newly generated IEC-C1galt1−/−; C3GnT−/− (DKO) mice. At age 8 – 9 months, gross analysis revealed thickening of the distal colon of both IEC C1galt1−/− and DKO vs. aged-match controls (WT and C3GnT−/− mice) (Figure 1A). This mucosal thickening progressed toward the middle colon only in DKO mice, where tumors of variable size were frequently observed protruding into the lumen (Figure 1A). Histologic characterization indicated that most tumors exhibited loss of epithelial polarity, high nuclear-to-cytoplasmic ratio, extensive stratification and nuclear atypia (Figure 1B and C). These highly dysplastic glands frequently invaded and/or were dispersed within the submucosa (Figure 1B and C, arrows), which are features of invasive adenocarcinomas. These adenocarcinomas occurred most frequently in distal colons where inflammation was most severe, especially in DKO mice (Figure 1B and D), but was never observed in the colon of WT or C3GnT−/− mice. The spontaneous colorectal cancer in DKO mice progressed more rapidly compared with IEC C1galt1−/− mice, as adenocarcinomas in the distal colon of 16-month-old DKO mice were more numerous, invaded deeper into the mucosal wall with extensive desmoplasia, and exhibited aggressive characteristics such as cribriform arrangements (Figure 1E) relative to that in IEC C1galt1−/− mice. Nevertheless, we did not find metastasis in caudal or mesenteric lymph nodes, liver, or lung.
Figure 1. Loss of colonic mucin-type O-glycans leads to spontaneous colorectal cancer.
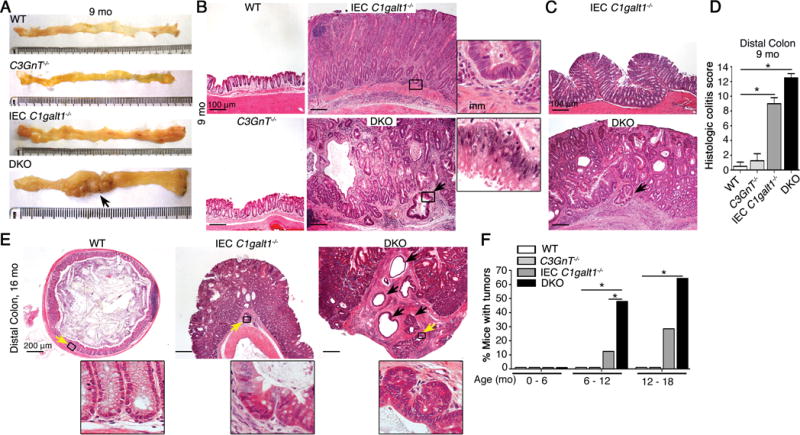
A. Representative gross morphology of murine colon of 9 month-old mice (n = 4 – 5 mice/group). Arrow, tumor. B and C. Representative H&E staining of colonic tissue (9 months). Arrow, invasive gland. Inset, magnified images of boxed regions. mm = muscularis mucosa. D. Histologic colitis score (mean ± SD, n = 4 – 5/group). E. Representative H&E staining of distal colon (n = 4 – 5 mice/group). Inset, magnified image of boxed regions (yellow arrow); black arrow, invasive glands. F. Tumor incidence (% mice with tumors; n = 8 – 10/group). *P < 0.05. Data are representative of at least 2 independent experiments.
Of the DKO mice in the age ranges 6–12 months and 12–18 months, 40% and 60%, respectively, exhibited spontaneous colon tumors, whereas IEC C1galt1−/− mice rarely developed tumors before 12 months and tumor incidence was only 20% during the period of 12–18 months (Figure 1F). Male and female IEC C1galt1−/− and DKO mice in a C57BL/6, 129/SvJ congenic, or mixed C57BL/6/129/SvJ background exhibited similar tumor incidence. These results revealed that the degree of loss of intestinal O-glycans is closely associated with initiation and progression of spontaneous colorectal cancer in our models.
Microbiota promote inflammation-associated colorectal cancer independent of Tn antigen exposure
DKO mice exhibited exacerbated colitis and early tumor formation suggesting that colonic inflammation contributes to tumor development. This likely relates to greater defects in mucus barrier function in DKO mice since complex glycans were greatly reduced in DKO vs. other groups including IEC C1galt1−/− mice, shown by Alcian Blue staining (Figure 2A). Yet, IEC C1galt1−/− and DKO mice also expressed high levels of Tn antigen in the colon (Figure 2B), which itself might influence tumorigenesis.12, 20, 21
Figure 2. Spontaneous tumor development is associated with inflammation but not Tn exposure.
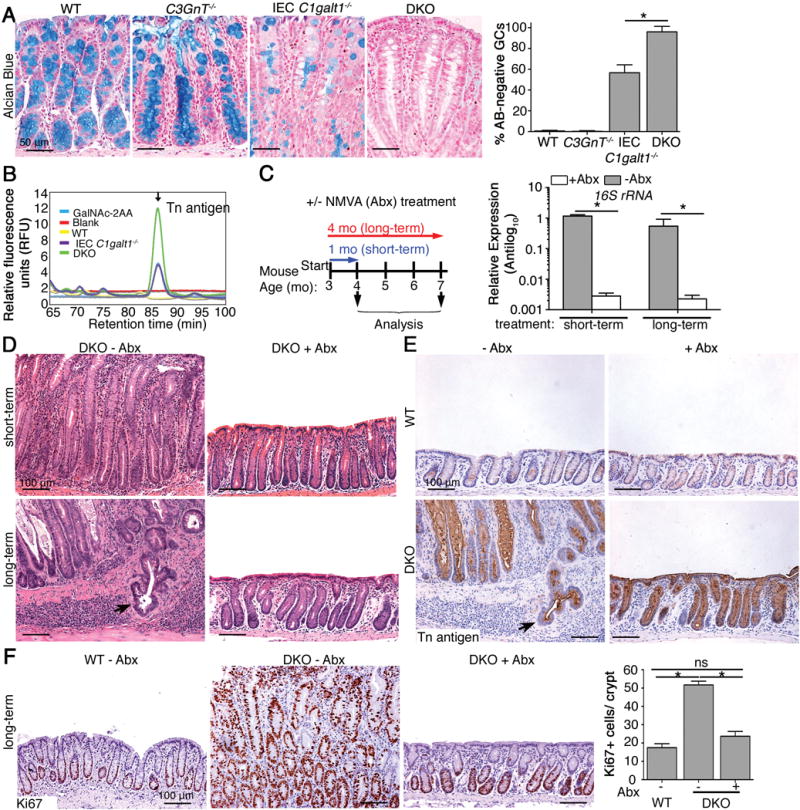
A. Representative Alcian blue (AB) staining of colonic tissues from 7 month-old mice. Graph: Quantitation of ABnegative cells in mucosal tissues (mean ± SD, n = 6/group). B. HPLC analysis of Tn levels of equal wet weights of colonic mucosal secretions from 2 month-old mice. GalNAc-2AA, positive control. C. Right: Schematic depiction of antibiotic-treatment regimen; Left: Representative qPCR for 16S rRNA gene from genomic DNA extracted from colon contents (n = 5 mice/group). D. Representative H&E staining of distal colons of 4 month-old (short-term) and 7 month-old (long term) mice. E and F. Representative IHC for Tn antigen (E) and Ki67 (F) in distal colons. Graph: Enumeration of proliferating nuclei in F (n = 5 mice/group). Arrows, invasive glands. *P < 0.05. For C – F, data are representative of 3 experiments.
To determine whether Tn antigen expression contributes to tumor development in our models, we treated 3-month-old DKO mice and WT littermates with short- and long-term regimens of broad-spectrum antibiotics to deplete the intestinal microbiota,8 and subsequently assessed colitis and carcinoma development (Figure 2C). Short-term (3 weeks) and long-term (17 weeks) antibiotic treatment led to a significant reduction in total bacteria, based on analysis of universal 16S rRNA gene expression in DNA isolated from feces (Figure 2C). Both short-and long-term antibiotic treatment dramatically reduced colitis in DKO mice compared with untreated controls (Figure 2D, upper panels). After long-term antibiotic treatment DKO mice exhibited a normal crypt architecture compared with adenocarcinoma observed in non-treated DKO controls (Figure 2D, lower panels). Antibiotic treatments did not change Tn antigen expression in the DKO colon (Figure 2E). Long-term antibiotic treatment significantly reduced epithelial proliferation in DKO mice to near WT levels, as assessed with Ki67 staining (Figure 2F). These results indicated that tumor development was promoted by bacteria-mediated colon inflammation and that Tn antigen exposure itself did not contribute significantly to colon tumorigenesis in DKO mice.
Chronic microbiota-driven colitis in DKO mice is inflammasome-dependent
We next sought to elucidate how bacteria promote inflammation and tumorigenesis in the absence of intestinal O-glycans. Colitis in IEC C1galt1−/− mice develops independently of adaptive immunity, and TLR4 and Myd88 signaling.8 We therefore hypothesized that microbial recognition through alternative mechanisms such as caspase 1– and/or 11–dependent inflammasomes is important to promote disease development in DKO mice.
To address this, we first analyzed whole colonic tissues of DKO mice with colitis (3 months old) by western blotting for caspase 1 activation.16 In WT mice, only a procaspase-1 (45 kDa) band was observed; in DKO mice, however, the active p10 subunit was also consistently detected (Figure 3A). We also analyzed expression of caspase 11, which mediates non-canonical inflammasome signaling and was recently implicated in modulating mucosal immunity.18 The level of caspase 11 in DKO mice was also increased as shown by the p38 and p43 doublet bands (Figure 3A). Densitometry of the caspase 1 p10 band at different time points revealed that caspase 1 was constitutively activated in DKO mice starting from at least 1 month of age, as revealed by a 3- to 16-fold induction over WT mice (Figure 3B). Caspase 11 expression was more transient, reverting to near baseline levels by 5 months (not shown). Consistent with caspase 1 activation, we detected increased levels of IL-1β and IL-18 in culture supernatants from colonic explants of DKO mice vs. WT mice at both 3 and 5 months of age (Figure 3C). Western analysis of tissues from antibiotic-treated vs. non-treated mice revealed caspase 1 activation and caspase 11 expression was markedly reduced in a microbiota-depleted environment (Figure 3D).
Figure 3. Microbiota-mediated chronic colitis in O-glycan-deficient mice is driven by mucosal inflammasomes.
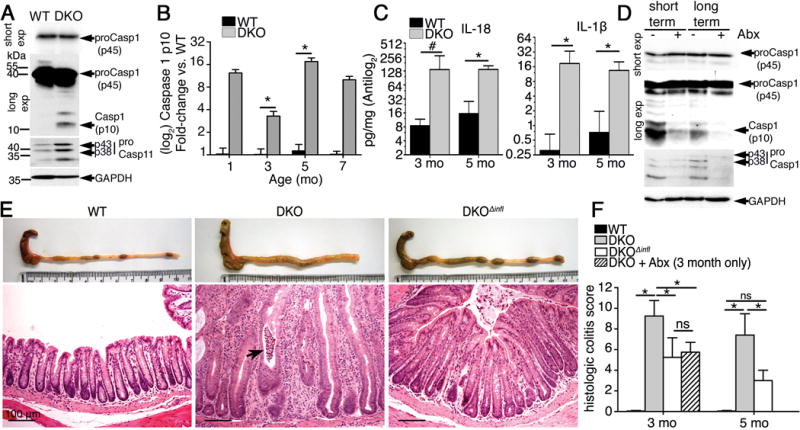
A. Representative western blot of colonic lysates derived from 3 month-old mice (n = 5/group). B. Densitometry of the caspase 1 p10 subunit visualized via western blot of colon lysates over time (n = 3 – 5/group). C. ELISA of colonic organ culture supernatants of 3 and 5 month-old mice (mean ± SD, n = 4 mice/group). D. Western blot of whole colon lysates (n = 5 mice/group). E. Representative macroscopic and histologic images (H&E distal colon) of 3 month-old mice (n = 5/group). Arrow, crypt abscess. F. Histologic colitis scores in distal colonic tissues (mean ± SD, n = 4 – 5/group). *P < 0.05. #P < 0.05 Mann-Whitney U-test. Data are representative at least 3 experiments.
To determine the biologic significance of inflammasomes in our model, we crossed DKO mice with mice lacking both caspase 1 and 11 (Casp1/11−/−, lacking major canonical and non-canonical inflammasomes) to generate DKO/Casp1/11−/− mice (DKOΔinfl) (Figure S2A). Starting from age 3 months, DKOΔinfl mice showed markedly reduced colonic thickening, hyperplasia, and inflammatory infiltrates as well as no crypt abscesses as compared with DKO mice (Figure 3E and F). Similar results were observed at age 5 months when comparing DKO and DKOΔinfl mice (Figure S2B, upper panel and 3F). These results indicate that mucosal inflammasomes mediate colitis in O-glycan-deficient mice.
Tumorigenesis in DKO mice is dependent on caspase 1 activation
To determine if inflammasome deficiency impacts colorectal cancer in the absence of O-glycans, we let WT, DKO, and DKOΔinfl mice reach 9 months of age. Macroscopic analysis revealed a lack of tumors in the colon of DKOΔinfl mice compared with DKO mice (Figure 4A). Histology confirmed the comparative lack of differentiated tumors in the DKOΔinfl colon (Figure 4B, S2B, lower panel). Overall, only ~20% of DKOΔinfl mice of age 6 – 12 months developed tumors compared with 80% of DKO mice (Figure 4B). Correspondingly, the number of invasive glands (carcinoma) was significantly elevated in DKO vs DKOΔinfl mice (Figure S2C). Inflammasome deficiency did not reduce Tn expression in the DKOΔinfl colon (Figure 4C), confirming that Tn antigen is not sufficient to cause colon tumorigenesis. Colorectal cancer in DKO mice was also associated with robust caspase 1 activation as well as inflammasome-dependent secretion of IL-1β and especially IL-18 (Figure S2D and E).
Figure 4. Mucosal inflammasomes promote colitis-associated tumorigenesis in O-glycan-deficient mice.
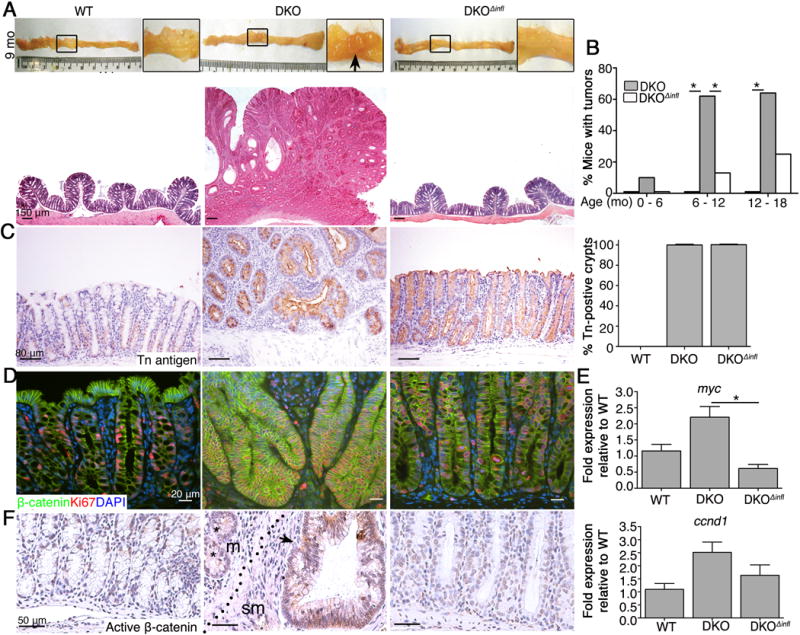
A. Upper panel: Representative picture of colons of 9 month-old mice. Box, magnified regions; Arrow, visible tumors; Lower panel: Representative H&E staining of middle colonic sections (n = 6 mice/group). B. Tumor incidence (% mice with tumors; n = 6 – 13 mice/group). C. Representative IHC for Tn antigen in 9 month-old mice (n = 5/group). Data are representative of 2 independent experiments. Graph on right: Percent Tn-positive crypts (mean ± SD, n = 6/group). D. Representative IF on colonic sections of 9 month-old mice (n = 4 mice/group). E. qPCR analysis of cDNA isolated from crypt cells form 8 – 9 month old mice (n = 5 mice/group). F. Representative IHC for Active (non phosphorylated) β-catenin in colon tissue (n = 4 mice/group). Arrow, positive gland; *, negatively stained gland; dotted line, muscularis mucosa; m, mucosa; sm submucosa. *P < 0.05. Data are representative of 2 (D-F) and 3 (A- C) independent experiments.
Overexpression of the oncogeneβ-catenin, a mediator of canonical Wnt signaling, is important in the pathogenesis of most colon cancers.1 To understand the relationship of the Wnt/β-catenin pathway to tumor development in our model, we first examined overall β-catenin expression in DKO vs. WT and DKOΔinfl tissue by IHC. We found total and activatedβ-catenin expression in DKO crypts was increased vs. other strains, but its expression pattern in DKO tumors was variegated, being expressed highly in some crypts but low in adjacent crypts (Figure S2F), suggesting tumor heterogeneity at the molecular level. However, proliferation was associated with crypts with elevated β-catenin levels in DKO tumor tissue (Figure 4D). Overall, expression of Wnt-target genes myc and ccnd1 were elevated in DKO vs. WT and DKOΔinfl crypts (Figure 4E). Active β-catenin staining was also seen in invasive glands relative to non-invasive glands in DKO mice (Figure 4F). In contrast, expression of the tumor suppressor p53 did not differ among these mice (not shown). These results suggest the Wnt/β-catenin pathway and likely others are associated with tumorigenesis in our model, but that Wnt may be important for tumor progression. Collectively, these results revealed that loss of O-glycans promotes bacterial-driven inflammation-associated spontaneous tumorigenesis and that this is largely inflammasome-dependent.
Because our DKOΔinfl mice were deficient in both caspases 1 and 11, we had to differentiate the roles of caspase 1 and 11 in colitis and cancer development. We thus analyzed 8-month-old DKO mice backcrossed (>10 generations) to a 129/Sv congenic background, which is deficient in caspase 11 owing to a spontaneous deleterious mutation in exon 7.17 DKO mice on the 129/Sv background exhibited activated caspase 1 and produced IL-1β and IL-18, but they did not express caspase 1l (Figure S3A–C, and not shown). The DKO mice (129/Sv) developed severe colitis and colon dysplasia at age 8 months (Figure S3D), similar to DKO mice on a mixed B6/129 (caspase 11–sufficient) background. These results support a primary role of caspase 1 but not caspase 11 in colitis development and colitis-associated tumorigenesis in our models.
Chronic inflammation driven by microbiota-dependent inflammasome activation orchestrates a protumorigenic environment in DKO mice
To better understand how the microbiota and mucosal inflammasomes promote tumor-initiating inflammation, we characterized the inflammatory milieu of 3-month-old (i.e. prior to tumor development in DKO mice) WT, DKO, DKOΔinfl, and antibiotic-treated DKO mice (short-term with broad spectrum antibiotics as in Figure 2C). FISH of colonic sections demonstrated that the microbiota was separated from the mucosa in WT mice and that the number of bacteria was reduced in antibiotic treated DKO mice as expected; however, in DKO and DKOΔinfl mice, the microbiota remained in direct contact with the mucosa, indicating that the observed reduction in colitis and tumorigenesis in DKOΔinfl mice was not due to reduced interactions of the microbiota with colonic tissues (Figure 5A). Myeloid cells including neutrophils are the primary immune cells initiating inflammation in IEC C1galt1−/− mice8 and can drive tumorigenesis.22 Consistent with this, immunostaining for myeloperoxidase (MPO), a marker for neutrophils, revealed robust infiltration of MPO+ cells into the mucosa of 3-month-old DKO mice, whereas an almost negligible difference in neutrophil infiltration occurred between WT, DKOΔinfl, and antibiotic-treated DKO mice (Figure 5B). In addition, total numbers of F4/80+ macrophages were increased in DKO mice relative to the other groups (not shown). Cytokine profiling by quantitative reverse transcription (RT)-qPCR revealed increased expression of cytokine genes that contribute to inflammation-associated cancer progression, including Il6 and Il17a (Figure 5C) and Tnf (Figure S5A). Consistent with neutrophil influx, expression of kc (Cxcl1) was also upregulated in DKO vs. DKOΔinfl mice (Figure S4A). Interestingly, expression of Nos2, which encodes inducible NOS (iNOS), was also upregulated ~10-fold in DKO mice relative to WT, DKOΔinfl, and antibiotic-treated DKO mice (Figure 5C), and this was confirmed by western blotting (Figure 5D). The cellular source of iNOS appeared to be neutrophils, although some iNOS could be detected in epithelia (Figure S4B).
Figure 5. Microbiota and inflammasomes orchestrate a highly protumorigenic environment in the absence of intestinal O-glycans.
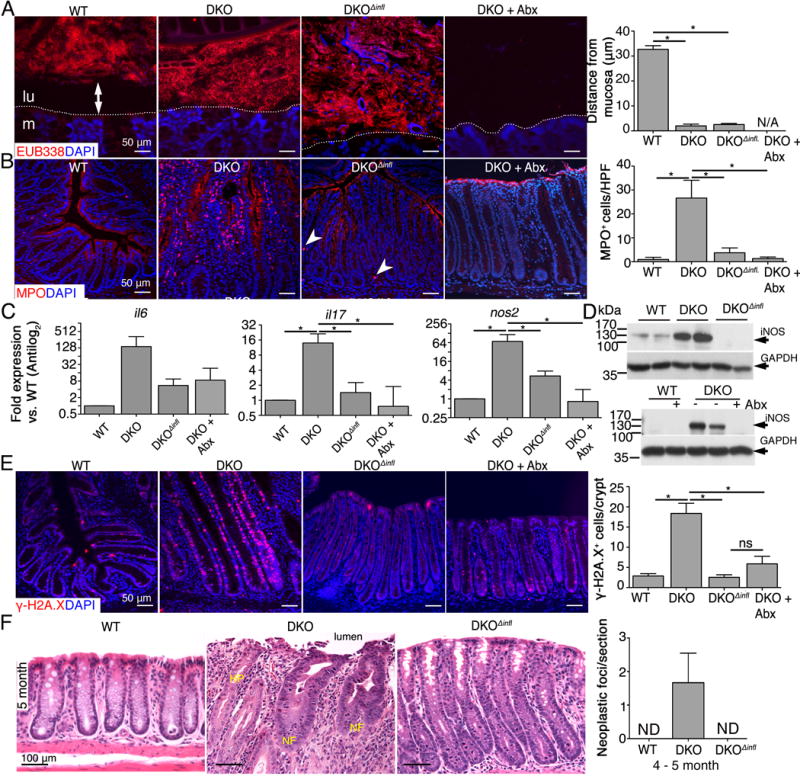
A. Representative FISH using EUB338 probe on Carnoy’s-fixed colonic sections of 3 month-old mice. Dashed line, interface between lumen (lu) and mucosa (m). Arrow, space between lumen and mucosa. Graph: Distance between luminal microbiota and epithelial surface (mean ± SD, n = 4 mice/group). B. Representative immunostaining for MPO in distal colon sections of 3 month-old mice. Arrowheads, MPO+ cell. Graph: Enumeration of MPO+ cells in mucosa (n = 4 mice/group). C. qPCR analysis in distal colons of 3 month-old mice (mean ± SD, n = 4 – 5 mice/group). D. Western blot of distal colon lysates of 3 month-old mice and 7 month-old antibiotic treated/nontreated mice; each lane = 1 animal. Results are representative of 4 mice/group. E. Immunostaining for γ-H2A.X (red) in distal colon sections. Graph: Quantification of γ-H2A.X+ nuclei in 3 month-old colons (n = 3 – 4/group). F. High power (20X) H&E staining of distal colon tissue. NF, neoplastic focus; HP, hyperplastic focus. *P < 0.05. Data are representative of 3 independent experiments.
Both inflammatory cell recruitment and iNOS expression indicated an environment rich in reactive oxygen and nitrogen species that can directly damage DNA.22, 23 Consistent with reduced expression of inflammatory mediators and producers of reactive nitrogen species, we observed dramatically reduced staining for phospho-histone 2A.X (γH2A.X), which marks damaged DNA,23, 24 in DKOΔinfl and antibiotic-treated DKO mice relative to DKO mice (Figure 5E). This correlated with significantly reduced epithelial proliferation in DKOΔinfl and antibiotic-treated DKO mice vs. DKO mice, determined by Ki67 staining (Figure S4C). Importantly, analysis of 5-month-old distal colonic tissue revealed the emergence of clear neoplastic foci characterized by nuclear hyperchromasia and pronounced atypia only within inflammatory stroma of DKO mice and especially near the mucosal surface (Figure 5F). Ki67 staining confirmed the hyperproliferative status of these lesions compared with adjacent non-neoplastic cells (Figure S4E and F). These results suggested that microbiota-mediated chronic colon inflammation via inflammasomes promotes the influx of immune cells, expression of DNA-damage mediators, and upregulation of cytokines, which drives dysregulated cell proliferation and subsequent neoplastic transformation.
Inflammasome activation in non-hematopoietic cells promotes inflammation-associated carcinogenesis induced by O-glycan deficiency
Inflammasomes are expressed in both hematopoietic and non-hematopoietic cells.16 By immunostaining, we detected the strongest caspase 1 signal in epithelial cells of both WT and DKO mice, but not in DKOΔinfl mice, confirming the specificity of the antibody. The adaptor ASC was detected in the epithelium of all strains (Figure 6A and Figure S5A). The endogenous caspase 1 inhibitor caspase 12 exhibited a modest expression relative to caspase 1 and ASC, but was more expressed in whole colonic tissue (Figure S5A), suggesting caspase 1 activity in epithelium is relatively unrestricted. These results point to epithelial cells as the major cell type in which inflammasomes are activated and drive disease in DKO mice.
Figure 6. Inflammasome function in non-hematopoietic cells drives colitis-associated carcinogenesis induced by O-glycan deficiency.
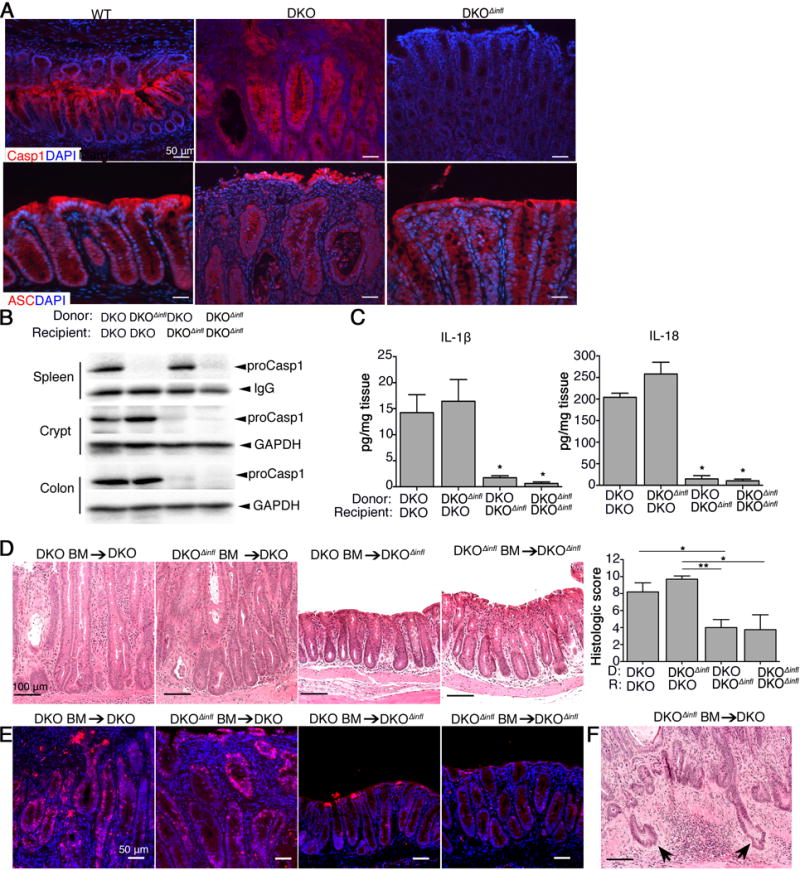
A. Representative immunostaining on PFA-fixed distal colon cryosections (CASP1) and FFPE section (ASC) of 3 month-old mice (n = 5/group). B. Representative western blot of spleen, crypt, and colon lysates from chimeric mice (n = 4 – 6 mice/group). IgG or GAPDH are internal controls. C. ELISA of IL1β and IL-18 from organ culture supernatants (n = 4 – 6 mice/group). D. Representative H&E staining of colonic tissues (n = 4 – 6/group). E. Representative immunostaining for γH2A.X (red) in distal colon sections (n = 4 – 6/group). F. H&E staining of distal colonic tissue showing presence of invasive glands (arrows). *P < 0.05 vs. each DKO recipient. Mice for BM-chimera experiments were 3 – 4 months-old at time of termination. Data are representative of 2 independent experiments.
We subsequently created reciprocal bone marrow (BM)-chimeric mice to address whether hematopoietic-derived vs. non-hematopoietic (i.e., epithelial or stromal)-derived inflammasomes were essential for disease. After lethal irradiation, DKO and DKOΔinfl mice were reconstituted with BM from either DKO or DKOΔinfl mice. At 9 weeks post-transplantation, the efficiency of hematopoietic reconstitution was confirmed by western blotting (Figure 6B). As expected, pro-caspase 1 was detected in the spleen (representing hematopoietic cells) but not in colon epithelia, and only weakly in colon tissues, of DKOΔinfl mice receiving DKO BM (DKO BM → DKOΔinfl); in contrast, DKOΔinfl BM → DKO mice expressed caspase 1 in the crypts but not the spleen (Figure 6B). Caspase 1 expressed in both compartments of DKO BM → DKO mice, and lacking in DKOΔinfl BM → DKOΔinfl mice (Figure 6B), demonstrating the efficiency and specificity of our BM chimera experiments. Consistent with the role for epithelial inflammasome activation we observed increased IL-1 β and IL-18 release from explants derived from DKO vs. DKOΔinfl recipients regardless of donor (Figure 6C). Analysis of colonic tissues revealed severe disease in DKO recipients receiving either DKO or DKOΔinfl BM, characterized by severe thickening, inflammatory infiltration, numerous abscesses, and massive hyperplasia, whereas DKOΔinfl recipients of either donor exhibited only mild inflammation by comparison (Figure 6D). γH2A.X staining revealed robust staining in DKO vs DKOΔinfl recipients (Figure 6E and S5E), where proliferation was also comparatively reduced (Figure S5E). Notably, we observed the presence of invasive crypts in DKO recipient mice (Figure 6F and S5F). Identical results were obtained with WT and Casp1/11−/− donors (Figure S5C–E). Collectively, these results indicate that inflammasome function in non-hematopoietic cells—most likely colonic epithelial cells—is instrumental in driving bacterial-mediated colitis-associated colorectal cancer in the absence of intact intestinal O-glycosylation.
Caspase 1-dependent inflammasomes are expressed in human UC and cancer epithelia and can be activated by colonic bacteria
Assembly of caspase 1-dependent inflammasomes is dependent upon various NLR family members, or the ALR family member Aim2.16 qPCR analysis revealed that Nlrc4, Aim2 and especially Nlrp6 were robustly expressed in both WT and DKO crypts, consistent with previous reports25, 26. In contrast, Nlrp1b, Nlrp3, Nlrp4, and Nlrp12 exhibited modest levels of expression (Figure 7A). No significant differences in levels of expression of these genes were observed between WT and DKO mice, suggesting that caspase 1 activation is unlikely mediated by differential expression of different receptors in WT relative to DKO colonic epithelium.
Figure 7. Inflammasome components are expressed in mouse and human epithelia.
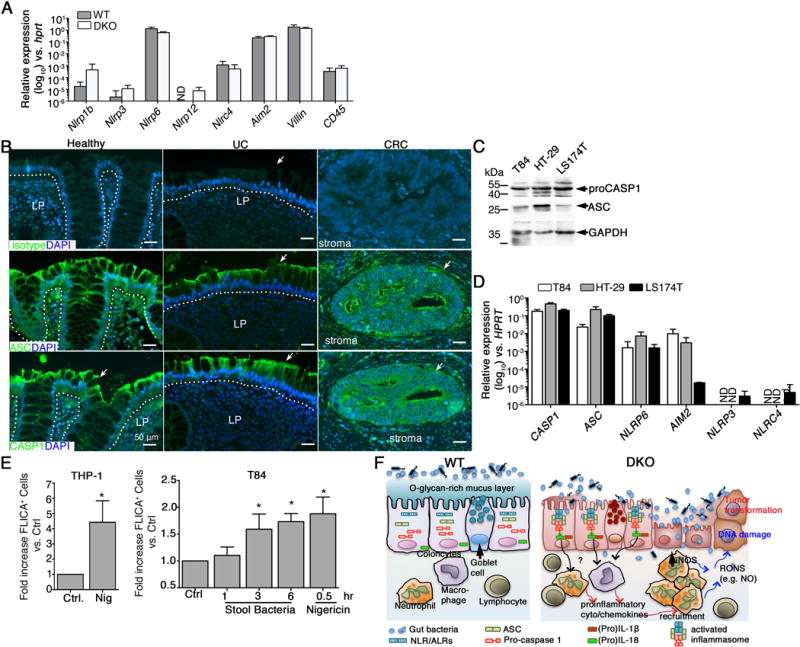
A. qPCR analysis in distal colon crypts of 3 – 4 month old mice (n = 5 – 8 mice/group). B. Representative IF of human colon (n = 3 patient samples/group). Dotted line divides epithelium from lamina propria (LP). Arrow, epithelium. C. Western blot of lysates derived from human colorectal cancer lines (representative of n = 3 cultures per line). D. qPCR analysis of human colorectal cell lines (n = 3 cultures/cell line). E. Quantification of Caspase 1 activation in human cell lines using FAM-FLICA probe. Nigericin treatment is used as a positive control. Data are representative of 3 independent experiments. * P < 0.05. F. Model: Mucin-type O-glycosylation maintains mucus barrier function limiting microbiota access to inflammasome-expressing epithelium (WT). Defective O-glycosylation compromises mucus barrier (DKO), leading to abnormal bacterial intrusion and activation of caspase 1-dependent epithelial inflammasomes. Chronic inflammation, characterized by immune cell infiltrates highly expressing DNA damage mediators like iNOS, initiates transformation of epithelium, driving tumorigenesis.
Previous studies show inflammasomes are activated in human tissues.13, 14, 27, 28 To characterize which inflammasomes are expressed in human UC and colon cancer, we performed immunostaining for Caspase 1 and ASC on colon sections from UC and cancer patients, and healthy controls (Figure 7B). Similar to mouse colon, ASC staining was similar in healthy, UC and colorectal cancer tissue (Figure 7B). Western blotting detected CASP1 and ASC proteins in three human epithelial CRC lines (T84, HT-29, and LS174T) (Figure 7C). Further qPCR analysis revealed inflammasome gene expression profiles in these cell lines mirrored that of mouse epithelial cells, with the exception of NLRC4, which showed lower expression (Figure 7D). To determine whether colon bacteria could activate caspsase 1 in human colon epithelia, we stimulated T84 cells with bacteria derived from DKO mice and analyzed caspase 1 activation by flow cytometry using FAM-FLICA (FAM-YVAD-fmk) assay. Exposure of T84 cells to bacteria (multiplicity of infection: ~10:1) led to a significant increase in FLICA+ cells in a time dependent manner (Figure 7E). These studies indicate in human tissues and human colon cancer cells lines, the inflammasome components are expressed and poised to be activated when the mucus barrier is compromised.
Discussion
Altered intestinal mucin-type O-glycosylation is associated with colorectal cancer,7,11,12 but whether and how defective O-glycosylation contributes to this disease is unclear. We now show that loss of major types of intestinal O-glycans leads to impaired mucus barrier function and colitis-associated colon cancer in which the microbiota promote inflammation and cancer via epithelial inflammasomes (Figure 7F).
Core 1– and core 3–derived O-glycans are two major types of intestinal O-glycans with core 1–derived O-glycans as the dominant form in mice.8 Loss of core 1–derived O-glycans is sufficient to cause spontaneous colon tumors, which is accelerated when core 3–derived O-glycans are also ablated in our models. This is most likely due to additional protective effects of core 3 O-glycans on mucus layer integrity in the absence of core 1, although a potential tumor suppressive function of C3GnT independent of the mucus barrier may also contribute to increased cancer invasiveness in DKO vs. IEC C1galt1−/− mice.29
Truncated O-glycan structures, such as Tn antigen, are associated with colorectal cancer,30 however their biological role in pathogenesis is unclear. Loss of C1GALT1 activity ascribed cancer cell-like characteristics to pancreatic and skin cell lines and aberrant expression of Tn antigen in the ER may promote the invasive potential of transformed cell lines.20,21 However, our in vivo results indicate that exposure of Tn itself does not significantly contribute to colon inflammation and tumorigenesis because either antibiotic treatment or loss of inflammasome activity significantly reduced these pathologies despite presence of high levels of Tn expression in the DKO colon. Tn can be also modified by sialyltransferase to form α2,6-sialylated Tn (sTn).11 Whether sTn plays a role in colon tumor development remains to be addressed.
Our results indicate that the microbiota is key for driving inflammation and tumorigenesis in the absence of core 1- and core 3-derived O-glycans largely through activation of epithelial inflammasomes. However, tumor development was significantly retarded but not completely obliterated in DKOinfl mice. This result suggests the TLR pathway, though not sufficient independently to mediate inflammation in the O-glycan-deficient mice,8 may collectively contribute to recognition of bacterial components in our models.
Much evidence supports a role for mucosal inflammasomes in human UC. Caspase 1 is activated in human IBD including CD and UC.13, 14 IL-1β and/or IL-18 is consistently produced in UC samples compared to non-IBD controls.13, 14, 27, 28 However, studies using chemically-induced IBD models (e.g. DSS) have yielded mixed results.31–34 In contrast, a more consistent picture emerges when using spontaneous models of IBD. For example, inhibition of caspase 1 reduces colitis in Il10−/− mice.35 Neutralization of IL-1β dramatically ameliorates severity of H. hepaticus-induced chronic colitis in Rag2−/− mice.36 Loss of the inflammasome adaptor ASC, or long-term pharmacologic inhibition reduced cecal inflammation and carcinogenesis in AHR−/− mice.37 Inflammasome-dependent IL-18 production is implicated in spontaneous tumor development in Apcmin/+ mice.38 These data support our conclusion that activation of caspase 1 promotes spontaneous chronic intestinal inflammation and colitis-associated tumorigenesis. Questions that remain to be addressed are which upstream activators (NLRs) in colonic epithelium is/are required for activating inflammasomes upon mucus barrier breach and the relative roles of caspase 1 effector functions (IL1β, IL18, pyroptosis) in mediating disease.
The current models for inflammation-associated colon cancer are typically on a background of immunodeficiency (il10−/−, Tbet/Rag2−/−, Gαi2−/−) and/or require an additional stimulus to induce the cancer (e.g., Helicobacter hepaticus in Rag1−/− mice, AOM in DSS-treated mice).4,39,40,41,42 While these models are important tools for understanding colitis-associated cancer, a considerable drawback of each of these interventions is their artificial nature that can confound potential translational efforts to identify novel strategies for prophylactic or therapeutic intervention in human patients. In contrast, our model is unique because it occurs on an immunocompetent background and develops cancer without an exogenous stimulus. Therefore, it provides a more clinically relevant opportunity to dissect how components of the microbiota or the immune system contribute to each stage of cancer pathogenesis.
In conclusion, our results underscore the essential role of intestinal mucin-type O-glycosylation in colonic homeostasis and protection from colitis-associated cancer. Our results also suggest that inhibition of key inflammatory pathways mediated by inflammasome activation (caspase 1-dependent) may reduce the incidence of carcinogenesis in patients with UC.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Rodger McEver for critical review of the data and Dr. Courtney Griffin for technical support. We also thank Dr. Jenny Ting for helpful suggestions for experimental approaches.
Funding
Supported by grants from National Institutes of Health (R01DK085691, GM103441, HL085607, HL093242, HL118676), Oklahoma Center for the Advancement of Science and Technology (HR13-160), National Natural Science Foundation of China (81470825, 31400692, 81172299), American Heart Association (SDG0835544N, 15EIA22210014), Crohn’s and Colitis Foundation of America Research Fellowship Award (RFA285148 to K. B.), and support from Oklahoma Center for Adult Stem Cell Research, the Stephenson Cancer Center, Oklahoma Center for Medical Glycobiology from the Vice-President for Research and the Department of Biochemistry & Molecular Biology of OUHSC.
Footnotes
Conflict of Interest Statement: The authors have declared that no conflict of interest exists.
Author contributions: L.X., K.B., J.F. conceived and designed the experiments, analyzed the data, and wrote the manuscript. K.B., X.L. and Y.Z. performed the experiments. Q.W., S. M., M.M., K.S., N.G., Y.C., Y.L performed some experiments and provided technical support; J.B., H.C., M.M.H., L.A.Z., W.C., C.W.H., C.M.W. helped analyze data and commented on the manuscript.
References
- 1.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 2.Ullman TA, Itzkowitz SH. Intestinal Inflammation and Cancer. Gastroenterology. 2011;140:1807–1816.e1. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- 3.Saleh M, Trinchieri G. Innate immune mechanisms of colitis and colitis-associated colorectal cancer. Nat Rev Immunol. 2011;11:9–20. doi: 10.1038/nri2891. [DOI] [PubMed] [Google Scholar]
- 4.Garrett WS, Punit S, Gallini CA, et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–19. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van der Sluis M, De Koning BA, De Bruijn AC, et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology. 2006;131:117–29. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 6.Johansson ME, Phillipson M, Petersson J, et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–9. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An G, Wei B, Xia B, et al. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med. 2007;204:1417–29. doi: 10.1084/jem.20061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu J, Wei B, Wen T, et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J Clin Invest. 2011;121:1657–1666. doi: 10.1172/JCI45538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ju T, Cummings RD. A unique molecular chaperone Cosmc required for activity of the mammalian core 1 beta 3-galactosyltransferase. Proc Natl Acad Sci U S A 2013. 2002;99:16613–8. doi: 10.1073/pnas.262438199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larsson JMH, Karlsson H, Crespo JGB, et al. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm Bowel Dis. 2011;17:2299–307. doi: 10.1002/ibd.21625. [DOI] [PubMed] [Google Scholar]
- 11.Itzkowitz SH, Young E, Dubois D, et al. Sialosyl-Tn antigen is prevalent and precedes dysplasia in ulcerative colitis: a retrospective case-control study. Gastroenterology. 1996;110:694–704. doi: 10.1053/gast.1996.v110.pm8608878. [DOI] [PubMed] [Google Scholar]
- 12.Springer GF. T and Tn, general carcinoma autoantigens. Science. 1984;224:1198–206. doi: 10.1126/science.6729450. [DOI] [PubMed] [Google Scholar]
- 13.Monteleone G, Trapasso F, Parrello T, et al. Bioactive IL-18 expression is up-regulated in Crohn’s disease. J Immunol. 1999;163:143–7. [PubMed] [Google Scholar]
- 14.Liu JJ, Davis EM, Wine E, et al. Epithelial cell extrusion leads to breaches in the intestinal epithelium. Inflamm Bowel Dis. 2013;19:912–21. doi: 10.1097/MIB.0b013e3182807600. [DOI] [PubMed] [Google Scholar]
- 15.Zaki MH, Lamkanfi M, Kanneganti T-D. Inflammasomes and Intestinal Tumorigenesis. Drug Discov Today Dis Mech. 2011;8:e71–e78. doi: 10.1016/j.ddmec.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Kayagaki N, Warming SR, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 18.Demon D, Kuchmiy A, Fossoul A, et al. Caspase-11 is expressed in the colonic mucosa and protects against dextran sodium sulfate-induced colitis. Mucosal Immunol. 2014 doi: 10.1038/mi.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dupaul-Chicoine J, Yeretssian G, Doiron K, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367–78. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 20.Gill DJ, Min K, Chia J, et al. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc Natl Acad Sci U S A. 2013;110:E3152–3161. doi: 10.1073/pnas.1305269110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radhakrishnan P, Dabelsteen S, Madsen FB, et al. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc Natl Acad Sci U S A. 2014;111:E4066–75. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jamieson T, Clarke M, Steele CW, et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J Clin Invest. 2012;122:3127–44. doi: 10.1172/JCI61067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaked H, Hofseth LJ, Chumanevich A, et al. Chronic epithelial NF-κβ activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc Natl Acad Sci U S A. 2012;109:14007–12. doi: 10.1073/pnas.1211509109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arthur JC, Perez-Chanona E, Mühlbauer M, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Man SM, Zhu Q, Zhu L, Liu Z, et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell. 2015;162:45–58. doi: 10.1016/j.cell.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wlodarska M, Thaiss CA, Nowarski R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell. 2014;156:1045–59. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leach ST, Messina I, Lemberg DA, et al. Local and systemic interleukin-18 and interleukin-18-binding protein in children with inflammatory bowel disease. Inflamm Bowel Dis. 2008;14:68–74. doi: 10.1002/ibd.20272. [DOI] [PubMed] [Google Scholar]
- 28.Andus T, Daig R, Vogl D, et al. Imbalance of the interleukin 1 system in colonic mucosa–association with intestinal inflammation and interleukin 1 receptor antagonist [corrected] genotype 2. Gut. 1997;41:651–7. doi: 10.1136/gut.41.5.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwai T, Kudo T, Kawamoto R, et al. Core 3 synthase is down-regulated in colon carcinoma and profoundly suppresses the metastatic potential of carcinoma cells. Proc Natl Acad Sci U S A. 2005;102:4572–7. doi: 10.1073/pnas.0407983102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itzkowitz SH, Marshall A, Kornbluth A, et al. Sialosyl-Tn antigen: initial report of a new marker of malignant progression in long-standing ulcerative colitis. Gastroenterology. 1995;109:490–7. doi: 10.1016/0016-5085(95)90337-2. [DOI] [PubMed] [Google Scholar]
- 31.Zaki MH, Boyd KL, Vogel P, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–91. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A. 2010;107:21635–40. doi: 10.1073/pnas.1016814108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aguilera M, Darby T, Melgar S. The complex role of inflammasomes in the pathogenesis of Inflammatory Bowel Diseases - Lessons learned from experimental models. Cytokine Growth Factor Rev. 2014;25:715–730. doi: 10.1016/j.cytogfr.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Siegmund B, Lehr HA, Fantuzzi G, et al. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc Natl Acad Sci U S A. 2001;98:13249–54. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Fu S, Sun S, et al. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol. 2014;7:1139–50. doi: 10.1038/mi.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coccia M, Harrison OJ, Schiering C, et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Ex Med. 2012;209:1595–609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikuta T, Kobayashi Y, Kitazawa M, et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis. 2013;34:1620–7. doi: 10.1093/carcin/bgt083. [DOI] [PubMed] [Google Scholar]
- 38.Huber S, Gagliani N, Zenewicz LA, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–63. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berg DJ, Davidson N, Kuhn R, et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010–20. doi: 10.1172/JCI118861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudolph U, Finegold MJ, Rich SS, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;10:143–50. doi: 10.1038/ng0695-143. [DOI] [PubMed] [Google Scholar]
- 41.Erdman SE, Rao VP, Poutahidis T, et al. Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc Natl Acad Sci U S A. 2009;106:1027–32. doi: 10.1073/pnas.0812347106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nature Prot. 2007;2:1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.