


Abstract
Gene cassettes are short, monogenic DNA elements that translocate between integrons through site-specific excision and integration. These events require that an integron-coded tyrosine recombinase forms a reactive complex with two sites, at least one of which belongs to the attC class. An attC site can be divided into two pairs of short repeats flanking a palindromic central region. The nucleotide sequence of attC among different cassettes varies extensively, implying that the site contains a structural recognition determinant with low sequence constraints. Oligonucleotides representing many different sequence modifications in either strand of the site were examined for integrase binding by using an electrophoresis mobility shift assay. The inner repeats, a central triplet and two single-nucleotide asymmetries in the site had the strongest influence on binding strength and strand choice. Our data show that the recombinase binds to a bulged hairpin in attC and that the hairpin distortion due to bulging could define the appropriate orientation of the otherwise symmetrical site. This is consistent with the strong bias for binding of recombinase to the bottom-strand oligonucleotides in vitro. Moreover, it was observed that the mobility-shifted complexes persisted under protein-denaturing assay conditions, indicating that a covalent link is indeed formed between integrase and DNA. Upon substitution of the presumed DNA-attacking residue, Y312, with a phenylalanine, DNA binding remained but there was no covalent linkage.
INTRODUCTION
Lateral DNA transfer is a widely employed strategy for genetic expansion and diversity among bacteria and, in particular, has been highlighted in connection with acquisition of virulence and resistance traits (1). Plasmids and phages that are efficient transfer vectors of laterally mediated DNA are often supplied with mechanisms for integration and excision of exogenous DNA by site-specific recombination. Recombination sites of most site-specific recombinases contain a core of short dyad sequences resembling repressor sites (2,3). However, it is not uncommon for the sites also to include nearby sequences or to be arranged in a more complex fashion to allow tight regulation and prevention of recombination between inappropriately disposed sites (4,5). The site-specific recombinases form characteristic phosphoprotein intermediates and one of the major enzyme families uses a tyrosine hydroxyl oxygen as the primary DNA-attacking reagent and acts similar to topoisomerases (6). Members of this tyrosine family of recombinases (7,8) are known to be involved in different mechanisms for integration and excision of DNA elements such as lysogenic phage or gene cassettes in integrons (9). Other functions of tyrosine recombinases include their role in safeguarding replicating bacterial chromosomes and plasmids (10–12).
DNA elements with a capacity for gene capture, integrons, were originally noticed on plasmids in human pathogens where they carry a broad variety of different antibiotic resistance genes (13–18). The organization of integrons is represented by the class 1 integron of Tn21 in Figure 1. The integron integrases form a subclass of the tyrosine recombinases with a characteristic motif (15,19,20). Recent observations suggest that plasmid-borne integrons stem from chromosomal integrons in mainly environmental and aquatic species on the gamma branch of the proteobacteria (21). It was revealed that the ancestral integrons contain a huge pool of cassette-borne open reading frames but only a minority of these are related to resistance genes. The integrons recruited to plasmids in commensal and pathogenic species have been retained in particular drug resistance genes, putatively because these hosts are more frequently exposed to antibiotics than the environmental source organisms.
Figure 1.
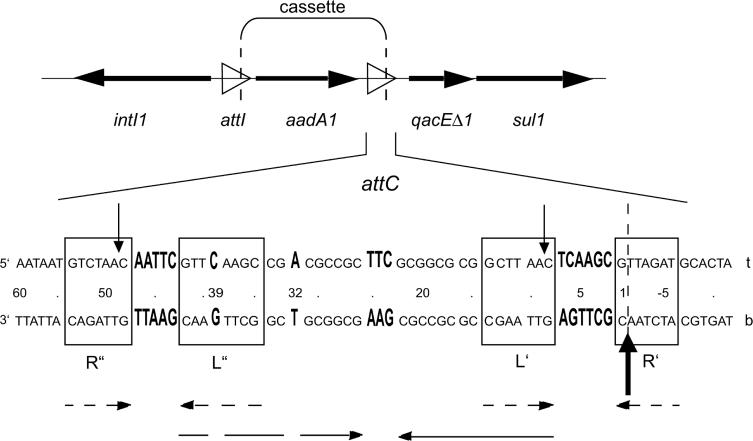
A diagram showing the organization of the integron in Tn21 (X12870), carrying the aadA1 cassette encoding streptomycin/spectinomycin resistance and the following qacEΔ1–sul1 combination downstream of attC (15,33). The integron integrase is encoded in the 5′-conserved sequence, to the left. Genes are shown in bold face horizontal arrows and recombination sites with an open arrowhead. Vertical dashed lines mark cassette borders. The lower panel shows the sequence of the attC site. The site is almost a perfect palindrome organized into two subsites of dyad symmetry (boxes R″ and L″ and boxes L′ and R′). The bold face vertical arrow denotes the primary crossing-over point and the thin vertical arrows secondary sites (27). The ‘t’ stands for top strand and ‘b’ for bottom strand. The nucleotides are numbered from the 3′ cassette border (vertical dashed line) in the 5′–3′ direction according to the bottom strand. Dashed horizontal arrows show the symmetry within each subsite and the thin horizontal arrows the palindromic appearance of the site that is disturbed by the five local interruptions (enlarged bases in bold face).
Bacterial species that carry a chromosomal integron encode their own type of integron integrase and contain a cognate recombinase site, attI, in the vicinity of the gene (22,23). The attI site is the preferred site for recombining with another site, attC, that occurs on each mobile gene cassette (17). Integrations of cassettes are balanced by excisions that occur by recombination between two flanking sites in direct orientation. The attC sites in chromosomal integrons are conserved within each bacterial species. The extensive sequence variation among plasmid-borne attC sites suggests that the cassettes must have been sampled from chromosomal integrons under multiple passages through different bacteria (21).
In contrast to the primary sites attC and attI, secondary sites are abundant in bacterial chromosomes and plasmids (22,24). Recombination at secondary sites must be restricted to maintain genome stability (23). The restrictions are assumed to rely both on a tightly regulated integrase expression and on strict recognition of primary sites by integrase. High recombinational specificity among attC sites has been reported despite the wide sequence variation. This implicates a structural protein recognition determinant in attC that is not sequence-constrained. Our study is a search for this recognition determinant. Reasonably, a favourable secondary structure could be generated by the long palindromic sequence that constitutes attC (15,25). The repeat organization has been further examined by multiple sequence alignments of different attC sites and found to include two pairs of short repeats [(26,27); Figure 1]. The inner of those repeats (L″ and L′), observed most recently, form imperfect inverted repeats of the flank repeats (R″ and R′) on either end (according to the nomenclature by Recchia and Sherratt [(28); Figure 1].
In order to produce a stronger and more specific binding, DNA-binding proteins commonly dimerize and tetramerize prior to or under cooperative binding to pairs of inverted repeats in the DNA (3,29). One example is the dimer resolution system Cre-loxP of bacteriophage P1 which has become the principal model for tyrosine-mediated site-specific recombination (30). The loxP site is a simple recombination site composed of two inverted repeats slightly apart to match a dimer of the tyrosine recombinase Cre (2). Sites of other site-specific recombinases are similar but may also include accessory protein-binding sequences (31). For complex sites such as the attP partner site of the lambda integration system, the middle portion has an analogous function to the simple sites attB and loxP (32). In the case of attC, it is uncertain how the four internal repeats are involved in formation of the expected equivalent to the loxP. The recombinational crossing-over occurs in one of the repeats, R′ (25), and it has been suggested to form an equivalent of a simple site with the neighbouring L′ repeat [(26–28); Figure 1].
We have searched the attC site of the aadA1–qacE cassette junction in Tn21 for features that influence its binding by integrase (Figure 1). The resistance cassette aadA1 encodes an adenylating enzyme mediating streptomycin resistance while the qacE cassette, represented in most integrons by its truncated form qacEΔ1, encodes a multidrug resistance efflux protein (33). The studied site in Tn21 belongs to a family of attC sites that is widespread among plasmid-borne aminoglycoside resistance cassettes, but seems to originate in cassettes borne on the chromosome of Xanthomonas campestris (34) (Figure 2). It was previously reported that integrase binds to the bottom DNA strand of attC in Tn21 but not to the top strand (35). Using oligo constructions we modified one DNA strand at a time in order to identify the determinants for strand-specific integrase binding. The strand-specific recognition could explain why recombination is preferential for the downstream, R′ end of attC.
Figure 2.

Sequence alignment of bottom strands of attC sites from the aadA family and related cassettes. Underlined positions differ from the site in Tn21. The boxed sequences highlight the organization of four inverted repeats. The five asymmetries found in the attC site in Tn21 and in the related attC sites, are shown in bold face. Deficient single nucleotide positions in the palindrome are marked with dashes. Sequences were from aadA1, accession no. X12870; aadA2, X68227; aadA3, AF047479; aadA5, AF137361; aadA6, AF140629; aadA7, AF224733; aadA9, AJ420072; Xcc8, NC003902.
MATERIALS AND METHODS
The mutagenesis strategy
In total, 57 mutations in attC of the aadA1 streptomycin resistance gene cassette in Tn21 were tested for their effects on the binding of integrase to oligodeoxynucleotides representing either the top- or bottom strands of the site. The nucleotide sequences of the bottom-strand oligonucleotides are given in Figure 3. The complementary top-strand oligonucleotides were also tested in most cases. All oligodeoxynucleotides were purchased from Interactiva, Germany, except those containing abasic nucleotides obtained from DNA Technology A/S, Denmark. Nucleotides are numbered in the 5′–3′ direction according to the bottom strand.
Figure 3.
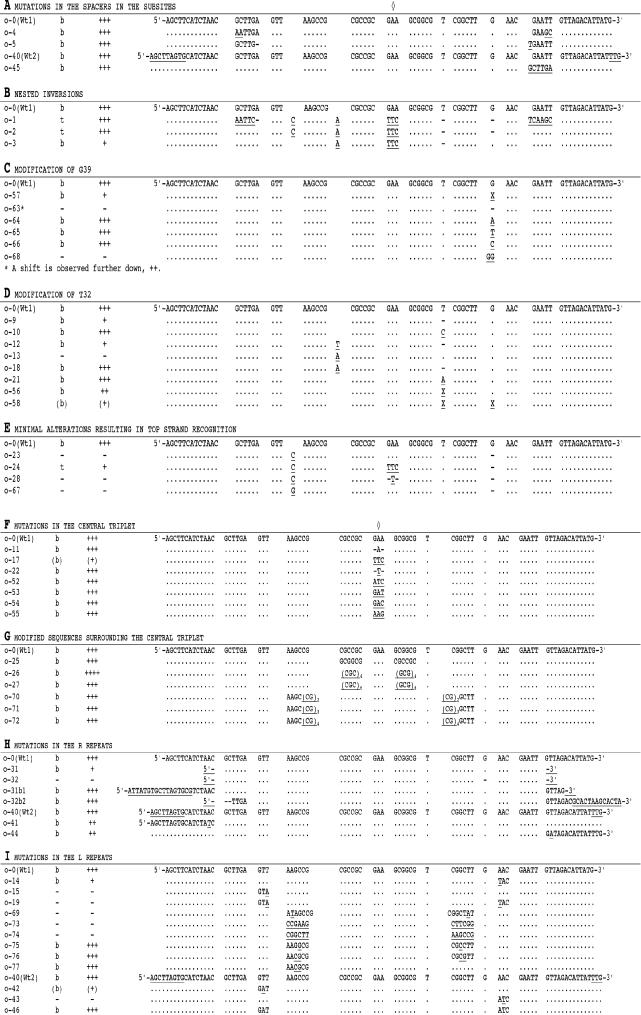
Aligned sequences of oligonucleotides of attC with the modifications studied in this work. The sequences of the bottom strands of the oligo pairs are shown. The symmetry centre is marked with separate diamonds for A–E and F–I respectively. Information for each oligonucleotide pair with respect to binding strand (t or b) and binding strength (highest score +++ to no binding marked with −) is given. The wild-type attC oligo o-0 is denoted wt-1 while wt-2 stands for wild-type attC oligo o-40 (see Materials and Methods). Underlined bases mark aberrations from wild type; hyphen, base deletion; dot, sequence identity to wild-type bottom strand; X, an abasic unit.
Construction of plasmids overexpressing IntI1 and IntI1Y312F
Plasmid p2350 was constructed in a previous study (M. Gullberg, K. Hansson, C. Johansson and L. Sundström, unpublished data) by ligation of a PCR-derived fragment of IntI1, from the class 1 integron of Tn21 (X12870), to the pET19b vector (Novagen). The PCR primer (5′-ATTACTAACAACATATGAAAACCGCCACTGCGCCGTTACC-3′, Scandinavian Gene Synthesis) binding to a site at the upstream end of intI1 was combined with a linker containing a recognition site for NdeI, a site which also occurs in the strong translational start signal in the vector. The PCR primer (5′-TATAGGATCCCTACCTCTCACTAGTGAGGGGCGGC-3′, Scandinavian Gene Synthesis) complementary to a region downstream of intI1 contained a linker site for BamH1. By vector ligation to NdeI- and BamH1-generated termini of the intI1 fragment, N-terminal fusion with the His•Tag™ sequence was obtained. Plasmid p2351 was made in this work through site-directed mutagenesis (QuikChange™ Site-Directed Mutagenesis Kit, Stratagene) of p2350 to replace tyrosine 312 in IntI1 with phenylalanine. The complementary primers used for mutagenesis were 5′-GTCTCTACGACGATGATTTTCACGCATGTGCTGAAAGTT-3′ and 5′-AACTTTCAGCACATGCGTGAAAACATCGTCGTAGAGAC-3′ (Interactiva, Germany), where the changed triplets are underlined. By sequencing the clones containing the replaced tyrosine, Y312F, a second mutation, Q255P, was observed. It was retrieved also in the parental clone, p2350, and was probably created during PCR amplification of the originally cloned fragment. The Q255P mutation was found here at an advanced stage of the experiments and was reverted to a glutamine by site-directed mutagenesis (p2352). In parallel, the same mutagenizing oligos (data not shown) were used to revert Q255P in p2351 to a glutamine in p2353. Resequencing of both p2352 and p2353 confirmed that His-tagged integrase from both these plasmids lacks the Q255P mutation as well as any further divergences from the original wild-type protein with the exception of Y312F in p2353. The effect of the Q255P mutation was modelled using XerD of Escherichia coli (accession no. P21891) as a master structure. This is the closest related protein to integron integrases among those for which the 3D structure has been solved. ClustalW Multiple Sequence Alignment of IntI1 with XerD resulted in a matching of Q255 with glutamine at position 221 in all known XerD recombinases. Q221 in Salmonella typhimurium XerD (U92525) has earlier been implicated to have a role in the binding specificity (36). According to our structure modelling of the IntI1 protein, Q255P was indeed suggested to be in the DNA-binding region. Control experiments in the presence of wild-type IntI1 from p2352 were therefore made with a broad selection of oligonucleotides that had been informative in the assay with IntI1Q255P (also named IntI1*) from p2350 (o-0t, o-0b, o-3t, o-3b, o-9t, o-9b, o-13b, o-17t, o-17b, o-23b, o-24t, o-24b, o-31b, o-32b2, o-44b, o-63b and o-69b). The results showed that the binding affinity of IntI1Q255P to all these oligos was qualitatively identical and quantitatively very similar when compared to that of the wild-type IntI1. These extensive experiments indicated that the results reported in this study are independent of the Q255P mutation.
Bacterial strains and growth media
The concentrations of antibiotic supplements (Sigma Chemical, Sweden) in growth media were for ampicillin, 100 μg/ml; chloramphenicol, 170 μg/ml. DH5α cells were used as a host for construction and maintenance of all plasmid clones. E.coli BL21(DE3) or E.coli BL21(DE3) pLysS were used for protein expression from pET19b clones.
Standard DNA procedures
Plasmids were isolated from E.coli and DNA fragments eluted from gels by using the Qiagen DNA Purification Systems (Qiagen, Germany). Enzymes were purchased from Roche Molecular Biochemicals, Scandinavia. Procedures were as described by Sambrook and Russel (37) or otherwise specifically described.
Integrase purification
Either of the E.coli strains BL21(DE3) or BL21(DE3) pLysS were transformed with one pET19b-clone of intI1 or intI1Y312F (plasmids p2350, p2351, p2352 or p2353). A loop of cells from each transformant colony was reinoculated in LB supplemented with ampicillin, in the case of BL21(DE3)pLysS also chloramphenicol was added, to be grown overnight. Portions of 4 ml from each overnight culture were harvested by centrifugation and resuspended in 4 ml of fresh medium. The resuspended cells were reinoculated in 400 ml LB with the same antibiotic supplements as in precultures and grown at 37°C to mid-exponential phase and cooled to 16°C on an ice bath. T7-polymerase-directed integrase expression was induced by addition of isopropyl-β-d-thiogalactopyranoside (IPTG) at 1 mM. After induction, retarded growth was continued at 16°C for 20 h. The cells were harvested by centrifugation and washed twice in phosphate buffer (20 mM [Na2HPO4–NaH2PO4] pH 7.2, 200 mM NaCl, 1 mM imidazole) supplemented with one-tenth of the volume of 1% Triton X-100 and 0.1 mM of the protease inhibitor phenylmethyl sulfonylflouride (PMSF). The cells were sonicated and insoluble material separated from soluble proteins by centrifugation at 15 000 r.p.m. for 30 min at 4°C. The supernatant was immediately loaded on a Hi-Trap Chelating column (Amersham Pharmacia Biotech, Sweden) charged with Ni2+ ions. Elution of His–IntI1 protein was obtained using a linear gradient of imidazole and NaCl ranging from 20 mM imidazole and 200 mM NaCl to 800 mM imidazole and 40 mM NaCl. The amount and purity of His–IntI1 protein was analysed using SDS–PAGE and the dye-binding method of Bradford (38).
Binding assay
Oligodeoxynucleotides representing either strand of attC were incubated with purified integrase. Protein–DNA complexes were assayed by reduction of their electrophoretic mobilities relative to control DNA incubated in the absence of protein. To accomplish autoradiographic detection, the dephosphorylated oligodeoxynucleotides were labelled at their 5′ termini with radioactive phosphate transferred from [γ-32P]ATP by polynucleotide kinase. The 20 μl binding mixtures were composed of 200–300 c.p.s. of ssDNA oligonucleotide (0–12 nM), 0–1900 nM of integrase, 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 1 mM CHAPS, 0.2 mM EDTA, 5% glycerol, 1 mM DTT, 1 μg poly(dI–dC)•poly(dI–dC), 0.7 μg BSA and ∼200 mM imidazole. Incubation for binding was for 15 min at 30°C and then one-tenth of the volume of loading dye was added (0.2% xylene cyanol, 0.2% bromophenol blue, 50% saccharose, 100 mM EDTA pH 7.9). The electrophoretic separation was performed on a 6% non-denaturing polyacrylamide gel [AccuGel™ 29:1 Acrylamide:Bis-Acrylamide (In vitro, Sweden)] at 4°C in 1× TBE buffer.
Assay for covalently linked integrase
The assay was performed as described above but under denaturing conditions due to the presence of SDS, with the following changes. The mixtures of 20 μl were incubated for 0, 15, 30 or 45 min. The reactions were stopped by addition of 4 μl of stop solution (Tris–HCl pH 7.5 50 mM and 1% SDS) before loading on a 6% polyacrylamide gel [AccuGel™ 29:1 Acrylamide:Bis-Acrylamide (In vitro, Sweden)] supplemented with 0.1% SDS. On this denaturing gel only protein that is covalently bound to the DNA causes a mobility shift. The running buffer contained 1× TBE and 0.1% SDS. The gel was run at room temperature.
Control excluding linker-design-related errors
Most complementary oligos in this study were prepared for having sticky ends after annealing to form double-stranded DNA fragments suitable for cloning in plasmid vectors. To check for design-related errors a prolonged wild-type oligo pair, o-40, was made (Figure 3). It is identical to attC of Tn21 and longer than o-0 although it lacks the linkers attached to the ends of the latter. Since the top- and bottom strands of the oligo pairs o-0 and o-40 gave the same assay results, the design-related modifications outside the R repeats were excluded from influencing the binding.
Proteinase K treatment
To verify that gel-shift responses were related to the binding of protein, complexes were subjected to proteolytic degradation by proteinase K. After the above incubation of the mixtures containing oligonucleotides and integrase, 0–100 μg of proteinase K was added. The buffer was optimized for the binding reaction but not for proteolytic treatment which is carried out by an excess amount of proteinase K. The mixtures were further incubated at 37°C for 30 min before analysis either by native or denaturing PAGE.
RESULTS
Assay of mobility shifts for mutant attC oligos
Oligonucleotides representing mutations in the attC site in Tn21 were tested for binding of integrase using the electrophoretic mobility shift assay, EMSA (Figure 3). In most instances oligos of both complementary DNA strands were tested and are therefore referred to as oligo pairs. This term does not mean that the oligos were annealed (unless stated). Our data confirmed the observation by Francia et al. (35) that the integrase, IntI1, binds to the bottom strand of wild-type attC and not to the top strand (Figure 4). Some oligo pairs failed to bind the protein at all while the majority of oligo pairs exhibited binding for either the top- or the bottom strand. The attC site in Tn21 is basically a palindromic sequence with five local interruptions (enlarged bases in Figure 1). These are the central triplet, a pentamer to the left, a hexamer to the right and finally, two single base pairs at positions 32 and 39. If attC is converted to a cruciform, either of the two hairpin arms will be bulged and the central triplets will form hairpin loops (for an illustration see Figure 7).
Figure 4.
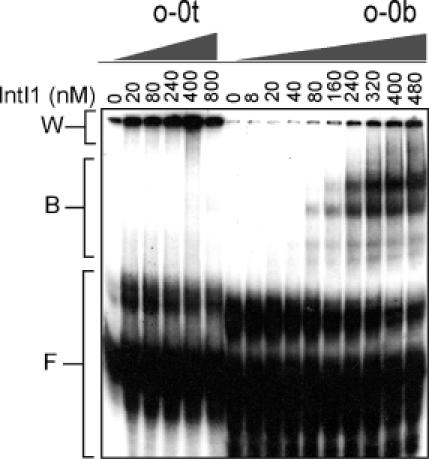
Gel retardation assay was done as described in Materials and Methods in the presence of the indicated amounts of IntI1. Oligomers comprising 72 nt of either top- or bottom strand of the wild-type attC (o-0t, o-0b) were labelled with 32P and used at a concentration of 8 and 4 nM, respectively, corresponding to 300 c.p.s. in each binding reaction. B, protein–DNA complexes; F, free single-stranded DNA; W, aggregated DNA that remained in the wells.
Figure 7.
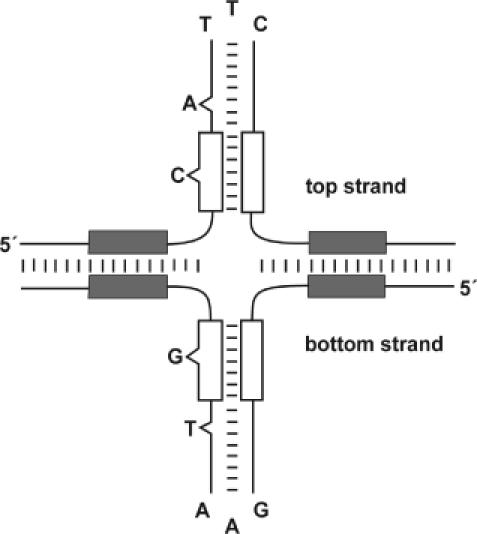
A sketch of the proposed cruciform drawn with the assumption that the structure is stabilized by the integrase. The L repeats (white boxes) are included in the hairpin arms since complementarity is observed to be important for binding (Results). The two spacers and the R repeats (grey boxes) are drawn outside the hairpin arms. The asymmetrical bases 23–25, 32, 39 are drawn as loops and bulges. Base pairing is visualized by short lines. Base pairing in the spacer region is assumed to be weakened upon extrusion of hairpins and therefore all base pairs are not shown.
Mutations in the spacers within the subsites
The R″L″ and L′R′ subsites, on either the 5′ or 3′ half of attC respectively, contain different sequences between the respective core repeats (Figure 1). The L′R′ subsite carries a hexamer spacer between the repeats and the R″L″ subsite, a pentamer. Extension or shortening of the spacers did not influence protein binding (o-5, Figure 3A). Neither did an exchange of nucleotides adjacent to the R″ and R′ repeats (o-4) nor substitution of the entire pentamer spacer with the hexamer spacer (o-45) affect binding. It was therefore concluded that the spacers in the subsites do not contain the main strand specificity (or site orientation) signal.
Nested inversions
A series of nested inversions was examined (Figure 3B). The longest inversion was that represented in o-1t (top strand) and o-1b (bottom strand). This inversion excluded R′ and R″ but comprised all sequences bracketed by these repeats. An intense electrophoresis mobility shift was observed for o-1t and no shift for o-1b, suggesting that the strand choice by integrase responded to the inversion and is independent of the R′ and R″ repeats (Figure 1). A shorter inversion that excluded also the hexamer and the pentamer (o-2t and o-2b), gave a very similar response: an intense retarded band appeared for the top-strand oligomer and no retardation for the bottom-strand oligomer. The next shorter inversion excluded also the two L repeats (o-3t and o-3b). Retardation was now observed for the bottom strand but a smaller amount of DNA was retarded than for the wild-type oligo (o-0b). The observed shifts with o-1t, o-2t and o-3b show that strand choice determinants are in either L′ or L″ (Figure 1).
Modification of G39
The inverted repeats L′ and L″ differ only by a single base pair in L″ that has no counterpart in L′ (a G at position 39 in the bottom strand, Figure 1). A consequence of the inversions in o-1 and o-2 that result in binding to the top strand (see previous paragraph) is that G39 is removed from L″ while it appears in L′. The asymmetric G39 could therefore be the principal determinant for directing integrase to the bottom strand. A semi-intense and weakly retarded shift was observed when G39 was deleted (o-63b; Figure 3C). The non-symmetrical nucleotide G39 is conserved among many different attC sites (26) (Figure 2). However, substitutions of G39 with any of the alternative bases (o-64b, o-65b and o-66b; Figure 3C) did not alter retardation by integrase. In oligonucleotide o-68b (Figure 3C) another G was inserted next to G39. No binding was observed to this oligo that most likely carries a disrupted secondary structure at G39, or a GG bulge. When an oligo with an abasic unit replacing G39 was tested (o-57b; Figure 3C), a weak retarded band appeared.
Modification of T32
Deletion of T32 (o-9b) did not change the bottom-strand specificity but the intensity of the shifted band was weak (Figure 3D). The asymmetry due to T32 was eliminated by the insertion of an A between positions 16 and 17 (Figure 1) to form an internal base pair with T32 in stem–loop DNA (o-13). Binding to neither strand was observed. In other oligo designs T32 was substituted with a C (o-10b) or an A (o-21b) but neither of these alterations affected binding of integrase. Finally, in oligos o-12 and o-18 the T32 was deleted while a T or an A was inserted between positions 16 and 17 (Figure 1) to generate a bulge on the opposite side of a potential stem–loop. Binding to the bottom strand now remained but the amount of retarded oligos responded to the choice of inserted nucleotide. Thus, T32 is a vital nucleotide for attracting integrase to the bottom strand and is needed to obtain full binding to the site but its positioning is less important than for the other asymmetrical nucleotide, G39. Oligos with abasic units replacing T32 or both T32 and G39 were tested. The oligo with an abasic ‘spacer’ at position 32 (o-56b) bound integrase stronger than the abasic ‘spacer’-39 oligo described earlier. The oligo o-58b with ‘spacer’ positions 32 and 39 combined, gave a barely detectable shift (Figure 3D).
Minimal alterations resulting in top-strand recognition
The minimal attC modification that turns over the integrase specificity to the top-strand oligo was a loop inversion between positions 23 and 25 in combination with a transfer of base 39 from L″ to L′ (o-24t; Figure 3E). However, the shifted o-24t band was relatively weak. To obtain full binding to o-24t, it seems possible that the two asymmetrical nucleotides need to be on the same side of the symmetry axis as in o-2t (Figure 3B). When the loop alteration was omitted while the change concerning transfer of nucleotide 39 was made, neither of the resulting complementary oligos in o-23 (C in L′) and o-67 (G in L′) bound integrase. Upon shortening of the inverted central triplet to 1 nt, again combined with repositioning of nucleotide 39 from L″ to L′, integrase bound to neither strand (o-28) (Figure 3E).
Mutations in the central triplet
Almost all tested top strands that did not bind integrase contained the central triplet sequence TTC. This infers a negative influence from TTC that was further observed in some bottom-strand designs such as o-17b (Figure 3F) and o-3b (Figure 3B). o-17 demonstrated a very weak binding to the bottom strand and no binding to the top strand. However, a bottom-strand oligo in which the central triplet (GAA) was shortened to an A or a T (o-11b, o-22b; Figure 3F) still gave full binding. The stability of hairpins formed at the strong topoisomerase II site in pBR322 has been reported to depend strongly on the loop sequence (39,40). These data suggest that the bottom strand of attC has a more stable conformation than the top strand due to the loop sequence. That in turn could promote binding of integrase to the bottom strand of attC. A few loop modifications that were reported to be responsive for the strong topoisomerase II site were introduced as substitutions for GAA in the bottom strand of the attC site and tested for integrase binding (Figure 3F). ATC (o-52b) and GAC (o-54b) were previously documented to promote hairpin stability and hence binding of topoisomerase II and gave the same effect on integrase binding to attC. However, GAT (o-53b) and AAG (o-55b) that reduced hairpin stability in the topoisomerase II site did not reduce integrase binding to the bottom strand of attC. Therefore, a simple correlation between loop stability and binding strength was not confirmed in the case of attC.
Modification of the sequences surrounding the central triplet
In the oligo o-25b, bases 17-22 and 26-31 (Figures 1 and 3G) were swapped. This did not affect binding. Due to the insertion of CGC (or GCG) units on either side of the central triplet, the length of the potentially formed hairpin increased by 6 (o-26) and 12 (o-27) intramolecular base pairs, respectively (Figure 3G). Finally, symmetrical insertions of one or several CG repeats were made between G39 and T32. This prolonged the hairpin by one CG repeat in o-70b, by two CG repeats in o-71b and by three repeats in o-72b. None of the five described extensions of the potential hairpin influenced the amount of shifted DNA (Figure 3G).
Mutations in the R repeats
The GTT/AAC motif is conserved among all four core repeats of attC in Tn21 (Figure 1) and also in most other sites within the site class (Figure 2). The AAC of R′ was changed to ATC (o-41b) and GTT of R″ was changed to GAT (o-44b; Figure 3H). Both modifications only slightly decreased the amount of integrase bound to the bottom strand. The bottom-strand oligo, o-31b (Figure 3H), with deletions comprising both R repeats, gave a weak shifted band. The band disappeared completely when the deletions of both R′ and R″ were combined with the deletion of G39 (o-32b; Figure 3H). By leaving R′ intact and truncating R″ to comprise only 5 nt (o-31b1; Figure 3H), an intense shifted band was observed. A reciprocal and more radical deletion of the entire R′ repeat and two bases of the hexamer was made in the 5′ end of the bottom-strand oligo o-32b2 (Figure 3H). This oligo still gave an intense shift.
Mutations in the L repeats
The GTT of L′ was modified to GTA in o-15b and to GAT in o-42b (Figure 3I). No shift was observed for o-15, and o-42b gave a barely discernable shifted band. The AAC in the L″ repeat was modified to TAC in o-14b and to ATC in o-43b. The results were a weak shifted band for o-14b and no band shift for o-43. Another oligo pair, o-19, carried the combined base changes in o-15 (L′) and o-14 (L″) and similarly the oligo pair o-46 combined the base alterations in o-42 (L′) and o-43 (L″). In the doubly mutated oligos, o-19 and o-46, base pairing was restored between altered bases. An intense shifted band for o-46b was observed but neither strand in o-19 was bound by the integrase (Figure 3I).
In the oligo pair o-73, internal bases (11-13 and 14-16 in L′; 33-35 and 36-38 in L″) were swapped within each repeat. By this reciprocal operation, base matching was maintained in the fold of a hairpin (Figures 1 and 3I). Although the swapped sequences did not overlap with the conserved GTT/AAC motif, neither o-73b nor o-73t bound IntI1. In o-74, the six bases (11–16) following GTT in L′ were replaced with the six bases (33–38) immediately preceding the G39 in the L″ tract, and vice versa. Neither o-74b nor o-74t shifted upon incubation with integrase. Thus, in addition to the conserved GTT/AAC motif and the G39 in L″, further bases of L′ and L″ are crucial for binding. The data show that these distal bases in the repeat pair have a function beyond that of creating base matching of L′ with L″. This is further confirmed by the oligo pair o-69 (Figure 3I) where one T was inserted between the fourth and fifth base in L′ and a complementary A was inserted between the fifth and sixth base in L″. Neither o-69t nor o-69b bound integrase, indicating that a minimal length of five (L′) or six (L″) bases is needed to specify the functional L repeats.
In o-76b, G13 and C14 in L′ were swapped, and base matching was retained by the alteration also for C36 and G35 in L″. Binding of integrase to o-76b was comparable to wild type, suggesting that the sixth nucleotide in the L repeats was exchangeable. In o-77b the base changes in L″ were omitted. Surprisingly, we observed a strong band of shifted o-77b, indicating that the requirement for matching between the distal parts of L′ and L″ is not absolutely essential for binding of integrase. To check the distal bases in the L repeats more directly, C14 in L′ was substituted with a G and G35 in L″ was changed to a matching C. The resulting oligo o-75b gave an intense integrase shift, equal to that of the wild-type oligo o-0b. Thus, the seventh nucleotide in the L′ repeat and the corresponding nucleotide in L″ did not influence binding of integrase. Our data suggest that the functional part of the L′ repeat comprises 5 nt while the L″ repeat is 1 nt longer because it contains the asymmetrical G39 (Figure 3I).
Phosphotyrosine linkage
The stability of the retarded IntI1–o-0b complexes was tested using EMSA under denaturing conditions (for details see Materials and Methods). A shifted band was observed further down the gel compared to the shift seen in the regular assay (Figure 5i). This reduced retardation is expected to be due to the denaturing effect of SDS on the integrase. The shifted band was recorded after all incubation times tested, except when the reaction had been stopped with an SDS-containing stop solution immediately after addition of integrase to the mixtures (Figure 5ii). The response to the addition of SDS indicates that a covalent linkage, probably a phosphotyrosine linkage, is established between DNA and protein. To confirm this hypothesis, integrase was modified by site-directed mutagenesis. In the modified integrase IntI1Y312F, the nucleophilic tyrosine was substituted with a phenylalanine. Using IntI1Y312F, the protein–DNA complex was not observed under denaturing conditions (Figure 5iii). However, this complex shifted as a normal wild-type IntI1–o-0b complex on a native gel (Figure 6), indicating that a covalent linkage is not necessary for binding of integrase to attC. However, the shifted amount of wild-type IntI1–o-0b complex is comparable under the two different conditions, suggesting that an attack on a phosphate in attC will be promoted if there is a tyrosine 312. The shifts observed on the denaturing gel (data not shown) and on the native gel disappeared when the samples were treated with proteinase K (Figure 6).
Figure 5.
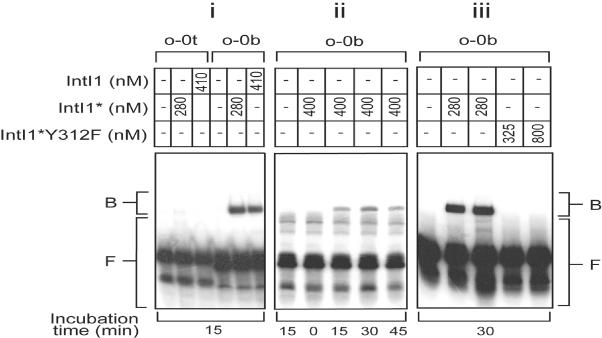
Gel retardation assay performed under denaturing conditions to detect the formation of a covalent linkage between oligomeric top- and bottom strands (o-0t, o-0b) of the wild-type attC and integrase. Protein fractions and concentrations as well as incubation times are indicated. The oligonucleotides were labelled with 32P and used at 1–6 nM, corresponding to ∼200–300 c.p.s. per binding mixture. The mutation in IntI1, Q255P, is abbreviated with an asterisk. For control experiments and more details see Materials and Methods. (i) A comparison of the binding of IntI1* and of IntI1 to top- and bottom strands of wild type attC in the presence of 0.42 μg poly(dI–dC)•poly(dI–dC). (ii) Assay at different incubation times before addition of SDS stop solution. (iii) Assay where the bottom strand of wild-type attC was mixed with IntI1* or IntI1*Y312F, in which the nucleophilic tyrosine is substituted with a phenylalanine. B, protein–DNA complexes; F, free DNA.
Figure 6.
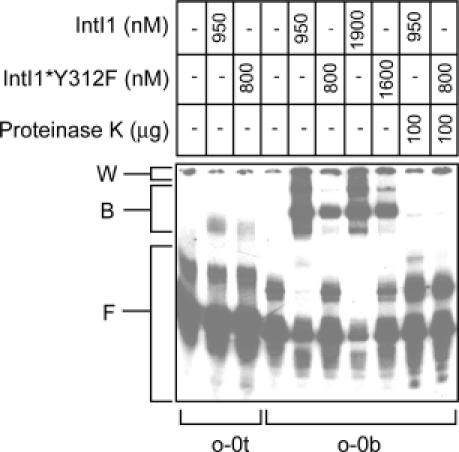
Gel retardation assay for comparing the binding of wild-type IntI1 versus IntI1*Y312F to top- and bottom-strand oligomers of attC. In the two lanes furthest right the effect of proteinase K treatment was investigated. Proteins and concentrations used are shown. The single-stranded oligomers o-0t and o-0b were labelled with 32P and used at a concentration of 8 and 12 nM respectively, corresponding to ∼250 c.p.s. per binding mixture. For more details and abbreviations, see Material and Methods. B, protein–DNA complexes; F, free DNA; W, aggregated DNA that remained in the wells.
DISCUSSION
In this work, the attC site, also known as a 59-base element, in Tn21 has been searched for features that could explain why it is specifically recognized by integron integrase. Our data imply that the complex repeat pattern of the site forms a robust structural recognition determinant that has allowed extensive sequence divergence under evolution. The attC site is a long, almost perfect palindrome composed of four internal repeats. The data presented here and earlier predictions (26,27) indicate that at least two of these repeats (L′ and L″) are involved in intramolecular base pairing that could promote the formation of cruciform DNA (Figure 7). Among the five features that diverge from the palindrome, we observed two single-nucleotide asymmetries in particular, that when deleted strongly influenced binding. At least one of these, T32, is predicted to bulge out from the hairpins of attC. We noticed that these nucleotides are also conserved among attC sites of generally low overall conservation [for alignments, see (26,27)] and give the hairpins a kinked structure that is implicated to be crucial for site recognition by integron integrase.
It was demonstrated by Francia et al. (35) that the integrase binds with a strong bias for the bottom strand of attC. By analysis of protein binding to the individual strands separately, it became possible to search attC for features that are related to the affinity of the integrase for the site. With minor alterations, we gained strong binding to the top strand with a reciprocal loss of binding to the bottom strand. In fact, we failed to find any pair of complementary oligonucleotides where integrase bound to both strands. Perhaps, this is a consequence of the specificity of integrase for the bottom strand being determined by the order, orientation and position of features in the hairpin arms. Naturally, these features are bound to appear differently in the two strands of a hairpin with its topological handedness.
Sites of various tyrosine recombinases are built on a core composed of two inverted repeats flanking a short central region. With its multiple repeat organization, attC exemplifies a unique type of site bound by a tyrosine recombinase. The multiplicity of repeats makes attC similar to sites of many resolving serine recombinases (4,41) but attC differs from those sites by its overall palindromic nature. The four repeats in attC are divided into two pairs, composed of L′ and R′ in one pair and R″ and L″ in the other (Figure 1). Either of these pairs could function as a simple site or subsite. The subsite on the upstream end of attC could be an accessory core site (K. Hansson and L. Sundström, unpublished data). We estimated the size for either subsite in attC to be about 16 bp, which is about half the size of loxP. Numerous secondary sites of integron integrases are scattered in plasmids and in bacterial chromosomes. These relatively inefficient sites have a simple site organization (22,24). Our data suggest that the much more efficiently recombining attC site has a flexible DNA conformation that may allow the four repeats to appear sequentially in alternative pairs or secondary structures. It is also possible that repeats that are remote from one another could constitute binding sites. For instance, the L′ and L″ repeats could form a simple site when extruded in top- and bottom-strand hairpins and the outer repeats might equally likely be moved close enough to form a functional binding site. We hypothesize the following scenario. The subsites R″L″ and L′R′, are weakly recognized to initiate hairpin extrusion that might be driven further by the stronger integrase binding to the paired L′ and L″ repeats. Finally, the site might adopt a mature, cruciform shape where the outer repeats (R′ and R″) are brought close enough to one another to be grasped by a postulated dimer of integrase. It seems very possible that the interaction studied in this work using oligonucleotides primarily reflects binding of integrase to the hybridized L repeats on at least one of the hairpin arms. In this step the complementarity between intrastrand L′ and L″ repeats is expected to be important. We have reported several sequence alterations in the L′ and L″ repeats that were deleterious for binding. The effects were not plainly explained by a reduced matching between the two repeats and imply other sequence contraints for the L′ and L″ repeats.
This work and very few previous articles pioneer detailed examination of how purified integrase binds to attC (35,42,43). The oligo-based experiments may only describe one conformational state of the site, while the differentiated appearance of the site in recombination complexes formed in vivo must be approached using other methods. Delicate transitions between different shapes of the site could sense local events such as transcription, replication and DNA topology and hence convey regulatory functions. All four repeats of attC are likely to be important components of the site but are now indicated to have different roles in recombination (K. Hansson and L. Sundström, unpublished data). The restricted viewpoint of our integrase-to-oligo EMSA could explain why point alterations in either of the R″ and R′ repeats were less responsive than those made in the L repeats. The R repeats are proven to be the most responsive parts of the site in a recombination assay in vivo [(27); K. Hansson and L. Sundström, unpublished data]. The R′ repeat, containing the primary crossing-over point, was reported to be highly responsive to mutations and unpublished results from this laboratory report a vital function also for R″. Removal of R′ in oligonucleotide o-32b2 did not reduce the intensity of the shifted band, but when both R′ and R″ were missing the band was much weaker. It is therefore likely that the L repeats are included in the extruded hairpins of the proposed cruciform while the two R repeats, the hexamer and the pentamer, are less likely included. The spacers in each subsite are not complementary and are hence deprived from secondary structure formation. Potentially these act as hinges where duplex DNA is transformed into a cruciform. The contracted form of attC would consist of the duplex forms of the two R repeats, the pentamer and the hexamer, spanning 21 bp in total. In addition, elongation will occur due to the hairpins, resulting in a size that could well approach a regular size of a tyrosine recombination core site.
We have reported in this study that the retarded complexes of integrase bound to oligomeric attC strands persist under denaturing conditions. This implicates the formation of a covalent linkage between oligomeric DNA and protein. Previous data have confirmed a crucial function of tyrosine 312 in recombination in vivo (44). When substituting the reactive tyrosine with the inert phenylalanine (IntI1Y312F), the capability of integrase to attack the DNA was lost. Consistently, the mutant integrase gave no shifts in denaturing gels. It is well established that the new bond, upon nucleophilic phosphodiester cleavage by tyrosine, is formed between Y312 in the integrase and the terminal 3′-phosphate group in the cleaved DNA (30,45). The cleavage reaction including the time course, is presently under further investigation. It seems unlikely that cleavage occurs in a non-folded single strand. If we do not consider oligo dimerization it seems most likely from stability calculations (data not shown) that hairpin extrusion progresses up to the L repeats. Cleavage coinciding with the regular crossover site in the R′ repeat is most likely. However, in the single-stranded and possibly folded state of the site, the tyrosine-attacked sequence might alternatively be L′ or R″, both of which have been reported to be fairly active secondary sites [Figure 1; (27)]. The L″ repeat, in contrast, is not reported to recombine in the literature. A very likely explanation is that L″ is silenced by the inserted nucleotide (G39) that is missing in the other three repeats.
Cruciform DNA and Holliday junctions are two related forms of branched DNA that are both substrates for tyrosine recombinases. We tentatively speculate over the exciting possibility of analogy with telomere resolution of replicating linear chromosomes, recently reported to depend on tyrosine recombinase-like proteins (12). This could mean that processing of a cruciform might occur in the course of recombination.
Recent data have proven that plasmid integrons stem from chromosomal integrons in a subclass of gram-negative bacteria in which their role is still only beginning to be understood (21). In each source bacterium the integron is of a distinct class and furthermore, its cassettes carry one class of attC. Remarkably, chromosomal attC sites in their broad variation are cross-recognized by plasmid integrase, such as that from Tn21 (46). The recently determined genome sequence of X.campestris reveals the presence of an integron with attC sites identical to that studied here (C. Norman-Setterblad, unpublished data; Figure 2). We conclude that our reported data on the Tn21 integron also holds true for cassettes borne on the chromosome of Xanthomonas spp. Clearly, integron integrases perform a new type of structural recognition, that with high precision directs recombination to the scissile phosphates in a very diverse family of sites.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank J. Karlsson, L. Skärberg for assistance, R. Robinson for help with structure modelling, O. Sköld, M. Rush, Y. Qvarnström and C. Norman-Setterblad for reading the manuscript. We are grateful for grants from the Swedish Research Council 12638, AFA Health Fund, Swedish Academy for Pharmaceutical Sciences, Anna-Maria Lundins Fund.
REFERENCES
- 1.Ochman H., Lawrence,J.G. and Groisman,E.A. (2000) Lateral gene transfer and the nature of bacterial innovation. Nature, 405, 299–304. [DOI] [PubMed] [Google Scholar]
- 2.Hoess R.H. and Abremski,K. (1984) Interaction of the bacteriophage P1 recombinase Cre with the recombining site loxP. Proc. Natl Acad. Sci. USA, 81, 1026–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pabo C.O. and Sauer,R.T. (1984) Protein–DNA recognition. Annu. Rev. Biochem., 53, 293–321. [DOI] [PubMed] [Google Scholar]
- 4.Stark W.M., Boocock,M.R and Sherratt,D.J. (1989) Site-specific recombination by Tn3 resolvase. Trends Genet., 5, 304–309. [DOI] [PubMed] [Google Scholar]
- 5.Echols H. (1990) Nucleoprotein structures initiating DNA replication, transcription, and site-specific recombination. J. Biol. Chem., 265, 14697–14700. [PubMed] [Google Scholar]
- 6.Mondragon A. (1997) Solving the cis/trans paradox in the Int family of recombinases. Nature Struct. Biol., 4, 427–429. [DOI] [PubMed] [Google Scholar]
- 7.Esposito D. and Scocca,J.J. (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res., 25, 3605–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunes-Düby S.E., Kwon,H.J., Tirumalai,R.S., Ellenberger,T. and Landy,A. (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res., 26, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nash H.A. (1996) Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology, 2nd edn. ASM Press, Washington, DC, pp. 2363–2376. [Google Scholar]
- 10.Austin S., Ziese,M. and Sternberg,N. (1981) A novel role for site-specific recombination in maintenance of bacterial replicons. Cell, 25, 729–736. [DOI] [PubMed] [Google Scholar]
- 11.Blakely G., Colloms,S., May,G., Burke,M. and Sherratt,D. (1991) Escherichia coli XerC recombinase is required for chromosomal segregation at cell division. New Biol., 3, 789–798. [PubMed] [Google Scholar]
- 12.Kobryn K. and Chaconas,G. (2002) ResT, a telomere resolvase encoded by the Lyme disease spirochete. Mol. Cell, 9, 195–201. [DOI] [PubMed] [Google Scholar]
- 13.Cameron F.H., Groot Obbink,D.J., Ackerman,V.P. and Hall,R.M. (1986) Nucleotide sequence of the AAD(2″) aminoglycoside adenylyltransferase determinant aadB. Evolutionary relationship of this region with those surrounding aadA in R538-1 and dhfrII in R388. Nucleic Acids Res., 14, 8625–8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouellette M., Bissonnette,L. and Roy,P.H. (1987) Precise insertion of antibiotic resistance determinants into Tn21-like transposons: nucleotide sequence of the OXA-1 beta-lactamase gene. Proc. Natl Acad. Sci. USA, 84, 7378–7382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundström L., Rådström,P., Swedberg,G. and Sköld,O. (1988). Site-specific recombination promotes linkage beween trimethoprim- and sulfonamide resistance genes. Sequence characterization of dhfrV and sulI and a recombination active locus of Tn21. Mol. Gen. Genet., 213, 191–201. [DOI] [PubMed] [Google Scholar]
- 16.Martinez E. and de la Cruz,F. (1990) Genetic elements involved in Tn21 site-specific integration, a novel mechanism for the dissemination of antibiotic resistance genes. EMBO J., 9, 1275–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall R.M. and Collis,C.M. (1995) Mobile gene cassettes and integrons: capture and spread of genes by site-specific recombination. Mol. Microbiol., 15, 593–600. [DOI] [PubMed] [Google Scholar]
- 18.Leverstein-van Hall M.A., M Blok,H.E., T Donders,A.R., Paauw,A., Fluit,A.C. and Verhoef,J. (2003) Multidrug resistance among Enterobacteriaceae is strongly associated with the presence of integrons and is independent of species or isolate origin. J. Infect. Dis., 187, 251–259. [DOI] [PubMed] [Google Scholar]
- 19.Ouellette M. and Roy,P.H. (1987) Homology of ORFs from Tn2603 and from R46 to site-specific recombinases. Nucleic Acids Res., 15, 10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Messier N. and Roy,P.H. (2001) Integron integrases possess a unique additional domain necessary for activity. J. Bacteriol., 183, 6699–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rowe-Magnus D.A., Guerout,A.M., Ploncard,P., Dychinco,B., Davies,J. and Mazel,D. (2001). The evolutionary history of chromosomal super-integrons provides an ancestry for multiresistant integrons. Proc. Natl Acad. Sci. USA, 98, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Recchia G.D., Stokes,H.W. and Hall,R.M. (1994) Characterization of specific and secondary recombination sites recognised by the integron DNA integrase. Nucleic Acids Res., 22, 2071–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansson K., Sköld,O. and Sundström,L. (1997) Non-palindromic attI sites of integrons are capable of site-specific recombination with one another and with secondary targets. Mol. Microbiol., 26, 441–453. [DOI] [PubMed] [Google Scholar]
- 24.Francia M.V., de la Cruz,F. and Garcia Lobo,J.M. (1993) Secondary sites for integration mediated by the Tn21 integrase. Mol. Microbiol., 10, 823–828. [DOI] [PubMed] [Google Scholar]
- 25.Hall R.M., Brookes,D.E. and Stokes,H.W. (1991) Site-specific insertion of genes into integrons: role of the 59-base element and determination of the recombination cross-over point. Mol. Microbiol., 5, 1941–1959. [DOI] [PubMed] [Google Scholar]
- 26.Francia M.V., Avila,P., de la Cruz,F. and Garcia Lobo,J.M. (1997) A hot spot in plasmid F for site-specific recombination mediated by Tn21 integron integrase. J. Bacteriol., 179, 4419–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stokes H.W., O'Gorman,D.B., Recchia,G.D., Parsekhian,M. and Hall,R.M. (1997) Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol. Microbiol., 26, 731–745. [DOI] [PubMed] [Google Scholar]
- 28.Recchia G.D. and Sherratt,D.J. (2002) Gene acquisition in bacteria by integron-mediated site-specific recombination. In Craig,N.L., Craigie,R., Gellert,M. and Lambowitz,A.M. (ed.), Mobile DNA II. ASM Press, Washington, DC, pp. 162–176. [Google Scholar]
- 29.Anderson W.F., Ohlendorf,D.H., Takeda,Y. and Matthews,B.W. (1981) Structure of the cro repressor from bacteriophage lambda and its interaction with DNA. Nature, 290, 754–758. [DOI] [PubMed] [Google Scholar]
- 30.Van Duyne G.D. (2001) A structural view of Cre-loxP site-specific recombination. Annu. Rev. Biophys. Biomol. Struct., 30, 87–104. [DOI] [PubMed] [Google Scholar]
- 31.Segall A.M. and Nash,H.A. (1993) Synaptic intermediates in bacteriophage lambda site-specific recombination: integrase can align pairs of attachment sites. EMBO J. 12, 4567–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dorgai L., Sloan,S. and Weisberg,R.A. (1998) Recognition of core binding sites by bacteriophage integrases. J. Mol. Biol., 277, 1059–1070. [DOI] [PubMed] [Google Scholar]
- 33.Paulsen I.T., Littlejohn,T.G., Radstrom,P., Sundstrom,L., Skold,O., Swedberg,G. and Skurray,R.A. (1993) The 3′ conserved segment of integrons contains a gene associated with multidrug resistance to antiseptics and disinfectants. Antimicrob. Agents Chemother., 37, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.da Silva A.C., Ferro,J.A., Reinach,F.C., Farah,C.S., Furlan,L.R., Quaggio,R.B., Monteiro-Vitorello,C.B., Van Sluys,M.A., Almeida,N.F., Alves,L.M. et al. (2002) Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature, 417, 459–463. [DOI] [PubMed] [Google Scholar]
- 35.Francia M.V., Zabala,J.C., de la Cruz,F. and Garcia Lobo,J.M. (1999) The IntI1 integron integrase preferentially binds single-stranded DNA of the attC site. J. Bacteriol., 181, 6844–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanya H.S., Arciszewska,L.K., Baker,R.A., Bird,L.E., Sherratt,D.J. and Wigley,D.B. (1997) Crystal structure of the site-specific recombinase, XerD. EMBO J., 16, 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambrook J. and Russel,D.W. (2001) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Bradford M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 39.Mauffret O., Amir-Aslani,A., Maroun,R.G., Monnot,M., Lescot,E. and Fermandjian,S. (1998) Comparative structural analysis by [1H,31P]-NMR and restrained molecular dynamics of two DNA hairpins from a strong DNA topoisomerase II cleavage site. J. Mol. Biol., 283, 643–655. [DOI] [PubMed] [Google Scholar]
- 40.El Amri C., Mauffret,O., Monnot,M., Tevanian,G., Lescot,E., Porumb,H. and Fermandjian,S. (1999) A DNA hairpin with a single residue loop closed by a strongly distorted Watson–Crick G × C base-pair. J. Mol. Biol., 294, 427–442. [DOI] [PubMed] [Google Scholar]
- 41.Sarkis G.J., Murley,L.L., Leschziner,A.E., Boocock,M.R., Stark,W.M. and Grindley,N.D. (2001) A model for the gamma delta resolvase synaptic complex. Mol. Cell, 8, 623–631. [DOI] [PubMed] [Google Scholar]
- 42.Gravel A., Fournier,B. and Roy,P.H. (1998) DNA complexes obtained with the integron integrase IntI1 at the attI1 site. Nucleic Acids Res., 26, 4347–4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collis C.M., Kim,M.J., Stokes,H.W. and Hall,R.M. (1998) Binding of the purified integron DNA integrase Intl1 to integron- and cassette-associated recombination sites. Mol. Microbiol., 29, 477–490. [DOI] [PubMed] [Google Scholar]
- 44.Gravel A., Messier,N. and Roy,P.H. (1998) Point mutations in the integron integrase IntI1 that affect recombination and/or substrate recognition. J. Bacteriol., 180, 5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y. and Rice,P.A. (2003) New insight into site-specific recombination from Flp recombinase-DNA structures. Annu. Rev. Biophys. Biomol. Struct., 32, 135–159. [DOI] [PubMed] [Google Scholar]
- 46.Mazel D., Dychinco,B., Webb,V.A. and Davies,J. (1998) A distinctive class of integron in the Vibrio cholerae genome. Science, 280, 605–608. [DOI] [PubMed] [Google Scholar]