


Abstract
Homing endonuclease genes (HEGs) are mobile DNA elements that are thought to confer no benefit to their host. They encode site-specific DNA endonucleases that perpetuate the element within a species population by homing and disseminate it between species by horizontal transfer. Several yeast species contain the VMA1 HEG that encodes the intein-associated VMA1-derived endonuclease (VDE). The evolutionary state of VDEs from 12 species was assessed by assaying their endonuclease activities. Only two enzymes are active, PI-ZbaI from Zygosaccharomyces bailii and PI-ScaI from Saccharomyces cariocanus. PI-ZbaI cleaves the Z.bailii recognition sequence significantly faster than the Saccharomyces cerevisiae site, which differs at six nucleotide positions. A mutational analysis indicates that PI-ZbaI cleaves the S.cerevisiae substrate poorly due to the absence of a contact that is analogous to one made in PI-SceI between Gln-55 and nucleotides +9/+10. PI-ZbaI cleaves the Z.bailii substrate primarily due to a single base-pair substitution (A/T+5 → T/A+5). Structural modeling of the PI-ZbaI/DNA complex suggests that Arg-331, which is absent in PI-SceI, contacts T/A+5, and the reduced activity observed in a PI-ZbaI R331A mutant provides evidence for this interaction. These data illustrate that homing endonucleases evolve altered specificity as they adapt to recognize alternative target sites.
INTRODUCTION
Homing endonuclease genes (HEGs) are mobile DNA elements that occur in numerous fungi, protists, bacteria and viruses (1,2). Similar to transposons, another class of mobile elements, HEGs inhabit a host organism in order to survive, but they do not transit through an extrachromosomal phase as part of their ‘life cycle’. HEGs have evolved mutually beneficial associations with other molecular elements possessing different activities to minimize their impact on host fitness. They are found situated within self-splicing RNA introns that excise the element at the RNA level or as integral components of inteins that are removed at the protein level by an autocatalytic protein splicing reaction. There is little evidence for any positive selection for intron- or intein-encoded HEGs by the host organism, yet excision of these elements from the host genome does not occur at high frequency because they are often inserted within critical regions of genes that are required for host viability where imprecise deletion would produce a non-functional host protein (3–5).
Phylogenetic analyses and biochemical studies indicate that HEGs ensure their persistence in nature by moving at high frequency among species through horizontal transmission (6–10). This lateral transfer between species genomes may be mediated by the HEG-encoded homing endonuclease that is responsible for propagating the element throughout the population of the new resident species. In HEG+/HEG− heterozygous individuals, homing endonucleases initiate a gene conversion event termed ‘homing’ by introducing a double-strand break in the allele of the host gene that lacks the HEG (11,12). DNA repair of the break using the HEG+ allele as template and host-encoded repair proteins results in the super-Mendelian inheritance of the HEG. Phylogenetic and biochemical evidences indicate that over evolutionary time, the absence of selection for HEGs leads to their degeneration through mutation and eventual elimination (7,13,14). Taken as a whole, the data support a model in which HEGs undergo recurrent cycles of (i) horizontal transmission to new genomes, (ii) spread and fixation in the recipient population by homing, (iii) degeneration due to the absence of intact recognition sequences and (iv) eventual loss (7,8). If invasion of a new species occurs prior to complete degradation of the homing endonuclease activity in the old species, the element continues to persist and the cycle begins anew.
An intein located within the VMA1 gene of several species of yeast exhibits many of the features of the proposed HEG life cycle. Autocatalytic protein splicing of the VMA1 intein generates the VMA1-derived endonuclease (VDE) (12,15,16), which is a member of the LAGLIDADG family of homing enzymes (2). A survey of 24 different yeast species revealed that 14 harbor VDEs that have diverged in amino acid sequence (average divergence was 40%) are inserted at identical locations in the VMA1 gene (8). The phylogeny of the VDEs is significantly different from those of their hosts, providing evidence for the central role of horizontal transmission in the dissemination of these elements (8,14). It is estimated that >120 gains and losses of VDEs have occurred in the 24 different yeast species over a period of about 800 million years (8). Whether a vector-based mechanism or other means transferred the VDE genes between yeast species remains unclear. Homing of the Saccharomyces cerevisiae VMA1 intein from donor HEG+ to recipient HEG− alleles occurs at high frequency in heterozygous diploids during meiosis when a double-strand break is created in the HEG− allele by PI-SceI, the S.cerevisiae VDE protein (12). The VMA1 intein behaves as a molecular parasite since it completes homing by using the same yeast DNA repair proteins that normally mediate Spo11p-induced meiotic recombination (17). It cannot be ruled out, however, that VDEs contribute to host fitness since it has been reported that the S.cerevisiae protein is involved in the expression of a high-affinity glutathione transporter (18). Evidence that the VMA1 intein degenerates during evolution has come from a detailed biochemical and mutational analysis of a yeast strain (DH1-1A) related to S.cerevisiae that contains both HEG+ and HEG− alleles (13). Although the DH1-1A protein differs from PI-SceI at only 11 amino acid positions, one of these substitutions significantly reduces the endonuclease activity of the enzyme. The protein splicing activity, unlike the endonuclease activity, does not appear to have degenerated during evolution. Loss of this activity would result in a large polypeptide insertion within an essential protein that would adversely affect host viability (17).
Elucidating the evolutionary history of PI-SceI has been facilitated by having detailed knowledge of the PI-SceI domain structure, the protein–substrate interaction and the active site architecture. PI-SceI consists of two domains, one that harbors the endonucleolytic catalytic center and the other that contains the active site for protein splicing (19,20). Within the endonuclease domain there are two subdomains that display 2-fold pseudosymmetry. Each of these is structurally related to the monomeric subunits of intron-encoded homing enzymes like I-CreI that are homodimeric and lack a splicing domain (21,22). It is speculated that gene duplication and fusion of a common ancestral endonuclease gene that encoded homodimeric enzymes gave rise to the monomeric proteins like PI-SceI with internal symmetry [for review see (1)]. Near the center of the symmetry axis within the endonuclease domain are two overlapping active sites that are comprised of two conserved aspartic acid residues that coordinate with the essential Mg2+ ion co-factors (20). Critical base-specific and phosphate backbone contacts are made using residues in both domains to a ∼31 bp target site that is unique in the 13 million base-pair genome (20,23).
In this work, VDEs from 12 different yeast species were cloned, expressed and purified in order to assess the frequency of degeneration of the VDE activity. All except two of the newly characterized proteins fail to cleave their own DNA substrate, which suggests that HEG degeneration is a common occurrence. Interestingly, the Zygosaccharomyces bailii VDE, PI-ZbaI, exhibits altered target site specificity since it cleaves its own substrate significantly faster than the S.cerevisiae target, which differs at six different base-pair positions. That homing endonucleases can evolve new DNA interactions has been demonstrated previously for the I-CreI and I-MsoI isoschizomers, where phylogenetic and structural analyses revealed that the proteins make different residue contacts to the same nucleotides (24,25). Here, a combined mutational and biochemical strategy was used to define some of the protein and DNA determinants that underlie the different specificities of PI-SceI and PI-ZbaI. Taken as a whole, the data reveal the inherent flexibility of the interface between VDEs and their targets that permits these enzymes to migrate efficiently between species.
MATERIALS AND METHODS
Materials
Synthetic oligonucleotides used for cloning of substrate plasmids, for mutagenesis and for PCR were purchased from Integrated DNA Technologies, Inc. or from MWG Biotech. DNA polymerases, restriction and DNA-modifying enzymes were obtained from New England Biolabs. Cobalt metal affinity resin (TALON) was purchased from CLONTECH, and SP-Sepharose was purchased from Amersham Pharmacia Biotech. All other chemicals were of reagent grade and were obtained from commercial sources.
Cloning of VDE alleles
Synthetic oligonucleotide primers complementary to the N- and C-termini of the intein alleles of the different yeast species were used to generate different PCR products from the genomic DNA. These were gel-purified, phosphorylated and ligated into the BsaBI site of pKSS (26). To facilitate cloning of the PCR products, NcoI and BamHI sites were introduced at the N- and C-termini of the open reading frame (ORF), respectively. The DNA sequence was confirmed by sequence analysis. The intein ORFs were subcloned between the NcoI and BamHI sites of pETPI-SceI C-his (NcoI), a derivative of pETPI-SceI C-His (27) containing an NcoI rather than an NdeI site at the position of the initiation codon. Those intein ORFs that contained internal NcoI sites were subcloned directly into the NdeI and BamHI sites of pETPI-SceI C-His.
Construction of PI-ZbaI A55Q and PI-SceI Q55A variants
PI-SceI Q55A and PI-ZbaI A55Q were constructed using duplex oligonucleotide cassette mutagenesis of pETPI-SceI C-His (27) and pETPI-ZbaI C-His, respectively. PI-ZbaI R331A and PI-ZbaI H329A were generated using mutagenic primers by a PCR mutagenesis strategy from pETPI-ZbaI C-His. All constructs were verified using automated DNA sequencing.
Generation of substrates for DNA cleavage and binding assays
Plasmids containing single VDE recognition sites were constructed by inserting 42 bp duplex cassettes into the EcoRI and HindIII sites of pBluescript as described previously (23). Clones were verified using automated DNA sequencing. A 1837 bp PCR product was generated from these plasmids using two synthetic primers (5′-GGCGCAGCGGTCGGGCTGAAC-3′ and 5′-GGGGCGAAAACTCTCAAGGATCTTACCGC-3′) for use in DNA cleavage reactions. DNA cleavage of the PCR substrate yields 782 and 1055 bp products. A 219 bp product used in DNA-binding assays was generated by PCR and 32P-endlabeled as described previously (27).
Protein purification
To express the different VDE proteins, the pET plasmid vectors were transformed into BL21 (DE3) cells. The proteins were overexpressed and purified using cobalt-metal affinity and ion-exchange chromatography as described previously for the purification of PI-SceI (27) except that the final storage buffer contained 300 mM KCl. The Saccharomyces dairenensis VDE protein was insoluble in the absence of imidazole; therefore, 300 mM imidazole was maintained in its storage buffer. Protein preparations were at least 95% purified with the exception of the Saccharomyces castellii protein, which had equimolar amounts of a low-molecular-weight contaminant. The Saccharomyces exiguus protein appeared as a tight doublet on SDS–PAGE, possibly due to limited proteolysis. The purified proteins were identified as VDEs by western blot analysis using monoclonal anti-histidine antibodies (Sigma).
Assay of DNA cleavage activity
To assay DNA cleavage activity, protein variants (100 nM) were incubated at 37°C with various 1.8 kb substrates (7 nM) as described previously (27). The reaction mixtures were analyzed by gel electrophoresis on 1% agarose gels. Digitized images of the gels were generated using a Kodak EDAS 290 system and the extents of DNA cleavage were quantified using Quantity One software (Bio-Rad Laboratories, Inc.).
Assay of DNA-binding activity by electrophoretic mobility shift assay
Gel-mobility shift assays were preformed as reported previously (27) using a radiolabeled 219 bp DNA duplex containing a single binding site.
RESULTS
Expression and purification of VDEs from different yeast species
A survey of 24 different species of saccharomycete yeasts from four closely related genera (Saccharomyces, Torulaspora, Zygosaccharomyces and Kluyveromyces) revealed the existence of VDEs within 14 species (S.cerevisiae, S.cariocanus, S.exiguus, S.castellii, S.dairenensis, S.unisporus, K.polysporus, Z.bailii, Z.bisporus, Z.rouxii, T.pretoriensis, T.globosa, K. lactis and C.tropicalis) (8). To investigate the endonuclease activity of these VDEs, their genes were amplified by PCR and cloned into a bacterial expression vector under the control of the T7 promoter. VDEs from each of two different K.lactis strains were purified, strains CBS 2896 and CBS 683. The Z.bisporus VDE gene was not pursued because a PCR product was not obtained. We obtained only a low yield of the C.tropicalis VDE, which precluded its analysis in this study. Recent studies have determined that this enzyme is inactive (28). The presence of the His6-tag was confirmed on each of the purified proteins by western blot analysis using anti-polyhistidine antibodies (data not shown). The different purified proteins are designated as PI-SceI (S.cerevisiae), PI-ScaIP (S.cariocanus), PI-SexIP (S.exiguus), PI-ScsIP (S.castellii), PI-SdaIP (S.dairenensis), PI-SunIP (S.unisporus), PI-KpoIP (K.polysporus). PI-ZbaIP (Z.bailii), PI-ZroIP (Z.rouxii), PI-TprIP (T.pretoriensis), PI-TglIP (T.globosa), PI-KlaIP1 (K.lactis strain CBS 2896) and PI-KlaIP2 (K.lactis strain CBS 683) in keeping with recently defined nomenclature, where the ‘P’ suffix indicates that endonuclease activity has not been demonstrated previously (29).
Endonucleolytic activity of VDE proteins from different yeasts on their respective substrates and the S.cerevisiae substrate
According to the model for the ‘life-cycle’ of VDEs, the endonuclease activity, but not the protein splicing activity, is expected to degenerate over evolutionary time once the elements become fixed within a genome (8). To address whether degeneration has occurred, the endonucleolytic activity of the purified proteins was assayed using DNA substrates containing the respective native target sequences. The reactions were performed using Mg2+ as a metal ion co-factor under conditions that had been optimized for PI-SceI. Only two VDE proteins, those from S.cariocanus and Z.bailii, cleaved their respective DNA substrates after 6 h of incubation at 37°C (Figure 1A). In accordance with nomenclature conventions, these active proteins are designated as PI-ScaI and PI-ZbaI, respectively. Surprisingly, although PI-ZbaI is less similar to PI-SceI than PI-ScaI (61 and 89% identity, respectively), it is more active. To increase the likelihood of observing DNA cleavage activity for the other enzymes, we relaxed the stringency of the cleavage reaction by substituting Mn2+ in the reaction buffer since it has been shown that this ion generally increases the DNA cleavage rate of PI-SceI and allows it to cleave DNA targets that are inactive in Mg2+ (23). Indeed, greater amounts of cleavage product were generated by PI-ScaI and PI-ZbaI in Mn2+ but no endonucleolytic activity was observed for any of the other VDEs under either condition (data not shown). Thus, these proteins may never have possessed an endonuclease activity or, more likely, they may have degenerated. Alternatively, the reaction conditions used here may not permit these VDEs to cleave their DNAs.
Figure 1.
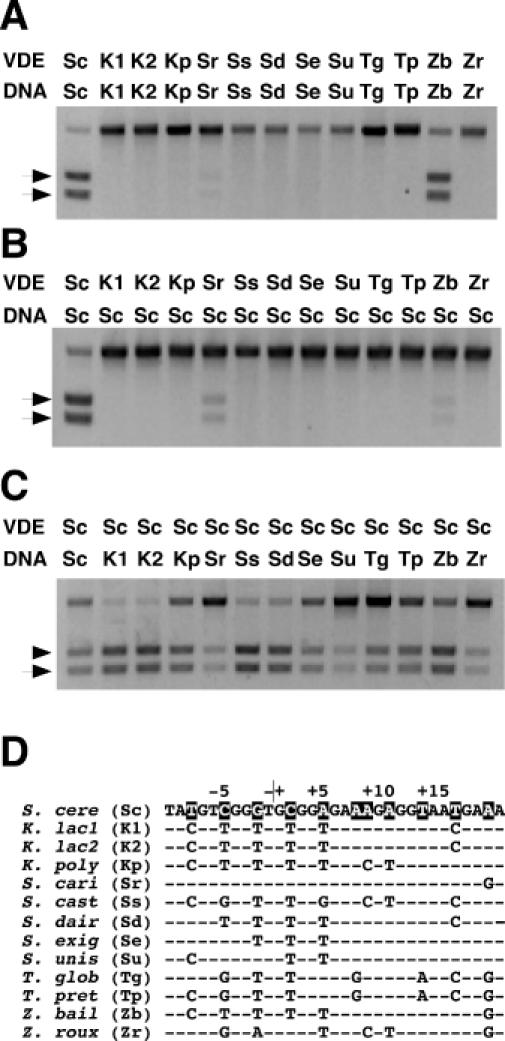
Assay of endonuclease cleavage activity of DNA substrates containing single recognition sites by VDE proteins. The reactions were performed by combining purified protein (100 nM) with duplex DNA substrates (7 nM) for various lengths of time at 37°C. (A) Cleavage of DNA substrates containing the predicted VDE target sites from the different yeast species after 6 h by their respective VDE proteins (species abbreviations are as follows: Sc, S.cerevisiae; Sr, S.cariocanus; Se, S.exiguus; Ss, S.castellii; Sd, S.dairensis; Su, S.unisporus; Kp, K.polysporus; Zb, Z.bailii; Zr, Z.rouxii; Tp, T.pretoriensis; Tg, T.globosa; K1, K.lactis strain CBS 2896; and K2, K.lactis strain CBS 683). Arrows indicate the position of the two DNA cleavage products. (B) Cleavage of a DNA substrate containing the S.cerevisiae recognition site after 6 h of incubation with different VDE proteins. (C) Cleavage of DNA substrates containing the different yeast VDE recognition sites after 1 h by PI-SceI. (D) A comparison of the DNA sequences of the different predicted target sites of each yeast species. The top strand of the S.cerevisiae recognition site is shown on the top line with the base-pair positions relative to the center of the 4 bp cleavage site overhang, as indicated above. Below are shown the aligned bases that differ from the S.cerevisiae sequence. The highlighted bases are those that differ in one or more of the yeast species.
Evidence that the degeneration of VDEs has occurred comes from aligning the residues in PI-SceI that are critical or important for catalysis with their analogs from the various VDEs (Figure 2). It is likely that a minimum criterion for the activity of any LAGLIDADG enzyme is the presence of conserved acidic residues that correspond to two essential PI-SceI amino acids (Asp-218 and Asp-326) that coordinate the metal ion co-factors (20,30,31). Given this assumption, it is predicted from Figure 2 that most of the VDEs analyzed here are inactive because one or both acidic residues are absent, while four proteins (PI-ZbaI, PI-ScaI, PI-TprIp and PI-TglIp) have the potential to be active. Unlike the acidic residues at the catalytic centers, the neighboring amino acids (Asp-229, Lys-301, Thr-341 and Lys-403 in PI-SceI) are not strictly conserved. Mutagenesis of these residues affects PI-SceI activity (27,32,33), and they likely contribute a key role in organizing an extensive network of water molecules at the active sites that is involved in catalysis (20,34). It cannot be predicted how residue substitutions at these positions affect hydration of the solvent pocket. Thus, of the four proteins predicted to be active based on the single criterion for activity, two were shown to be functional in biochemical assays (Figure 1A).
Figure 2.
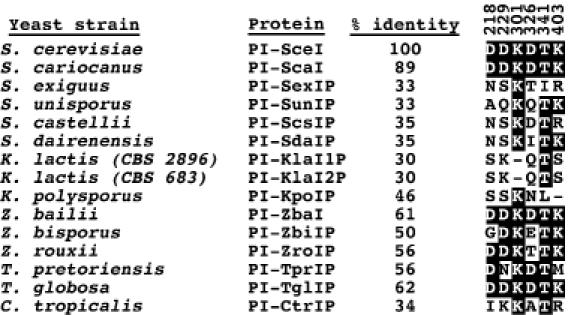
Sequence alignment of active site residues in PI-SceI with those of other yeast VDEs. The percent identity between the different VDE sequences and PI-SceI is given. Active site residues that are identical at the aligned positions are highlighted. The positions of the PI-SceI residues in the primary sequence are indicated above each position. Asp-218 and Asp-326 are two conserved amino acids that coordinate the essential divalent metal ions and are critical for activity. The exact roles of Asp-229, Lys-301, Thr-341 and Lys-403 in catalysis have not been defined, but their substitution with other amino acids decreases the PI-SceI catalytic activity (27).
The different VDE proteins were also assayed on the S.cerevisiae substrate to assess their DNA cleavage activity. Only PI-ScaI and PI-ZbaI were active using this substrate, and their cleavage activity was lower than PI-SceI (Figure 1B). One observation from Figure 1 is that PI-ScaI, which is significantly less active than PI-SceI or PI-ZbaI, prefers the S.cerevisiae target to its own. Alignment of the PI-ScaI sequence with that of PI-SceI reveals that only 11% of the residues differ between the two proteins (data not shown). We hypothesize that the low PI-ScaI activity may be due to the substitution of Lys-336 and Lys-376 of PI-SceI, which have been shown to make important phosphate backbone contacts (20,27), with threonine and serine, respectively. What is most interesting is that PI-ZbaI cleaves the Z.bailii substrate significantly faster than the S.cerevisiae target. In contrast, PI-SceI exhibits similar activity toward both substrates (Figure 1C and data not shown). Based on these data, we speculate that PI-ZbaI lacks important contacts used by PI-SceI to bind the S.cerevisiae target or that steric clashes or electrostatic repulsions prevent PI-ZbaI from binding to the substrate. Second, we hypothesize that contact(s) present in PI-ZbaI permit it to cleave the Z.bailii DNA target. Elucidating why PI-ZbaI cleaves the S.cerevisiae target poorly and how it recognizes its own target is the focus of the remainder of this work.
Endonucleolytic activity of PI-SceI on the different yeast substrates
VDEs have adapted for horizontal transfer by targeting to a gene (VMA1, the catalytic subunit of the vacuolar H+-ATPase) that is well conserved among yeasts and to a highly conserved recognition sequence located within that gene (8). Moreover, VDE proteins may contact many of the same nucleotides within the recognition sequence since the 11 bp within the PI-SceI recognition sequence identified as being more important for activity (23,35) are conserved in the VDE target sites (8). These important base pairs may be conserved because mutations at these positions change the amino acid sequence of the ATPase near its catalytic center. We assayed each of the different yeast VDE target sequences with PI-SceI to examine the effect of the various nucleotide substitutions on DNA cleavage activity by this enzyme (Figure 1C). The nucleotide differences among the targets do not occur at the 11 important positions (Figure 1D), with the exception of those with +9 alterations, and it was predicted that all of the substrates would be active. Indeed, PI-SceI cleaves all of the different VDE targets, with significantly faster cleavage evident for the K.lactis, S.castellii, S.dairenensis targets relative to the S.cerevisiae substrate (Figure 1C). Thus, none of the target sequences has diverged to such a degree that it cannot be cleaved using any homing enzyme. The site recognition flexibility exhibited by PI-SceI is consistent with the hypothesis that a VDE can home to similar target sites in closely related species (8). Previously, it was demonstrated that PI-SceI cleaves a S.cerevisiae substrate containing a G/C base pair at position +20 ∼40% slower than the wild-type substrate (23). This sequence is also identical to the S.cariocanus target, which may explain why PI-ScaI is more active on the S.cerevisiae target than on its own.
Identification of a PI-SceI contact absent in PI-ZbaI that is important for cleavage of the S.cerevisiae substrate
To elucidate why PI-ZbaI cleaves the S.cerevisiae substrate poorly, we aligned the PI-ZbaI and PI-SceI protein sequences and used the Swiss-PDB viewer program (36) to generate a model of the PI-ZbaI/DNA interaction using the PI-SceI/DNA X-ray structure as a template (data not shown). All of the PI-ZbaI residues that make putative base-specific contacts are identical to their PI-SceI counterparts, except for Ala-55, which occurs as Glu-55 in PI-SceI (Table 1). In the PI-SceI/DNA complex, Glu-55 makes the only minor groove contacts and hydrogen bonds to both G+10 on the top strand and T+9 on the bottom strand (20). Substitutions for these nucleotides decrease the cleavage activity (Figure 3A). In a complementary experiment, a Q55A PI-SceI variant cleaves both the Z.bailii and S.cerevisiae substrates 4-fold slower than the wild-type protein after 30 min (Figure 3A). The decreased activity of a PI-SceI Q55A mutant was demonstrated previously (37). The finding that the Q55A mutation does not further decrease DNA cleavage activity of the +9 and +10 mutant substrates relative to wild-type PI-SceI (Figure 3A) is consistent with the existence of the Gln-55/+9/+10 interaction. Unlike other mutations in PI-SceI and its homing site (27,37), neither the Q55A PI-SceI mutation nor the +9/+10 substrate mutations completely eliminate cleavage activity, suggesting that the Gln-55 contact contributes less binding energy than others.
Table 1. PI-SceI residues involved in catalysis and in binding the DNA target site are listed together with the corresponding residues in PI-ZbaI and PI-ScaI.
| PI-SceI residue | Contact type | Nucleotide position | PI-ZbaI residue | PI-ScaI residue |
|---|---|---|---|---|
| Lys-53 | P | +11A/+12G | Lys-53 | Lys-53 |
| His-56 | P | +7T/+8T | His-56 | His-56 |
| Arg-57 | P | +6C/+7T | Arg-57 | Arg-57 |
| Ser-127 | P | +19T/+20T | Ser-128 | Ser-127 |
| Ser-129 | P | +20T/+21T | Ser-130 | Ser-129 |
| Ser-169 | P | +17A/+18C | Ala-170 | Ser-169 |
| Lys-173 | P | +11A/+12G | Lys-172 | Lys-173 |
| Ser-227 | P | −2C/−3C | Ser-228 | Ser-227 |
| Ala-261 | P | −2C/−1A | Ala-262 | Lys-261 |
| Tyr-267 | P | −8T/−7G | Tyr-268 | Tyr-267 |
| Asn-274 | P | −11G/−10A | Asn-275 | Asn-273 |
| Gly-275 | P | −11G/−10A | Asp-276 | Gly-274 |
| Arg-277 | P | −11G/−10A | Arg-278 | Arg-276 |
| Leu-280 | P | −9A/−8T | Leu-281 | Leu-279 |
| Asn-281 | P | −9A/−8T | Asn-282 | Asn-280 |
| Thr-282 | P | −9A/−8T | Tyr-283 | Thr-281 |
| Tyr-328 | P | +4G/+5A | His-329 | Tyr-327 |
| Lys-336 | P | +8T/+9T | Ser-337 | Thr-335 |
| Ser-362 | P | +7T/+8T | Ser-362 | Ser-361 |
| Asn-364 | P | +6C/+7T | Asn-365 | Asn-363 |
| Lys-376 | P | −1T/−2G | Asn-377 | Ser-375 |
| Lys-378 | P | −1T/+1G | Arg-379 | Lys-377 |
| Tyr-384 | P | +8T/+9T | Tyr-385 | Tyr-383 |
| Gln-55 | P, m, m | +7T/+8T, +10G, +9T | Ala-55 | Gln-55 |
| Arg-90 | M | +15A | Arg-91 | Arg-90 |
| Arg-94 | M | +18G | Arg-95 | Arg-94 |
| His-170 | M | +16T | His-171 | His-170 |
| Arg-223 | M, M | −7G, −8T | Arg-224 | Arg-223 |
| Lys-340 | M, M, M | +3G, +4G, +4G | Lys-341 | Lys-339 |
| Glu-366 | M, M | +3C, +4C | Glu-367 | Glu-365 |
| His-377 | M | +1G | His-378 | His-376 |
| Asp-218 | Mg | — | Asp-219 | Asp-218 |
| Asp-229 | Mg | — | Asp-230 | Asp-229 |
| Lys-301 | C | — | Lys-301 | Lys-300 |
| Asp-326 | Mg | — | Asp-327 | Asp-325 |
| Thr-341 | Mg | — | Thr-342 | Thr-340 |
| Lys-403 | C | — | Lys-404 | Lys-402 |
Residues that differ are shown in boldface. Phosphate contacts are represented with ‘P’, minor groove contacts with ‘m’, major groove contacts with ‘M’, catalytic residues with ‘C’ and metal binding residues with ‘Mg’. The Ala-261 and Gly-275 contacts to DNA are through the main peptide chain, and therefore, should be unaffected by side chain substitutions.
Figure 3.
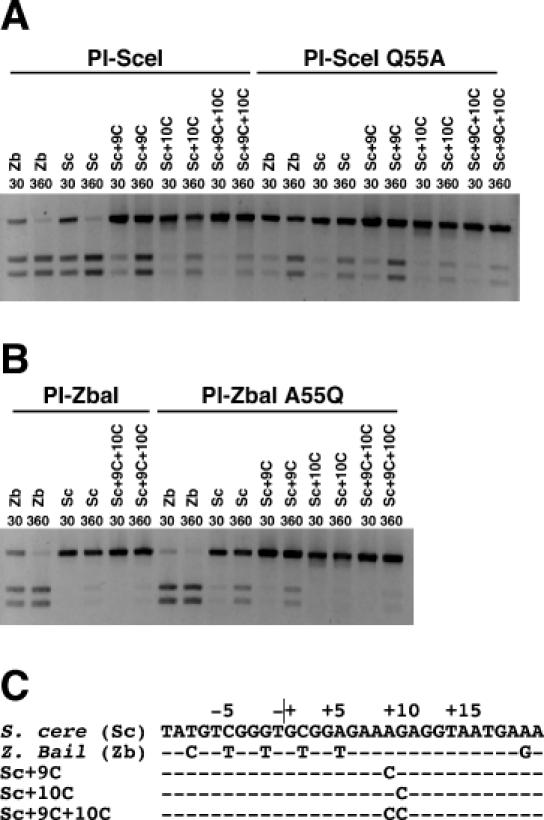
Assay of endonuclease cleavage activity of wild-type PI-SceI, PI-SceI Q55A, wild-type PI-ZbaI and PI-ZbaI A55Q proteins on DNA substrates containing a single recognition site. In each reaction, the protein (100 nM) was incubated with DNA substrate (7 nM) in Mg2+ cleavage buffer at 37°C. (A) DNA cleavage of wild-type and mutant substrates (indicated above each lane) by wild-type PI-SceI and PI-SceI Q55A proteins. DNA substrates containing the S.cerevisiae and Z.bailii recognition sites are denoted as Sc and Zb, respectively, followed by any introduced substitutions. (B) DNA cleavage assay of wild-type PI-ZbaI and PI-ZbaI A55Q proteins using wild-type and mutant DNA substrates. (C) The sequences of the different DNA substrates.
The absence of Gln-55 in wild-type PI-ZbaI contributes to its low cleavage of the S.cerevisiae substrate since the introduction of a glutamine at this position significantly increases the cleavage rate of the protein [38% (A55Q) versus 13% (wild-type) substrate cleavage after 6 h; Figure 3B]. However, other differences between PI-SceI and PI-ZbaI are also responsible since PI-ZbaI A55Q does not cleave the S.cerevisiae substrate as well as PI-SceI (92% cleaved). The introduction of Gln-55 into PI-ZbaI also increases its activity toward the Z.bailii substrate, presumably because this residue makes similar DNA contacts as in PI-SceI (Figure 3B and data not shown).
Definition of nucleotides within the Z.bailii substrate that enable DNA cleavage by PI-ZbaI
We reasoned that one or more of the six base-pair differences between the S.cerevisiae and Z.bailii substrates account for the ability of PI-ZbaI to cleave its own target (Figure 4). A ‘minus-ones/plus-ones’ analysis (38) was used to identify the critical positions by selectively adding or removing the six different base pairs in the S.cerevisiae and Z.bailii targets, respectively (Figure 4C). In our biochemical analysis of the ‘plus-ones’ substrates, insertion of a T/A+5 base pair (Sc+5T) into the S.cerevisiae target contributes more to DNA cleavage by PI-ZbaI than any other replacement and its removal from the Z.bailii substrate dramatically reduces DNA cleavage activity (Figure 4A). This is not the only important nucleotide as base-pair changes at the −8, −5 and −2 positions reproducibly increase activity when present in the S.cerevisiae target [–C/T−8 (Sc-8C), T/A−5 (Sc-5T) and T/A−2 (Sc-2T)] and decrease activity when removed from the Z.bailii substrate [Zb-8T, Zb-5C and Zb-2G]. Thus, our initial experiments using singly substituted mutations suggest that four of the six base pairs contribute positively to the cleavage activity of the Z.bailii target.
Figure 4.
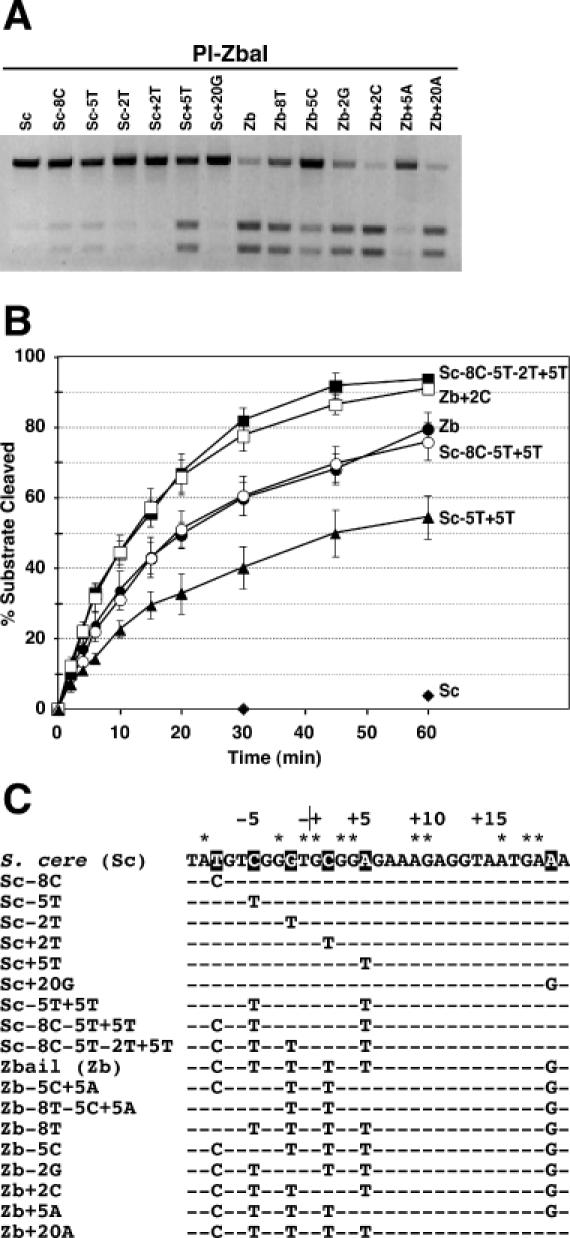
Identification of the base pairs in the Z.bailii substrate that contribute to PI-ZbaI activity. (A) Plus-ones/minus-ones analysis of the PI-ZbaI activity. DNA substrates (7 nM) containing individually substituted Z.bailii base pairs within the S.cerevisiae recognition site (plus-ones) or containing individually substituted S.cerevisiae base pairs within the Z.bailii recognition site (minus-ones) were incubated at 37°C for 6 h with PI-ZbaI (100 nM). (B) Time course of DNA cleavage of wild-type and mutant substrates by PI-ZbaI. The percentages of product formed by digestion of the S.cerevisiae (filled diamonds), Sc-5T+5T (filled triangles), Z.bailii (filled circles), Sc-8C-5T+5T (open circles), Sc-8C-5T-2T+5T (filled squares) and Zb+2C (open squares) substrates are plotted as a function of time. (C) The DNA sequences of the different DNA substrates used in this study. The S.cerevisiae target base pairs indicated by an asterisk are those that are important for substrate cleavage by PI-SceI. The base pairs in the VDE target sequences that are investigated in this experiment are highlighted.
Different plus-one base-pair substitutions were combined to assess their contribution toward the total PI-ZbaI activity and to reconstitute the activity of the Z.bailii substrate. A substrate containing both the −5T and +5T mutations, which individually contribute the most activity, is cleaved slower than the Z.bailii target (∼54% of the −5T+5T substrate is cleaved after 1 h compared with 79% for the Z.bailii substrate; Figure 4B). When the −8C substitution is combined with the other two mutations, cleavage of the triple mutant substrate is equal to that of the Z.bailii target (Figure 4B). Surprisingly, addition of the last of the four substitutions that individually affect activity increases the level of activity above that of the Z.bailii substrate. This observation raised the possibility that either or both the +2T or +20G substitutions negatively affect DNA cleavage. Indeed, the +2T substitution has a small negative effect on the cleavage of the Z.bailii substrate (Figure 4B). Unexpectedly, the Zb+20A substrate was cleaved at a slower rate than the Z.bailii substrate (data not shown). Taken together, these data demonstrate that +5T contributes the most toward the activity to the Z.bailii substrate of the six base-pair differences, with the −5T, −8C and −2T substitutions playing lesser roles.
Identification of PI-ZbaI amino acid residues that make putative contacts to the T/A+5 base pair
To elucidate the role of the T/A+5 position within the Z.bailii substrate, we inspected the model of the PI-ZbaI/DNA complex for potential protein contacts to this base pair. Arg-331 is predicted to be ∼2.2 Å distant from the adenine base of the T/A+5 base pair in the Z.bailii substrate (data not shown), and we speculate that its basic side chain makes base-specific contacts to the DNA. The analogous residue in PI-SceI, Thr-330, is not involved in DNA binding (20). His-329, which is also proximal to the +5 position, is analogous to Tyr-328 in PI-SceI, which makes a phosphate contact between the +4 and +5 positions (27,35).
Mutant PI-ZbaI proteins containing individual alanine substitutions at the Arg-331 and His-329 positions were purified and their DNA cleavage activities were assayed. After 6 h of incubation in the presence of Mg2+, only trace levels of DNA cleavage of a T/A+5 → A/T+5 mutant substrate are evident (Figure 5). Similarly low levels of activity are observed when the PI-ZbaI R331A protein is incubated with the wild-type Z.bailii substrate, consistent with the idea that Arg-331 contacts T/A+5. His-329 also appears to play a critical role in the protein–DNA interactions since alanine substitution at this position abolishes DNA cleavage activity (Figure 5). Substitution of Mn2+ for Mg2+ dramatically rescues not only the activity of the Zb+5A substrate, but also the activities of the R331A and H329A mutant proteins. The ability of Mn2+ to restore function to inactive or partially active DNA substrates and proteins has been observed previously (23,27,39). These data indicate that His-329 and Arg-331 are critical for the cleavage of the Z.bailii substrate.
Figure 5.
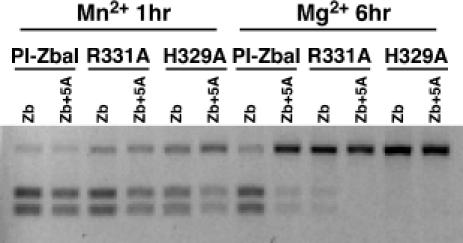
Cleavage of DNA substrates containing the wild-type or Zb+5A recognition site by wild-type PI-ZbaI, PI-ZbaI R331A and PI-ZbaI H329A proteins. Protein (100 nM) was incubated with substrate (7 nM) at 37°C for either 1 h in buffer containing Mn2+ or 6 h in buffer containing Mg2+. The DNA sequences of these substrates are indicated in Figure 4C.
DISCUSSION
In the proposed ‘life cycle’ for HEGs, it is suggested that homing endonuclease activity degenerates over evolutionary time once all HEG− alleles in a population have been converted into HEG+ alleles due to the absence of selection for function (7). However, if, prior to complete degeneration, homing endonucleases evolve new DNA contacts and lose others to acquire a changed specificity, HEGs have the potential to invade new species that harbor variant target site sequences (7–10). To investigate whether these types of events have occurred during evolution, we purified and biochemically characterized 12 homing endonucleases from a diverse set of yeast species that had previously been shown to harbor analogous VDE homing elements (8). The data support the idea that degeneration occurs at high frequency since we find that most of the yeast homing endonucleases are inactive, which may be due to sequence drift of essential catalytic or DNA-binding residues. It cannot be ruled out, however, that activity was not detected simply because special reaction conditions are required that were not tested. During our survey of yeast VDEs, we discovered that the Z.bailii PI-ZbaI enzyme is significantly less active on the S.cerevisiae target site than on the Z.bailii site, which differs at six positions. A detailed molecular dissection of this large observed difference in target site specificity between related homing enzymes suggests that different protein/DNA contacts may have evolved in the PI-ZbaI and PI-SceI enzymes.
A myriad of different combinations of mutations could potentially have inactivated the degenerate homing endonucleases during evolution, and we did not attempt to define the specific defects of each enzyme. We determined previously that a degenerate VDE from yeast strain DH1-1A differs from PI-SceI at 11 amino acid positions, and we showed that a single substitution accounted for a 10-fold reduction in activity while other changes exerted little effect (13). Given the larger sequence divergence between PI-SceI and the VDEs characterized here, identifying the residues that abolish activity would be more difficult. Several enzymes, including PI-SexIP, PI-ScsIP, PI-SdaIP, PI-SunIP, PI-KpoIP and PI-ZroIP, lack one or both of the conserved acidic residues [Asp-218 and Asp-326 in PI-SceI (20)] that coordinate the essential metal ion co-factors (Figure 2), and it was not unexpected that these enzymes are inactive for this reason. The other enzymes may be inactive due to binding defects or for other reasons. Two of the enzymes analyzed in this report, PI-SunIP and PI-KlaP, have been shown previously to be inactive (14). The extensive degeneration of the VDE elements parallels that observed in a survey of the ω intron HEGs that occur inserted in a group I self-splicing intron within the yeast mitochondrial large subunit (LSU) rRNA gene (7). In this case, it was speculated that two of five enzymes were inactive based on the presence of sequence insertions that disrupted the reading frame. In contrast to the VDE and ω elements, four intron-encoded LSU homing endonucleases from green algae (I-CreI, I-MsoI, I-PakI and I-CvuI) are all active in biochemical assays (24). Moreover, all 15 members of the enzymes related to I-CreI contain potential catalytic residues, suggesting that these proteins are active as well (24). Why there is less evidence of degeneration of the I-CreI-related HEGs compared with the VDE or ω elements is unclear, but it may reflect a more recent transmission of active I-CreI elements into the different species, or the existence of an undefined positive selection for activity.
The divergence of the PI-ZbaI and PI-SceI target site specificities arose due to variations that permit PI-ZbaI to recognize and cleave the Z.bailii sequence and others that inhibit it from cleaving the S.cerevisiae target. Here, we show that the absence from PI-ZbaI of Gln-55, which contacts base pairs +9 and +10 in PI-SceI, partially accounts for why PI-ZbaI is inactive toward the S.cerevisiae cleavage site. Gln-55 is present in only a minority of the surveyed VDEs, including PI-SceI, PI-ScaI and PI-ScsIP (data not shown). Since PI-ScsIP is unrelated to the other two enzymes in a sequence-based lineage analysis (8), and since there is no apparent evidence of co-evolution of Gln-55 and the A/T+9 G/C+10 base pairs (data not shown), we speculate that the introduction of the Gln-55 interaction occurred randomly in a limited number of the different VDEs and was probably not present in the common ancestor of these proteins. The new minor groove contact that is made by Gln-55 in PI-SceI may underlie its ability to cleave a wide variety of related VDE recognition sequences. As to why PI-ZbaI cleaves the Z.bailii target when it is inactive against the S.cerevisiae sequence, we provide evidence for an important contact between Arg-331 and T/A+5 that is absent in PI-SceI. Over one-half of the surveyed VDEs contain an arginine or lysine at the position 331 and have the potential to make a similar contact (data not shown). Many of these are distantly related to PI-ZbaI, suggesting that the putative Arg-331 contact was present in a common VDE ancestor, but was lost by PI-SceI and other enzymes.
This finding that homing endonucleases evolve new contacts while losing others has been well-established in the case of the related intron-encoded homing endonucleases I-CreI and I-MsoI, which display only 33% sequence identity, yet recognize very similar target sites. By comparing the sequences of I-CreI and its homologs, it was predicted that similar contacts mediate subunit assembly, but that new contacts have arisen at the protein–DNA interaction surface (24). Indeed, elucidation of the X-ray crystal structure of the I-MsoI homing endonuclease revealed that the extent of the divergence between the enzymes was even greater than predicted since only one-fifth of the I-CreI residues that contact the DNA substrate are conserved in I-MsoI (25). The conclusion that alternative contacts can be made by homing endonucleases to identical base pairs has also been suggested by the recent demonstration that active chimeric VDEs can be constructed from PI-SceI and the C.tropicalis PI-CtrP enzymes (28), which share little identity. Our results go one step further by showing for the first time that in the process of evolving contacts to one substrate, homing endonucleases can lose the ability to interact with related recognition sites. This finding supports the idea that homing endonuclease sequences diversify at high rates, which may increase the frequency of their invasion into new genomes, and that there is weak selective pressure to maintain identical contacts (24,25). Counterbalancing the evolution of broader recognition that facilitates invasion of related species is the selection for narrower specificity that reduces the frequency of potentially deleterious ectopic cleavage.
Does the endonucleolytic activity of PI-ZbaI imply that homing occurs in Z.bailii yeast? Unlike S.cerevisiae, Z.bailii tolerates low pH and is stress resistant, and it has been frequently associated with food spoilage (40–42). Substantial evidence from genetic and flow cytometry studies indicates that Z.bailii is diploid (41,42). An unusual feature of the yeast is that it produces tetrads with mitotic rather than meiotic spores (41,42). Thus, it would be expected that opportunities for homing would be limited due to the absence of nuclear fusion during sporulation. This suggests that the VDE may have spread through the Z.bailii population by homing when the yeast still included a haploid phase in its life cycle. The high activity of the homing endonucleolytic activity indicates that little degeneration of the enzyme has occurred, either because it confers a selective advantage to the organism or because its entry into Z.bailii is recent.
This study demonstrates that homing endonucleases have evolved new target specificities in nature as they adapt to inhabit new host genomes. There is now considerable interest in more rapidly generating altered specificity enzymes in the laboratory. The ability of homing endonucleases to introduce double-strand breaks at the defined loci in complex genomes has been successfully recruited to stimulate gene targeting in mammalian systems through homologous recombination (43–45). As these methods currently require the pre-existence of the homing endonuclease recognition sequence at or near the gene of interest, their utility would be expanded if it were possible to generate altered specificity enzymes that cleave at endogenous targets. Recently, a two-hybrid genetic strategy that selects for variants of PI-SceI that binds to target sites with different base substitutions has resulted in proteins with markedly shifted specificities (46). Other genetic methods have screened for variant homing endonucleases that gain the ability to cleave at altered recognition sequences (47). These studies have been complemented by rational design strategies that have engineered chimeric homing enzymes that cleave chimeric target sequences derived from the recognition sequences of two parent enzymes (48,49). Elucidating the sources of altered specificity that arise in nature in the PI-ZbaI and PI-SceI proteins may yield new insights that will aid in further development of these novel reagents in the laboratory.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Hongye Li for performing the western analysis of the purified VDE proteins. We thank Dr Carmen Moure and Dr Florante Quiocho for valuable discussions. This work was supported by a grant from the National Science Foundation (MCB-0321550) and from the Welch Foundation (BE-1452).
REFERENCES
- 1.Gimble F.S. (2000) Invasion of a multitude of genetic niches by homing endonuclease genes. FEMS Microbiol. Lett., 185, 99–107. [DOI] [PubMed] [Google Scholar]
- 2.Chevalier B.S. and Stoddard,B.L. (2001) Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res., 29, 3757–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrett R.A., Dalgaard,J., Larsen,N., Kjems,J. and Mankin,A.S. (1991) Archaeal rRNA operons. Trends Biochem. Sci., 16, 22–26. [DOI] [PubMed] [Google Scholar]
- 4.Dalgaard J.Z., Moser,M.J., Hughey,R. and Mian,I.S. (1997) Statistical modeling, phylogenetic analysis and structure prediction of a protein splicing domain common to inteins and hedgehog proteins. J. Comput. Biol., 4, 193–214. [DOI] [PubMed] [Google Scholar]
- 5.Edgell D.R., Belfort,M. and Shub,D.A. (2000) Barriers to intron promiscuity in bacteria. J. Bacteriol., 182, 5281–5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho Y., Qiu,Y.L., Kuhlman,P. and Palmer,J.D. (1998) Explosive invasion of plant mitochondria by a group I intron. Proc. Natl Acad. Sci. USA, 95, 14244–14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goddard M.R. and Burt,A. (1999) Recurrent invasion and extinction of a selfish gene. Proc. Natl Acad. Sci. USA, 96, 13880–13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koufopanou V., Goddard,M.R. and Burt,A. (2002) Adaptation for horizontal transfer in a homing endonuclease. Mol. Biol. Evol., 19, 239–246. [DOI] [PubMed] [Google Scholar]
- 9.Edgell D.R., Stanger,M.J. and Belfort,M. (2003) Importance of a single base pair for discrimination between intron-containing and intronless alleles by endonuclease I-BmoI. Curr. Biol., 13, 973–978. [DOI] [PubMed] [Google Scholar]
- 10.Landthaler M. and Shub,D.A. (2003) The nicking homing endonuclease I-BasI is encoded by a group I intron in the DNA polymerase gene of the Bacillus thuringiensis phage Bastille. Nucleic Acids Res., 31, 3071–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacquier A. and Dujon,B. (1985) An intron-encoded protein is active in a gene conversion process that spreads an intron into a mitochondrial gene. Cell, 41, 383–394. [DOI] [PubMed] [Google Scholar]
- 12.Gimble F.S. and Thorner,J. (1992) Homing of a DNA endonuclease gene by meiotic gene conversion in Saccharomyces cerevisiae. Nature, 357, 301–306. [DOI] [PubMed] [Google Scholar]
- 13.Gimble F.S. (2001) Degeneration of a homing endonuclease and its target sequence in a wild yeast strain. Nucleic Acids Res., 29, 4215–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okuda Y., Sasaki,D., Nogami,S., Kaneko,Y., Ohya,Y. and Anraku,Y. (2003) Occurrence, horizontal transfer and degeneration of VDE intein family in Saccharomycete yeasts. Yeast, 20, 563–573. [DOI] [PubMed] [Google Scholar]
- 15.Kane P.M., Yamashiro,C.T., Wolczyk,D.F., Neff,N., Goebl,M. and Stevens,T.H. (1990) Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H+-adenosine triphosphatase. Science, 250, 651–657. [DOI] [PubMed] [Google Scholar]
- 16.Hirata R., Ohsumi,Y., Nakano,A., Kawasaki,H., Suzuki,K. and Anraku,Y. (1990) Molecular structure of a gene, VMA1, encoding the catalytic subunit of H+-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem., 265, 6726–6733. [PubMed] [Google Scholar]
- 17.Fukuda T., Nogami,S. and Ohya,Y. (2003) VDE-initiated intein homing in Saccharomyces cerevisiae proceeds in a meiotic recombination-like manner. Genes Cells, 8, 587–602. [DOI] [PubMed] [Google Scholar]
- 18.Miyake T., Hiraishi,H., Sammoto,H. and Ono,B. (2003) Involvement of the VDE homing endonuclease and rapamycin in regulation of the Saccharomyces cerevisiae GSH11 gene encoding the high affinity glutathione transporter. J. Biol. Chem., 278, 39632–39636. [DOI] [PubMed] [Google Scholar]
- 19.Duan X., Gimble,F.S. and Quiocho,F.A. (1997) Crystal structure of PI-SceI, a homing endonuclease with protein splicing activity. Cell, 89, 555–564. [DOI] [PubMed] [Google Scholar]
- 20.Moure C.M., Gimble,F.S. and Quiocho,F.A. (2002) Crystal structure of the intein homing endonuclease PI-SceI bound to its recognition sequence. Nature Struct. Biol., 9, 764–770. [DOI] [PubMed] [Google Scholar]
- 21.Jurica M.S., Monnat,R.J.,Jr and Stoddard,B.L. (1998) DNA recognition and cleavage by the LAGLIDADG homing endonuclease I-CreI. Mol. Cell, 2, 469–476. [DOI] [PubMed] [Google Scholar]
- 22.Heath P.J., Stephens,K.M., Monnat,R.J.,Jr and Stoddard,B.L. (1997) The structure of I-CreI, a group I intron-encoded homing endonuclease. Nature Struct. Biol., 4, 468–476. [DOI] [PubMed] [Google Scholar]
- 23.Gimble F.S. and Wang,J. (1996) Substrate recognition and induced DNA distortion by the PI-SceI endonuclease, an enzyme generated by protein splicing. J. Mol. Biol., 263, 163–180. [DOI] [PubMed] [Google Scholar]
- 24.Lucas P., Otis,C., Mercier,J.P., Turmel,M. and Lemieux,C. (2001) Rapid evolution of the DNA-binding site in LAGLIDADG homing endonucleases. Nucleic Acids Res., 29, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chevalier B., Turmel,M., Lemieux,C., Monnat,R.J. and Stoddard,B.L. (2003) Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J. Mol. Biol., 329, 253–269. [DOI] [PubMed] [Google Scholar]
- 26.Kast P. (1994) pKSS—a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene (Amst.), 138, 109–114. [DOI] [PubMed] [Google Scholar]
- 27.He Z., Crist,M., Yen,H.-C., Duan,X., Quiocho,F.A. and Gimble,F.S. (1998) Amino acid residues in both the protein splicing and endonuclease domains of the PI-SceI intein mediate DNA binding. J. Biol. Chem., 273, 4607–4615. [DOI] [PubMed] [Google Scholar]
- 28.Steuer S., Pingoud,V., Pingoud,A. and Wende,W. (2004) Chimeras of the homing endonuclease PI-SceI and the homologous Candida tropicalis intein: a study to explore the possibility of exchanging DNA-binding modules to obtain highly specific endonucleases with altered specificity. ChemBioChem, 5, 206–213. [DOI] [PubMed] [Google Scholar]
- 29.Roberts R.J., Belfort,M., Bestor,T., Bhagwat,A.S., Bickle,T.A., Bitinaite,J., Blumenthal,R.M., Degtyarev,S., Dryden,D.T., Dybvig,K. et al. (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res., 31, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gimble F.S. and Stephens,B.W. (1995) Substitutions in conserved dodecapeptide motifs that uncouple the DNA binding and DNA cleavage activities of PI-SceI endonuclease. J. Biol. Chem., 270, 5849–5856. [DOI] [PubMed] [Google Scholar]
- 31.Schöttler S., Wende,W., Pingoud,V. and Pingoud,A. (2000) Identification of Asp218 and Asp326 as the principal Mg2+ binding ligands of the homing endonuclease PI-SceI. Biochemistry, 39, 15895–15900. [DOI] [PubMed] [Google Scholar]
- 32.Gimble F.S., Hu,D., Duan,X. and Quiocho,F.A. (1998) Identification of Lys403 in the PI-SceI homing endonuclease as part of a symmetric catalytic center. J. Biol. Chem., 273, 30524–30529. [DOI] [PubMed] [Google Scholar]
- 33.Christ F., Schoettler,S., Wende,W., Steuer,S., Pingoud,A. and Pingoud,V. (1999) The monomeric homing endonuclease PI-SceI has two catalytic centres for cleavage of the two strands of its DNA substrate. EMBO J., 18, 6908–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chevalier B.S., Monnat,R.J.,Jr and Stoddard,B.L. (2001) The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nat. Struct. Biol., 8, 312–316. [DOI] [PubMed] [Google Scholar]
- 35.Moure C.M., Gimble,F.S. and Quiocho,F.A. (2003) The crystal structure of the gene targeting homing endonuclease I-SceI reveals the origins of its target site specificity. J. Mol. Biol., 334, 685–695. [DOI] [PubMed] [Google Scholar]
- 36.Guex N. and Peitsch,M.C. (1997) SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis, 18, 2714–2723. [DOI] [PubMed] [Google Scholar]
- 37.Wende W., Schottler,S., Grindl,W., Christ,F., Steuer,S., Noel,A.J., Pingoud,V. and Pingoud,A. (2000) Analysis of binding and cleavage of DNA by the gene conversion PI-SCEI endonuclease using site-directed mutagenesis. Mol. Biol. (Mosk), 34, 1054–1064. [PubMed] [Google Scholar]
- 38.Naumann T.A. and Reznikoff,W.S. (2002) Tn5 transposase with an altered specificity for transposon ends. J. Bacteriol., 184, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu D., Crist,M., Duan,X. and Gimble,F.S. (1999) Mapping of a DNA binding region of the PI-SceI homing endonuclease by affinity cleavage and alanine-scanning mutagenesis. Biochemistry, 38, 12621–12628. [DOI] [PubMed] [Google Scholar]
- 40.Cheng L., Moghraby,J. and Piper,P.W. (1999) Weak organic acid treatment causes a trehalose accumulation in low-pH cultures of Saccharomyces cerevisiae, not displayed by the more preservative-resistant Zygosaccharomyces bailii. FEMS Microbiol. Lett., 170, 89–95. [DOI] [PubMed] [Google Scholar]
- 41.Mollapour M. and Piper,P. (2001) Targeted gene deletion in Zygosaccharomyces bailii. Yeast, 18, 173–186. [DOI] [PubMed] [Google Scholar]
- 42.Rodrigues F., Ludovico,P., Sousa,M.J., Steensma,H.Y., Corte-Real,M. and Leao,C. (2003) The spoilage yeast Zygosaccharomyces bailii forms mitotic spores: a screening method for haploidization. Appl. Environ. Microbiol., 69, 649–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rouet P., Smih,F. and Jasin,M. (1994) Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol., 14, 8096–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smih F., Rouet,P., Romanienko,P.J. and Jasin,M. (1995) Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res., 23, 5012–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choulika A., Perrin,A., Dujon,B. and Nicolas,J.-F. (1995) Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol. Cell. Biol., 15, 1968–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gimble F.S., Moure,C.M. and Posey,K.L. (2003) Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J. Mol. Biol., 334, 993–1008. [DOI] [PubMed] [Google Scholar]
- 47.Seligman L.M., Chisholm,K.M., Chevalier,B.S., Chadsey,M.S., Edwards,S.T., Savage,J.H. and Veillet,A.L. (2002) Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res., 30, 3870–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chevalier B.S., Kortemme,T., Chadsey,M.S., Baker,D., Monnat,R.J.,Jr and Stoddard,B.L. (2002) Design, activity, and structure of a highly specific artificial endonuclease. Mol. Cell, 10, 895–905. [DOI] [PubMed] [Google Scholar]
- 49.Epinat J.C., Arnould,S., Chames,P., Rochaix,P., Desfontaines,D., Puzin,C., Patin,A., Zanghellini,A., Paques,F. and Lacroix,E. (2003) A novel engineered meganuclease induces homologous recombination in yeast and mammalian cells. Nucleic Acids Res., 31, 2952–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]