

ABSTRACT
Polyester polyurethane (PU) coatings are widely used to help protect underlying structural surfaces but are susceptible to biological degradation. PUs are susceptible to degradation by Pseudomonas species, due in part to the degradative activity of secreted hydrolytic enzymes. Microorganisms often respond to environmental cues by secreting enzymes or secondary metabolites to benefit their survival. This study investigated the impact of exposing several Pseudomonas strains to select carbon sources on the degradation of the colloidal polyester polyurethane Impranil DLN (Impranil). The prototypic Pseudomonas protegens strain Pf-5 exhibited Impranil-degrading activities when grown in sodium citrate but not in glucose-containing medium. Glucose also inhibited the induction of Impranil-degrading activity by citrate-fed Pf-5 in a dose-dependent manner. Biochemical and mutational analyses identified two extracellular lipases present in the Pf-5 culture supernatant (PueA and PueB) that were involved in degradation of Impranil. Deletion of the pueA gene reduced Impranil-clearing activities, while pueB deletion exhibited little effect. Removal of both genes was necessary to stop degradation of the polyurethane. Bioinformatic analysis showed that putative Cbr/Hfq/Crc-mediated regulatory elements were present in the intergenic sequences upstream of both pueA and pueB genes. Our results confirmed that both PueA and PueB extracellular enzymes act in concert to degrade Impranil. Furthermore, our data showed that carbon sources in the growth medium directly affected the levels of Impranil-degrading activity but that carbon source effects varied among Pseudomonas strains. This study uncovered an intricate and complicated regulation of P. protegens PU degradation activity controlled by carbon catabolite repression.
IMPORTANCE Polyurethane (PU) coatings are commonly used to protect metals from corrosion. Microbiologically induced PU degradation might pose a substantial problem for the integrity of these coatings. Microorganisms from diverse genera, including pseudomonads, possess the ability to degrade PUs via various means. This work identified two extracellular lipases, PueA and PueB, secreted by P. protegens strain Pf-5, to be responsible for the degradation of a colloidal polyester PU, Impranil. This study also revealed that the expression of the degradative activity by strain Pf-5 is controlled by glucose carbon catabolite repression. Furthermore, this study showed that the Impranil-degrading activity of many other Pseudomonas strains could be influenced by different carbon sources. This work shed light on the carbon source regulation of PU degradation activity among pseudomonads and identified the polyurethane lipases in P. protegens.
INTRODUCTION
Microbiologically induced deterioration of polyurethane (PU) and other military-relevant coatings has been recognized as a critical sustainability and sustainment issue for several decades (reviewed in references 1 to 3). A wide variety of fungi and Gram-positive and Gram-negative bacteria have been shown to secrete enzymes capable of degrading PUs. Among bacteria, some of the most well-studied PU-degrading organisms are pseudomonads—Gram-negative, heterotrophic bacteria (2, 4–8). Several Pseudomonas species including Pseudomonas chlororaphis, Pseudomonas aeruginosa, and Pseudomonas fluorescens have been shown to degrade a model polyester PU, Impranil DLN (Impranil) (4–6). Three PU-degrading enzymes have been purified and characterized from some of these strains, including polyurethane esterases A (PueA) and B (PueB) from Pseudomonas chlororaphis (5, 9) and polyurethane lipase (PulA) from P. fluorescens (5, 9–11). Although they are often referred to as “polyurethanases,” these enzymes are more accurately classified as extracellular lipases and esterases.
Enzyme secretion in bacteria is often dictated by environmental conditions. It has been observed that the expression of the extracellular proteome of bacteria can be affected by nutrient conditions as well as by growth phases (12–19). However, very little is known about how enzymes that effect polymer degradation are physiologically regulated in pseudomonads. Several studies have proposed that polyurethanase expression is constitutive (11), while others have reported that PU deterioration by both Gram-positive and Gram-negative bacteria is affected by the presence or absence of organic nutrients in the form of yeast extract (6, 20, 21). The biotechnology industry has investigated regulatory elements for PU-degrading enzymes in pseudomonads to optimize lipase production (reviewed in reference 22). For example, Makhzoum et al. published an extensive study on the factors affecting growth and extracellular lipase production by P. fluorescens 2D, a psychrotrophic bacterium that causes milk spoilage (18). Growth and lipase production in this strain were stimulated by organic acid carbon sources such as pyruvate. On the other hand, glucose (at 0.5%) completely inhibited lipase production, while supporting more growth than any other mono- or disaccharides. This was also true for Pseudomonas fragi cultures grown in ammonium sulfate-glucose medium formulations (23). In contrast, addition of glucose stimulated lipase activity in P. aeruginosa cultures (24).
In addition to saccharides and organic acids, iron(III) and other transition metals, triglycerides, fatty acids, growth phase, and the presence of the siderophore pyoverdin all have been reported to affect lipase production in P. fluorescens (18, 25, 26). Organic solvents and detergents, such as hexadecane and Tween 80, respectively, in low concentrations also have been shown to enhance lipase secretion in Burkholderia glumae (13). Collectively, these reports suggest that multiple factors and routes of extracellular lipase regulation, ranging from transcription to secretion, exist among different microorganisms.
The goal of our research was 2-fold: to understand how regulation of PU degradation occurs at the molecular level in Pseudomonas protegens Pf-5 and then to determine if a common regulatory mechanism of PU degradation exists among pseudomonads. We characterized the role of carbon sources and carbon catabolite repression (CCR) in the well-characterized P. protegens strain Pf-5 (27, 28), by identifying its two key PU-degrading enzymes, polyurethane esterases A (PueA) and B (PueB) and examining the phenotypic effects of mutations in these genes. We further determined how carbon sources influenced polyurethane degradation in pueA and pueB knockout strains. Lastly, we demonstrated the impact of medium conditions such as carbon sources and their concentrations on polyurethane degradation by various Pseudomonas strains.
MATERIALS AND METHODS
Bacterial strains and materials.
The strains and plasmids used in this study are listed in Table 1. Pseudomonas strains were cultured in lysogeny broth-Miller (LB) or minimal medium M9 at 27°C with agitation at 200 rpm. Escherichia coli strain S17-1/λpir was cultured in LB at 37°C. Media were supplemented with carbon sources and the PU dispersion agent Impranil DLN (Impranil) (Bayer Materials Science, PA) as indicated. If not specifically indicated, M9 was supplemented with 20 mM sodium citrate (M9-citrate) or 20 mM glucose (M9-glucose). To prepare agar plates, M9 and LB media were solidified with 1.5% agar. p-Nitrophenyl palmitate (4-NPP) was purchased from Sigma-Aldrich, MO. Sequences of DNA primers used to create and verify Pf-5 deletion mutants (Integrated DNA Technologies, Inc., IA) are listed in Table 2.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Bacterial strains | ||
| P. aeruginosa PAO1 | Wild type | 53 |
| P. fluorescens A506 | Wild type | 54 |
| P. fluorescens SBW25 | Wild type | 55 |
| P. fluorescens 204 | Wild type (ATCC 17571) | 56 |
| P. fluorescens Pf0-1 | Wild type | 57 |
| P. protegens pv. fluorescens Pf-5 | Wild type | 27 |
| P. protegens CHA0 | Wild type | 58 |
| P. protegens pv. chlororaphis BC2-12 | Mutant of wild-type BC2; enhanced polyurethane degrading activity (ATCC 55729) | 29 |
| P. putida KT2440 | Wild type | 59 |
| E. coli S17-1/λpir | K-12 strain; used for conjugative transfer of plasmids into Pf-5 | 60 |
| Plasmid | ||
| pMQ30 | Allelic replacement vector | 33 |
TABLE 2.
Sequences of primers used to generate pueA and pueB deletion mutants
| Primer name | Primer sequence (5′ to 3′)a |
|---|---|
| Allelic replacement primers | |
| Pf5pueALfor | tcgactgagcctttcgttttatttgatgcctggcagttccGAAGTCACTGAACGCAAGCG |
| Pf5pueALrev | ACAACAGAAGAGGCAATACCTGCCGGCGCGGCGCGAGGCC |
| Pf5pueARfor | GGCCTCGCGCCGCGCCGGCAGGTATTGCCTCTTCTGTTGT |
| Pf5pueARrev | ggaattgtgagcggataacaatttcacacaggaaacagctAAGGCGCCTGGTTCAAGGTC |
| Pf5pueBLfor | tcgactgagcctttcgttttatttgatgcctggcagttccGTTGTTCTTGTCCAGGCCCG |
| Pf5pueBLrev | AATAAAAAAGAGGAATGAGCCCCTCACCGATATCCAGCGC |
| Pf5pueBRfor | GCGCTGGATATCGGTGAGGGGCTCATTCCTCTTTTTTATT |
| Pf5pueBRrev | ggaattgtgagcggataacaatttcacacaggaaacagctGAGTTGCTGGTGCGGGTGCT |
| Knockout mutant verification primers | |
| Pf5pueATestF | AATTCGTTACGCACCTGCT |
| Pf5pueATestR | CTTCGCTAACCTGGCTTATGT |
| Pf5pueBTestF | CGTAGACCTGGGCATTGAAG |
| Pf5pueBTestR | CGGGAAACCCTGATTGAACT |
| pueAF | CGCTGGCGATCACCCTGTACTCC |
| pueAR | CTGGGAATTGCCGCTGCCGATCAG |
| pueBF | GCACCCACGACAAGCCCCAGG |
| puber | GTAGTCCACCGCACTATGGGCCG |
| rpoDF | GCCAACCTGCGTCTGGTGATCTCC |
| rpoDR | GCCGCGACGGTATTCGAACTTGTCC |
Uppercase letters correspond to sequences in the Pf-5 genome; lowercase letters correspond to sequences in the cloning vector pMQ30.
Polyurethane degradation assays.
To test the PU-degrading activity of cultures in agar plate assays, cells from 1.0-ml aliquots of overnight cultures grown in LB were pelleted by centrifugation, washed, and resuspended in 1.0 ml of 50 mM sodium potassium phosphate buffer (pH 7.4). Twenty microliters of the resultant cell suspension were placed onto M9 agar plates containing 3 g/liter Impranil and carbon sources as indicated. Plates were incubated for 48 h at 27°C. Zones of clearing around areas of bacterial growth indicated Impranil degradation.
A liquid-based assay using an Impranil dispersion was developed to quantitatively measure PU-degrading activity in cell-free supernatants. Culture samples were collected periodically during 14 h of growth. For each time point, cultures were centrifuged for 10 min at 12,800 × g, and supernatants were filter sterilized immediately using a 0.1-μm polyvinylidene difluoride (PVDF) filter (Millipore). Cell-free filtrates were stored at 4°C until further use. Similar to the assay described in reference 29, 20 μl of a 40 g/liter Impranil stock solution was added to 980 μl of the filtered supernatant samples. Clearing of the Impranil dispersion was monitored at 600 nm with a spectrophotometer (SpectraMax M3; Molecular Devices, CA). Impranil clearing rates, an indication of PU degradation, during exponential and early-stationary growth phases of the cultures were calculated over four time points within the first 45 min of the experiment.
Endpoint assays of Impranil clearing by late-stationary-phase cultures (22 to 24 h growth) were set up as described above without filtration. Clearing of the Impranil dispersion was measured as above periodically over a 24-hour period. Impranil clearing appeared to have approached maximum by 3 h postincubation (data not shown). The decrease in optical density at 600 nm (OD600) after a 3-h incubation was used to calculate the levels of Impranil clearing as an indication of Impranil degradation. The OD of some samples, particularly those of supernatants of M9-glucose cultures, increased over time. These samples did not show any Impranil degradation based on 1H nuclear magnetic resonance (NMR) studies (as described in Results); therefore, the change in OD was set to 0.
Chemical characterization of Impranil degradation by NMR spectroscopy.
Although Impranil clearing appeared to have approached a maximum by 3 h postincubation, the clearing reaction was carried out to 24 h before 1H NMR analysis was performed (30). At the end of the incubation period, Impranil-containing samples and control samples that did not contain Impranil were centrifuged in microcentrifuge tubes at 14,000 rpm for 30 min. Clarified supernatants were transferred to new microcentrifuge tubes and stored at −80°C until analysis. Samples for NMR measurements of hydrolysis products were prepared using 490 μl of sample and 10 μl of a 0.28 M 3-(trimethylsilyl)propionic-2,2,3,3-d4 acid sodium salt aqueous solution as an internal standard (30). A sealed capillary filled with D2O was added for NMR lock. The concentrations of the soluble hydrolysis products from Impranil were calculated using the average integrated area of two different 1H NMR signals resulting from a single experiment. Each experiment was performed in biological triplicates, and the concentration was calculated using the average concentrations from all three replicates. The first NMR signal (if not obscured by signals from metabolic products) used to calculate the concentration was the background-subtracted triplet at δH 3.5 ppm, and the second signal was the collective area of the 3 singlets between δH 1.0 and 0.50 ppm. The average integrated area of the peak at δH 3.5 ppm (and supported by the area from the signals between δH 1.0 to 0.5 ppm) was standardized to the signal for the internal standard (δH of −0.25 ppm), which had a known concentration. The use of two different signals from the same NMR spectrum allowed redundancy in the calculation of the concentration of soluble hydrolysis products.
HPLC analysis of glucose and citrate consumption by liquid-phase cultures.
Aliquots of cell-free culture supernatants were analyzed using high-performance liquid chromatography (HPLC) for the amount of carbon source consumed during the growth of P. protegens Pf-5 in M9 medium. A Varian Prostar HPLC system equipped with a refractive index (RI) detector and diode array detector (DaD) was used for each sample. The flow rate was 0.2 ml/min at 40°C using water as the mobile phase. The chromatography column was a conditioned 8 μm (300 × 7.7 mm) Agilent PL Hi-Plex column (H+). The concentrations of citrate and glucose were determined using external standards monitored at 213 nm with the DaD detector and with the RI detector, respectively.
General lipase activity assay.
Extracellular lipase activity was assessed using 4-NPP as the substrate. Bacteria were grown in minimal medium M9 supplemented with 20 mM succinate at 27°C with agitation for 22 h. Cells were pelleted at 12,000 × g for 30 min at 4°C to obtain clarified supernatants. For the assay, 495 μl of clarified supernatant was mixed with 5 μl of 25 mM 4-NPP (dissolved in dimethyl formamide [DMF]). The reactions were carried out at room temperature and monitored at 405 nm over 30 min. The rate of 4-NPP hydrolysis was calculated based on the extinction coefficient of 1.78 × 104 M−1 cm−1 (31).
Protein assays.
Protein concentrations were determined with a Bradford-based assay (Bio-Rad, CA) using the microassay procedure for microtiter plates. A bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, MA) was employed to measure protein concentrations in concentrated culture supernatants. Pierce bovine serum albumin standards (Thermo Fisher Scientific) were used to generate standard curves for determination of protein concentrations in samples.
PAGE and zymogram analysis.
To identify the proteins responsible for PU biodeterioration in Pf-5, a combination of nondenaturing (i.e., native) PAGE, zymogram analysis, and standard SDS-PAGE was conducted. Cell-free supernatants from overnight cultures grown in M9 medium containing 20 mM citrate were found to contain PU-degrading activity. An overnight culture (16 ml) was centrifuged (10 min at 12,800 × g), and the resultant supernatant was filtered through a 0.1-μm PVDF filter. The filtrate was supplemented with 2 mM dithiothreitol (DTT) and concentrated approximately 130-fold in an Amicon Centricon concentrator with a molecular mass cutoff of 50 kDa that had been passivated with 5% Triton X-100. Concentrates were washed with 8 ml of 50 mM Tris-HCl buffer (pH 8.0) containing 2 mM DTT and 0.1% Triton X-100. Samples were subjected to native PAGE using a 3 to 12% NativePAGE Novex bis-Tris gel according to the manufacturer's instructions (Invitrogen). The resulting protein gels were stained with colloidal blue or used for zymograms; proteins from the NativePAGE gel slices were also extracted for further analysis by standard denaturing SDS-PAGE.
For zymograms, a native protein gel containing samples of interest was washed with 50 mM Tris-HCl buffer (pH 8.0) for 15 min and subsequently placed on top of the zymogram gel (50 mM Tris-HCl [pH 8.0], 0.7% agarose, 1.5 g/liter Impranil, 0.1% Triton X-100, and 0.1 mM CaCl2). The zymogram was incubated overnight at room temperature and analyzed for clearing the following day.
To resolve proteins within the native protein gel that corresponded to zones of clearing in zymogram analysis, gel slices were excised and incubated in 100 μl of 50 mM Tris-HCl (pH 8.0) containing 2 mM DTT and 0.1% Triton X-100 overnight to elute proteins in these regions. The resulting eluates were concentrated by overnight precipitation with ice-cold acetone (80% final concentration) at −20°C. Precipitated proteins were resuspended in 25 μl of LDS sample buffer containing reducing agent (Invitrogen). SDS-PAGE was performed using a 4 to 12% bis-Tris gel and morpholineethanesulfonic acid (MES) buffer according to the manufacturer's instructions (Invitrogen). After electrophoresis, the gel was stained using colloidal blue (Invitrogen).
Protein identification.
Identification of proteins in the SDS-PAGE gel bands was performed by the University of Cincinnati Proteomics Laboratory according to the protocol described elsewhere (32). In brief, selected protein bands were excised from an SDS-PAGE gel and subjected to tryptic digests. The resulting peptides were analyzed using matrix-assisted laser desorption ionization–time of flight mass spectrometry and tandem mass spectrometry (MALDI-TOF-TOF MS/MS) for sequence determination. Protein identification was performed using the MASCOT search algorithm (Matrix Science, Boston, MA).
Bioinformatic analysis.
Protein sequences were obtained from the protein database at the National Center for Biotechnology Information (NCBI) (www.ncbi.nlm.nih.gov). Homologous proteins were identified using the Basic Local Alignment Search Tool (BLAST) on the NCBI website (PSI-BLAST) (blast.ncbi.nlm.nih.gov/Blast.cgi). Protein sequence alignments and percent identity determinations were performed using EMBOSS Stretcher software (EMBL-EBI) (www.ebi.ac.uk).
Creation of genetic knockout mutants.
In-frame deletions of pueA and pueB genes were created according to published protocols with minor modification (33). Briefly, 1,000 bp of both up- and downstream regions of either pueA or pueB were PCR amplified. The 5′ region of the upstream PCR product and the 3′ region of the downstream PCR product contained sequences overlapping portions of the cloning vector pMQ30 into which they would be inserted. In addition, the 3′ region of the upstream PCR product and the 5′ region of the downstream PCR product contained sequences overlapping each other. The PCR products were cloned into linearized pMQ30 via homologous recombination with the aid of a Gibson assembly kit (New England BioLabs, MA). The resulting allelic replacement constructs (ARCs) were introduced into Pf-5 by conjugation through E. coli strain S17-1/λpir. The exoconjugants were selected in the presence of 30 μg/ml gentamicin (to select for Pf-5 merodiploids) and 20 μg/ml nalidixic acid (to eliminate S17-1/λpir). Subsequently, the Pf-5 merodiploids were grown in the presence of 50 mg/ml sucrose to force the removal of integrated ARC, which in turn resulted in deletion of the pueA or pueB gene and the gentamicin-resistant cassette. To create the pueAB double-knockout mutant, the pueB single-knockout mutant was subjected to another round of deletion protocol to remove the pueA gene from the chromosome.
RESULTS
Induction and repression of polyurethane-degrading activity of P. protegens strain Pf-5.
We previously reported that a zone of clearing around the P. protegens strain Pf-5 colony was visible when this strain was grown on minimal medium M9 agar supplemented with 10 mM citrate and Impranil, an indication of polyurethane hydrolysis (34). We further investigated the impact of carbon sources on polymer degradation by Pf-5 using the Impranil agar plate-clearing assays (Fig. 1). Impranil clearing was mostly promoted when Pf-5 was grown on a minimal medium M9 agar plate in the presence of 20 mM citrate or succinate, while little to no clearing was observed when 20 mM glucose was supplied. Supplementation with other carbon sources resulted in an intermediate phenotype. To further define the effects of carbon sources that might be involved in pathways for the degradation of polyester polyurethanes, we focused on citrate and glucose due to their clear relationship with degradation. We first examined the effects of carbon source concentrations on Impranil clearing. In M9-Impranil agar plate assays with various concentrations of citrate, Impranil clearing was observed at all citrate concentrations with the greatest degree of clearing observed on plates containing 20 mM or 40 mM citrate (see Fig. S1A in the supplemental material). In contrast, no zones of clearing were observed when Pf-5 was grown on plates containing 20 mM or higher glucose levels (see Fig. S1B in the supplemental material). Insignificant levels of Impranil clearing were visible from Pf-5 cultured on M9-Impranil plates containing 5 mM or 10 mM glucose after 48 h of incubation.
FIG 1.

Effects of carbon sources on polyurethane degradation by P. protegens strain Pf-5. P. protegens strain Pf-5 was grown on M9 agar plates supplemented with Impranil and different carbon sources for 48 h. Degradation of Impranil resulted in visible zones of clearing.
It is possible that the effects of carbon sources on Pf-5 Impranil-clearing activity might be due to differences in the level of growth when Pf-5 is cultured on different carbon sources on agar plates. To address this possibility, we monitored the growth and polyurethane-degrading activity of strain Pf-5 in planktonic cultures containing either 20 mM citrate (M9-citrate) or 20 mM glucose (M9-glucose) as a carbon source (Fig. 2). Strain Pf-5 grew at similar rates in citrate (0.532 ± 0.056 h−1) and glucose (0.521 ± 0.006 h−1) and reached comparable optical densities (Fig. 2A and B). A liquid-based assay was used to determine the Impranil-clearing activity of cell-free culture supernatants using Impranil colloid suspensions (Fig. 2A and B). The Impranil-clearing activity started to increase at the end of the exponential growth and reached higher levels in supernatants from citrate-grown cultures than in those from glucose-grown cultures. Supernatants of citrate-grown cultures also contained higher protein concentrations than glucose cultures. Elevated extracellular protein concentrations correlated with the increase of Impranil-clearing activity as the culture transitioned into stationary phase (Fig. 2C).
FIG 2.
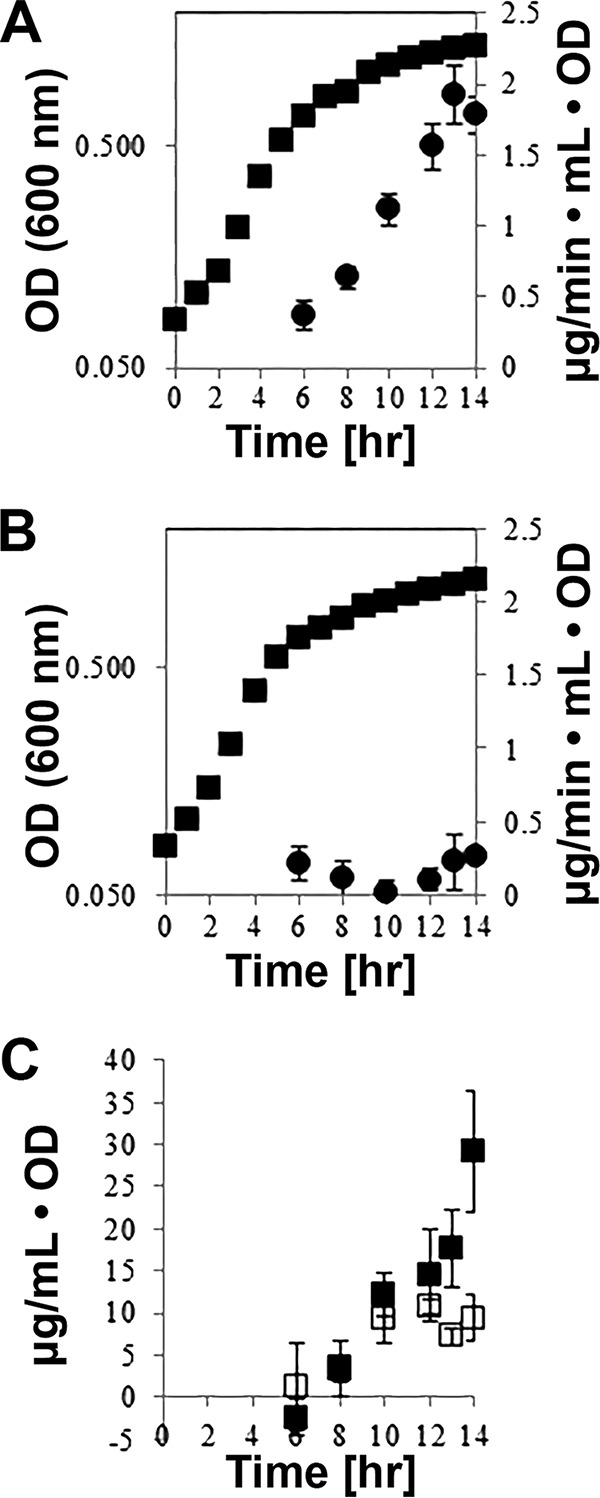
Relationship between growth state, protein secretion, and Impranil-clearing activities. (A and B) Growth (■) and Impranil-clearing activity (●) in cultures of P. protegens strain Pf-5 grown in M9 medium containing 20 mM citrate (A) or 20 mM glucose (B) over 14 h. Cell-free supernatants from culture samples were assayed for Impranil clearing as a measure of PU degradation. PU degradation rates were calculated over four time points within the first 45 min of the Impranil-clearing assay. Cultures were grown in triplicate, and individual supernatant samples were assayed twice. (C) Protein concentrations in supernatant samples from cultures of strain Pf-5 grown in M9 medium with 20 mM citrate (■) or 20 mM glucose (□) normalized to the OD of the cultures.
Both agar plate- and liquid Impranil-clearing assays are rapid and straightforward methods to determine if an organism can affect the light-diffractive properties of the Impranil colloids. To confirm that clearing of Impranil suspensions equates to hydrolysis of polyurethane polymers, we used 1H NMR to detect the presence of Impranil hydrolysis products in Impranil-clearing assay samples (30). The Impranil-degrading activities of late-stationary-phase (22 h) cell-free culture supernatants were examined. Supernatants of Pf-5 grown in M9-citrate contained substantial Impranil-clearing activity (0.131 ± 0.005 OD600 change/OD culture), while those from M9-glucose medium did not exhibit any activity. Similar to clearing results, Pf-5 M9-citrate but not M9-glucose culture supernatants produced detectable hydrolysis products (0.52 ± 0.04 mM) at the end of the Impranil-clearing assays (see Fig. S2 in the supplemental material). These results confirm that the disappearance of Impranil colloids was the consequence of PU polymer hydrolysis.
We suspected that the preferential metabolism of one carbon source over another might affect the amount of Impranil degradation by Pf-5. Therefore, the concentrations of glucose and citrate remaining in the culture supernatants containing 20 mM citrate with increasing levels of glucose were assessed by HPLC to determine how much of each carbon source was metabolized. As more glucose was supplemented into M9-citrate cultures, the amount of glucose utilization increased (see Fig. S3A in the supplemental material). Conversely, an inverse relationship was observed between citrate consumption and glucose addition (see Fig. S3B in the supplemental material). Most of the citrate was depleted in cultures containing 0 or 1.25 mM glucose. Although citrate utilization was reduced in cultures containing ≥2.5 mM glucose, very little citrate was consumed in cultures containing 10 and 20 mM glucose.
These results strongly indicate that glucose is the preferential carbon source in planktonic cultures. We hypothesized that the presence of glucose would repress the Impranil-degrading activity by Pf-5 induced by citrate. To test this hypothesis, we cultured strain Pf-5 in M9 medium containing 20 mM citrate supplemented with various amounts of glucose. Addition of increasing concentrations of glucose to M9-citrate resulted in a dose-dependent decrease of Impranil-degrading activity in the culture supernatants (Fig. 3A). We also observed a positive correlation between citrate consumption and the levels of Impranil clearing (Fig. 3B). These results indicate that glucose actively suppresses the induction of Impranil-degrading activity in P. protegens strain Pf-5.
FIG 3.

Glucose-inhibited induction of PU-degrading activity in strain Pf-5 late-stationary-phase culture supernatants. (A) Cultures of strain Pf-5 were grown for 22 h in M9-citrate medium supplemented with various concentrations of glucose. Cell-free supernatants of these cultures were subsequently incubated with Impranil to determine the levels of PU degradation. (B) The levels of citrate consumption were correlated with the degrees of Impranil clearing in Pf-5 cultures from panel A to assess correlations between citrate utilization and Impranil clearing. *, P < 0.05; **, P = 0.0001; ***, P < 0.0001.
Polyurethane-degrading enzymes in P. protegens strain Pf-5.
To identify PU-degrading extracellular enzymes from strain Pf-5, sterile-filtered supernatant from an overnight culture of strain Pf-5 grown in M9-citrate medium was concentrated and subjected to nondenaturing (i.e., native) PAGE (Fig. 4A). The resulting gel was used to obtain a zymogram that revealed two zones of polyurethane clearing (Fig. 4B). Proteins in the clearing zones and regions in between were eluted from a native gel and analyzed using SDS-PAGE (Fig. 4C). Each region from the native PAGE gel contained distinct protein banding patterns. The major bands (bands 1A, 1B, 6A, and 6B in Fig. 4C) from regions corresponding to the clearing zones identified by the zymogram were subjected to mass spectrometry analyses. Four proteins were identified in these regions (Table 3). Six tryptic peptides from protein band 6A were identical to sequences of the annotated polyurethane esterase A (pueA) gene products from P. chlororaphis strain O6 and P. protegens strain Pf-5 (see Table S1 in the supplemental material). Similarly, 6 tryptic peptides from protein band 1B were found to be identical to sequences of the annotated pueB gene product from P. protegens strain Pf-5 (see Table S2 in the supplemental material).
FIG 4.
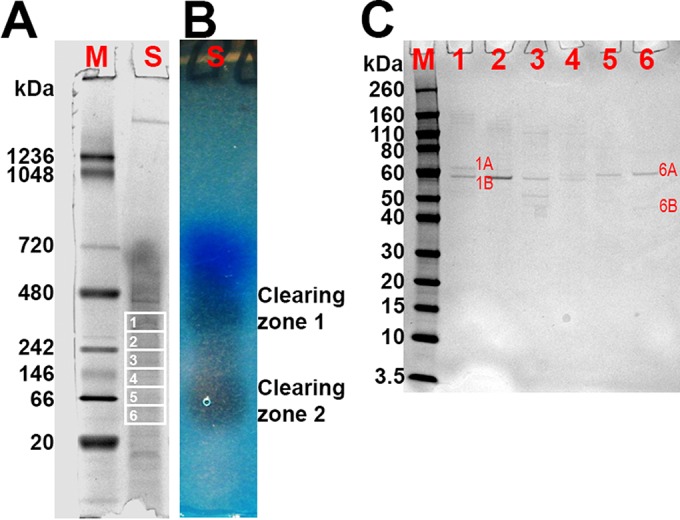
Zymogram analysis and identification of proteins with PU degradation activity. (A and B) NativePAGE with concentrated supernatant from a Pf-5 culture containing 103 μg of protein (A) and corresponding zymogram (B). M, NativeMARK protein standard; S, supernatant sample. (C) SDS-PAGE analysis of eluates 1 to 6 obtained from NativePAGE gel slices 1 to 6. M, Novex Sharp protein standard. Proteins in bands 1A, 1B, 6A, and 6B were identified using mass spectrometry analysis.
TABLE 3.
Identification of proteins in SDS-PAGE by mass spectrometry
To better understand the prevalence of these proteins among Pseudomonas species, we searched the NCBI nonredundant protein database for proteins homologous to both Pf-5 PueA and PueB in pseudomonads (Table 4). We also compared the amino acid sequences of PueA and PueB from strain Pf-5 with these homologs, including known polyurethanases PueA and PueB from P. chlororaphis (Table 4). Pf-5 PueA showed the highest amino acid sequence identity with P. protegens strain CHA0 lipase, class 3 (99.5%), P. fluorescens strain Pf0-1 lipase, class 3 (88.5%), and P. chlororaphis O6 polyurethanase A (82.0%). Pf-5 PueB showed the highest amino acid sequence identity with CHA0 lipase (100%), Pf0-1 triacylglycerol lipase (77.1%), and O6 triacylglycerol lipase (67.4%). The PueA homologs in P. fluorescens strains A506 and SBW25 and the PueB homolog in strain SBW25 were shorter than their counterparts in strain Pf-5 based on primary sequences. No homologs were identified in P. aeruginosa strain PAO1 or Pseudomonas putida strain KT2440.
TABLE 4.
Amino acid sequence comparison of PueA (YP_260307, 617 aa) and PueB (YP_260310, 561 aa) of P. protegens strain Pf-5 with derived protein sequences from known genomes of strains investigated in our studiesa
| Pseudomonas strain | Identity with PueAb | %b | Identity with PueBb | %b |
|---|---|---|---|---|
| P. protegens CHA0 | Lipase, class 3 (617 aa, YP_008000506) | 99.5 | Lipase (561 aa, YP_008000509) | 100 |
| P. fluorescens Pf0-1 | Lipase, class 3 (617 aa, YP_348417) | 88.5 | Triacylglycerol lipase (562 aa, YP_348416) | 77.1 |
| P. chlororaphis O6 | Polyurethanase A (617 aa, WP_009049212) | 82.0 | Triacylglycerol lipase (566 aa, EIM14910) | 67.4 |
| P. fluorescens A506 | Lipase (474 aa, YP_006324104) | 66.1 | Lipase (569 aa, YP_006323649) | 69.1 |
| P. fluorescens SBW25 | Lipase (469 aa, YP_002872716) | 65.9 | Lipase (469 aa, YP_002872716) | 48.3 |
| P. aeruginosa PAO1 | Alkaline metalloprotease (479 aa, NP_249940) | 31.1 | Alkaline metalloprotease (479 aa, NP_249940) | 30.3 |
| P. putida KT2440 | No significant identity (<10 %) | NAc | Hemolysin-type calcium binding bacteriocin (3,619 aa, NP_744706) | 10 |
Genome analysis in P. protegens strain Pf-5.
The impact of carbon sources on the Impranil-degrading activity by Pf-5 might be due to the levels of pueA and pueB gene expression. Reverse transcription-quantitative PCR (qRT-PCR) analyses of RNA from Pf-5 grown in either M9-citrate or M9-glucose were performed. While the expression of both pueA and pueB transcripts was detected, the levels of pueA and pueB expression were similar under both growth conditions (data not shown).
We subsequently analyzed the genome sequence of strain Pf-5 in an attempt to uncover the putative regulatory mechanisms for these genes. Adenine-rich sequence motifs located upstream of both pueA (5′ AACAACAG 3′) and pueB (5′ AATAAAAA 3′) genes in the Pf-5 genome matched the consensus binding site (5′ AANAANAA 3′) of Hfq/Crc, key regulatory proteins of the catabolite control system found in pseudomonads. The presence of cbrA and cbrB genes in the Pf-5 genome supports the existence of the CbrB/Hfq/Crc catabolite control system in this strain. Furthermore, we were able to identify a potential Crc homologue, exodeoxyribonuclease III (YP_263115.1), among the Pf-5 coding sequences. In addition, two regions of noncoding sequences in the Pf-5 genome that exhibited high sequence identities (69% and 86%) to the crcZ gene from P. aeruginosa PAO1 were identified, suggesting the presence of regulatory RNA encoding genes (see the “Identification of Putative Cbr/crc Catabolite Repression System in P. protegens Pf-5” in the supplemental material).
Determination of the roles of Pf-5 PueA and PueB in polyurethane biodeterioration in strain Pf-5.
To determine if pueA and pueB are solely responsible for Impranil-degrading activity in Pf-5, we created in-frame deletion mutants of pueA (Pf5ΔpueA), pueB (Pf5ΔpueB), and both genes (Pf5ΔpueAB). Deletion of the respective genes in each knockout mutant was verified by PCR (see Fig. S4 in the supplemental material). None of the deletion mutants exhibited any defects in their viability or growth compared to those for the wild-type strain (data not shown). Mutants of strain Pf-5 were grown in M9-citrate medium, and the cell-free culture supernatants were tested for Impranil-degrading activity (Fig. 5A). Deletion of the pueA gene (Pf5ΔpueA) resulted in a greater than 50% reduction in Impranil-degrading activity in culture supernatants based on clearing. Deletion of the pueB gene (Pf5ΔpueB) did not affect the ability of strain Pf-5 to degrade Impranil. Removal of both genes from the chromosome (Pf5ΔpueAB) almost completely abolished the Impranil-degrading activity of Pf-5 culture supernatants. We further examined the mutants in M9-glucose medium to assess regulation of PueA or PueB activities by glucose (Fig. 5A). Similar to the wild-type Pf-5, none of the mutants expressed Impranil-degrading activities when cultured in M9 minimal medium with glucose as the sole carbon source.
FIG 5.
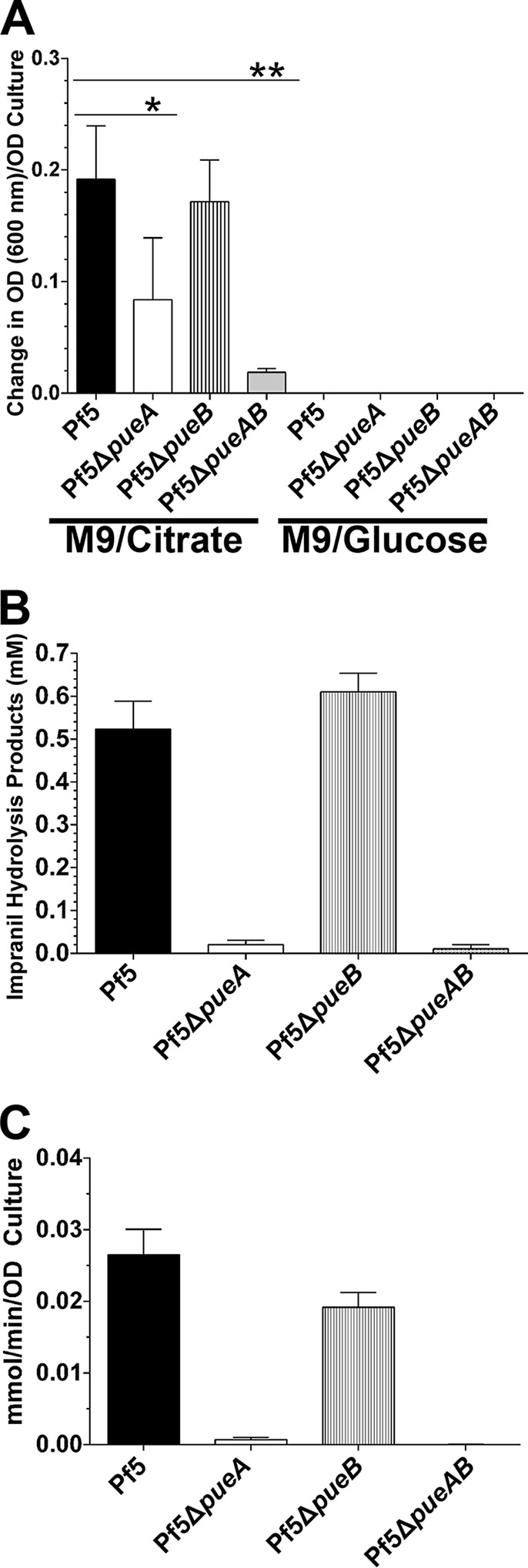
Involvement of polyurethane lipase PueA and PueB in PU-degrading activity and their regulation. (A) Cell-free supernatants of Pf-5 and pueA or pueB mutants (Pf5ΔpueA, Pf5ΔpueB, and Pf5ΔpueAB) grown in M9-citrate or M9-glucose medium were assayed for their abilities to clear Impranil. Results shown are average of 3 experiments with triplicate samples per experiment. (B) Impranil colloids were added to cell-free supernatants of Pf-5 and pueA or pueB mutants (Pf5ΔpueA, Pf5ΔpueB, and Pf5ΔpueAB) grown in M9-citrate medium. After 24 h of incubation, clarified samples were analyzed by 1H NMR for the presence of hydrolysis products. (C) Cell-free supernatants of M9-citrate cultures were also incubated with 4-NPP to assess general lipase activities of the strains. *, P < 0.001; **, P < 0.0001.
The Impranil-clearing reactions of Pf-5 mutants grown in M9-citrate culture supernatants were also analyzed by 1H NMR to assess for the levels of detectable Impranil hydrolysis products (Fig. 5B). Comparable to the Impranil-clearing results, the culture supernatant of the pueB deletion mutant (Pf5ΔpueB) hydrolyzed Impranil to a level similar to that of wild-type Pf-5. Abolishment of PueA expression in Pf5ΔpueA severely affected Impranil hydrolysis. Deletion of both pueA and pueB genes completely abrogated the ability of this strain to hydrolyze Impranil.
In addition to the PU activity assays from above, we also determined general extracellular lipase activity of strains of Pf-5 and its mutants using the 4-NPP hydrolysis assays (Fig. 5C). Cell-free culture supernatants of the wild-type strain of Pf-5 and the Pf5ΔpueB mutant showed approximately 28 and 20 nmol/min/OD culture lipase activity, respectively. However, deletion of pueA (Pf5ΔpueA) or both genes (Pf5ΔpueAB) resulted in an almost complete loss of extracellular lipase activity in the culture supernatants (0.67 and 0 nmol/min/OD culture, respectively).
Screening of carbon sources for impact on polyurethane-degrading ability of Pseudomonas strains.
After revealing how different carbon sources affected the PU-degrading ability of P. protegens strain Pf-5, Impranil agar plate-clearing assays were subsequently used to investigate the impact of carbon sources on various Pseudomonas species (Fig. 6). While P. putida KT2440 and P. fluorescens SBW25 showed no Impranil clearing during the time frame of the investigation, all other strains exhibited Impranil-clearing activity to various degrees. P. fluorescens strains A506 and Pf0-1 and P. aeruginosa strain PAO1 showed minimal Impranil clearing. Extensive Impranil clearing was observed with P. protegens strains Pf-5, CHA0, and BC2-12. The type of carbon source used in the growth medium had a strong impact on the ability of the strains to degrade polyurethane. In P. protegens strains, Impranil clearing was mostly promoted when they were grown in the presence of citrate and succinate, while little to no clearing was observed when glucose was supplied. The lack of Impranil clearing in the presence of glucose was also observed with P. fluorescens strains A506 and P. aeruginosa PAO1. With the exception of P. fluorescens SBW25 and P. putida KT2440, growth in pyruvate-containing medium resulted in Impranil clearing in all strains tested.
FIG 6.

Effects of carbon sources on polyurethane degradation. Various Pseudomonas strains were grown on M9 agar plates supplemented with Impranil and different carbon sources for 48 h. Degradation of Impranil resulted in visible zones of clearing. Strain Pf-5 was used as an internal reference on all plates.
DISCUSSION
In order to thrive and survive, microorganisms often secrete enzymes or secondary metabolites in response to environmental conditions. Many secreted enzymes from fungal and bacterial organisms also possess the ability to degrade PUs of various formulations (4, 6, 7, 35–38). Genes that encode enzymes capable of degrading PU ex vivo from some of these organisms have also been identified and cloned (5, 9, 11). Comparison of these gene sequences revealed that the gene products from these diverse organisms all contain a lipase domain and are predicted to be secreted. We report here that strain Pf-5 also secretes lipases capable of degrading Impranil.
Although several PU-degrading lipases have been characterized on a biochemical level, to date, only the pueA and pueB genes in P. chlororaphis have been formally demonstrated through insertional mutation analysis to be responsible for Impranil degradation in the context of an organism (39). Using zymogram techniques and gene deletion mutants, we identified the products of pueA and pueB genes, annotated as polyurethanase A and polyurethanase B (28), respectively, in P. protegens strain Pf-5 to be responsible for the degradation of the polyester PU Impranil.
Using 3 different assays, we showed that PueA protein plays a major role in Impranil clearing, while PueB, although also able to degrade Impranil, contributes less to these activities. However, deletion of both genes was required to abolish the Impranil-degrading activity of strain Pf-5. We previously reported that Pf-5 preferentially hydrolyzes the ester components of Impranil in a solidified film in a manner similar to 4-NPP hydrolysis (34). Of note is that Pf5ΔpueA, which presumably still secretes PueB, did not hydrolyze 4-NPP, a prototypic substrate for lipases. It is conceivable that PueA has higher intrinsic activity for hydrolyzing Impranil and 4-NPP than does PueB. Alternatively, PueA might be synthesized and secreted at higher levels than PueB in wild-type Pf-5. Our results demonstrated a critical role for PueA and PueB in Impranil degradation, but it must be noted that we still observed a low level of Impranil-clearing activity by the culture supernatant of the pueAB double-knockout mutant (Pf5ΔpueAB) grown in citrate-containing medium. This might be due to a small degree of cell lysis in the culture, releasing other intracellular enzymes that attack PU. We have observed that metabolism of citrate by both wild-type Pf-5 and the mutants leads to an alkaline pH shift in the growth medium (ranges from 7.7 to 8.3), and thus it was possible that the residual Impranil clearing observed in Pf5ΔpueAB culture supernatants might be due, in part, to base hydrolysis (34). Alternatively, this activity might be due to other unidentified extracellular enzymes that were not identified through zymogram analysis. The Pf-5 genome also contains a P. aeruginosa PAO1 lipA homolog (PFL0617) which encodes a putative secreted triacylglycerol lipase unrelated to PueA and PueB. It is possible that Pf-5 LipA contributed to the residual activity in Impranil clearing.
Pseudomonas spp. are heterotrophic bacteria that are well adapted to living in diverse environments. They possess versatile cellular physiologies that permit metabolic adjustments, allowing them to utilize a large variety of carbon and nitrogen sources and to degrade many types of organic compounds. Carbon catabolic repression allows pseudomonads to adapt their metabolism to a specific carbon source in order to maximize assimilation of that specific carbon source (40). It is also known that carbon catabolite repression controls enzyme secretion (41, 42) and various metabolic pathways in Pseudomonas. It has been found that the Cbr/Hfq/Crc system, Cyo terminal oxidase, and the PTSNtr system are involved in such regulation (40, 43, 44). The Cbr/Hfq/Crc regulatory system involves small RNAs that scavenge the mRNA binding protein complex, Hfq/Crc, resulting in a block of translation of their targeted mRNA (45–52). The identification of putative Hfq/Crc binding sites upstream of pueA and pueB genes, the lack of differences in pueA and pueB transcript levels (data not shown), and the effects of carbon source types on Pf-5 Impranil degradation suggest that the expression of PueA and PueB might be regulated at the posttranscriptional level by carbon catabolite repression. This mode of regulation may be common to many pseudomonads, as indicated by this study as well as those of other authors (6, 20, 21). Further study on the regulation and biochemical modes of action for these proteins might greatly benefit our understanding of microbiologically induced polymer coatings biodeterioration.
Supplementary Material
ACKNOWLEDGMENTS
We thank Virginia Stockwell, Oregon State University, Gary Banowetz, U.S. Department of Agriculture, and George O'Toole, Dartmouth Medical School, for providing Pseudomonas strains A506, SBW25, and Pf0-1, respectively. We also thank Gregory Anderson, Indiana University-Purdue University Indianapolis, and Robert Shanks, University of Pittsburgh School of Medicine, for their guidance in the creation of Pseudomonas genetic mutants. Additionally, we thank Robert Shanks for providing the Pseudomonas genetic knockout system, including the pMQ30 used in this study. Protein mass spectrometry analysis was performed by the Ken Greiss research group at the Proteomics Laboratory, University of Cincinnati College of Medicine.
Author contributions were as follows. S.Z., C.-S.H., D.B., J.N.R., L.J.N., and W.J.C.-G. conceived and designed the experiments. S.Z., C.-S.H., L.J.N., and J.C.B. performed the experiments. S.Z., C.-S.H., L.J.N., J.C.B., and W.J.C.-G. analyzed the data. S.Z., C.-S.H., L.J.N., C.A.D., A.L.C., J.C.B., D.B., J.N.R., and W.J.C.-G. discussed, interpreted, and guided the research direction. S.Z., C.-S.H., L.J.N., J.C.B., and W.J.C.-G. wrote the manuscript. S.Z., C.-S.H., L.J.N., C.A.D., A.L.C., J.C.B., D.B., J.N.R., and W.J.C.-G. edited the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01448-16.
REFERENCES
- 1.Cregut M, Bedas M, Durand MJ, Thouand G. 2013. New insights into polyurethane biodegradation and realistic prospects for the development of a sustainable waste recycling process. Biotechnol Adv 31:1634–1647. doi: 10.1016/j.biotechadv.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 2.Howard G. 2012. Polyurethane biodegradation, p 371–394. In Singh SN. (ed), Microbial degradation of xenobiotics. Springer, Berlin, Germany. [Google Scholar]
- 3.Zafar U, Houlden A, Robson GD. 2013. Fungal communities associated with the biodegradation of polyester polyurethane buried under compost at different temperatures. Appl Environ Microbiol 79:7313–7324. doi: 10.1128/AEM.02536-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard GT, Blake RC. 1998. Growth of Pseudomonas fluorescens on a polyester-polyurethane and the purification and characterization of a polyurethanase-protease enzyme. Int Biodeter Biodegr 42:213–220. doi: 10.1016/S0964-8305(98)00051-1. [DOI] [Google Scholar]
- 5.Howard GT, Crother B, Vicknair J. 2001. Cloning, nucleotide sequencing and characterization of a polyurethanase gene (pueB) from Pseudomonas chlororaphis. Int Biodeter Biodegr 47:141–149. doi: 10.1016/S0964-8305(01)00042-7. [DOI] [Google Scholar]
- 6.Kay MJ, Morton LHG, Prince EL. 1991. Bacterial degradation of polyester polyurethane. Int Biodeter 27:205–222. doi: 10.1016/0265-3036(91)90012-G. [DOI] [Google Scholar]
- 7.Peng YH, Shih YH, Lai YC, Liu YZ, Liu YT, Lin NC. 2014. Degradation of polyurethane by bacterium isolated from soil and assessment of polyurethanolytic activity of a Pseudomonas putida strain. Environ Sci Pollut Res Int 21:9529–9537. doi: 10.1007/s11356-014-2647-8. [DOI] [PubMed] [Google Scholar]
- 8.Spontón M, Casis N, Mazo P, Raud B, Simonetta A, Ríos L, Estenoz D. 2013. Biodegradation study by Pseudomonas spp. of flexible polyurethane foams derived from castor oil. Int Biodeter Biodegr 85:85–94. doi: 10.1016/j.ibiod.2013.05.019. [DOI] [Google Scholar]
- 9.Stern RV, Howard GT. 2000. The polyester polyurethanase gene (pueA) from Pseudomonas chlororaphis encodes a lipase. FEMS Microbiol Lett 185:163–168. doi: 10.1111/j.1574-6968.2000.tb09056.x. [DOI] [PubMed] [Google Scholar]
- 10.Nomura N, Shigeno-Akutsu Y, Nakajima-Kambe T, Nakahara T. 1998. Cloning and sequence analysis of a polyurethane esterase of Comamonas acidovorans TB-35. J Ferment Bioeng 86:339–345. doi: 10.1016/S0922-338X(99)89001-1. [DOI] [Google Scholar]
- 11.Ruiz C, Howard GT. 1999. Nucleotide sequencing of a polyurethanase gene (pulA) from Pseudomonas fluorescens. Int Biodeter Biodegr 44:127–131. doi: 10.1016/S0964-8305(99)00074-8. [DOI] [Google Scholar]
- 12.Allison SD, Vitousek PM. 2005. Responses of extracellular enzymes to simple and complex nutrient inputs. Soil Biol Biochem 37:937–944. doi: 10.1016/j.soilbio.2004.09.014. [DOI] [Google Scholar]
- 13.Boekema BK, Beselin A, Breuer M, Hauer B, Koster M, Rosenau F, Jaeger KE, Tommassen J. 2007. Hexadecane and Tween 80 stimulate lipase production in Burkholderia glumae by different mechanisms. Appl Environ Microbiol 73:3838–3844. doi: 10.1128/AEM.00097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns RG, DeForest JL, Marxsen J, Sinsabaugh RL, Stromberger ME, Wallenstein MD, Weintraub MN, Zoppini A. 2013. Soil enzymes in a changing environment: current knowledge and future directions. Soil Biol Biochem 58:216–234. doi: 10.1016/j.soilbio.2012.11.009. [DOI] [Google Scholar]
- 15.Casas-Godoy L, Duquesne S, Bordes F, Sandoval G, Marty A. 2012. Lipases: an overview, p 3–30. In Sandoval G. (ed), Lipases and phospholipases, vol 861 Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 16.Fairbairn DJ, Law BA. 1987. The effect of nitrogen and carbon sources on proteinase production by Pseudomonas fluorescens. J Appl Bacteriol 62:105–113. doi: 10.1111/j.1365-2672.1987.tb02387.x. [DOI] [PubMed] [Google Scholar]
- 17.Kulkarni N, Gadre RV. 1999. A novel alkaline, thermostable, protease-free lipase from Pseudomonas sp. Biotechnol Lett 21:897–899. doi: 10.1023/A:1005591009596. [DOI] [Google Scholar]
- 18.Makhzoum A, Knapp JS, Owusu RK. 1995. Factors affecting growth and extracellular lipase production by Pseudomonas fluorescens 2D. Food Microbiol 12:277–290. doi: 10.1016/S0740-0020(95)80108-1. [DOI] [Google Scholar]
- 19.Voigt B, Schweder T, Sibbald MJ, Albrecht D, Ehrenreich A, Bernhardt J, Feesche J, Maurer KH, Gottschalk G, van Dijl JM, Hecker M. 2006. The extracellular proteome of Bacillus licheniformis grown in different media and under different nutrient starvation conditions. Proteomics 6:268–281. doi: 10.1002/pmic.200500091. [DOI] [PubMed] [Google Scholar]
- 20.Kay MJ, McCabe RW, Morton LHG. 1993. Chemical and physical changes occurring in polyester polyurethane during biodegradation. Int Biodeter Biodegr 31:209–225. doi: 10.1016/0964-8305(93)90006-N. [DOI] [Google Scholar]
- 21.Klausmeier RE. 1996. The effect of extraneous nutrientees on the biodeterioration of plastics, p 232−243. In Microbiological deterioration in the tropics, vol 23 Society of Chemical Industry, London, United Kingdom. [Google Scholar]
- 22.Gupta R, Gupta N, Rathi P. 2004. Bacterial lipases: an overview of production, purification and biochemical properties. Appl Microbiol Biotechnol 64:763–781. doi: 10.1007/s00253-004-1568-8. [DOI] [PubMed] [Google Scholar]
- 23.Alford JA, Pierce DA. 1963. Production of lipase by Pseudomonas fragi in a synthetic medium. J Bacteriol 86:24–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadkarni SR. 1971. Studies on bacterial lipase. I. Nutritional requirements of Pseudomonas aeruginosa for production of lipase. Enzymologia 40:286–301. [PubMed] [Google Scholar]
- 25.Fernandez L, San José C, Cholette H, McKellar RC. 1988. Characterization of a pyoverdine-deficient mutant of Pseudomonas fluorescens impaired in the secretion of extracellular lipase. Arch Microbiol 150:523–528. doi: 10.1007/BF00408243. [DOI] [PubMed] [Google Scholar]
- 26.McKellar RC, Shamsuzzaman K, San Jose C, Cholette H. 1987. Influence of iron(III) and pyoverdine on extracellular proteinase and lipase production by Pseudomonas fluorescens B52. Arch Microbiol 147:225–230. doi: 10.1007/BF00463479. [DOI] [PubMed] [Google Scholar]
- 27.Howell CR, Stipanovic RD. 1979. Control of Rhizoctonia solani on cotton seedlings with Pseudomonas fluorescens and with an antibiotic produced by the bacterium. Phytopathology 69:480–482. doi: 10.1094/Phyto-69-480. [DOI] [Google Scholar]
- 28.Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GS, Mavrodi DV, DeBoy RT, Seshadri R, Ren Q, Madupu R, Dodson RJ, Durkin AS, Brinkac LM, Daugherty SC, Sullivan SA, Rosovitz MJ, Gwinn ML, Zhou L, Schneider DJ, Cartinhour SW, Nelson WC, Weidman J, Watkins K, Tran K, Khouri H, Pierson EA, Pierson LS III, Thomashow LS, Loper JE. 2005. Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23:873–878. doi: 10.1038/nbt1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montgomery MT, Campbell JR, Crabbe JR, Walz SE, Thompson L. 1998. Pseudomonas chlororaphis microorganism, polyurethane degrading enzyme obtained therefrom, and method of use. US patent 5,714,378.
- 30.Biffinger JC, Barlow DE, Cockrell AL, Cusick KD, Hervey WJ, Fitzgerald LA, Nadeau LJ, Hung CS, Crookes-Goodson WJ, Russell JN Jr. 2015. The applicability of Impranil DLN for gauging the biodegradation of polyurethanes. Polym Degrad Stab 120:178–185. doi: 10.1016/j.polymdegradstab.2015.06.020. [DOI] [Google Scholar]
- 31.Lu J, Brigham C, Rha C, Sinskey A. 2013. Characterization of an extracellular lipase and its chaperone from Ralstonia eutropha H16. Appl Microbiol Biotechnol 97:2443–2454. doi: 10.1007/s00253-012-4115-z. [DOI] [PubMed] [Google Scholar]
- 32.Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M, Edwards MJ, Greis KD, Shertzer HG, Fisher AB, Lentsch AB. 2009. Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice. Am J Physiol Gastrointest Liver Physiol 296:G266−G274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biffinger JC, Barlow DE, Pirlo RK, Babson DM, Fitzgerald LA, Zingarelli S, Nadeau LJ, Crookes-Goodson WJ, Russell JN Jr. 2014. A direct quantitative agar-plate based assay for analysis of Pseudomonas protegens Pf-5 degradation of polyurethane films. Int Biodeter Biodegr 95(Pt B):311–319. doi: 10.1016/j.ibiod.2014.09.005. [DOI] [Google Scholar]
- 35.Bentham RH, Morton LHG, Allen NG. 1987. Rapid assessment of the microbial deterioration of polyurethanes. Int Biodeter 23:377–386. doi: 10.1016/0265-3036(87)90026-1. [DOI] [Google Scholar]
- 36.Cosgrove L, McGeechan PL, Robson GD, Handley PS. 2007. Fungal communities associated with degradation of polyester polyurethane in soil. Appl Environ Microbiol 73:5817–5824. doi: 10.1128/AEM.01083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Howard GT, Ruiz C, Hilliard NP. 1999. Growth of Pseudomonas chlororaphis on a polyester-polyurethane and the purification and characterization of a polyurethanase-esterase enzyme. Int Biodeter Biodegr 43:7–12. doi: 10.1016/S0964-8305(98)00057-2. [DOI] [Google Scholar]
- 38.Russell JR, Huang J, Anand P, Kucera K, Sandoval AG, Dantzler KW, Hickman D, Jee J, Kimovec FM, Koppstein D, Marks DH, Mittermiller PA, Nunez SJ, Santiago M, Townes MA, Vishnevetsky M, Williams NE, Vargas MP, Boulanger LA, Bascom-Slack C, Strobel SA. 2011. Biodegradation of polyester polyurethane by endophytic fungi. Appl Environ Microbiol 77:6076–6084. doi: 10.1128/AEM.00521-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Howard GT, Mackie RI, Cann IKO, Ohene-Adjei S, Aboudehen KS, Duos BG, Childers GW. 2007. Effect of insertional mutations in the pueA and pueB genes encoding two polyurethanases in Pseudomonas chlororaphis contained within a gene cluster. J Appl Microbiol 103:2074–2083. doi: 10.1111/j.1365-2672.2007.03447.x. [DOI] [PubMed] [Google Scholar]
- 40.Rojo F. 2010. Carbon catabolite repression in Pseudomonas: optimizing metabolic versatility and interactions with the environment. FEMS Microbiol Rev 34:658–684. doi: 10.1111/j.1574-6976.2010.00218.x. [DOI] [PubMed] [Google Scholar]
- 41.Boethling RS. 1975. Regulation of extracellular protease secretion in Pseudomonas maltophilia. J Bacteriol 123:954–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reales-Calderón JA, Corona F, Monteoliva L, Gil C, Martínez JL. 2015. Quantitative proteomics unravels that the post-transcriptional regulator Crc modulates the generation of vesicles and secreted virulence determinants of Pseudomonas aeruginosa. J Proteomics 127(Pt B):352−364. doi: 10.1016/j.jprot.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 43.Deutscher J. 2008. The mechanisms of carbon catabolite repression in bacteria. Curr Opin Microbiol 11:87–93. doi: 10.1016/j.mib.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Görke B, Stulke J. 2008. Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat Rev Microbiol 6:613–624. doi: 10.1038/nrmicro1932. [DOI] [PubMed] [Google Scholar]
- 45.Marzi S, Romby P. 2012. RNA mimicry, a decoy for regulatory proteins. Mol Microbiol 83:1–6. doi: 10.1111/j.1365-2958.2011.07911.x. [DOI] [PubMed] [Google Scholar]
- 46.Moreno R, Hernández-Arranz S, La Rosa R, Yuste L, Madhushani A, Shingler V, Rojo F. 2015. The Crc and Hfq proteins of Pseudomonas putida cooperate in catabolite repression and formation of ribonucleic acid complexes with specific target motifs. Environ Microbiol 17:105–118. doi: 10.1111/1462-2920.12499. [DOI] [PubMed] [Google Scholar]
- 47.Sonnleitner E, Bläsi U. 2014. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet 10:e1004440. doi: 10.1371/journal.pgen.1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonnleitner E, Haas D. 2011. Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl Microbiol Biotechnol 91:63–79. doi: 10.1007/s00253-011-3332-1. [DOI] [PubMed] [Google Scholar]
- 49.Browne P, Barret M, O'Gara F, Morrissey JP. 2010. Computational prediction of the Crc regulon identifies genus-wide and species-specific targets of catabolite repression control in Pseudomonas bacteria. BMC Microbiol 10:300. doi: 10.1186/1471-2180-10-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lapouge K, Perozzo R, Iwaszkiewicz J, Bertelli C, Zoete V, Michielin O, Scapozza L, Haas D. 2013. RNA pentaloop structures as effective targets of regulators belonging to the RsmA/CsrA protein family. RNA Biol 10:1030–1041. doi: 10.4161/rna.24771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lapouge K, Schubert M, Allain FH, Haas D. 2008. Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol Microbiol 67:241–253. [DOI] [PubMed] [Google Scholar]
- 52.Sonnleitner E, Abdou L, Haas D. 2009. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 106:21866–21871. doi: 10.1073/pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holloway BW. 1955. Genetic recombination in Pseudomonas aeruginosa. J Gen Microbiol 13:572–581. [DOI] [PubMed] [Google Scholar]
- 54.Stockwell VO, Stack JP. 2007. Using Pseudomonas spp. for integrated biological control. Phytopathology 97:244–249. doi: 10.1094/PHYTO-97-2-0244. [DOI] [PubMed] [Google Scholar]
- 55.Silby MW, Cerdeno-Tarraga AM, Vernikos GS, Giddens SR, Jackson RW, Preston GM, Zhang XX, Moon CD, Gehrig SM, Godfrey SA, Knight CG, Malone JG, Robinson Z, Spiers AJ, Harris S, Challis GL, Yaxley AM, Harris D, Seeger K, Murphy L, Rutter S, Squares R, Quail MA, Saunders E, Mavromatis K, Brettin TS, Bentley SD, Hothersall J, Stephens E, Thomas CM, Parkhill J, Levy SB, Rainey PB, Thomson NR. 2009. Genomic and genetic analyses of diversity and plant interactions of Pseudomonas fluorescens. Genome Biol 10:R51. doi: 10.1186/gb-2009-10-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stanier RY, Palleroni NJ, Doudoroff M. 1966. The aerobic pseudomonads: a taxonomic study. J Gen Microbiol 43:159–271. [DOI] [PubMed] [Google Scholar]
- 57.Compeau G, Al-Achi BJ, Platsouka E, Levy SB. 1988. Survival of rifampin-resistant mutants of Pseudomonas fluorescens and Pseudomonas putida in soil systems. Appl Environ Microbiol 54:2432–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stutz EW, D G, Kern H. 1986. Naturally occurring fluorescent pseudomonasds involved in suppression of black root rot of tobacco. Phytopathology 76:181–185. doi: 10.1094/Phyto-76-181. [DOI] [Google Scholar]
- 59.Timmis KN. 2002. Pseudomonas putida: a cosmopolitan opportunist par excellence. Environ Microbiol 4:779–781. doi: 10.1046/j.1462-2920.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- 60.Simon R, Priefer U, Puhler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram negative bacteria. Nat Biotechnol 1:784–791. doi: 10.1038/nbt1183-784. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.