

Supplemental Digital Content is available in the text.
Keywords: alternative splicing, confusion, dilated cardiomyopathy, human, mutation
Abstract
Background—
Truncating mutations in the giant sarcomeric gene Titin are the most common type of genetic alteration in dilated cardiomyopathy. Detailed studies have amassed a wealth of information about truncating variant position in cases and controls. Nonetheless, considerable confusion exists as to how to interpret the pathogenicity of these variants, hindering our ability to make useful recommendations to patients.
Methods and Results—
Building on our recent discovery of a conserved internal promoter within the Titin gene, we sought to develop an integrative statistical model to explain the observed pattern of Titin truncation variants in patients with dilated cardiomyopathy and population controls. We amassed Titin truncation mutation information from 1714 human dilated cardiomyopathy cases and >69 000 controls and found 3 factors explaining the distribution of Titin mutations: (1) alternative splicing, (2) whether the internal promoter Cronos isoform was disrupted, and (3) whether the distal C terminus was targeted (in keeping with the observation that truncation variants in this region escape nonsense-mediated decay and continue to be incorporated in the sarcomere). A model using these 3 factors had strong predictive performance with an area under the receiver operating characteristic curve of 0.81. Accordingly, individuals with either the most severe form of dilated cardiomyopathy or whose mutations demonstrated clear family segregation experienced the highest risk profile across all 3 components.
Conclusions—
We conclude that quantitative models derived from large-scale human genetic and phenotypic data can be applied to help overcome the ever-growing challenges of genetic data interpretation. Results of our approach can be found at http://cvri.ucsf.edu/~deo/TTNtruncationvariant.html.
Truncation mutations in the giant sarcomeric gene Titin result in cardiac and skeletal myopathies.1–9 In contrast with many disease genes, the distribution of truncation mutations in Titin patients is not uniform, with a preponderance of mutations in the C-terminal two thirds of the protein.5,9 This region of the protein corresponds to the distal I-band, A-band, and M-line regions, named after distinct portions of the sarcomere visualized on electron micrographs.10 Titin truncation mutations are seen in ≤25% of patients with dilated cardiomyopathy (DCM), and such variants may be found in as much as 1% of the general population.9,11 Given this widespread prevalence, clarity in how to interpret the significance of these variants is needed.
Editorial see p 392
Clinical Perspective on p 425
We recently described the phenotype of 6 zebrafish lines with truncating mutations in the orthologous zebrafish ttna gene.12 Although homozygous mutations of all 6 targeted exons resulted in severe cardiac phenotypes, skeletal muscle phenotypes differed dramatically, with N-terminal mutations (proximal one third of ttna) having a phenotype indistinguishable from wild type, whereas C-terminal mutations (distal two third of ttna) had severe sarcomeric disarray and resulting inability to swim. Through a mixture of systematic gene disruption and transcriptome/epigenome analysis, we were able to map a novel internal promoter in Titin in the distal I-band, which is active in mouse and human hearts and, when disrupted, resulted in the more severe phenotype seen in C terminal mutants. We named the resulting protein isoform Cronos. Given its role in sarcomere development and the striking coincidence of the location of this internal promoter with the observed distribution of human Titin mutations, we concluded that this was a likely contributor to the more severe disease phenotype seen in cardiomyopathy patients with C-terminal Titin mutations.
Because all of the ttna exons we targeted were constitutive, alternative splicing was not relevant to the phenotypic differences we observed with mutation position. Nonetheless, alternative splicing has long been known to modulate the severity of some Mendelian diseases—perhaps most notably with the distinctions between the Becker and Duchenne forms of muscular dystrophy, where mutations in the milder Becker form sometimes target exons in the Dystrophin gene that are at least partially excluded from transcripts by splicing.13 Titin itself has many alternatively spliced exons, mostly located in the I-band, and variable inclusion of these exons seems to regulate passive tensile properties of the muscle fiber.14 Mutations in unaffected individuals tend to map to these alternatively spliced areas,9 and recently, in induced pluripotent cell-derived cardiomyocytes, homozygous disruption of an alternatively spliced I-band exon resulted in some retained ability to generate systolic force, in contrast with disruption of a constitutive exon in the A-band.15
Despite awareness of 2 sources of variability (the internal Cronos isoform and alternative splicing) in explaining how mutation position might affect risk and severity of disease in Titin truncation mutation patients, considerable disagreement remains among the cardiovascular genetics counselor community on how to advise patients with such mutations. Part of the challenge resides in the need for a quantitative, nonbinary approach to mutation classification, which, in the case of truncating mutations, is not adequately addressed in current variant interpretation guidelines.16 Given that publicly available efforts have sequenced over tens of thousands of controls and nearly 2000 DCM cases for Titin, I reasoned it might be feasible to build statistical models that more effectively classify patient mutations.
Methods
Genetic Variant Analysis
I compiled Titin truncation variant data from 4 sequencing efforts5,8,9,17 of DCM cases (1714 in total) and control information from 3 additional efforts (≈69 000 in total) to yield 1143 individuals with Titin truncation variants (ie, causing premature nonsense codons, frameshifts, or mutations in the canonical±1,2 splice position). The exact location of Titin truncation variants was obtained from supplementary tables of previously published work5,8,9,17 in the case of patients with DCM or from control databases: Exome Variant Server (http://evs.gs.washington.edu), 1000 Genomes (http://browser.1000genomes.org/), and Exome Aggregation Consortium (http://exac.broadinstitute.org). All variants were mapped to the inferred complete isoform ENST00000589042, which is an isoform that includes all 363 Titin exons (with the exception of the Novex-3 exon found exclusively in a single isoform). I used genotype quality scores when possible to exclude variants called with lower confidence. Where such quality scores were provided, I restricted the analysis to variants with a PASS designation, which implies that all user quality filters were met. I did not consider mutations mapping to the Novex-3 exon because these seem unlikely to contribute to disease and are in fact seen in senior competitive athletes.12
Many of these reported variants were observed in >1 individual. I counted multiple instances of a variant as separate data points—under the assumption that these individuals were unrelated (this is true for Exome Aggregation Consortium and 1000 Genomes and stated explicitly for Akinrinade et al17 and a portion of Roberts et al9). I excluded what seemed to be a spurious splice variant identified exclusively in the Exome Variant Server data set (chr2, position 179563642, CT>C), not present in dbSNP146, 1000 Genomes, or the much larger Exome Aggregation Consortium database, and yet stated within Exome Variant Server to have a minor allele frequency close to 4% with over 374 alleles observed. Although there may be other false positives, as all other observed alleles are rare, these are unlikely to skew results. Finally, as Cohort B in Herman et al5 seemed to describe the same 71 individuals as found in Roberts et al,9 including 17 overlapping mutations, I excluded this group from analysis.
The resulting count of (non-Novex 3) Titin (TTN) truncations is as follows (Figure I in the Data Supplement):
Exome Aggregation Consortium database: 639 truncation variants (390 unique)
Exome Variant Server database: 214 truncation variants (50 unique)
1000 Genomes database: 43 truncation variants (28 unique)
The corresponding count of TTN truncations in DCM cases is as follows:
Human RNA-Sequencing Analysis
This analysis required assessment of the extent of alternative splicing of each Titin exon in cardiac tissue. To compute this, I downloaded fastq format files for transcriptomic data from heart tissue from patients with DCM (from GEO series GSE57344) and healthy controls (from GSE57344 and GTEx project) corresponding to Short Read Archive accessions SRR1272187 (control), SRR1272188 (control), SRR1272190 (DCM), SRR1272191 (DCM), SRR598148 (GTEx), SRR600474 (GTEx), SRR600852 (GTEx), SRR601986 (GTEx), SRR598589 (GTEx), and SRR599249 (GTEx). These provided a range of individuals sequenced at high read depth for the estimation of the extent of alternative splicing. Reads were mapped to the hg19 build of the human genome using TopHat.18 I exclusively used junction reads (ie, reads that directly span an exon–exon junction) for computation of percent spliced in (PSI) values because these have been shown to overcome inaccuracies arising from variability in read depth at different exons.19 PSI is a metric of the fraction of a gene’s transcripts (in a particular tissue) that include the exon of interest. It can be estimated from RNA-Seq data by the ratio of the number of reads that support inclusion of a particular exon versus the total number of reads that support either its inclusion or its exclusion. I computed a read depth-weighted mean PSI for each exon across all 10 samples and used these for subsequent analyses. Splicing estimates are in good agreement with those reported at https://cardiodb.org/titin/.
Logistic Regression Models of the Distribution of TTN Truncation Variants
All statistical analysis was performed in R (3.1.1). The primary goal of this work was to understand how variants found in cases differ from those found in controls, according to the characteristics of the variant (where it is found in the protein, is it alternatively spliced, etc.). My starting point was a list of variants found in cases and a corresponding list found in controls. I annotated each variant according to its location in the protein (taking into account regions of the sarcomere and the position of the internal promoter), as well as the extent of alternative splicing for the exon in which it is found. The data of the 1143 truncation variants (247 from DCM cases and 896 from controls) were analyzed using logistic regression to identify the factors that characterize mutations found in cases and controls.
The same 1143 data points were used for every analysis described below. For transparency, I have also provided an R Markdown file that describes all analyses performed and plots generated here (File I in the Data Supplement).
Exploring the Variation of TTN Truncation Variant Distribution With Alternative Splicing
I first generated a scatter plot comparing the distribution of PSI between DCM cases and controls (Figure II in the Data Supplement). Visually, it is clear that patients with DCM are more likely to have mutations with high PSI values, an observation consistent with previous reports.9 The distribution of PSI values was not uniform across the range of 0 to 1 and appeared to cluster into discrete bins. To simplify subsequent analyses, including developing of a classification model (see below), I created 4 bins for PSI: very low (PSI between 0 and 0.399), low (0.400 and 0.649), medium (0.65 and 0.749), and high (0.75 and 1). As described above, logistic regression was used to estimate odds ratios (OR) for whether individual PSI bins differ in their distribution of case versus control mutations. The reference bin for this analysis is the very low category (arbitrarily set to an OR of 1).
Exploring the Impact of Cronos Disruption on the Case–Control Distribution
We had previously demonstrated that the position of the Titin internal promoter in the terminal I-band coincided sharply with the position of mutations seen in end-stage DCM.12 I estimated the contribution of disruption of Cronos on the distribution of mutations by fitting a logistic regression model with 2 predictors: PSI and whether it would disrupt Cronos. As a parallel analysis, I focused only on exons that are constitutive (PSI>0.95). The adjusted Cronos disruption OR is illustrated graphically, both for the full protein (Figure 1B) and for the I-band alone (Figure III in the Data Supplement).
Figure 1.
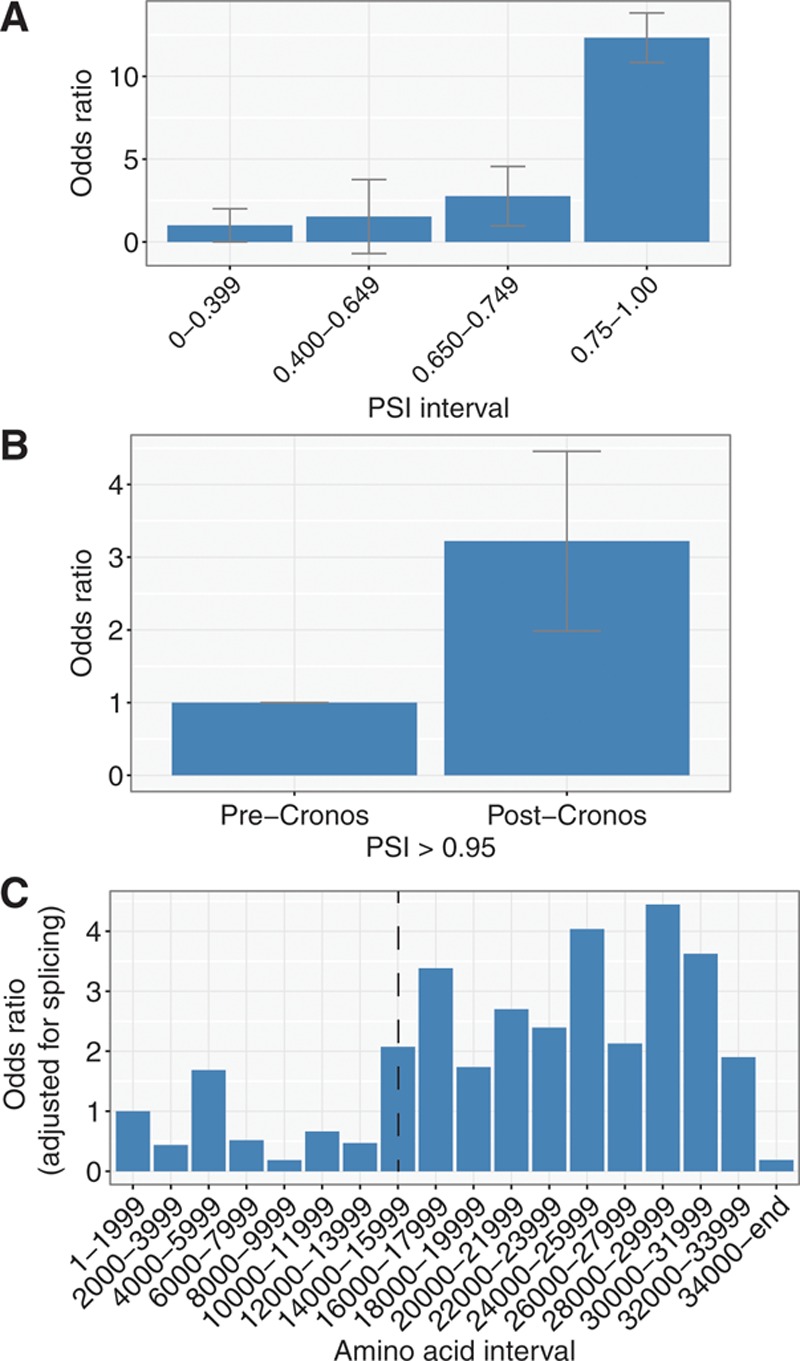
Analysis of Titin truncation variant data from 1714 dilated cardiomyopathy cases and 69 210 controls reveals 3 primary determinants of mutation distribution. A, The odds that mutations are found in cases vs controls increases with percent spliced in (PSI). Odds ratios were computed with logistic regression models, controlling for whether the Cronos isoform is disrupted. B, Within constitutive exons (PSI>0.95), there is a 3.1-fold increased odds of mutations being found in cases vs controls for mutations that disrupt the Cronos isoform (P=8×10−8). C, Analysis of odds ratios across bins of 2000 amino acids from N terminus to C terminus reveals 2 primary sources of variation: an increase in risk for mutations that disrupt Cronos (shown by position of dotted line) and a sharp drop in risk for those affecting the distal C terminus. For all plots, error bars, when shown, correspond to the SE.
Exploring the Variation of TTN Truncation Variant Distribution With Amino Acid Position
To understand how the distribution of TTN truncation variants varies along the length of the protein, I divided the protein into bins of 2000 amino acids and plotted a histogram displaying the number of mutations found for cases and controls in each bin (Figures 1C and 2). I then fit a logistic regression model, including amino acid bin and PSI as predictors. The reference for the computation of ORs is the first amino acid bin, located at the N terminus (ie, amino acids 1–1999). Two trends are obvious in this plot—an increase in OR at the position of Cronos and a drop at the distal C terminus.
Figure 2.
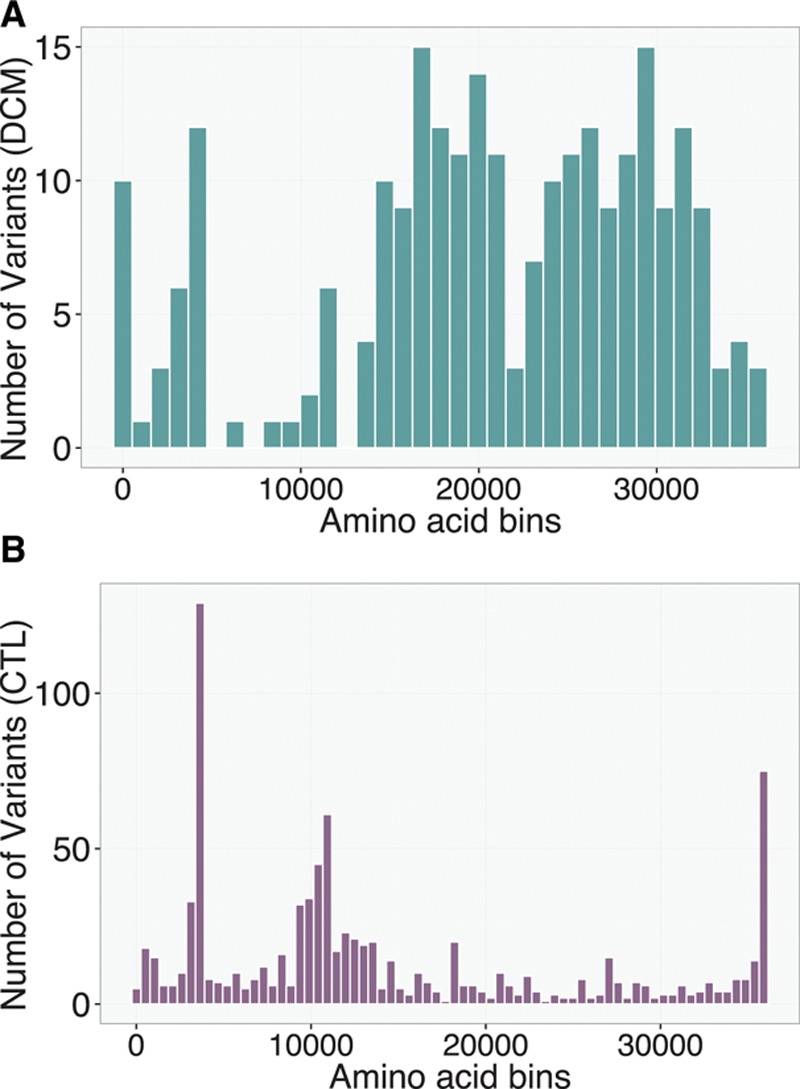
Distribution of TTN truncation variants in bins of amino acids from N terminus to C terminus in the (A) dilated cardiomyopathy (DCM) and (B) population control (CTL) groups. A smooth increase in number of variants is seen at the distal C terminus in the CTL group, whereas a corresponding drop is seen in the DCM group. Each bin corresponds to 1000 amino acids for the DCM plot and 500 amino acids for the CTL plot.
Estimating the Relative Contributions of Individual Predictors
The logistic regression analyses described here focus on the extent to which characteristics of a protein variant can help distinguish whether it has arisen from a case or a control population. ORs derived from these analyses estimate the influence of a unit increase in a given predictor on this discriminating ability, assuming that all other predictors are fixed. The concept of a unit increase is not straightforward to interpret across categories measured on different scales. In such cases, it can be helpful to standardize predictors by dividing each value by the SD for that predictor across the sample. The interpretation is then the impact on odds for a standard unit change, which is more readily interpretable. This becomes important when answering questions, such as does variation in alternative splicing matter more than whether Cronos is disrupted? To give another example, although the OR of case versus control status for variants mapping to the extreme C terminus may be low, whether there are few controls observed with these mutations, the overall contribution of this predictor to the distribution of mutations may not be substantial.
Cronos disruption and C-term mutation location are categorical variables with only 1 level and are encoded as dummy variables with 2 integer values (0 and 1). One can, thus, compute a mean and SD for each of these dummy variables and standardize accordingly. I treated PSI as a continuous variable for this analysis. I then repeated the logistic regression analysis and plotted the resulting ORs in a caterpillar plot (Figure 3).
Figure 3.
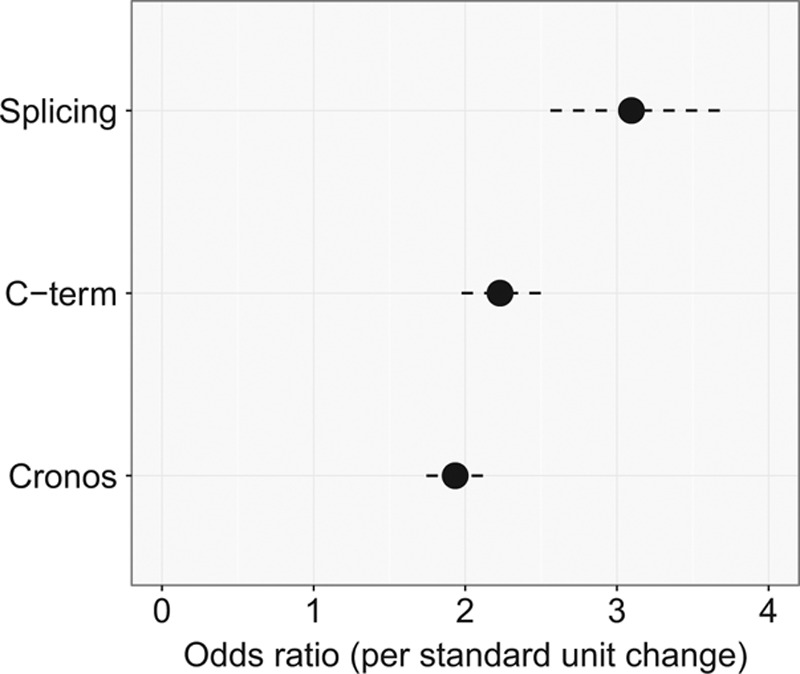
Comparison of 3 factors reveals their relative contribution to the observed distribution of TTN truncation variants. By plotting the increase in odds that a truncating variant is found in cases vs controls per 1 standard unit change for different predictors, one can perform an approximate relative comparison of variable importance.
Analysis of Model Performance
I next focused on assessing the performance of a model to evaluate how well the identified characteristics distinguish whether a truncation variant was found in cases versus controls. This analysis was again performed using only carriers of TTN truncations. To assess the performance of a model for assessing case versus control status based on input predictors, I used an area under the receiver operating characteristic curve (AUROC) analysis with the help of the ROCR package. Predictors in the logistic model were those identified above in univariate analysis: 4 PSI groupings (very low, low, medium, and high), whether the Cronos isoform is disrupted, and whether the mutation resides in the distal 1899 amino acids. I first divided data into training and test sets corresponding to 2 of 3 and 1 of 3 of the data.20 Coefficients for the final fitted model for variant classification were derived using the training data set and the AUROC computed on the test set. To deal with sampling variation in defining training and test data, this process was repeated 100× (including fitting a new model on the training data and evaluating it on the test data) and the AUROC averaged (see Figure 4 for representative plot and a distribution of AUROC values in the simulation).
Figure 4.
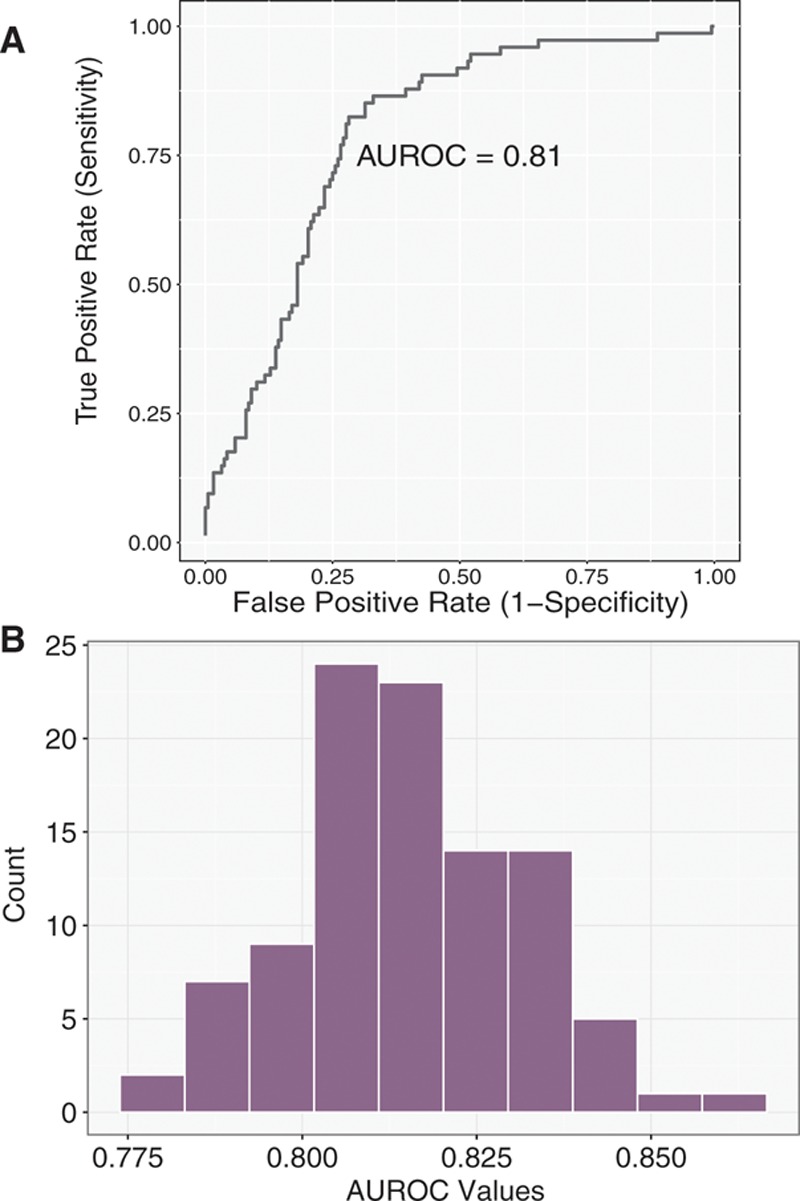
Receiver operating characteristic curve for ability to discriminate mutations as belonging to cases or controls using 3 predictors is shown in Figure 3. A model was derived from a training set corresponding to 2 of 3 of the data and then evaluated on a test set of the remaining data. A, Representative plot. B, Distribution of area under the receiver operating characteristic curve (AUROC) values in 100 simulations.
I explored the sensitivity of the model to different PSI groupings, PSI as a continuous variable, different threshold definitions of the distal C terminus, and treatment of the distal C terminus as a continuous variable, allowing risk to vary linearly with amino acid number past some threshold (ie, a knot in a piecewise regression). None of these approaches improved the AUROC, at least within the limits of the available data.
Truncation Variant Categories
Values of the discrete predictors (Cronos disruption, whether the distal C terminus is mutated, and very low/low/medium/high splicing classes) were used to classify previously reported TTN truncations into 6 groups. Although these 3 predictors would yield 16 possible groups, only 6 of these had >1 individual. For each of these, a tally of the number of variants observed in DCM cases and controls was performed, and a disease OR (and 95% confidence interval) estimated from a 2×2 contingency table assuming 1714 cases and 69 210 controls. The null hypothesis, evaluated with a 2-sided Fisher exact test, was that membership in a given bin did not affect the odds of having a diagnosis of DCM. The referent category for each comparison was the set of all individuals without a TTN truncation mutation in the exons defined by that bin. This included individuals with no TTN truncations and those with TTN truncations in other categories of exons.
Because I do not have access to individual data, I cannot exclude that there are (cryptically) related individuals within some of these cohorts, or whether the sequencing depth and variant calling across the protein, which was done at many different centers, was uniform. Such sources of error could affect the ORs. All data were plotted using the ggplot2 package.21
Results
After compiling TTN truncation variant data from patients with DCM and population controls, I fit a series of simple logistic regression model with the goal of explaining the distribution of truncating variants. Initial features included a quantitative estimate of exon inclusion in the heart (PSI), whether the Cronos protein product is disrupted, and mutation position (in 2000 amino acid bins). Focusing first on splicing, I found (controlling for whether Cronos is disrupted) a steady but nonlinear increase in the risk of mutations being found in cases rather than controls, with very low (PSI=0–0.399), low (0.4–0.649), medium (0.65–749), and high (0.75–1.00) risk bins (Figure 1A). Although a relationship between PSI and case/control status had previously been demonstrated,9 the much larger sample size looked at here allows more precise estimation of risk with PSI variation allowing this data-driven grouping of exons into discrete bins (note that the very low class has been arbitrarily chosen as the reference, and thus has an OR of 1). Next focusing on Cronos and restricting my analysis to constitutive exons (PSI>0.95) or controlling for PSI as a continuous variable, I found that mutations that further disrupt Cronos in addition to the full-length transcript were 3.2× more likely to be found in cases than in controls (P=3×10−8; Figure 1B). This result persisted even when focusing solely on the I-band region (OR=4.8; P=0.006; Figure II in the Data Supplement).
I next looked to see whether there were any additional factors, beyond these 2 that could explain case–control mutation distribution. Controlling for PSI, I found 2 shifts in risk profile one involving elevated risk at the position of Cronos (Figure 1C, dashed line) and another involving a drop in risk with mutations in the C-terminal 1992 amino acids (ie, starting at amino acid 34 000). Although the cause of this latter effect is unclear, it is most likely consistent with the observation that distal C-terminal homozygote truncation mutants still demonstrate Titin protein incorporation in the sarcomere, as has been observed both in a mouse model22 and in multiple human patients.4 Presumably, these mutations evade nonsense-mediated decay and allow production of a stable truncated protein. Examining the distribution of mutations in patients and controls reveals a relatively smooth increase in mutation frequency in these terminal 1992 amino acids because one moves toward the C terminus and a corresponding drop in mutation frequency in cases (Figure 2). This region would correspond approximately to the terminal 5 exons of the Titin ENST00000589042 transcript and a portion of the 6th (MEX-1 exon). If the boundary is moved a little more downstream at amino acid 34 094 (ie, terminal 1899 amino acids), we would completely spare the Titin kinase domain, which is preserved in the only described homozygote TTN truncation mutant patients4 and causes early lethality when deleted in mice.23,24 Although the sparsity of mutation data does not allow pinpointing the exact position of this C-terminal boundary, it seemed sensible to use this position, which both fit the data well and has a reasonable biological basis.
To assess the relative contribution of these 3 factors, I standardized them and assessed the additional risk as a function of a 1 SD increase in each predictor.25 Increased exon inclusion had the greatest risk of predicting case versus control status, with a 3.1-fold increase in risk per standard unit change (P=3×10−9), followed by the effect of not disrupting the C terminus (P=2×10−11), at 2.2-fold increased risk per standard unit and the effect of disrupting Cronos (P=2×10−10) at 1.9-fold (Figure 3). Collectively, a model incorporating all 3 of these was able to classify TTN mutations as belonging to cases versus controls with an AUROC of 0.81 (Figure 4A and 4B). I explored the sensitivity of the performance to changes in definition of the distal C terminus (eg, C-terminal 2500 amino acids, 2000 amino acids, and 1500 amino acids) and also allowed a continuous linear variation in risk but found no clear improvement in the model. I also modeled PSI as a continuous variable but again saw no improvement in AUROC.
Given that these predictors are discrete (eg, disrupt Cronos or not), I next classified patients into 6 groups (Table) according to mutually exclusive combinations of predictor values. The highest risk groups, which involved exons with high transcript inclusion, Cronos disruption, and no involvement of the distal C terminus, had the highest OR for disease, at 43-fold increased risk. Importantly, all 30 previously reported families with segregation of mutation with disease,1,3,5–7 and 31 of 32 previously described end-stage DCM cases9 mapped to this patient class (Figure IV in the Data Supplement). Additional variant classes showed elevated ORs of mapping to a case rather than a control (eg, high PSI, no Cronos involvement, OR=12) but had, to our knowledge, no previously published evidence of segregation or end-stage disease. Given their low ORs, it is unlikely that TTN truncation variants affecting predicted lower-risk exons (ie, nongroup I) will show convincing disease segregation in any kindreds, but I would anticipate that as more and more patients with DCM are sequenced, some variants in end-stage DCM will map to these exons, either by chance or perhaps in conjunction with other genetic or environmental risk factors.
Table.
Titin Truncation Variants Can Be Categorized Into Discrete Bins on the Basis of Alternative Splicing (Very Low, Low, Middle, and High Percent Spliced In Values), Whether the Cronos Isoform Is Disrupted, and Whether the Variant Falls Within the Distal C-Terminal Region (Last 1899 Amino Acids, Immediately Downstream of the Titin Kinase Domain)
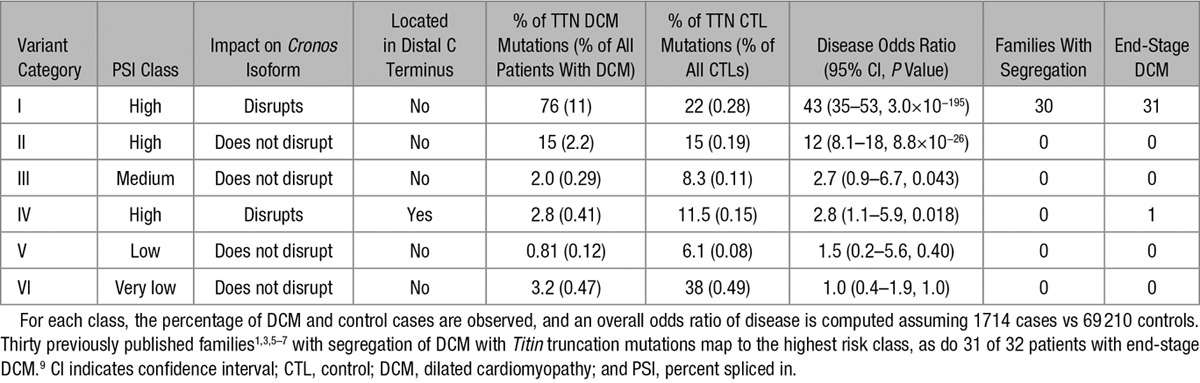
To assist in clinical variant interpretation, I have compiled a categorization of TTN exons for the 5 major isoforms (RefSeq and Ensembl identifiers) at http://cvri.ucsf.edu/~deo/TTNtruncationvariant.html.
Conclusions
I derive 2 main conclusions from this work. The first conclusion is the need for quantitative models for variant classification in complex situations, such as this one and the resulting importance of large human datasets for building these, thus allowing careful model calibration and sensitivity analyses. Patterns, such as the drop in disease risk with mutations in the distal C terminus, were not obvious from analyses of smaller sample sizes.5,9 Moreover, precise estimates of ORs for variant classes would not be possible without the tens of thousands of individuals considered here.
The second conclusion is the realization that even with this multifactorial model, there is a lack of determinism with predicting outcomes of even the highest risk class of Titin truncations. Although nearly 80% of DCM cases map to this region, so do 22% of controls, the most obvious explanation is that many of these controls may develop disease with age, a motivating factor for our previous work sequencing senior athletes.12 Nonetheless, there may be other modulating factors at play,7,26,27 and identification of these, if possible, will be needed to build even more accurate models.
This work differs from previous studies categorizing TTN truncating variants9,28 in its primary focus on building a quantitative classification model for clinical use. This required, in part, including knowledge of the position of the Cronos promoter in this analysis as well as the use of data-driven grouping of exons based on the extent of alternative splicing. Importantly, quantitative estimates were made for all splicing classes, rather than restricting to a subset of exons with high inclusion. Sensitivity analyses and exploration of alternative models were important to derive confidence in the final patient classification, and standardized predictors were used to measure relative importance of different inputs. This approach also differs in our prioritization of biological information (Cronos isoform and kinase domain) to guide interpretation of mutation patterns rather than electron micrograph–based divisions (eg, A-band) because the former would be expected to provide more robust models with lasting predicting value. Finally, this work is, to my knowledge, the first that emphasizes the decrease in risk seen with variants in the distal C terminus, which has an established biological basis22 and clear relevance for the prediction of pathogenicity. In terms of future improvements, model accuracy will further increase with larger sample size, knowledge of which individuals, if any, are related, and identification of any genotyping error. I acknowledge that such factors, especially variability in genotype calling sensitivity and specificity across studies, would affect the final ORs. However, these should not affect any of the primary conclusions of this article.
This work also highlights the challenges of how to counsel patients with mutations with more modest ORs of disease, such as those seen in groups II to IV, which have failed to show familial segregation or progress to end-stage disease in most cases. These ORs would be more in keeping with a multifactorial model for disease and are consistent with those of other familial disorders, such as some inherited NOD gene mutations in inflammatory bowel disease29 or a low-frequency variant in the MODY-3 (maturity onset-diabetes mellitus of the young) gene HNF1A with type 2 diabetes mellitus.30 Although such profiles of risk fall short of certainty, they are still stronger than many of the nongenetic risk factors routinely used in clinical decision-making (particularly, group II) and so warrant careful integration into clinical practice.
Sources of Funding
This work is funded by National Institutes of Health/National Heart, Lung, and Blood Institute grants DP2 HL123228.
Disclosures
None.
Supplementary Material
Footnotes
The Data Supplement is available at http://circgenetics.ahajournals.org/lookup/suppl/doi:10.1161/CIRCGENETICS.116.001513/-/DC1.
CLINICAL PERSPECTIVE
Variants that truncate (ie, prematurely terminate) the giant sarcomeric protein Titin are the leading genetic cause of dilated cardiomyopathy. However, they are also seen in control populations. As a result of advances in sequencing technologies, detailed studies have now amassed a wealth of information about truncating variant position in cases and controls. Nonetheless, considerable confusion exists as to how to interpret the pathogenicity of these variants, hindering our ability to make useful recommendations to patients. Part of the challenge arises from the historical approach taken to treat all truncating variants in a given gene as the same: that is, either disease causing or not. Such an approach may lack the subtlety needed to characterize their impact on a large multifunctional protein, such as Titin. In this article, I have used statistical modeling to identify and estimate the contribution of 3 primary factors that characterize those mutations found in cases compared with those found in controls. Such characteristics deal with whether the heart selectively removes the region of the protein where the variant is found, whether the variant also disrupts an alternative form of Titin, and whether the mutation falls within the distal end of the protein, where it is likely to have little consequence. Collectively, such characteristics accurately describe in which group a given mutation is likely to be found—though still fall short of certainty. We have combined these 3 factors into a classification system for TTN truncation variants and have included the results at http://cvri.ucsf.edu/~deo/TTNtruncationvariant.html.
References
- 1.Gerull B, Gramlich M, Atherton J, McNabb M, Trombitás K, Sasse-Klaassen S, et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–204. doi: 10.1038/ng815. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 2.Itoh-Satoh M, Hayashi T, Nishi H, Koga Y, Arimura T, Koyanagi T, et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun. 2002;291:385–393. doi: 10.1006/bbrc.2002.6448. doi: 10.1006/bbrc.2002.6448. [DOI] [PubMed] [Google Scholar]
- 3.Gerull B, Atherton J, Geupel A, Sasse-Klaassen S, Heuser A, Frenneaux M, et al. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med (Berl) 2006;84:478–483. doi: 10.1007/s00109-006-0060-6. doi: 10.1007/s00109-006-0060-6. [DOI] [PubMed] [Google Scholar]
- 4.Carmignac V, Salih MA, Quijano-Roy S, Marchand S, Al Rayess MM, Mukhtar MM, et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61:340–351. doi: 10.1002/ana.21089. doi: 10.1002/ana.21089. [DOI] [PubMed] [Google Scholar]
- 5.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, et al. National Heart, Lung, and Blood Institute GO Exome Sequencing Project and the Exome Sequencing Project Family Studies Project Team. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. doi: 10.1161/CIRCGENETICS.111.000062. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, et al. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur Heart J. 2014;35:2165–2173. doi: 10.1093/eurheartj/ehu050. doi: 10.1093/eurheartj/ehu050. [DOI] [PubMed] [Google Scholar]
- 8.Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, et al. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–1135a. doi: 10.1093/eurheartj/ehu301. doi: 10.1093/eurheartj/ehu301. [DOI] [PubMed] [Google Scholar]
- 9.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. doi: 10.1126/scitranslmed.3010134. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fürst DO, Osborn M, Nave R, Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol. 1988;106:1563–1572. doi: 10.1083/jcb.106.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akinrinade O, Koskenvuo JW, Alastalo TP. Prevalence of titin truncating variants in general population. PLoS One. 2015;10:e0145284. doi: 10.1371/journal.pone.0145284. doi: 10.1371/journal.pone.0145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou J, Tran D, Baalbaki M, Tang LF, Poon A, Pelonero A, et al. An internal promoter underlies the difference in disease severity between N- and C-terminal truncation mutations of Titin in zebrafish. Elife. 2015;4:e09406. doi: 10.7554/eLife.09406. doi: 10.7554/eLife.09406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shiga N, Takeshima Y, Sakamoto H, Inoue K, Yokota Y, Yokoyama M, et al. Disruption of the splicing enhancer sequence within exon 27 of the dystrophin gene by a nonsense mutation induces partial skipping of the exon and is responsible for Becker muscular dystrophy. J Clin Invest. 1997;100:2204–2210. doi: 10.1172/JCI119757. doi: 10.1172/JCI119757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res. 2004;94:967–975. doi: 10.1161/01.RES.0000124301.48193.E1. doi: 10.1161/01.RES.0000124301.48193.E1. [DOI] [PubMed] [Google Scholar]
- 15.Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, et al. Heart disease. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349:982–986. doi: 10.1126/science.aaa5458. doi: 10.1126/science.aaa5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36:2327–2337. doi: 10.1093/eurheartj/ehv253. doi: 10.1093/eurheartj/ehv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pervouchine DD, Knowles DG, Guigó R. Intron-centric estimation of alternative splicing from RNA-seq data. Bioinformatics. 2013;29:273–274. doi: 10.1093/bioinformatics/bts678. doi: 10.1093/bioinformatics/bts678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deo RC. Machine learning in medicine. Circulation. 2015;132:1920–1930. doi: 10.1161/CIRCULATIONAHA.115.001593. doi: 10.1161/CIRCULATIONAHA.115.001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wickham H. ggplot2. New York, NY: Springer Science & Business Media; 2009. [Google Scholar]
- 22.Weinert S, Bergmann N, Luo X, Erdmann B, Gotthardt M. M line-deficient titin causes cardiac lethality through impaired maturation of the sarcomere. J Cell Biol. 2006;173:559–570. doi: 10.1083/jcb.200601014. doi: 10.1083/jcb.200601014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gotthardt M, Hammer RE, Hübner N, Monti J, Witt CC, McNabb M, et al. Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem. 2003;278:6059–6065. doi: 10.1074/jbc.M211723200. doi: 10.1074/jbc.M211723200. [DOI] [PubMed] [Google Scholar]
- 24.Peng J, Raddatz K, Labeit S, Granzier H, Gotthardt M. Muscle atrophy in titin M-line deficient mice. J Muscle Res Cell Motil. 2005;26:381–388. doi: 10.1007/s10974-005-9020-y. doi: 10.1007/s10974-005-9020-y. [DOI] [PubMed] [Google Scholar]
- 25.Gelman A, Hill J. Data Analysis Using Regression and Multilevel/Hierarchical Models. New York, NY: Cambridge University Press; 2006. [Google Scholar]
- 26.Hazebroek MR, Moors S, Dennert R, van den Wijngaard A, Krapels I, Hoos M, et al. Prognostic relevance of gene-environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol. 2015;66:1313–1323. doi: 10.1016/j.jacc.2015.07.023. doi: 10.1016/j.jacc.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 27.Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, et al. IMAC-2 and IPAC Investigators. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374:233–241. doi: 10.1056/NEJMoa1505517. doi: 10.1056/NEJMoa1505517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90:49–54. doi: 10.1111/cge.12741. doi: 10.1111/cge.12741. [DOI] [PubMed] [Google Scholar]
- 29.Karban A, Waterman M, Panhuysen CI, Pollak RD, Nesher S, Datta L, et al. NOD2/CARD15 genotype and phenotype differences between Ashkenazi and Sephardic Jews with Crohn’s disease. Am J Gastroenterol. 2004;99:1134–1140. doi: 10.1111/j.1572-0241.2004.04156.x. doi: 10.1111/j.1572-0241.2004.04156.x. [DOI] [PubMed] [Google Scholar]
- 30.Estrada K, Aukrust I, Bjørkhaug L, Burtt NP, Mercader JM, García-Ortiz H, et al. SIGMA Type 2 Diabetes Consortium. Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA. 2014;311:2305–2314. doi: 10.1001/jama.2014.6511. doi: 10.1001/jama.2014.6511. [DOI] [PMC free article] [PubMed] [Google Scholar]