

Abstract
This study aimed to determine protein expression levels of fibroblast growth factor receptors (FGFR) 1, 2 and 3 in early stage non‐small cell lung cancer (NSCLC). Additionally, a screen to define the frequency of FGFR3‐TACC3 translocation and FGFR3 amplification was performed. Archived tissues from 653 NSCLC samples (adenocarcinoma (AC), squamous cell carcinoma (SCC) and large cell carcinoma (LCC)) were analysed with immunohistochemistry (IHC) for expression of FGFR1, 2 and 3. Expression levels of FGFR1, 2 and 3 were correlated with clinicopathological features. The presence of FGFR3‐TACC3 translocation was detected by RT‐PCR and FGFR3 amplification was detected by fluorescence in situ hybridization. FGFR1, 2 and 3 proteins were highly expressed in 64 (10.6%), 76 (12.9%) and 20 (3.3%) NSCLC tumour samples, respectively. Protein expression of FGFR1 was significantly related to worse overall survival in NSCLC. Furthermore, FGFR1 protein expression was associated with light smoking and histological subtype (AC), FGFR2 protein expression with female gender, younger age, histological subtype (AC) and lower tumour stage, and FGFR3 protein was significantly overexpressed in tumours of older patients and SCC histology. The FGFR3‐TACC3 fusion was detected in 3.0% (6/200) of NSCLC samples and the FGFR3 gene was amplified in 4.7% of IHC positive NSCLC samples (2/43). FGFR1, 2 and 3 proteins are expressed in a high number of early stage NSCLC and FGFR1 protein expression may serve as a prognostic biomarker. Recurrent translocations and amplifications in FGFR3 can be found in NSCLC. This study shows that FGFR family members are frequently aberrant in NSCLC and could be interesting therapeutic targets for the treatment of NSCLC.
Keywords: non‐small cell lung cancer (NSCLC), fibroblast growth factor receptor (FGFR), immunohistochemistry (IHC), FGFR3‐TACC3 translocation, FGFR3 amplification
Introduction
Lung cancer is the leading cause of cancer‐related deaths in the western world. Non‐small cell lung cancer (NSCLC) accounts for over 75% of all lung cancer cases and consists of adenocarcinoma (AC), squamous cell carcinoma (SCC) and large cell carcinoma (LCC). Approximately half of the patients presenting with NSCLC have curable disease, but around 30% of them will eventually develop an incurable metastatic stage 1. Insights into genomic aberrations in cancer cells and the development of targeted therapies have led to significant improvements in treatment and survival. For patients with advanced NSCLC with tumours harbouring an EGFR mutation or ALK rearrangement, a tyrosine kinase inhibitor has replaced chemotherapy as the first choice of treatment ie, erlotinib/gefitinib/afatinib and crizotinib respectively 2, 3. However, particularly in the western population the incidence of these aberrations is rare and they occur mainly in the AC histological subtype, so only a minority of patients have benefited so far. Thus, there is still a great need for new targets to augment treatment options and improve prognosis for advanced NSCLC, especially in squamous cell lung carcinoma.
Recently, a novel potential therapeutic target has been identified in cancer: the fibroblast growth factor receptor (FGFR) family. The FGFR family members play a physiological role in stimulating cell proliferation and migration as well as in promoting survival of various types of cells 4. In the FGFR pathway, 18 ligands (fibroblast growth factors, FGFs) can bind to four homologous receptor tyrosine kinases FGFR1‐4. Binding of FGF ligand activates FGFR1‐4, which subsequently leads to a highly complex signalling response through multiple pathways – the MAPK signalling cascade and the PI3K/AKT pathway, among others 5, 6. In cancer, different FGFR aberrations have been identified that constitutively activate these downstream pathways and contribute to tumour development. These include receptor overexpression through gene amplification or post‐transcriptional regulation, FGFR mutations, FGFR translocations, alternative splicing of FGFR, isoform switching and paracrine/autocrine activation through overexpression of FGFs in cancer or stromal cells 7. Apart from NSCLC, aberrant FGFR signalling has been observed in many cancers, such as breast, ovarian, bladder, gastric, prostate, endometrial, head and neck, colorectal cancer and glioblastoma 7, 8, 9. In tumours with FGFR1‐3 aberrations, inhibition with several small molecules known to target the FGF/FGFR‐pathway has shown anti‐tumour activity in vitro 10, 11, 12, 13, 14, 15, 16. Clinical trials with FGFR inhibitors in advanced NSCLC are ongoing, whether combined with chemotherapy or not, and partial responses have been observed 17, 18.
Recently we developed a high‐throughput platform for systematically profiling kinase fusions, which enabled the identification of a recurrent FGFR3‐TACC3 translocation in a cohort of 95 early stage NSCLC samples. Both samples carrying this fusion were of squamous histology and showed FGFR3 protein overexpression. We also identified two SCC samples that carried an activating mutation in FGFR3 19. The mutation causes a serine‐to‐cysteine substitution at position 249 and is the most common FGFR3 activating mutation identified in bladder cancer. FGFR3‐TACC3 translocations and FGFR3 mutations may become an interesting target for therapy in SCC, but many aspects regarding the FGFR‐signalling pathway in NSCLC remain unknown. Insights into the frequency of protein (over)expression of FGFR1, 2 and 3 may assist in a better understanding of the role of this pathway in NSCLC and help to identify patients who may benefit from targeted therapy.
Here, we assessed a large cohort of 653 early stage NSCLC samples for FGFR1, 2 and 3 protein expression using immunohistochemistry (IHC). In light of our recent finding of a recurrent FGFR3‐TACC3 translocation in the squamous subgroup of NSCLC and because of the lack of targeted therapy options for SCC of the lung, we subsequently screened a subset of FGFR3 IHC positive samples for additional FGFR3 molecular aberrations.
Material and methods
Sample collection and patient cohort
Inclusion criteria for this cohort were patients who had undergone a lung resection between 1990 and 2013 at one of four Dutch medical centres. Exclusion criteria were non‐NSCLC histology eg, SCLC, metastatic non‐NSCLC, a synchronous primary tumour, unavailability of tumour tissue and unavailability of patient follow‐up data. The cohort included 768 samples with adequate patient and tumour characteristics. For all these patients, formalin‐fixed, paraffin‐embedded (FFPE) tumour samples were collected. After a second pathology review where samples without sufficient viable tumour material or histological subtype other than AC, SCC and LCC were excluded, 635 samples were eligible for further analysis. The cohort consisted of 329 AC, 278 SCC and 28 LCC samples. Median follow‐up time was 96.0 months (95% CI: 86–103). All tumours were classified according to the 2004 WHO classification system. TNM classification was redefined for resections that were done before 2010 according to the 7th lung cancer TNM classification and staging system. Light smokers were defined as having fewer than 10 pack years (PY) including never smokers. The Translational Research Board of the Netherlands Cancer Institute‐Antoni van Leeuwenhoek hospital (NKI‐AVL) approved the use of patient material in this study.
All FFPE tissues were constructed into tissue microarrays (TMAs) using a TMA Grand Master instrument (3D HISTECH, Budapest, Hungary). From each FFPE tissue block, three cores (0.6 mm) were punched from tumour areas that had been marked by a pathologist on HE slides, and arrayed into a recipient TMA donor block.
A subset of 200 samples for which RNA was available was screened for the FGFR3‐TACC3 translocation. Of these 200 samples, 44 were kinome capture sequenced and analysed for fusion detection together with 15 matched Fresh Frozen (FF) samples previously reported by Majewski and colleagues 19. The FGFR3 IHC positive samples of this 200‐sample‐subset were selected for FGFR3‐amplification screening by fluorescence in situ hybridization (FISH). In addition, we screened for the S249C FGFR3 activating mutation previously found by Majewski and colleagues in the SCC subgroup.
Immunohistochemical staining for FGFR1, 2 and 3
Immunohistochemical staining was performed using the Ventana Benchmark Ultra stainer (Ventana Medical Systems, Tucson, AZ). In short, for FGFR1 and FGFR2 IHC staining 3 µm TMA slides were deparaffinized using EZ prep buffer (Ventana Medical Systems) and pretreated for 24 min at 95°C using CC1 Buffer (Ventana Medical Systems). 100 µl antibody was incubated for 32 min at 36°C. (FGFR1: ab10646, Abcam, Cambridge, UK, dilution 1:2000) (FGFR2: ab10648, Abcam, Cambridge, UK, dilution 1:2000). For FGFR3 IHC staining 3 µm TMA slides were deparaffinized using EZ prep buffer (Ventana Medical Systems) and pretreated for 64 min at 95°C using CC1 Buffer (Ventana Medical Systems). 100 µl antibody was incubated for 1 h at 36°C. (FGFR3 Clone B‐9, sc‐13121, Santa Cruz Biotechnology, Dallas, TX, dilution 1/50). Signal was amplified using the Amplification Kit (Ventana Medical Systems, 760‐080). Detection for all three FGFRs was performed with the UltraView universal DAB kit (Ventana Medical Systems, 760‐500). Slides were then counterstained with Haematoxylin II for 8 min, and Bluing Reagent for 4 min (Ventana Medical systems), after which slides were dehydrated and cover slipped. Slides were scored by two observers (S.W. and W.T.). Protein expression of FGFR1 and FGFR2 was quantified as a percentage (range 0–100%) of positive cells present among all tumour cells present in the TMA cores. Stain intensity was not scored because it was homogenous among all tumours. A mean score was calculated over one to three available measurements for each patient. Previous studies with FGFR IHC all used different arbitrary cutoffs 13, 20, 21, 22, 23, 24, so we used FGFR1 and 2 as continuous variables. When assessing the prognostic value of FGFR expression for survival, FGFR1 and 2 were used both as a continuous and ordinal four‐level variable, where <10% was considered negative (N), 10–50% low (L) expression, 50–80% median (M) and 80% and above was considered high (H). Expression of FGFR3 was semi‐quantitatively scored based on the intensity: 0 (negative, N), 1+ (low, L), 2+ (high, H). The percentage of cells was not scored because it was homogenous among all tumours, in all of them it was 100%. The highest score among all three measurements of the same tumour was used for further analysis. FGFR1, FGFR2 and FGFR3 IHC scores were unavailable for 34 (5.4%), 45 (7.1%) and 23 (3.6%) of the samples, respectively. This was due to the lack of viable tumour in any of the three cores or the loss of all three cores on the TMA after staining. Samples were left out of the analysis only when all three FGFRs scores were unavailable. To verify the extent to which tumour heterogeneity would influence the FGFR IHC scoring we also scored some whole slides (n = 22), but in none of these cases did this lead to a different score (ordinal four‐level variable for FGFR1 and 2 or intensity score for FGFR3).
Fusion detection and validation
Patient material was available from FFPE tissues. Sequencing libraries were constructed with a TruSeq mRNA library preparation kit using Ribo‐ZeroTM (Epicentre). Capture enrichment was performed with the human kinome DNA capture baits (Agilent Technologies, Santa Clara, CA). Captured libraries were sequenced on an Illumina HiSeq2000 platform with a paired‐end 51 base protocol. Sequences were aligned to the human genome (Hg19) with TopHat 25. Two pipelines were used to identify and rank candidate fusion genes: TopHat‐fusion 26 and de novo transcript assembly with Trinity 27. We selected candidate fusion transcripts with at least one spanning read and one spanning pair. Detailed selection criteria were previously described 19. We excluded fusions also identified in an unrelated normal sample.
Each fusion transcript was assessed to determine if the transcript would produce an in‐frame fusion. The Maxima First Strand cDNA Synthesis Kit was used to produce input cDNA for RT‐PCR (Thermo Scientific, Newigton, NH). PCR primers were designed to amplify across the fusion breakpoints and products were confirmed by capillary sequencing using the Big Dye Terminator V3.1 Sequencing Kit (Applied Biosystems, Foster City, CA). A complete list of the validated fusion transcripts and the primers used for the validation is reported in supplementary material, Table S1. The FGFR3‐TACC3 fusion transcript was screened with two different primer pairs previously reported 19, 28.
Mutation screening
The activating mutation in FGFR3 was verified by PCR amplification from cDNA (F: 5′‐CATTGGAGGCATAAGCTG; R: 5′‐AGCACGGTAACGTAGGGTGT) and capillary sequencing using the Big Dye Terminator V3.1 Sequencing Kit (Applied Biosystems, Foster City, CA).
Fluorescence in situ hybridization
Fresh cut 4 µm sections of formalin‐fixed paraffin embedded tissue were submitted to dual‐colour FISH analysis using an IGH/FGFR3 translocation dual fusion FISH probe developed at Cytocell (Cambridge, UK). One probe set was designed in the short arm of chromosome 4 (red signal), where lies the FGFR3 gene and the other one in the long arm of chromosome 14 (green signal), where lies the IGH gene. FISH was performed according the standard method with some minor modifications. The slides were counterstained in phosphate‐buffered saline containing 4′,6 diamidino‐2 phenylindole dihydrochloride (DAPI) and mounted with Vectashield. Since there are no standard criteria to evaluate FGFR3 FISH assays in NSCLC yet, we decided to follow the strategy suggested by Schildhaus HU et al for scoring FGFR1 amplifications. We counted 20 tumour cells from three areas, resulting in a total of 60 nuclei. We called high level amplification if the FGFR3/IGH ratio was equal to or above 2 or the average gene copy per nucleus was equal to or above 6. When a sample showed a FGFR3/IGH ratio of 1.5, so not sufficient for a high‐level amplification status but different from an unamplified status, the sample was defined as having a low level amplification or FGFR3 gain, and below 1.5 was defined as a normal FGFR3 copy‐number 29. We performed the FISH on a whole slide (in addition to the TMA) if samples were of low quality on the TMA or they were scored as positive for FGFR3 amplification on the TMA.
Statistical analysis
All analyses were carried out using IBM SPSS statistics 22 and R version 3.2. The data were summarized using standard descriptive statistics and frequency tabulations. FGFR1 and 2 expression were treated as continuous variables whereas FGFR3 was treated as an ordinal variable with three levels: negative, low, high.
Univariate associations between the FGFR protein expression and the patients’ clinical demographic variables age, gender, smoking history, histological type and pathological stage were assessed using a linear by linear association test and Spearman rank correlation for ordinal variables, Fisher's exact test for categorical variables and Wilcoxon‐Mann‐Whitney or Kruskal‐Wallis test for continuous variables. Multivariate associations between FGFR protein expression and clinical variables were assessed using linear regression for FGFR1 and 2 and logistic regression for FGFR3 after dichotomizing the latter into negative versus positive (low and high).
For survival analysis, overall survival (OS) was estimated from time of surgery until death or end of follow‐up. Survival curves were estimated using the Kaplan‐Meier method and compared between patients groups by four‐level (FGFR1 and 2) or three‐level (FGFR3) IHC scores by Logrank tests. The association between OS and FGFR expression was further analysed with univariable and multivariable Cox proportional hazard models. To correct for confounding variables, expression of all three FGFRs and patient characteristics were included in the multivariable model as cofounders and the model was stratified for tumour stage. Median follow‐up time was estimated by a reversed Kaplan‐Meier method. Also, in the multivariate models FGFR3 was entered as a two level factor (negative versus positive). Two‐sided p‐values below 0.05 were considered significant throughout all statistical computations.
Results
FGFR1, FGFR2 and FGFR3 protein are highly expressed in a subset of NSCLC
Patient characteristics and adequate tumour material were available for 635 samples. The median age at surgery was 64 years and 84.1% were early stage (≤stage II). Of the 48 light smokers 33 were never smokers. Median OS was 148 months (95% CI: >103 months). Table 1 summarizes the clinicopathological characteristics of our patient cohort.
Table 1.
Patient and tumour characteristics of the non‐small cell lung cancer cohort. PY = pack years
| Frequencies (n = 635) | |
|---|---|
| Gender | |
| Male | 363 (57.2%) |
| Female | 272 (42.8%) |
| Median age at surgery (years, range) | 64 (30–84) |
| Neo‐adjuvant therapy | |
| Chemotherapy | 25 (3.9%) |
| Concurrent chemo radiotherapy | 14 (2.2%) |
| Sequential chemo radiotherapy | 3 (0.5%) |
| Erlotinib [52] | 31 (4.9%) |
| Radiotherapy | 3 (0.5%) |
| No adjuvant therapy | 532 (84.8%) |
| Smoking | |
| Light smokers <10 PY | 48 (7.6%) |
| Heavy smokers ≥10 PY | 497 (78.3%) |
| Unknown | 90 (14.2%) |
| Packyears (median, range) | 38 (0–150) |
| Histology | |
| Adenocarcinoma | 329 (51.8%) |
| Squamous cell carcinoma | 278 (43.8%) |
| Large cell carcinoma | 28 (4.4%) |
| Tumour stage at resection | |
| Tis | 1 (0.2%) |
| Stage I | 352 (55.4%) |
| Stage II | 181 (28.5%) |
| Stage III | 82 (12.9%) |
| Stage IV | 17 (2.7%) |
| Unknown | 2 (0.3%) |
| Median overall survival (months, range) | 49.0 (0–289) |
High protein expression of FGFR1, FGFR2 and FGFR3 was found in 64 (10.6%), 76 (12.9%) and 20 (3.3%) of NSCLC tumour samples, respectively (Table 2, Figure 1). Of all samples, 21.3% showed high‐level expression of at least one of the three FGFRs. No samples showed high expression of all three FGFRs. There was a positive correlation between FGFR1 and FGFR2 expression (R = 0.20 (95% CI: 0.13–0.28)). No association was seen between expression of FGFR1 or 2 with expression of FGFR3. Nor could mutual exclusivity be determined, because there were samples showing overlap of high FGFR1 or 2 expression and high FGFR3 expression.
Table 2.
FGFR1, 2 and 3 immunohistochemical expression in non‐small cell lung cancer
| FGFR1a | FGFR2a | FGFR3b | |
|---|---|---|---|
| N | 601 | 590 | 612 |
| Negative | 117 (19.5%) | 269 (45.6%) | 505 (82.5%) |
| Low | 267 (44.4%) | 166 (28.1%) | 87 (14.2%) |
| Medium | 153 (25.5%) | 79 (13.4%) | NA |
| High | 64 (10.6%) | 76 (12.9%) | 20 (3.3%) |
Negative <10%, low 10 < 50%, median 50 < 80%, high ≥80%.
Negative 0, low 1+, high 2+.
Figure 1.
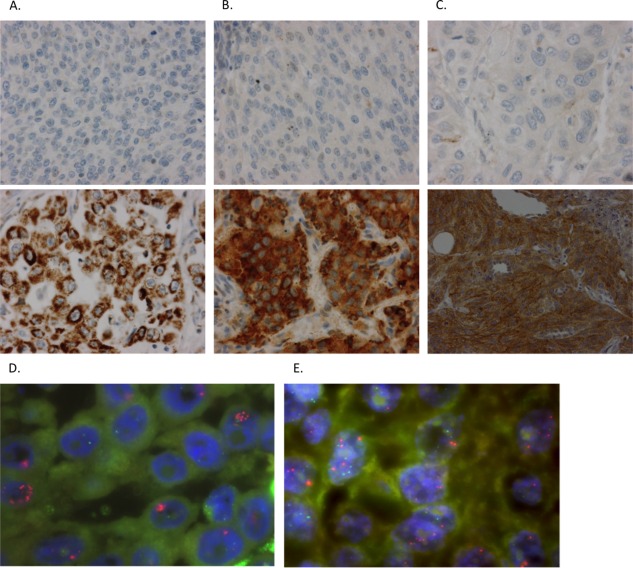
FGFR1, 2 and 3 protein expression by immunohistochemistry and FGFR3 copy numbers by FISH in non‐small cell lung cancer. (A, B, C) Representative immunohistochemical staining of FGFR1, 2 and 3 negative samples versus FGFR1, 2 and 3 positive (high expression) samples, respectively. FGFR1: ab10646, abcam; FGFR2: ab10648, abcam; FGFR3: Clone B‐9, sc‐13121, Santa Cruz Biotechnology. (D, E) FGFR3 amplification and FGFR3 gain, respectively, assessed by dual‐colour FISH analysis using an IGH/FGFR3 translocation dual fusion FISH probe (Cytocell).
FGFR expression differs significantly between histological subtypes
We correlated gender, age, smoking status, histology and tumour stage with FGFR expression scores.
Interestingly, FGFR expression differed significantly between histological subtypes: FGFR1 and FGFR2 were significantly associated with AC histological subtype (p < 10−6 and p < 10−15 respectively), whereas FGFR3 was significantly associated with SCC (p < 10−12) (Figure 2, supplementary material, Table S2).
Figure 2.
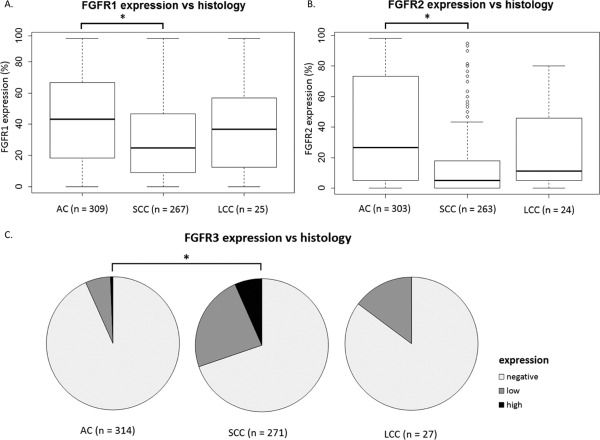
FGFR protein expression differs significantly between histological subtypes in non‐small cell lung cancer. (A, B) The asterisks (*) represents statistical significance. FGFR1 and 2 protein expression used as a continuous variable in a Wilcoxon‐Mann‐Whitney test. (C) The asterisks (*) represents statistical significance. FGFR3 protein expression used as a categorical variable in a Fisher's exact test. AC, adenocarcinoma; SCC, squamous cell carcinoma; LCC, large cell carcinoma.
There was a significant difference in FGFR2 expression between genders, with higher FGFR2 expression in females (mean 34.1% versus 23.2%, p‐value <0.0001). FGFR2 correlated negatively with age for the whole cohort (R = −0.16 (95% CI: −0.24 to −0.8)) and for the female patients only (R = −0.16 (95% CI −0.28 to −0.04)). There was a negative correlation between tumour stage and FGFR2 expression (rho = −0.12, p = 0.002). FGFR3 correlated positively with age (rho = 0.16, p < 0.001). FGFR1 expression was associated with light smoking (p = 0.02) and FGFR3 expression was associated with heavy smoking (higher PY, p < 0.0001). All univariate associations are summarized in supplementary material, Table S3.
In the multivariate models, the associations of FGFR expression with their distinct histological subtype remained statistically significant. FGFR1 was still associated with FGFR2 expression. FGFR2 remained associated with gender and FGFR3 with a rising number of pack years (Table 3).
Table 3.
Multi‐variate analysis: linear regression analysis for FGFR1 and 2 protein expression and logistic regression for FGFR3 protein expression with clinicopathological variables in non‐small cell lung cancer
| Variable | FGFR1a | FGFR2a | FGFR3b |
|---|---|---|---|
| FGFR1a | NA | pos, p = 0.0005 | pos p = 0.24 |
| FGFR2a | pos, p = 0.0005 | NA | neg p = 0.83 |
| FGFR3b | pos, p = 0.21 | neg, p = 0.61 | NA |
| Genderc | pos, p = 0.46 | pos, p = 0.01 | neg, p = 0.69 |
| Age at diagnosisd | pos, p = 0.50 | neg, p = 0.26 | pos, p = 0.17 |
| Smoking statuse | neg, p = 0.17 | pos, p = 0.58 | pos, p = 0.008 |
| SCC versus AC | neg, p = 0.002 | neg, p < 10−7 | pos, p < 10−6 |
| LCC versus AC | neg, p = 0.47 | neg, p = 0.10 | pos, p = 0.15 |
| Tumour stage | pos, p = 0.31 | neg, p = 0.30 | neg, p = 0.98 |
Direction of association and adjusted p‐values are given.
Per 10%‐point increase.
Dichotomized into negative (0) and positive (1+/2+).
Female versus male.
Per year.
FGFR1 and 2 as heavy versus light smoking; FGFR3 as PY.
SCC, squamous cell carcinoma; AC, adenocarcinoma; LCC, large cell carcinoma.
FGFR1 protein expression is related to poor overall survival in NSCLC
Higher expression of FGFR1 was associated with worse OS: HR 1.06 per 10%‐point increase in FGFR1 level (95% CI 1.02–1.11, p = 0.01) (Figure 3). When including gender, age, smoking status, histology as cofounders into the cox model and stratifying for tumour stage, the prognostic effect of FGFR1 remains statistically significant (p = 0.006) (supplementary material, Table S4). Expression of FGFR2 (p = 0.51) and FGFR3 (p = 0.13) were not associated with a difference in OS (Figure 3).
Figure 3.
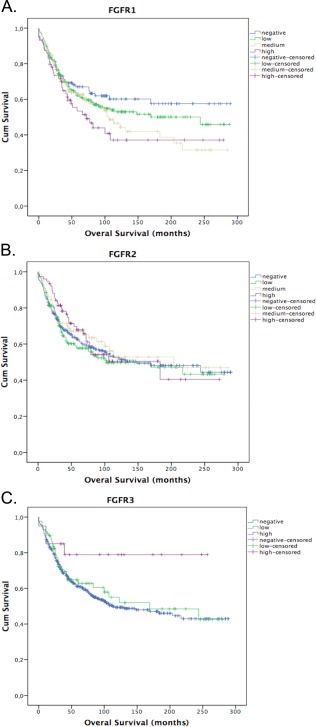
FGFR1 protein expression is related to poor overall in non‐small cell lung cancer. Survival curves by Kaplan‐Meier method. (A, B) FGFR1 and 2 compared as an ordinal four‐level variable by Logrank test. (C) FGFR3 compared as an ordinal three‐level variable by Logrank test. FGFR3 high protein expression is a rare event (n = 20), which might have influenced the statistical significance of this test.
FGFR3‐TACC3 translocations and FGFR3 gene amplification occur in a small subset of NSCLC
RNA was available for 200 out of 635 samples in the cohort at the time of the study (111 (55.5%) AC, 76 (38.0%) SCC and 13 (6.5%) LCC). These samples were screened by RT‐PCR for the presence of the FGFR3‐TACC3 fusion. We detected six samples (3.0%) in which FGFR3 was fused with TACC3. FGFR3‐TACC3 fusions occurred in 4/76 (5.3%) SCC and in 2/111 (1.8%) AC. Further details of IHC, histology and expression of FGFR1 and FGFR2 are summarized in Table 4 for these six samples. All patients had over 25 pack‐years, three were male, four had stage I and two had stage II disease and all six patients were still alive at the end of follow‐up.
Table 4.
Histological features of the FGFR3‐TACC3 translocated and FGFR3 amplified non‐small cell lung cancer samples
| Sample | FGFR3‐TACC3 positive 3.0% (6/200) | FGFR3 amplification 4.7% (2/43) | FGFR3 IHC | Histology | FGFR1 IHC | FGFR2 IHC |
|---|---|---|---|---|---|---|
| 1 | yes | no | High | SCC | Low | Medium |
| 2 | yes | no | High | SCC | Low | Negative |
| 3 | yes | no | Low | SCC | Low | Negative |
| 4 | yes | no | Low | AC | Low | Medium |
| 5 | yes | no | Negative | SCC | Low | Negative |
| 6 | yes | no | Negative | AC | Medium | Medium |
| 7 | no | yes | High | SCC | High | Negative |
| 8 | no | yes | Low | SCC | Low | Low |
Sample #2 and #6 showed a FGFR3 gain by FISH.
In order to understand whether high FGFR3 levels by IHC could be explained by FGFR3 amplification we screened 43 samples (36 SCCs, 6 ACs, 1 LCC) with FGFR3 IHC1+ or 2+ using FISH. FGFR3 was amplified in two samples (4.7%) (FGFR3/IGH ratio ≥2) (Figure 1D) and two samples showed an intermediate pattern that we could define as low level amplification or gain (Figure 1E). Interestingly, both samples with the FGFR3 gain were positive for the FGFR3‐TACC3 translocation by RT‐PCR. Also, we screened the 141 FFPE samples of the 200 samples for which RNA was available that were not previously screened for the S249C FGFR3 activating mutation in the Majewski paper, but we did not find any new cases.
Discussion
This study shows that high protein expression of FGFR1, 2 and 3 occurs frequently in NSCLC and that recurrent FGFR3‐TACC3 fusions and FGFR3 amplifications are found. Also, FGFR1 protein expression may serve as a prognostic biomarker. These results suggest that the FGFR pathways play an important role in growth and malignant progression of NSCLC.
Histological subtype seems to be an important determinant of which FGFR protein is expressed. FGFR1 expression is significantly associated with AC, as was previously reported by Seo et al 30. The same association was seen for FGFR2 protein expression, similarly to Behrens et al 22. In contrast, FGFR3 protein expression was predominantly seen in SCC samples. To our knowledge, no data have been published concerning FGFR3 IHC and histology. Higher FGFR1 protein expression was associated with worse OS, which is in concordance with other studies highlighting the possible prognostic effect of FGFR1 protein expression 22, 31. We did not detect an association of FGFR2 and FGFR3 with OS.
It is an interesting finding that FGFR2 is associated with female gender in our cohort. We did not have data about the menopausal state of the female patients, but age and FGFR2 expression correlated negatively in the female cohort alone. Also, FGFR2 expression in the youngest quartile of women (age 31–53 year) was significantly higher compared to the upper three quartiles (44.5% versus 30.6%). These findings suggest an effect of hormonal status on FGFR2 expression or a synergy between oestrogen receptor and FGFR signalling in lung carcinogenesis. In NSCLC, there is indeed evidence of cross‐talk between oestrogen and other growth factor pathways 32. FGFR2 mutations have been found in approximately 10% of endometrial cancers 33 and FGFR2‐amplification in 4% of triple‐negative breast cancer 34. Remarkably, there was no statistically significant association between FGFR1 expression and gender, although FGFR2 expression was significantly associated with both. However, the association between FGFR1 expression and gender does exist when comparing dichotomized (above versus below median) FGFR1 scores between genders instead of using FGFR1 as a continuous variable, but the latter method was deemed more appropriate for there is no consensus about cut‐offs and this way less information is lost. Also, the proportion of FGFR2 positive FGFR1 negative samples in females was slightly higher than in males (cut‐off by median score) and the association between FGFR1 and gender is much weaker and indeed disappears when compensating for FGFR2 in a bivariate model.
The strong association of histological subtype and FGFR protein expression suggests that FGFR3 positive tumours are a distinct subgroup within NSCLC. Because of the lack of targeted therapy options in the squamous sub‐group of NSCLC and because of our recent finding regarding FGFR3 molecular aberrations 19, we screened a subset of our cohort for additional FGFR3 molecular aberrations depending on DNA/RNA availability. We detected a frequency of 5.3% of FGFR3‐TACC3 fusions in our SCC sample group, which is higher than previously reported (1.8–3.0%) 16, 35. However, it has to be taken into account that this subset was enriched for FGFR3 IHC positive samples. We also found two AC samples carrying the FGFR3‐TACC3 fusion (1.8% in the AC group only). The translocation has been described in AC previously 35, 36. Only four out of six samples carrying the FGFR3‐TACC3 fusion showed positive FGFR3 IHC staining. This discrepancy may show that the FGFR3‐TACC3 fusion does not always lead to protein (over)expression. The expression levels of the FGFR3‐TACC3 transcript by RT‐qPCR confirmed that samples with negative FGFR3 IHC staining but carrying the FGFR3‐TACC3 fusion (Table 4, samples #5 and #6) showed very low FGFR3‐TACC3 mRNA expression (data not shown). In addition, diagnostic and technical issues regarding IHC staining must also be taken into consideration: tumour heterogeneity, unclear standards on FGFR IHC scoring, and the quality of the FGFR3 antibody 13, 20, 21, 22, 23, 24, 35. This will need further investigation.
Because FGFR3 protein overexpression may also be explained by increased FGFR3 copy number (GCN, Gain Copy Number), we assessed the amplification status of FGFR3 using FISH. We found two samples with FGFR3 amplification (4.7%). FGFR3 amplifications are rare events in bladder cancer (3.4% of cases) 37, but no studies have so far investigated the amplification status of FGFR3 in NSCLC specifically. The Cancer Genome Atlas (TCGA) reported a lower frequency of FGFR3 amplification (1% in SCC and AC populations together), but our selection of FGFR3 IHC+ samples for the FISH screening could explain this difference. Furthermore, our FISH assay was specifically designed to detect FGFR3 amplification, which makes it a more sensitive assay than the CGH technology used by the TCGA. This finding shows that FGFR3 amplification is a rare event in our sample set, similar to bladder cancer. We found two different patterns of high level amplification: a cluster‐pattern which consists of a tight accumulation of more than six FGFR3 signals in the nuclei (IHC 2+, Figure 1D); and FGFR3 signals spread in the nuclei. The low level amplification (gain) found in two of the samples positive for the FGFR3‐TACC3 translocation (Figure 1E) might be explained by the formation of this fusion via tandem duplication 9, 38.
Still, we could not detect an underlying molecular aberration in the majority of the screened samples with high FGFR3 protein expression. On the one hand, diagnostic issues may play a role. FFPE archival methods lead to chemical modifications (methylene dimerization or monomethylolation) and partial degradation of the RNA, which makes RNA extraction, reverse transcription and quantitation a difficult process. Therefore, fusion‐transcript detection in such poor quality material can be very challenging and lead to false negative results. Unfortunately, FF material was available for only a very limited number of samples in our cohort (16%, 33/210) to confirm the absence/presence of the event by screening both FF/FFPE samples. In addition, the result that the FGFR3‐TACC3 frequency in our cohort is even higher than previously reported highlights the reliability of our screening procedure. On the other hand, aberrant pathway activation needs to be considered. Marek and colleagues described coexpression of FGF2 and FGFRs (mRNA and IHC) in NSCLC cell lines without underlying molecular aberrations 39. These findings, which suggest a FGFR autocrine and/or paracrine loop, were seen not only in NSCLC but also in other solid tumours 5, 6, 20, 22, 40, 41, 42, 43, 44, 45, 46, 47.
There is evidence that FGFR3‐TACC3 is an oncogenic driver in several cancer types, such as bladder cancer, glioblastoma and nasopharyngeal carcinoma. In these cancers, responses to FGFR inhibition have been observed in FGFR3‐TACC3 translocated cell lines 16, 28, 38, 48, 49. Similarly, Ba/F3 cells expressing the FGFR3‐TACC3 fusion were sensitive to FGFR3 inhibition 36. Direct correlation of FGFR3‐TACC3 translocation and increased sensitivity to FGFR inhibition in NSCLC specifically is still under investigation, but response rates to FGFR inhibition as shown by recent phase II clinical trials with the FGFR inhibitors AZD4547 and BGJ398 in FGFR1 amplified lung cancers have been unsatisfactory 50, 51. It is not clear at present whether this lack of clinical response is due to issues with detection of those tumours in which FGFR is a true driver of the oncogenic phenotype for the reasons mentioned above, or whether feedback regulation of signalling limits the effects of FGFR inhibition in lung cancer. We are currently studying the latter possibility using functional genetic approaches.
In conclusion, FGFR1, 2 and 3 proteins are often overexpressed in early stage NSCLC with distinctive expression according to histological subtype. Also, recurrent molecular aberrations such as FGFR3 amplification and FGFR3‐TACC3 translocation are present, making these pathways an interesting target for the treatment of NSCLC. Our results contribute to the refinement of selection of patients who may benefit from FGFR inhibition.
Author contributions
WT involved in writing the protocol synopsis, conceived and carried out experiments, data collection, analysed the data, generated the figures and tables; LM conceived and carried out experiments, data collection and analysed the data; SW involved in writing the protocol synopsis, sample collection, conceived and carried out experiments; AJ conceived and carried out experiments; DP sample collection, conceived and carried out experiments; VvdN analysed the data, generated the figures and tables; EJ data and sample collection; KK conceived and carried out experiments; TP conceived and carried out experiments; TS conceived and carried out experiments; JB data and sample collection; CvN sample collection; RB involved in writing the protocol synopsis; MvdH involved in writing the protocol synopsis; All authors were involved in writing the paper and had final approval of the submitted and published versions.
Supporting information
SUPPLEMENTARY MATERIAL ONLINE
This supplementary file contains Tables S1 to S4:
Table S1. List of the validated fusion transcripts and the primers used
Table S2. FGFR1, 2 and 3 immunohistochemical expression in non‐small cell lung cancer by histological subtype
Table S3. The association between FGFR1, 2 and 3 protein expression with clinicopathological features
Table S4. Cox‐regression analysis of overall survival on clinicopathological variables stratified for tumour stage
Acknowledgements
The authors would like to thank Ian Majewski, Nadia M Davidson, Alicia Oshlack, Jeroen de Jong, Mark van Lanen and Bart Wittgen for their support in the data collection. We would like to acknowledge the NKI‐ AVL Core Facility Molecular Pathology & Biobanking (CFMPB) for supplying NKI‐AVL Biobank material and/or lab support. This work was supported by the European Union FP7 grant ‘Eurocanplatform’.
All authors declare that they have no conflicts of interest.
Contributor Information
Willemijn SME Theelen, Email: w.theelen@nki.nl.
Michel M van den Heuvel, Email: m.vd.heuvel@nki.nl.
References
- 1. Kelsey CR, Marks LB, Hollis D, et al Local recurrence after surgery for early stage lung cancer: an 11‐year experience with 975 patients. Cancer 2009; 115: 5218–5227. [DOI] [PubMed] [Google Scholar]
- 2. Lee CK, Brown C, Gralla RJ, et al Impact of EGFR inhibitor in non‐small cell lung cancer on progression‐free and overall survival: a meta‐analysis. J Natl Cancer Inst 2013; 105: 595–605. [DOI] [PubMed] [Google Scholar]
- 3. Shaw AT, Kim DW, Nakagawa K, et al Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013; 368: 2385–2394. [DOI] [PubMed] [Google Scholar]
- 4. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov 2009; 8: 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer 2010; 10: 116–129. [DOI] [PubMed] [Google Scholar]
- 6. Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J 2011; 437: 199–213. [DOI] [PubMed] [Google Scholar]
- 7. Dienstmann R, Rodon J, Prat A, et al Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol 2014; 25: 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dieci MV, Arnedos M, Andre F, et al Fibroblast growth factor receptor inhibitors as a cancer treatment: from a biologic rationale to medical perspectives. Cancer Discov 2013; 3: 264–279. [DOI] [PubMed] [Google Scholar]
- 9. Parker BC, Engels M, Annala M, et al Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J Pathol 2014; 232: 4–15. [DOI] [PubMed] [Google Scholar]
- 10. Dutt A, Ramos AH, Hammerman PS, et al Inhibitor‐sensitive FGFR1 amplification in human non‐small cell lung cancer. PLoS One 2011; 6: e20351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weiss J, Sos ML, Seidel D, et al Frequent and focal FGFR1 amplification associates with therapeutically tractable FGFR1 dependency in squamous cell lung cancer. Sci Transl Med 2010; 2: 62ra93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gozgit JM, Wong MJ, Moran L, et al Ponatinib (AP24534), a multitargeted pan‐FGFR inhibitor with activity in multiple FGFR‐amplified or mutated cancer models. Mol Cancer Ther 2012; 11: 690–699. [DOI] [PubMed] [Google Scholar]
- 13. Zhang J, Zhang L, Su X, et al Translating the therapeutic potential of AZD4547 in FGFR1‐amplified non‐small cell lung cancer through the use of patient‐derived tumor xenograft models. Clin Cancer Res 2012; 18: 6658–6667. [DOI] [PubMed] [Google Scholar]
- 14. Greenman C, Stephens P, Smith R, et al Patterns of somatic mutation in human cancer genomes. Nature 2007; 446: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liao RG, Jung J, Tchaicha J, et al Inhibitor‐sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res 2013; 73: 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu YM, Su F, Kalyana‐Sundaram S, et al Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov 2013; 3: 636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reck M, Kaiser R, Mellemgaard A, et al Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non‐small‐cell lung cancer (LUME‐Lung 1): a phase 3, double‐blind, randomised controlled trial. Lancet Oncol 2014; 15: 143–155. [DOI] [PubMed] [Google Scholar]
- 18. Okamoto I, Yoshioka H, Takeda K, et al Phase I clinical study of the angiogenesis inhibitor TSU‐68 combined with carboplatin and paclitaxel in chemotherapy‐naive patients with advanced non‐small cell lung cancer. J Thorac Oncol 2012; 7: 427–433. [DOI] [PubMed] [Google Scholar]
- 19. Majewski IJ, Mittempergher L, Davidson NM, et al Identification of recurrent FGFR3 fusion genes in lung cancer through kinome‐centred RNA sequencing. J Pathol 2013; 230: 270–276. [DOI] [PubMed] [Google Scholar]
- 20. Donnem T, Al‐Shibli K, Al‐Saad S, et al Prognostic impact of fibroblast growth factor 2 in non‐small cell lung cancer: coexpression with VEGFR‐3 and PDGF‐B predicts poor survival. J Thorac Oncol 2009; 4: 578–585. [DOI] [PubMed] [Google Scholar]
- 21. Pros E, Lantuejoul S, Sanchez‐Verde L, et al Determining the profiles and parameters for gene amplification testing of growth factor receptors in lung cancer. Int J Cancer 2013; 133: 898–907. [DOI] [PubMed] [Google Scholar]
- 22. Behrens C, Lin HY, Lee JJ, et al Immunohistochemical expression of basic fibroblast growth factor and fibroblast growth factor receptors 1 and 2 in the pathogenesis of lung cancer. Clin Cancer Res 2008; 14: 6014–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kohler LH, Mireskandari M, Knosel T, et al FGFR1 expression and gene copy numbers in human lung cancer. Virchows Arch 2012; 461: 49–57. [DOI] [PubMed] [Google Scholar]
- 24. Guddo F, Fontanini G, Reina C, et al The expression of basic fibroblast growth factor (bFGF) in tumor‐associated stromal cells and vessels is inversely correlated with non‐small cell lung cancer progression. Hum Pathol 1999; 30: 788–794. [DOI] [PubMed] [Google Scholar]
- 25. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics 2009; 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim D, Salzberg SL. TopHat‐fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol 2011; 12: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Grabherr MG, Haas BJ, Yassour M, et al Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat Biotechnol 2011; 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Williams SV, Hurst CD, Knowles MA. Oncogenic FGFR3 gene fusions in bladder cancer. Hum Mol Genet 2013; 22: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schildhaus H‐U, Nogova L, Wolf J, et al FGFR1 amplifications in squamous cell carcinomas of the lung: diagnostic and therapeutic implications. Translat Lung Cancer Res 2013; 2: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seo AN, Jin Y, Lee HJ, et al FGFR1 amplification is associated with poor prognosis and smoking in non‐small‐cell lung cancer. Virchows Arch 2014; 465: 547–558. [DOI] [PubMed] [Google Scholar]
- 31. Volm M, Koomagi R, Mattern J, et al Prognostic value of basic fibroblast growth factor and its receptor (FGFR‐1) in patients with non‐small cell lung carcinomas. Eur J Cancer 1997; 33: 691–693. [DOI] [PubMed] [Google Scholar]
- 32. Stabile LP, Lyker JS, Gubish CT, et al Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non‐small cell lung cancer shows enhanced antiproliferative effects. Cancer Res 2005; 65: 1459–1470. [DOI] [PubMed] [Google Scholar]
- 33. Byron SA, Gartside M, Powell MA, et al FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One 2012; 7: e30801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andre F, Bachelot T, Campone M, et al Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 2013; 19: 3693–3702. [DOI] [PubMed] [Google Scholar]
- 35. Wang R, Wang L, Li Y, et al FGFR1/3 tyrosine kinase fusions define a unique molecular subtype of Non‐small Cell Lung Cancer. Clin Cancer Res 2014; 20: 4107–14. [DOI] [PubMed] [Google Scholar]
- 36. Capelletti M, Dodge ME, Ercan D, et al Identification of recurrent FGFR3‐TACC3 fusion oncogenes from lung adenocarcinoma. Clin Cancer Res 2014; 20: 6651–8. [DOI] [PubMed] [Google Scholar]
- 37. Fischbach A, Rogler A, Erber R, et al Fibroblast growth factor receptor (FGFR) gene amplifications are rare events in bladder cancer. Histopathology 2015; 66: 639–649. [DOI] [PubMed] [Google Scholar]
- 38. Parker BC, Annala MJ, Cogdell DE, et al The tumorigenic FGFR3‐TACC3 gene fusion escapes miR‐99a regulation in glioblastoma. J Clin Invest 2013; 123: 855–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marek L, Ware KE, Fritzsche A, et al Fibroblast growth factor (FGF) and FGF receptor‐mediated autocrine signaling in non‐small‐cell lung cancer cells. Mol Pharmacol 2009; 75: 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berger W, Setinek U, Mohr T, et al Evidence for a role of FGF‐2 and FGF receptors in the proliferation of non‐small cell lung cancer cells. Int J Cancer 1999; 83: 415–423. [DOI] [PubMed] [Google Scholar]
- 41. Wang Y, Becker D. Antisense targeting of basic fibroblast growth factor and fibroblast growth factor receptor‐1 in human melanomas blocks intratumoral angiogenesis and tumor growth. Nat Med 1997; 3: 887–893. [DOI] [PubMed] [Google Scholar]
- 42. Poon RT, Fan ST, Wong J. Clinical implications of circulating angiogenic factors in cancer patients. J Clin Oncol 2001; 19: 1207–1225. [DOI] [PubMed] [Google Scholar]
- 43. Presta M, Dell'Era P, Mitola S, et al Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005; 16: 159–178. [DOI] [PubMed] [Google Scholar]
- 44. Guagnano V, Kauffmann A, Wohrle S, et al FGFR genetic alterations predict for sensitivity to NVP‐BGJ398, a selective pan‐FGFR inhibitor. Cancer Discov 2012; 2: 1118–1133. [DOI] [PubMed] [Google Scholar]
- 45. Fischer H, Taylor N, Allerstorfer S, et al Fibroblast growth factor receptor‐mediated signals contribute to the malignant phenotype of non‐small cell lung cancer cells: therapeutic implications and synergism with epidermal growth factor receptor inhibition. Mol Cancer Ther 2008; 7: 3408–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kono SA, Marshall ME, Ware KE, et al The fibroblast growth factor receptor signaling pathway as a mediator of intrinsic resistance to EGFR‐specific tyrosine kinase inhibitors in non‐small cell lung cancer. Drug Resist Updat 2009; 12: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuhn H, Kopff C, Konrad J, et al Influence of basic fibroblast growth factor on the proliferation of non‐small cell lung cancer cell lines. Lung Cancer 2004; 44: 167–174. [DOI] [PubMed] [Google Scholar]
- 48. Singh D, Chan JM, Zoppoli P, et al Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012; 337: 1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan L, Liu ZH, Lin ZR, et al Recurrent FGFR3‐TACC3 fusion gene in nasopharyngeal carcinoma. Cancer Biol Ther 2014; 15: 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nogova L, Sequist LV, Cassier P, et al Targeting FGFR1‐amplified lung squamous cell carcinoma with the selective pan‐FGFR inhibitor BGJ398. J Clin Oncol 2014; 32: 5s (suppl; abstract 8034). [Google Scholar]
- 51. Smyth EC, Nicholas C, Popat S, et al Proof‐of‐concept study of AZD4547 in patients with FGFR1 or FGFR2 amplified tumours. J Clin Oncol 2013; 31 (suppl; abstract TPS2626). [Google Scholar]
- 52. Schaake EE, Kapper I, Codrington HE, et al Tumor response and toxicity of neoadjuvant erlotinib in patients with early stage non‐small‐cell lung cancer. J Clin Oncol 2012; 30: 2731–2738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTARY MATERIAL ONLINE
This supplementary file contains Tables S1 to S4:
Table S1. List of the validated fusion transcripts and the primers used
Table S2. FGFR1, 2 and 3 immunohistochemical expression in non‐small cell lung cancer by histological subtype
Table S3. The association between FGFR1, 2 and 3 protein expression with clinicopathological features
Table S4. Cox‐regression analysis of overall survival on clinicopathological variables stratified for tumour stage